

Abstract
Benzylsuccinate synthase catalyzes the first step in the anaerobic metabolism of toluene through an unusual reaction in which toluene is added to fumarate to produce (R)-benzylsuccinate. We have exploited the broad substrate range of the enzyme to demonstrate that the enzyme can catalyze the exchange of p-cresol with the benzyl portion of benzylsuccinate to form (4-hydroxybenzyl)-succinate, indicating that the reverse reaction (disproportionation of benzylsuccinate to toluene and fumarate) must have occurred. Even though the equilibrium constant for the reverse reaction is extremely unfavorable, Keq ∼ 8 × 10-11 M at 4 °C, the enzyme catalyzes the reverse reaction at rate only 250-fold slower than the forward reaction. Furthermore, using deuterium-labeled benzylsuccinate we observe partial exchange of deuterium with the solvent. This provides the first direct evidence that the migrating hydrogen is transferred to a labile site on the protein during catalysis, which is consistent with the participation of the proposed active site cysteine residue in the mechanism of BSS.
Aromatic compounds such as toluene are an important class of long-lived pollutants that have relatively high water solubility. They are a particular problem in environments where oxygen is scarce, such as ground water supplies, because most pathways to degrade such compounds require molecular oxygen to functionalize otherwise inert aromatic hydrocarbons. Recently, however, various denitrifying and sulfate-reducing bacteria such as Thauera aromatica and Desulfobacula toluolica have been discovered that can live on toluene and related aromatic compounds as their sole source of carbon and energy under anaerobic conditions.1,2 This discovery has aroused much interest, both for the potential such bacteria offer for bioremediation in situations where oxygen is scarce, and because of the novel chemistry involved.3,4
The first step in the anaerobic metabolism of toluene is its reaction with fumarate to produce (R)-benzylsuccinate.5 This remarkable reaction is catalyzed by benzylsuccinate synthase (BSS), which is a member of the glycyl radical family of enzymes that include pyruvate formate lyase and anaerobic ribonucleotide reductase.6,7 In the presumed mechanism (Figure 1) the radical is first transferred to an active site cysteine that then abstracts a hydrogen atom from the methyl group of toluene to generate a benzylic radical. The benzyl radical undergoes addition to the double bond of fumarate to form a benzylsuccinyl radical and in the last step the hydrogen is replaced from cysteine and the glycyl radical regenerated.
Figure 1.
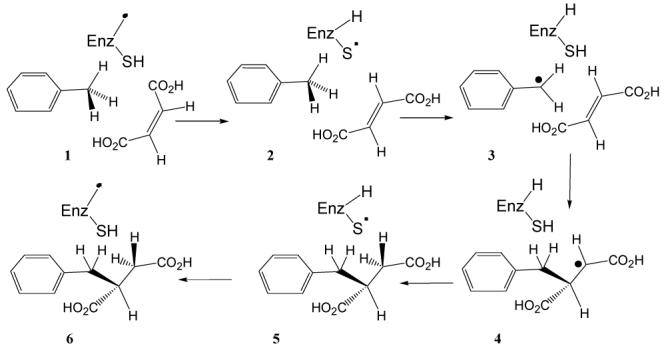
Proposed mechanism for the reaction catalyzed by BSS
There is good evidence that the BSS-catalyzed reaction involves radical intermediates, but its extreme oxygen sensitivity and lability8 have hampered efforts to investigate the mechanism of this unusual enzyme. We have determined the stereochemistry of hydrogen transfer from toluene to benzylsuccinate and provided evidence for the intermediacy of a radical at C-3 of benzylsuccinate.9 We have recently undertaken a kinetic study of the reaction and through isotope effect measurements provided evidence that the formation of the benzylic radical (3) is most likely the rate-determining step in the forward reaction.10
To gain further insight into the mechanism of the BSS-catalyzed reaction, we have now investigated the ability of BSS to catalyze the reverse reaction, i.e. the formation of toluene and fumarate from benzylsuccinate. The forward reaction, driven by the formation of a strong C–C σ-bond at the expense of the weaker C–C π-bond, is calculated to be exothermic by about 13 kcal/mol.11 This is comparable to the highly exothermic hydrolysis of phosphoenolpyruvate to phosphate and pyruvate (ΔG ° = -14.8 kcal/mol) that is generally considered irreversible in the biochemical context. The exothermic nature of this reaction raises the interesting question of how the kinetics of the BSS-catalyzed reaction are changed in the reverse direction.
We have exploited the broad substrate range of the enzyme12to demonstrate that BSS can catalyze the exchange of p-cresol with the benzyl portion of benzylsuccinate (BS) to form (4-hydroxybenzyl)-succinate (HBS), indicating that the reverse reaction (disproportionation of BS to toluene and fumarate) must have occurred. Even though the equilibrium constant for the reverse reaction is extremely unfavorable, Keq ∼ 8 × 10-11 M at 4 °C, the enzyme catalyzes the reverse reaction at rate only 250-fold slower than the forward reaction. Furthermore, using deuterium-labeled BS we observe partial exchange of deuterium with the solvent. This provides the first direct evidence that the migrating hydrogen is transferred to a labile site on the protein during catalysis, which is consistent with the participation of the proposed active site cysteine residue in the mechanism of BSS.
Measuring the reverse reaction
It has previously been shown that BSS can catalyze the addition of various substituted toluene derivatives, including p-cresol, to fumarate.12 We developed a quantitative HPLC-based assay to detect the formation of HBS catalyzed by BSS in cell free extracts of T. aromatica (Figure 2). The assay is very similar to that which we recently described for assaying BSS activity with its normal substrate, toluene.10
Figure 2.
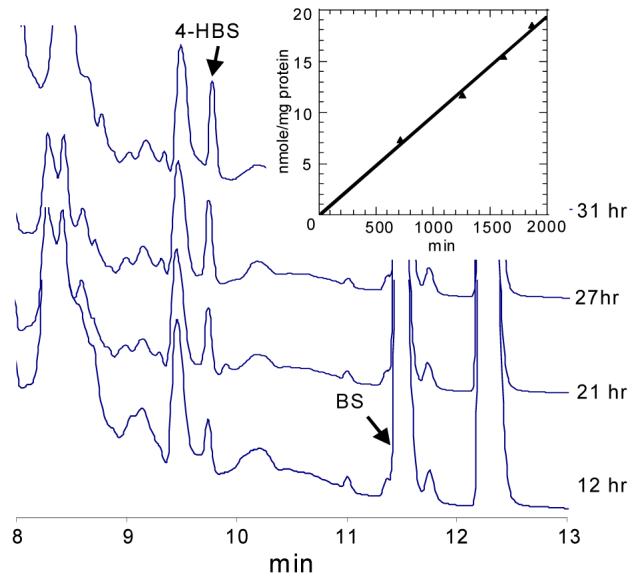
Representative HPLC chromatograms demonstrating the formation of HBS from BS and p-cresol. Inset: plot of reaction rate derived from integration of HBS peak areas.
In initial experiments, HPLC analysis clearly revealed the presence of small quantities of HBS in cell-free extracts after ∼24 h incubation at 4 °C with p-cresol (16 mM) and BS (10 mM, racemic material). The identity of HBS was verified by mass spectroscopy. This result implied that BSS had catalyzed the disproportionation of BS to toluene and fumarate, which was then trapped by reaction with p-cresol to produce HBS. A similar result was obtained when cell-free extracts were incubated with toluene and HBS (synthesized enzymatically and purified by HPLC); in this case BS was recovered from the reaction. Exposure of the cell-free extract to air abolished the exchange activity. This is consistent with the exchange reactions being catalyzed by BSS, which, like all glycyl radical enzymes, is very rapidly inactivated by O2.
In subsequent experiments it was found that the rate at which HBS was formed by the enzyme from BS and p-cresol could be significantly increased by rapidly stirring the reaction solution under a layer of hexane. This is because the hexane extracts the toluene formed from the aqueous phase, thereby preventing it from competing with p-cresol, which is not very soluble in hexane. This allowed us to reliably measure the rate at which HBS was formed from BS and p-cresol. As shown in Figure 2, the formation of HBS was linear with time and its rate of formation was 9.7 pmol/min/mg of protein. Control experiments (see supporting information) in which BS was lacking eliminated the possibility that HBS could have been formed by the reaction of p-cresol with small amounts of residual fumarate in the cell-free extract.
We next compared the rate of the exchange reaction with the rate at which the enzyme catalyzes the formation HBS directly from saturating concentrations of p-cresol (16 mM) and fumarate (10 mM). Under these conditions HBS was formed at a rate of 420 pmol/min/mg of protein. This is over 40-fold faster than the exchange reaction with measured with p-cresol and BS, indicating that it is the reverse reaction to form fumarate and toluene that is the rate-limiting step in the exchange reaction. For toluene reacting under the same assay conditions the rate of BS formation was 2400 pmol/min/mg of protein, about 6-fold faster than p-cresol. Therefore the ratio of the rates for the forward and reverse reactions, with the substrates at saturating concentrations, is ∼250 : 1. We note that this does not allow one deduce anything about the equilibrium constant for the reaction since the Km s for the substrates are not known.
Evidence for thiyl radical intermediate in BSS
To directly confirm the reversibility of the BSS reaction we aimed to isolate the toluene produced in the reaction by using BS deuterium labeled in the toluene portion as the substrate. d8-BS was synthesized enzymatically from d8-toluene and fumarate and isolated by HPLC. A reaction containing d8-BS and p-cresol was set up under the conditions described above and overlaid with hexane. After 24 h, analysis of the hexane layer by GC-MS clearly identified the presence of d8-toluene (peaks in the MS with m/z = 100 and 98 amu), which could only have derived from d 8-BS (Figure 3). Indeed, even in the absence of p-cresol, deuterated toluene could be detected in the hexane layer, although in much lower abundance. Thus even though the chemical equilibrium strongly favors the formation of BS, removing toluene from the aqueous solution is sufficient to detectably shift the equilibrium towards the reverse reaction.
Figure 3.
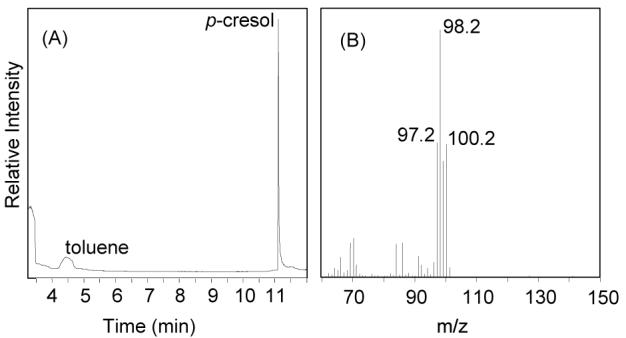
Detection of d8- and d7-labelled toluene from the BSS-catalyzed exchange reaction of d8-labeled BS with p-cresol. (A) GC trace; (B) mass spectrum of toluene peak.
Most interestingly, analysis of the recovered toluene from these reactions revealed a significant amount of d7-toluene as evidenced by characteristic peaks at m/z = 99 and 97 amu. This implies that during the reaction there is a certain probability of a deuterium exchanging with a proton on the protein, or from the solvent. This observation provides the first experimental evidence that the migrating hydrogen at C-3 of BS is transferred to a solvent exchangeable residue on BSS before its subsequent transfer to the methyl group of toluene. This residue is most likely Cys-492, which along with Gly-828 is conserved in other glycyl radical enzymes such as pyruvate formate-lyase and anaerobic ribonucleotide reductase.13
In the forward reaction, which occurs much more rapidly, we observed no noticeable loss of isotope when the reaction was conducted with deuterium-labeled toluene.10 This suggests that in the reverse direction conversion of 4 to 3 (Figure 1) occurs relatively slowly, allowing the active site cysteine time to exchange a proton with the solvent or other labile site on the protein. In contrast, in the forward direction, once 3 is formed the remaining steps in the reaction proceed relatively rapidly, so that exchange of protons with the solvent does not have time to occur.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. Peter Coschigano (Ohio University) for advice on the culture of T. aromatica and Christel Fox and Dr. Mou-Chi Cheng for assistance. This work was supported by NIH grant GM 59227 to E.N.G.M.
Footnotes
Supporting Information Available: Details of the preparation of cell-free extracts, enzyme assay, control experiments, HPLC chromatographs and mass spectra of compounds.
REFERENCES
- (1).Biegert T, Fuchs G, Heider F. Eur. J. Biochem. 1996;238:661–668. doi: 10.1111/j.1432-1033.1996.0661w.x. [DOI] [PubMed] [Google Scholar]
- (2).Rabus R, Heider J. Arch. Microbiol. 1998;170:377–384. [Google Scholar]
- (3).Spormann AM, Widdel F. Biodegradation. 2000;11:85–105. doi: 10.1023/a:1011122631799. [DOI] [PubMed] [Google Scholar]
- (4).Heider J, Spormann AM, Beller HR, Widdel F. Fems Microbiol. Rev. 1998;22:459–473. [Google Scholar]
- (5).Beller HR, Spormann AM, Sharma PK, Cole JR, Reinhard M. Appl. Environ. Microbiol. 1996;62:1188–1196. doi: 10.1128/aem.62.4.1188-1196.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Marsh ENG, Huhta MS, Patwardhan A. Bioorganic Chem. 2004;32:326–340. doi: 10.1016/j.bioorg.2004.06.001. [DOI] [PubMed] [Google Scholar]
- (7).Frey PA, Hegerman AD, Reed GH. Chem. Rev. 2006;106:3302–3316. doi: 10.1021/cr050292s. [DOI] [PubMed] [Google Scholar]
- (8).Leuthner B, Leutwein C, Schulz H, Horth P, Haehnel W, Schiltz E, Schagger H, Heider J. Mol. Microbiol. 1998;28:615–628. doi: 10.1046/j.1365-2958.1998.00826.x. [DOI] [PubMed] [Google Scholar]
- (9).Qiao CH, Marsh ENG. J. Am. Chem. Soc. 2005;127:8608–8609. doi: 10.1021/ja051972f. [DOI] [PubMed] [Google Scholar]
- (10).Li L, Marsh ENG. Biochemistry. 2006 [Google Scholar]
- (11).Himo F. J. Phys. Chem. B. 2002;106:7688–7692. [Google Scholar]
- (12).Beller HR, Spormann AM. Fems Microbiology Letters. 1999;178:147–153. doi: 10.1111/j.1574-6968.1999.tb13771.x. [DOI] [PubMed] [Google Scholar]
- (13).Coschigano PW, Wehrman TS, Young LY. Appl. Environ. Microbiol. 1998;64:1650–1656. doi: 10.1128/aem.64.5.1650-1656.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.