

Abstract
Arylsulfonates of hindered secondary alcohols are converted to the corresponding alkyl chlorides very rapidly and in good yields in the presence of titanium tetrachloride at low temperatures. These reactions proceed with exclusive retention of configuration.
We are interested in the development of highly efficient leaving groups containing chelating units capable of attracting incoming nucleophiles. We envision that electrophiles containing sulfonate leaving groups could be rendered more reactive by modifying them to contain a metal chelating moiety.1 In this chelated form, the nucleophilic portion of a salt or Lewis acid is localized near the electrophilic center thus decreasing the entropic barrier relative to intermolecular reactions. The chelated metal would also be expected to stabilize the developing negative charge on the oxygens of the sulfonate leaving group in the transition state.2 We have recently demonstrated that metal halides react at greatly accelerated rates with arylsulfonate-based nucleophile assisting leaving groups (NALGs) that contain a polyether unit (including macrocyclic) attached to the aryl ring ortho to the sulphonate.3 In an attempt to further explore the generality of the NALG technology, we discovered a highly efficient method to convert NALG sulfonates of hindered alcohols to the corresponding chlorides using TiCl4 at low temperatures.
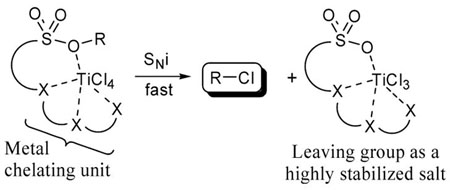
While a related chlorination procedure with alkyl tosylates has been reported,4 in this note, we demonstrate, for the first time, the stereospecificity and utility of the sulfonate/TiCl4 technique in the synthesis of a variety of alkyl chlorides.5
A series of alcohols were reacted with aryl sulfonyl chlorides containing a diethylene glycol moiety to give sulfonate esters 1 (Table 1). In general, the treatment of sulfonate esters 1 with TiCl4 in methylene chloride at −78 °C led to high yields of the corresponding alkyl chlorides. In each case, the reaction was complete in less than 2 minutes. With 1,3-diphenyl-2-propanol, we have observed that its NALG and tosyl esters primarily give the elimination product in the presence of metal chloride agents.6 To our knowledge this chloride has only been prepared by radical chlorodecarboxylative methods.7 However, upon treatment of the NALG ester with TiCl4, the respective chloride was obtained exclusively in 91% yield (entry 1).
Table 1.
Reaction of various sulphonate esters with TiCl4 leading to alkyl chlorides
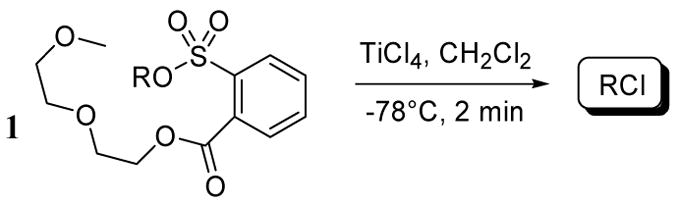
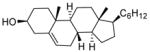
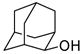
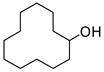
Isolated yields. All chloride products have been previously reported
Elimination product also observed (21%, 1.5:1 E/Z).
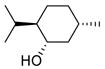
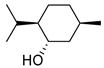
With sulfonates of chiral secondary alcohols (Table 1, entries 2–4), the chloride products were obtained as single diastereomers with complete retention of configuration.8 Thus, the NALG sulfonates of highly hindered menthol and isomenthol were both converted to the corresponding chlorides of the same configuration in excellent yields by exposure to TiCl4 for two minutes. Similarly, the NALG sulfonate of cholesterol (entry 4) was converted to the corresponding chloride almost instantaneously at −78 °C. Proton NMR confirms the formation of one stereoisomer with retention of configuration
In the 2-adamantyl system, we observed a 90% conversion to the chloride with no side product arising from rearrangement of the adamantine nucleus (entry 5).9 Backside nucleophilic displacement of 2-adamantyl sulfonate is essentially precluded due to steric crowding,10 thus the chlorination reaction likely proceeds via a front-side SNi-type mechanism. While reactions involving the SNi mechanism are generally mediated by four-centered cyclic transition states,11,12 the chlorination of the various sulfonate substrates in Table 1 is one of the first examples of an SNi transition state stabilized by intramolecular chelation which may account for the very rapid conversion rates.3 Neopentyl NALG substrates also reacted very rapidly with TiCl4 leading to the rearrangement products (Scheme 1).
Scheme 1.
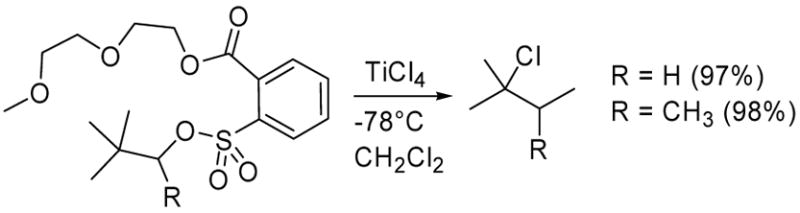
Reaction of neopentyl NALG sulfonates with TiCl4 resulting in rearrangement product.
In conclusion, we believe that the method described in this note offers an important alternative to existing techniques for the synthesis of secondary alkylchlorides with retention of configuration.
Experimental Section
Representative Procedure: preparation of 1,3-diphenylpropyl 2-(2,5-dioxoheptylcarboxy)-1-benzo-sulfonate and conversion to 2-chloro-1,3-diphenylpropane (Table 1, entry 1)
Step 1
A mixture of o-sulfobenzoic acid anhydride (1.23 g, 6.6 mmol) and phosphorous pentachloride (3.68 g, 13.2 mmol) was heated at 90 °C for 6 h. The oil was allowed to cool, dissolved in ether, and rinsed with ice water to remove unreacted phosphorous pentachloride. The solvent was evaporated in vacuo leaving 2.1 g of crude oil. The crude oil (1.5 g, 6.2 mmol) was then dissolved in excess methoxyethoxyethanol (2.4 g, 20 mmol) and heated to 60 °C for 20 h. The reaction mixture was purified by flash chromatography by eluting with a hexane/acetone (10% v/v) to yield 2,5-dioxoheptyl 2-(chlorosulfonyl)-benzoate (1.9 g, 95%): IR (neat) 1731 (C=O), 1353, 1196 (SO2) cm−1; 1HNMR (400 MHz, CDCl3) δ 8.14 (dd, 1H, J = 0.9 and 7.74 Hz), 7.80-7.69 (m, 2H), 4.57-4.54 (m, 2H), 3.86-3.84 (m, 2H), 3.67-3.65 (m, 2H), 3.55-3.53 (m, 2H), 3.36 (s, 3H); 13CNMR (100.75 MHz, CDCl3) δ 166.0, 141.6, 135.4, 132.4, 131.6, 130.4, 129.2, 72.0, 70.6, 68.7, 65.9, 59.2; HRMS (EI) [M+H]+: calcd for C12H16ClO6S 323.0356, found 323.0350.
Step 2
To a dichloromethane solution (50 mL) of 2,5-dioxoheptyl 2-(chlorosulfonyl)-benzoate (2.0 g, 6.2 mmol) was added DMAP (0.9 g, 7.4 mmol) under argon. The reaction mixture was cooled to 0 °C followed by the addition of 1,3-diphenyl-2-propanol (2.6 g, 12.4 mmol). The resulting mixture was warmed to room temperature and stirred for 15 h. The solvent was evaporated in vacuo and the resulting crude oil was then purified by flash chromatography by eluting with hexane/acetone (30% v/v) to give 1,3-diphenylpropyl 2-(2,5-dioxoheptylcarboxy)-1-benzosulfonate as a gummy material (2.7 g, 88 %): IR (neat) 1730 (C=O), 1357, 1201 (SO2) cm−1; 1HNMR (400 MHz, CDCl3) δ 7.51-7.43 (m 3H), 7.27-7.22 (m, 1H), 7.18-7.09 (m, 10H), 5.12 (p, 1H, J = 6.25 Hz), 4.54-4.52 (m, 2H), 3.82-3.79 (m, 2H), 3.63-3.61 (m, 2H), 3.54-3.51 (m, 2H), 3.35 (s, 3H), 3.01-2.91 (m, 4H); 13CNMR (100.75 MHz, CDCl3) δ 167.2, 136.3, 134.3, 133.0, 130.8, 129.8, 129.1, 129.0, 128.6, 126.9, 86.3, 72.0, 70.6, 68.9, 65.5, 59.2, 40.7. HRMS (EI) [M+H]+: calcd for C27H31O7S 499.1790, found 499.1813.
Step 3
To a dichloromethane solution (10 mL) of 1,3-diphenylpropyl 2-(2,5-dioxoheptylcarboxy)-1-benzosulfonate (200 mg, 0.46 mmol) cooled to −78 °C was added TiCl4 (100 mg, 0.91 mmol). The resulting mixture was stirred for 1 min, quenched with water (50 mL) and diluted with additional dichloromethane (50 mL). The organic phase was collected and passed through a silica gel plug followed by solvent evaporation in vacuo to give pure 2-chloro-1,3-diphenylpropane (98 mg, 0.42 mmol, 91 %).
Supplementary Material
Copies of 1H NMR spectra for NALG sulfonate and alkyl chloride products of Table 1 and chloride products depicted in Scheme 1. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank the National Institute of Mental Health (66963-01) and the Florida Center of Excellence in Marine Biotechnology (contribution P200412) for financial support. An acknowledgment is also made to the National Science Foundation Division of Undergraduate Education (0311369) for the 400 MHz NMR used in this study.
References
- 1.(a) Cacciapaglia R, Di Stefano S, Mandolini J Org Chem. 2002;67:521. doi: 10.1021/jo016149q. [DOI] [PubMed] [Google Scholar]; b Cacciapaglia R, Mandolini L. Pure and App Chem. 1993;65:533. [Google Scholar]; c Alfimov MV, Gromov SP, Fedorov YuV, Fedorova OA, Vedernikov AI, Churakov AV, Kuz’mina LG, Howard JAK, Bossmann S, Braun A, Woerner M, Sears DF, Jr, Saltiel J. J Am Chem Soc. 1999;121:4992. [Google Scholar]
- 2.Gobbi A, Landini D, Maia A, Secci D. J Org Chem. 1995;60:5954. [Google Scholar]
- 3.Lepore SD, Bhunia AK, Cohn PC. J Org Chem. 2005;70:8117. doi: 10.1021/jo051241y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vizgert RV, Polyakova EV, Chervinski A. Yu Zhurnal Obshchei Khimi. 1984;54:905. [Google Scholar]
- 5.Other complementary chlorination techniques have also proved successful with intransigent substrates. Yasuda M, Yamasaki S, Onishi Y, Baba A. J Amer Chem Soc. 2004;126:7186. doi: 10.1021/ja048688t.Kozikowski AP, Lee J. Tetrahedron Lett. 1988;29:3053.
- 6.Lepore SD, Bhunia AK, Cohn PC. unpublished results. [Google Scholar]
- 7.(a) Okada K, Okamoto K, Oda M. J Chem Soc Chem Comm. 1989;21:1636. [Google Scholar]; b Barton DHR, Crich D, Motherwell WB. Tetrahedron. 1985;41:3901. [Google Scholar]
- 8.The stereochemistry of all chloride products was established by comparison of their NMR spectra with those previously reported. For the chloride products in Table 1 see the following references: entry 1 (ref. 7), entries 2–4 (ref. 5b), entries 5 and 6 (ref. 5a).
- 9.Sinnott ML, Storesund HJ, Whiting MC. Chem Commun. 1969:1000. [Google Scholar]
- 10.Kozikowski AP, Lee J. Tet Lett. 1988;29:3053. and references cited therein. [Google Scholar]
- 11.Lee CC, Clayton JW, Lee DG, Finlayson AJ. Tetrahedron. 1962;18:1395. [Google Scholar]; b Lewis ES, Herndon WC, Duffy DC. J Am Chem Soc. 1961;83:1959. [Google Scholar]
- 12.Moss RA, Fu X, Tian J, Sauers R, Wipf P. Org Lett. 2005;7:1371. doi: 10.1021/ol050185k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Copies of 1H NMR spectra for NALG sulfonate and alkyl chloride products of Table 1 and chloride products depicted in Scheme 1. This material is available free of charge via the Internet at http://pubs.acs.org.