

Abstract
Iron regulatory protein 2 (IRP2) is a key iron sensor that post-transcriptionally regulates mammalian iron homeostasis by binding to iron-responsive elements (IREs) in mRNAs that encode proteins involved in iron metabolism (e.g. ferritin and transferrin receptor 1). During iron deficiency, IRP2 binds IREs to regulate mRNA translation or stability, whereas during iron sufficiency IRP2 is degraded by the proteasome. Here, we identify an iron-independent IRP2 phosphorylation site that is regulated by the cell cycle. IRP2 Ser-157 is phosphorylated by Cdk1/cyclin B1 during G2/M and is dephosphorylated during mitotic exit by the phosphatase Cdc14A. Ser-157 phosphorylation during G2/M reduces IRP2 RNA-binding activity and increases ferritin synthesis, whereas Ser-157 dephosphorylation during mitotic exit restores IRP2 RNA-binding activity and represses ferritin synthesis. These data show that reversible phosphorylation of IRP2 during G2/M has a role in modulating the iron-independent expression of ferritin and other IRE-containing mRNAs during the cell cycle.
Iron is essential for cellular growth due to its role as a cofactor in proteins involved in mitochondrial respiration and DNA synthesis (1, 2). Proliferating cells are particularly sensitive to iron depletion, because the R2 subunit of ribonucleotide reductase, the rate-limiting enzyme for DNA synthesis, requires iron as a cofactor (3). In addition to inhibiting ribonucleotide reductase, iron depletion alters the expression of cell cycle proteins, including cyclin D (4, 5), cyclin-dependent kinase 2 (Cdk2)3 (5), and the cyclin-dependent kinase inhibitor p21CIP1/WAF (6), which are involved in cell cycle progression from G1 to S phase. Because the cellular response to iron depletion is G1/S cell cycle arrest, iron chelators are used clinically as anti-proliferative agents (7, 8). The mechanisms, however, that regulate iron levels specifically during the cell cycle are not known.
Although iron is essential for cellular proliferation, excess iron can be toxic due to its ability to generate reactive oxygen species. Cellular iron levels are therefore tightly regulated to maintain a balance between insufficient and excessive iron. IRP1 and IRP2 are the primary iron sensors in mammalian cells (1, 9, 10). IRPs are cytosolic RNA-binding proteins that post-transcriptionally regulate iron uptake by transferrin receptor 1 (TfR1) and divalent metal transporter 1, iron export by ferroportin, and iron storage by ferritin. When iron is limited, IRPs bind with high affinity to IREs located in the 5′-untranslated regions of ferritin and ferroportin, mRNAs and in the 3′-untranslated regions of TfR1 and divalent metal transporter 1 mRNAs. IRP binding during iron deficiency inhibits the translation of 5′-IRE-containing mRNAs (e.g. ferritin) and stabilizes 3′-IRE-containing mRNAs (e.g. TfR1). During iron sufficiency, IRPs lose affinity for IREs, stimulating the translation of 5′-IRE-containing mRNAs and destabilizing 3′-IRE-containing mRNAs. By regulating iron transport and sequestration, IRPs directly modulate the amount of bioavailable iron. IRPs also regulate other IRE-containing mRNAs that encode proteins involved in the citric acid cycle (mitochondrial aconitase) (11) and heme biosynthesis (erythroid aminolevulinate acid synthase) (12). Hypoxia-inducible factor-2α (13), the cell cycle phosphatase Cdc14A (14), and myotonic dystrophy kinase-related Cdc42 binding kinase α (15) mRNAs each contain an IRE that binds IRPs in vitro, but the physiological roles of these IREs in vivo have not been entirely determined. The expanding number of IRE-containing mRNAs suggests that IRPs have more extensive functions than originally thought.
IRP1 and IRP2 share 64% sequence identity, but their RNA-binding activities are regulated by different mechanisms. Iron regulates IRP1 primarily by stimulating the assembly of an [4Fe-4S] cluster, which converts it from an RNA-binding protein into a cytosolic aconitase (16). In contrast, IRP2 does not assemble an [4Fe-4S] cluster during iron sufficiency (17) but is rapidly degraded by the proteasome (18, 19). The exact mechanism for IRP2 iron-mediated degradation has remained elusive. In addition to iron, IRP RNA-binding activities are also regulated by hypoxia. IRP1 RNA-binding activity is decreased during hypoxia due to stabilization of the [4Fe-4S] cluster in the aconitase form of the protein (20). Conversely, hypoxia increases IRP2 RNA-binding activity due to protein stabilization (21–23). The regulation of IRP RNA-binding activities by oxygen deserves significant consideration due to the hypoxic nature of most tissues. At physiological oxygen concentrations (3–6%), IRP1 exists primarily in its c-aconitase form, whereas IRP2 is stabilized and functions solely as an RNA-binding protein (23). In accordance, mouse models of IRP1 and IRP2 deficiency have revealed that IRP1 cannot fully compensate for a lack of IRP2 (24–27). Consequently, IRP2 has been proposed to be the predominant RNA-binding protein and regulator of iron homeostasis in vivo.
IRP1 and IRP2 are also regulated by phosphorylation. Treatment of HL-60 and HEK293 cells with the protein kinase C activator phorbol 12-myristate 13-acetate (PMA) stimulates IRP1 phosphorylation at Ser-138 and Ser-711 (28–31). Phosphorylation of IRP1 at Ser-711 inhibits the forward citrate to isocitrate aconitase reaction without altering RNA-binding activity (30). Phosphorylation at Ser-138 destabilizes the [4Fe-4S] cluster (32), which increases RNA-binding activity and enhances the iron-mediated degradation of IRP1 (31). IRP2 is also phosphorylated in PMA-treated HL-60 cells (29). PMA increases IRP2 RNA binding by activating a latent pool of redox-sensitive IRP2. In that study, the identity of the phosphorylated residue(s) and the mechanism of IRP2 phosphorylation were not determined.
Here, we identify an iron-independent IRP2 phosphorylation site that is regulated by the cell cycle. IRP2 Ser-157 is phosphorylated during G2/M by Cdk1/cyclin B1 and dephosphorylated during mitotic exit by Cdc14A. Ser-157 phosphorylation regulates IRP2 RNA-binding activity and ferritin expression during mitosis. Our data provide evidence for a mechanism to modulate iron bioavailability during the cell cycle.
EXPERIMENTAL PROCEDURES
Plasmids and Site-directed Mutagenesis—The WT rat IRP2 cDNA was C-terminally tagged with 5×-Myc and cloned into pcDNA3.1/Zeo(+) (Invitrogen) with NheI and XbaI. IRP2-Myc was cloned into pcDNA5/FRT/TO (Invitrogen) by engineering KpnI and ApaI restriction sites on the N and C termini, respectively, and was subsequently used to generate the S503A, S407A, and S157A IRP2-Myc mutants by site-directed mutagenesis (Stratagene QuikChange). The WT human IRP2 cDNA was N-terminally tagged with 2×-FLAG and cloned into pcDNA5/FRT/TO with XhoI and ApaI. The S157A FLAG-IRP2 mutant was generated from WT FLAG-IRP2 by site-directed mutagenesis. A TEV-10×-His tag and an ApaI restriction site were engineered onto the C terminus of 2×-FLAG-IRP2. 2×-FLAG-IRP2-TEV-10×-His was cloned into pcDNA5/FRT/TO with XhoI and ApaI. The Myc-Cdc14A pcDNA3 plasmid was a kind gift from Jiri Lukas (Institute of Cancer Biology, Danish Cancer Society) (33) and was used to generate the catalytically inactive C278S Cdc14A mutant by site-directed mutagenesis. All nucleotide changes generated by site-directed mutagenesis were verified by sequencing.
Cell Culture—HEK293T, HEK293, and HeLa cells were cultured in Dulbecco's modified Eagle's medium containing 5% fetal bovine serum. Flp-In T-REx-293 cells (Invitrogen) were cultured in Dulbecco's modified Eagle's medium containing 5% fetal bovine serum, 100 μg/ml Zeocin (Invitrogen), and 15 μg/ml blasticidin (Invitrogen). Transient transfections were performed with Lipofectamine 2000 (Invitrogen). Cells stably expressing WT FLAG-IRP2, S157A FLAG-IRP2, or FLAG-IRP2-TEV-His were generated by transfecting Flp-In T-REx-293 cells with the appropriate FLAG-IRP2 pcDNA5/FRT/TO plasmid and the Flp recombinase plasmid pOG44. Stable isogenic integrants were selected with 50 μg/ml hygromycin B (Invitrogen), and expression was induced with 1 μg/ml tetracycline (Sigma).
Cell Synchronization and Flow Cytometry—To inhibit Cdk1 activity, cells were treated with 10 μm purvalanol A (Calbiochem). To enrich for cells in G2/M, HEK293 or HeLa cells were treated with 1 μm nocodazole (Sigma) for 18 h. For nocodazole release experiments, nocodazole was removed by washing cells three times with phosphate-buffered saline (PBS). Cells were harvested at the indicated times after release. Double-thymidine blocks were performed by treating HeLa cells with 2 mm thymidine (Sigma) for 16 h. Cells were washed in PBS, split, and released into thymidine-free media for 10 h. Thymidine was added for an additional 16 h, and then cells were washed and released into thymidine-free media. At the indicated times, duplicate plates of cells were lysed with Nonidet P-40 lysis buffer (see below) for Western blot analysis or fixed with 70% ethanol for flow cytometry. After fixation, cells were rehydrated in PBS, stained with propidium iodide (Sigma), and analyzed for DNA content by flow cytometry (FACScan, BD Biosciences). Cell cycle distribution was modeled using ModFit software (Verity Software House).
Antibodies—Antibodies used in this study include anti-Myc (9E10) (sc-40, Santa Cruz Biotechnology), anti-FLAG (F3165, Sigma), anti-IRP2 (18), anti-β-tubulin (69126, MP Biomedicals), anti-phospho-histone H3 (Ser-10) (06-570, Upstate), anti-cyclin B1 (MAB3684, Chemicon), anti-ferritin (F5012, Sigma), anti-transferrin receptor (13-6800, Invitrogen), anti-phospho-Cdk1-Tyr15 (PA1–4617, Affinity BioReagents), and anti-Cdk1 (PSTAIR) (06-923, Upstate). The anti-phospho IRP2-Ser-157 antibody (IRP2-pS157) was generated by Covance (Denver, PA). Rabbits were immunized with the keyhole limpet hemocyanin-conjugated phosphopeptide CKAGKLS(p)PLKVQ (amino acids 152–162) that was synthesized at the DNA/Peptide Facility at the University of Utah. IRP2-pSer-157 was purified by applying serum to a SulfoLink column (Pierce) coupled with the phosphopeptide. Eluted fractions were then applied to a SulfoLink column coupled with the unphosphorylated peptide. The unbound fractions containing IRP2-pSer-157 were assayed for specificity by dot blotting with the phosphopeptide and nonphosphopeptide. Specificity was also confirmed by Western blotting with WT and S157A FLAG-IRP2 expressing cellular extracts.
Cytosolic Extract Preparation for Western Blotting and Immunoprecipitations—Protein extracts were prepared by lysing PBS-washed cells in Nonidet P-40 lysis buffer (20 mm Hepes, pH 7.5, 25 mm KCl, 0.5% Nonidet P-40, 10 mm NaF, and 25 mm β-glycerol phosphate) or Triton lysis buffer (20 mm Hepes, pH 7.4, 25 mm KCl, 1% Triton X-100, 10 mm NaF, and 25 mm β-glycerol phosphate) for Cdc14A experiments. Complete Protease Inhibitor Mixture tablets (Roche Applied Science) were included in the lysis buffer for immunoprecipitations. Lysates were clarified by centrifugation at 18,000 × g for 15 min, and protein concentrations were determined by Coomassie Plus reagent (Pierce). Immunoprecipitations were performed in PBS with 6 μg of anti-Myc (Santa Cruz) or 6 μgof anti-FLAG (Sigma) for 1 h at 4 °C. Recombinant protein G-agarose (Invitrogen) was added for 1 h at 4 °C. Beads were washed four times with PBS containing 0.05% Nonidet P-40, boiled in SDS-loading buffer, and loaded onto an 8% SDS-PAGE or 4–12% NuPAGE Bis-Tris gel (Invitrogen).
[32P]Orthophosphate Labeling and Phosphopeptide Mapping—HEK293T cells were transfected with IRP2-Myc. Twenty-four hours post-transfection, one plate of cells was treated with 100 μm deferoxamine mesylate (DFO) for 16 h, and one plate was left untreated. After 16 h, cells were prelabeled for 2 h with 2.5 mCi/ml [32P]orthophosphate in phosphate-free Dulbecco's modified Eagle's medium containing 10% Tris-dialyzed fetal bovine serum and 50 μm MG132 (Sigma). Because the DFO was removed during media exchange, 100 μm DFO was added back to the original DFO-treated plate. After the 2 h prelabel, 100 μg/ml ferric ammonium citrate (FAC) was added to the untreated plate, and cells were labeled for an additional 8 h in the presence of DFO or FAC. Cells were harvested with Nonidet P-40 lysis buffer, and IRP2-Myc was immunoprecipitated from 2 mg of cellular extract with 10 μg of anti-Myc antibody and protein G-agarose. The immunoprecipitates were separated by 8% SDS-PAGE, and the gel was destained, dried, and analyzed with a PhosphorImager (Molecular Dynamics). Radiolabeled IRP2-Myc was excised from the gel and digested with 10 μg of trypsin (Roche Applied Science) or 2.5 μgof Lys-C (Roche Applied Science) in 50 mm ammonium bicarbonate, pH 8.0, at 37 °C overnight. The supernatant was recovered from the gel slices and lyophilized to dryness. Phosphopeptides were separated by two-dimensional peptide mapping as described (34). Briefly, peptides were suspended in electrophoresis buffer (2.5% formic acid, 7.8% acetic acid, pH 1.9) and spotted onto 20 × 20 cm cellulose TLC plates. Electrophoresis was carried out at 1300 V for 1 h. The plates were dried and subjected to ascending chromatography in phospho-chromatography buffer (62.5% isobutyric acid, 1.9% n-butanol, 4.8% pyridine, 2.9% acetic acid). Phosphopeptides were visualized with a PhosphorImager, scraped from the TLC plate, and eluted with electrophoresis buffer. Eluted phosphopeptides were subjected to phosphoamino acid analysis (34) and Edman 32P-release sequencing, which was performed by the DNA/Peptide Facility (University of Utah) using previously described methods (35).
In Vitro Kinase Assays—WT or S157A FLAG-IRP2 was immunoprecipitated from tetracycline-induced T-REx-293 stable cells using 6 μg of anti-FLAG antibodies and protein G-agarose. The agarose beads were washed three times in PBS followed by a wash in Cdk kinase buffer (50 mm Tris-HCl, 10 mm MgCl2, 2 mm dithiothreitol, 1 mm EGTA, 0.01% Brij 35). Beads were then incubated with the indicated amounts of recombinant Cdk1-cyclin B1 or Cdk2-cyclin A (New England Biolabs), 200 μm ATP, and 5 μCi of [γ-32P]ATP in 1× Cdk kinase buffer for 30 min at 30 °C. Immunoprecipitates were resolved on a 4–12% NuPAGE Bis-Tris gel (Invitrogen) and transferred to nitrocellulose. Radioactivity was quantified with a PhosphorImager prior to immunoblotting with anti-FLAG antibodies.
Quantitative Real-time RT-PCR—Total RNA was extracted from HeLa cells using TRIzol reagent (Invitrogen). First strand cDNA synthesis was performed with 1 μg of total RNA, random primers, and SuperScript III (Invitrogen). Quantitative real-time RT-PCR (qRT-PCR) was performed using SYBR GreenER qPCR supermix (Invitrogen) and an ABI Prism 7900HT Sequence Detection System. Primer sequences were previously described (14), and relative amplification efficiencies were validated prior to using the comparative CT method to determine mRNA -fold change. The data represent the average ± S.E. of three independent experiments performed in triplicate. Statistical significance of the differences between time points within an individual transcript was determined by a one-way analysis of variance: *, p ≤ 0.0027.
[35S]Methionine/Cysteine Labeling and Immunoprecipitations—HeLa cells synchronized by a double thymidine block were washed with PBS and cultured for the last 1.5 h in Dulbecco's modified Eagle's medium (5% fetal bovine serum, Cys-, and Met-free) containing 100 μCi/ml (1175 Ci/mmol) Tran35S-Label (MP Biomedicals, Inc.). For immunoprecipitations, 300 μg of cell lysate was incubated with either 6 μl of anti-ferritin or 2 μl of anti-transferrin receptor antibodies for 1 h at 4 °C. Recombinant protein A-agarose (Invitrogen) was added for an additional 1 h at 4 °C. Beads were washed four times with PBS containing 0.05% Nonidet P-40, boiled in SDS-loading buffer, and resolved on 8% or 15% SDS-PAGE gels. Gels were dried and analyzed by phosphorimaging.
RNA Electrophoretic Mobility Shift Assay—A [32P]GTP-labeled ferritin-L probe (100,000 cpm) was incubated with 12 μg of protein extract for 10 min at 25 °C followed by the addition of 20 units of RNase T1 (Roche Applied Science) and 100 μgof sodium heparin (Sigma) for 10 min each (36). IRP2 was supershifted with anti-IRP2 for 20 min, and RNA complexes were resolved by 5% non-denaturing PAGE. Band intensity was quantified with a PhosphorImager (Molecular Dynamics).
RESULTS
Identification of an in Vivo Iron-independent IRP2 Phosphorylation Site—To identify in vivo IRP2 phosphorylation sites, HEK293T cells expressing IRP2-Myc were labeled with [32P]orthophosphate. Immunoprecipitated radiolabeled IRP2-Myc was analyzed by two-dimensional tryptic phosphopeptide mapping. The IRP2 tryptic peptide map yielded one major phosphorylated peptide (1) and two minor phosphorylated peptides (2 and 3), none of which were significantly altered by treatment with the iron chelator DFO or FAC (Fig. 1A). Phosphoamino acid analysis of peptide 1 revealed that IRP2 was phosphorylated on a serine(s) (Fig. 1B).
FIGURE 1.

Identification of an in vivo iron-independent IRP2 phosphorylation site. A, HEK293T cells transfected with IRP2-Myc were treated with DFO or FAC and labeled with 2.5 mCi/ml [32P]orthophosphate. Immunoprecipitated IRP2-Myc was separated by SDS-PAGE, excised from the dried gel, eluted, digested with trypsin, and analyzed by two-dimensional phosphopeptide mapping. The results are representative of four independent experiments. B, phosphopeptide 1 from FAC-treated cells was eluted, hydrolyzed, and electrophoresed in two-dimensions with phosphoserine (pSer), phosphothreonine (pThr), and phosphotyrosine (pTyr) standards that were visualized by ninhydrin staining. C, phosphopeptide 1 from trypsin or Lys-C digestion was subjected to solid-phase Edman degradation. Radioactive phosphate release was detected by Cerenkov counting. D, sequence analysis of IRP2 yielded three conserved candidate phosphopeptides with a serine (S) in position 5 of a complete (▾) or incomplete (▿) digest. E, HEK293T cells transfected with WT, S503A, S407A, or S157A IRP2-Myc were labeled with 1 mCi/ml [32P]orthophosphate. Immunoprecipitated IRP2-Myc was analyzed by SDS-PAGE. The results in B, C, and E are representative of two independent experiments.
To identify the major iron-independent IRP2 phosphorylation site (peptide 1), a strategy was devised employing Edman phosphate release sequencing and bioinformatics (37). Candidate phosphoamino acids were identified from the cleavage of radiolabeled phosphoprotein analysis (37) and the cycle of Edman phosphate release. Tryptic peptide 1 released phosphate in Edman cycle 5 (Fig. 1C). Because bioinformatics indicated that four IRP2 tryptic peptides contained a serine in the fifth position, in vivo phosphorylated IRP2-Myc was subjected to Lys-C phosphopeptide mapping (data not shown) to narrow the number of candidate peptides. Edman phosphate release sequencing of the major Lys-C peptide also released phosphate in cycle 5 (Fig. 1C). Assuming complete digestion, this narrowed the candidate IRP2 phosphorylation sites to Ser-503 and Ser-735, while partial digestion also implicated Ser-407 and Ser-157 (Fig. 1D). Ser-735 is not conserved in IRP2 orthologues, so it was eliminated as a potential candidate. [32P]Orthophosphate labeling of HEK293T cells transfected with WT, S503A, S407A, or S157A IRP2-Myc indicated that S157A IRP2-Myc incorporated ∼70% less 32P than WT IRP2-Myc (Fig. 1E).
These data identify Ser-157 as the major IRP2 phosphorylation site but show that IRP2 can be phosphorylated at other sites. Mass spectrometry of FLAG-IRP2-His purified from DFO- and FAC-treated HEK293 cells confirmed that Ser-157 is phosphorylated during iron deficiency and sufficiency (Table 1). Additionally, Ser-157 phosphorylation was not modulated by DFO or FAC treatment in HEK293 cells, as determined by Western blotting with IRP2 Ser-157 phosphospecific antibodies (IRP2-pS157) (“Experimental Procedures” and supplemental Fig. S1), verifying that IRP2 Ser-157 phosphorylation is iron-independent (supplemental Fig. S2).
TABLE 1.
Detection of IRP2 Ser-157 phosphorylation by mass spectrometry
Recombinant 2×-FLAG-IRP2-TEV-10×-His was purified from DFO- or
FAC-treated cells and digested with either trypsin or chymotrypsin prior to
phosphopeptide enrichment using immobilized metal affinity chromatography
(supplemental “Experimental Procedures”). Electrospray
ionization/MS/MS analysis identified multiple peptides phosphorylated at
Ser-157 (underlined) in both DFO- and FAC-treated samples. The theoretical
molecular mass is listed for each peptide, which includes +80 Da for the
phosphate. Observed mass-to-charge (m/z) ratios are listed
for the indicated ion charge state (+1 = MH+,
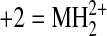 , and
, and
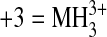 ). Mass errors for all ions were
<4 ppm. The data were generated from four independent experiments.
). Mass errors for all ions were
<4 ppm. The data were generated from four independent experiments.
|
Sequence |
Theoretical molecular mass |
DFO |
FAC |
||
|---|---|---|---|---|---|
| MH+ | 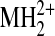 |
MH+ | 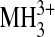 |
||
| KLSPL | 636.325 | 637.332 | 637.334 | ||
| AGKLSPL | 764.383 | 765.393 | |||
| KAGKLSPL | 892.478 | 447.248 | |||
| AGKLSPLK | 892.478 | 447.247 | |||
| AGKLSPLKVQPK | 1344.753 | 449.259 | |||
IRP2 Ser-157 Is Phosphorylated during Mitosis by Cdk1/Cyclin B1—Ser-157 is conserved in IRP2 orthologues and is located within a canonical Cdk motif (SpPLK), which suggests that Ser-157 phosphorylation may be cell cycle-regulated. To determine whether IRP2 Ser-157 was a Cdk phosphorylation site, stably expressed FLAG-IRP2 was immunoprecipitated from asynchronous Flp-In T-REx 293 cells and used as a substrate for in vitro kinase assays with recombinant Cdk1/cyclin B1 or Cdk2/cyclin A. FLAG-WT IRP2 but not FLAG-S157A IRP2 was phosphorylated by Cdk1/cyclin B1 and Cdk2/cyclin A in a dose-dependent manner (Fig. 2, A and B). Radiolabeled FLAG-IRP2 was not detected in the absence of exogenously added kinase, indicating that the endogenous Ser-157 kinase was not immunoprecipitated with FLAG-IRP2 (Fig. 2, A and B, lanes 2 and 6). Another proline-directed serine/threonine kinase that recognizes the minimum S/TP sequence, mitogen-activated protein kinase, failed to phosphorylate FLAG-IRP2 (data not shown). These data indicate that IRP2 Ser-157 is a substrate for Cdk1 and Cdk2 in vitro.
FIGURE 2.

IRP2 Ser-157 is phosphorylated by Cdk1 during mitosis. Stably expressed WT or S157A FLAG-IRP2 was immunoprecipitated from Flp-In T-REx-293 cells and incubated with recombinant Cdk1/cyclin B1 (A) or Cdk2/cyclin A (B) and [γ-32P]ATP. Phosphorylated FLAG-IRP2 was separated by SDS-PAGE and analyzed by phosphorimaging and anti-FLAG Western blot. C, HeLa cells were grown asynchronously (Asyn) or treated with nocodazole for 18 h. Nocodazole was removed, and cells were released and harvested at the indicated times for Western blot analysis with anti-IRP2-pS157, anti-IRP2, anti-H3-pS10, anti-cyclin B1, and anti-β-tubulin antibodies. D, HEK293 cells were grown asynchronously (Asyn) or arrested in mitosis by treatment with nocodazole (Noc) for 18 h. The Cdk inhibitor purvalanol A (PA) (10 μm) was added for the last 2 h of treatment. Lysates were separated by SDS-PAGE and analyzed by Western blotting with anti-IRP2-pS157, anti-IRP2, anti-Cdk1-pY15, anti-Cdk1, anti-cyclin B1, and anti-β-tubulin antibodies. E, HEK293 cells were treated with 10 μm purvalanol A for the last 30, 60, or 120 min of an 18-h nocodazole treatment. Lysates were separated by SDS-PAGE and analyzed by Western blotting with anti-IRP2-pS157, anti-IRP2, and anti-β-tubulin antibodies. All results are representative of at least two independent experiments.
It is not surprising that both Cdk1 and Cdk2 can phosphorylate IRP2 in vitro, because both kinases recognize the same sequence motif. In vivo, however, the kinase activities of Cdk1 and Cdk2 differ throughout the cell cycle. Cdk2/cyclin A is active during the S/G2 transition, whereas Cdk1/cyclin B1 is active during the G2/M transition and throughout mitosis (38). To determine whether Cdk1/cyclin B1 phosphorylates IRP2 Ser-157 in vivo, HeLa cells were arrested in mitosis by treatment with the microtubule depolymerizing agent nocodazole. IRP2 Ser-157 phosphorylation increased in nocodazole-treated cells, whereas total IRP2 protein was unchanged (Fig. 2C, lanes 1 and 2). Accumulation of cyclin B1 and phosphorylation of histone H3-S10, which occurs during chromosome condensation (39), confirmed that nocodazole-treated cells were mitotic (Fig. 2C, lanes 1 and 2). To determine whether IRP2 Ser-157 was dephosphorylated during mitotic exit, cells were released from nocodazole arrest and Ser-157 phosphorylation was assessed. Two hours after nocodazole removal, IRP2 Ser-157 phosphorylation decreased (Fig. 2C, lane 5) and was eventually reduced to the level seen in asynchronous cells. The reduction in IRP2 Ser-157 phosphorylation coincides with the degradation of cyclin B1 and diminished histone H3-S10 phosphorylation, which are markers of mitotic exit. These results show that IRP2 Ser-157 is phosphorylated during mitosis and dephosphorylated during mitotic exit, which suggests the involvement of the mitotic Cdk, Cdk1.
To determine if Cdk1 was the IRP2 Ser-157 kinase, asynchronous and nocodazole-treated cells were treated with the Cdk inhibitor purvalanol A. Purvalanol A treatment decreased Ser-157 phosphorylation in asynchronous cells and to a greater extent in nocodazole-treated cells (Fig. 2D, lanes 2 and 4). Inhibition of Cdk1 by purvalanol A was confirmed by increased inhibitory phosphorylation of Cdk1 at Tyr-15 in asynchronous cells and decreased cyclin B1 protein in nocodazole-treated cells (Fig. 2D). Purvalanol A inhibition of Cdk1 was rapid and decreased IRP2 Ser-157 phosphorylation in nocodazole-treated cells within 30 min (Fig. 2E, lane 2). Taken together, these data indicate that Cdk1/cyclin B1 is the likely kinase that phosphorylates IRP2 at Ser-157 during mitosis.
IRP2 Ser-157 Phosphorylation during Mitosis Is Associated with Increased Ferritin Expression and Reduced RNA-binding Activity—To monitor IRP2 Ser-157 phosphorylation throughout the cell cycle, HeLa cells were synchronized at the G1/S border by a double-thymidine block and released into the cell cycle. Flow cytometry showed that cells reached mid-S phase at 6 h, entered mitosis at 8 h and exited mitosis by 14 h (Fig. 3A). Accumulation of cyclin B1 and phosphorylation of histone H3 at Ser-10, confirmed that cells were mitotic (8–12 h) (Fig. 3B). IRP2 Ser-157 phosphorylation was minimal until cells entered mitosis (8 h), elevated during mitosis (8–12 h), and diminished during mitotic exit (14 h) (Fig. 3B). Because IRP2 regulates ferritin translation and TfR1 mRNA stability, their protein levels were measured throughout the cell cycle. Ferritin increased during late G2 to M (11–12 h) and decreased during mitotic exit (14 h), whereas TfR1 expression was relatively constant until 11–14 h post-thymidine release, when a subtle but consistent decrease in expression was observed (Fig. 3B). Because IRP2 protein levels are unaltered throughout the cell cycle (Fig. 3B) and Ser-157 phosphorylation does not affect IRP2 stability (supplemental Fig. S3), these alterations in ferritin and TfR1 expression suggest that IRP RNA-binding activity may be reduced during mitosis. IRP RNA-binding activity was therefore measured by electrophoretic mobility shift assay in HeLa cell extracts from a double-thymidine block. IRP2 RNA-binding activity decreased as cells progressed into G2/M (8–12 h) and increased as cells exited mitosis (14 h), whereas IRP1 RNA-binding activity was unchanged during mitosis (Fig. 3C). Quantification of the data shows that IRP2 RNA-binding activity is reduced by 16.9% as cells enter S phase and by 58.7% as cells enter G2/M (Fig. 3D). The reduction in IRP2 RNA-binding activity during G2/M correlates with IRP2 Ser-157 phosphorylation and increased ferritin expression (Fig. 3B). The increase in ferritin occurred 11 h after thymidine release, which was 3 h after Ser-157 phosphorylation was first detected. This timing would allow for newly synthesized ferritin to accumulate to a detectable level. These data indicate that Ser-157 phosphorylation during G2/M coincides with reduced IRP2 RNA-binding activity, leading to increased ferritin and decreased TfR1 abundance.
FIGURE 3.

IRP2 Ser-157 phosphorylation during mitosis is associated with reduced RNA-binding activity and altered ferritin and TfR1 expression. HeLa cells were synchronized at the G1/S border by a double-thymidine block or left asynchronous (Asyn). A, cells were released for the indicated times and fixed for flow cytometric analysis of DNA content. G1 phase is indicated by the 2N peak (black), S phase is indicated by the cross-hatched peak (gray), and G2/M phases are indicated by the 4N peak (black). B, duplicate plates of cells were harvested for Western blot analysis with anti-IRP2-pS157, anti-IRP2, anti-cyclin B1, anti-H3-pS10, anti-ferritin, anti-TfR1, and anti-β-tubulin antibodies. The results in A and B are representative of five independent experiments. A cross-reacting band is indicated by an asterisk. C, extracts were used in an electrophoretic mobility shift assay with a 32P-labeled ferritin-L IRE probe and supershifted with anti-IRP2 antibody. RNA complexes were separated by non-denaturing PAGE and visualized by phosphorimaging. A representative gel from three independent experiments is shown. D, IRP2 RNA-binding activity was normalized to IRP2 protein, which was normalized to β-tubulin protein, and plotted as the percentage of RNA binding in G1 (0 h). The average Ft-L IRP2 RNA-binding activity was 83.1% (±7.1%) during S phase (6 h) and 41.3% (±6.6%) during G2/M (10 h). The bar graph is the result of three independent experiments ± S.D. *, p < 0.01; **, p < 0.02, as determined by unpaired two-tailed Students t test.
Regulation of Ferritin and TfR1 mRNAs during the Cell Cycle—To determine whether increased ferritin abundance during G2/M was due to translational regulation by IRP2, ferritin synthesis was measured during the cell cycle by immunoprecipitating 35S-Met/Cys-labeled ferritin during a double-thymidine release. Ferritin synthesis increased during mitosis (9–12 h) and decreased during mitotic exit (15 h) (Fig. 4A). As a positive control, ferritin synthesis increased in cells treated with FAC for 3 h, as expected. To eliminate the possibility that increased ferritin synthesis during G2/M was due to increased transcription, we measured ferritin mRNA levels by qRT-PCR. Ferritin-H and ferritin-L mRNA amounts were not significantly altered during cell cycle progression (Fig. 4B), indicating that the increase in ferritin abundance during G2/M was due to increased synthesis.
FIGURE 4.

Regulation of IRE-containing mRNAs during the cell cycle. Ferritin (A) and TfR1 (C) synthesis were measured throughout the cell cycle by labeling cells with 35S-Met/Cys for the last 1.5 h of double thymidine release. FAC is cells treated (50 μg/ml) for 3 h and labeled with 35S-Met/Cys for the last 1.5 h. Immunoprecipitated ferritin or TfR1 was separated by SDS-PAGE and analyzed by phosphorimaging. Control immunoprecipitation lacking antibody is indicated by (–). FTH and FTL (B) and TfR1 (D) mRNA levels during the cell cycle were determined by real-time qRT-PCR. Triplicate reactions were performed for each double thymidine release time point and data were normalized to glyceraldehyde-3-phosphate dehydrogenase mRNA levels. The graphs were generated from three independent experiments and represent fold increase relative to 0 h after double thymidine release. Data are presented as mean ± S.E. Statistical significance of the differences between time points within an individual transcript was determined by a one-way analysis of variance: *, p ≤ 0.0027.
Our data indicate that TfR1 abundance decreases during G2/M (Fig. 3B). To determine the mechanism for decreased TfR1 expression, TfR1 synthesis and mRNA levels were measured during a double-thymidine release. TfR1 synthesis was unaltered during the cell cycle (Fig. 4C), whereas mRNA levels were constant until 12 h when a small, but significant, increase occurred that was maintained as cells exited mitosis by 15 h (Fig. 4D). The decrease in TfR1 protein 11–14 h after double-thymidine release (Fig. 3B) did not correspond with decreased mRNA levels (Fig. 4D). These results may be explained by the cessation of mRNA degradation during mitosis, which leads to global mRNA stabilization (43) and potentially masks TfR1 mRNA destabilization.
Cdc14A Associates with and Dephosphorylates IRP2 in Vivo—The dephosphorylation of IRP2 Ser-157 during mitotic exit (Figs. 2C and 3B) points to the existence of a phosphatase that recognizes phosphorylated Ser-157. A potential candidate Ser-157 phosphatase is Cdc14A. In yeast, Cdc14 regulates mitotic exit by dephosphorylating Cdk1 substrates (40, 41). The human homologue, Cdc14A, can dephosphorylate Cdk1 substrates, including hCdh1 and can activate the anaphase promoting complex (APCCdh1) in vitro (42, 43), suggesting a role for Cdc14A during mitotic exit. To determine if Cdc14A associated with IRP2 in vivo, a co-immunoprecipitation was performed. Flp-In T-REx 293 cells stably expressing WT or S157A FLAG-IRP2 were transfected with catalytically inactive (CI) Myc-Cdc14A, which has been shown to increase the stability of the transient Cdc14A/substrate interaction (43, 44). Immunoprecipitated Myc-Cdc14A-CI associated with WT FLAG-IRP2 but not S157A FLAG-IRP2 (Fig. 5A). To determine if Ser-157 was a substrate for Cdc14A in vivo, HEK293 cells were transfected with Myc-Cdc14A-WT or Myc-Cdc14A-CI and extracts were analyzed for Ser-157 phosphorylation by Western blotting. Overexpression of Myc-Cdc14A-WT caused a decrease in endogenous IRP2 Ser-157 phosphorylation, whereas Myc-Cdc14A-CI expression did not alter IRP2 phosphorylation as compared with the mock-transfected control (Fig. 5B). These results indicate that the IRP2 Ser-157 Cdk1 phosphorylation site is antagonized by Cdc14A and identify a novel in vivo Cdc14A substrate.
FIGURE 5.

Cdc14A associates with and dephosphorylates IRP2. A, Flp-In T-REx-293 cells stably expressing WT or S157A FLAG-IRP2 were transiently transfected with catalytically inactive (CI) Myc-Cdc14A-CI. Cells were treated with nocodazole for 18 h, and lysates were immunoprecipitated with anti-Myc antibody. Immunoprecipitates and lysates were resolved by SDS-PAGE and analyzed by Western blotting with anti-FLAG or anti-Myc antibodies. The results are representative of two independent experiments. B, HEK293 cells were transiently transfected with Myc-Cdc14A-WT or Myc-Cdc14A-CI and treated with nocodazole for 18 h. Lysates were separated by SDS-PAGE and analyzed by Western blotting with anti-IRP2-pS157, anti-IRP2, anti-Myc, and anti-β-tubulin antibodies. The results are representative of three independent experiments. C, Cdc14A IRE and non-IRE mRNA levels during the cell cycle were determined by real-time qRT-PCR as described in Fig. 4.
Regulation of Cdc14A IRE and Non-IRE mRNAs during the Cell Cycle—Given that the phosphatase Cdc14A has an IRE in a mRNA splice variant (14) and has roles during the cell cycle (42, 43, 45), we determined whether Cdc14A mRNA was regulated during the cell cycle by IRP2. Our identification of Cdc14A as an IRP2 Ser-157 phosphatase also suggests that an intriguing regulatory feedback mechanism may exist between IRP2 and Cdc14A. The human Cdc14A mRNA has three splice variants that differ in their 3′ sequence (46) and two splice variants that differ in their 5′ sequence (14). Only one of the 3′ mRNA splice variants has an IRE in the 3′-untranslated region (accession number NM_003672), which has been shown to bind IRP1 and IRP2 in vitro (14). Quantitative RT-PCR was performed with primers that were specific for the Cdc14A IRE mRNA splice variant and one of the Cdc14A non-IRE mRNA splice variants (accession number NM_033312). Both Cdc14A IRE and non-IRE mRNAs increased 2-fold during G1/S (2–7 h), decreased to approximately initial levels (0 h) as cells entered G2/M (8 h), and then remained relatively constant throughout G2/M and during mitotic exit (14–15 h) (Fig. 5C). These data show that Cdc14A IRE and non-IRE mRNAs levels are similarly regulated during the cell cycle and suggest that the IRE variant is not preferentially regulated by IRP2.
DISCUSSION
Iron is essential for cellular growth and proliferation. Although it is well known that iron chelation arrests cells in G1/S (47), less is known about the regulation of iron homeostasis during the cell cycle. Here, we provide evidence for a direct link between IRP2 and the cell cycle. IRP2 Ser-157 is phosphorylated by Cdk1 during G2/M and dephosphorylated during mitotic exit by Cdc14A. IRP2 Ser-157 phosphorylation disrupts its interaction with ferritin mRNA, leading to the derepression of ferritin translation during G2/M. The cell cycle regulation of ferritin and possibly other IRE-mRNAs, such as TfR1, may provide a mechanism to precisely modulate the labile intracellular iron pool during cellular proliferation.
Regulation of IRP2 Phosphorylation—Although IRP2 was previously shown to be phosphorylated in HL60 cells stimulated with PMA (29), the phosphoamino acid and the kinase were not identified. In our study, IRP2 phosphorylation was not increased in HEK293 cells treated with PMA (data not shown). Moreover, unlike the Schalinske and Eisenstein study (29) where PMA increased RNA-binding activity by activating a pool of redox-sensitive IRP2, we found that Ser-157 phosphorylation led to decreased RNA-binding activity. Although Ser-157 was identified as the major in vivo IRP2 phosphorylation site, we observed two additional phosphorylation sites that were below the detection limits of Edman phosphate release sequencing and mass spectrometry. Whether phosphorylation of one of these sites is stimulated by PMA remains to be determined.
Both in vitro and in vivo data indicate that IRP2 is a likely Cdk1 target. We found that IRP2 phosphorylation was enhanced during nocodazole-induced mitotic arrest and blocked by the Cdk1 inhibitor purvalanol A. In addition, IRP2 was phosphorylated in vitro by Cdk1/cyclin B1. Cdk1 is critical for triggering mitosis and has been shown to phosphorylate more than 70 proteins involved in processes such as nuclear envelope breakdown, chromosome condensation, centrosome separation, spindle assembly, and Golgi fragmentation (48, 38, 49). To our knowledge, IRP2 is the first Cdk1 substrate that is involved in iron homeostasis.
Given that Cdc14A has a potential role during mammalian mitotic exit (42, 43, 45) and a 3′-IRE in one mRNA splice variant (14), we considered Cdc14A as a candidate IRP2 phosphatase. We found that IRP2 and Cdc14A interact in cells and that overexpression of Cdc14A during mitosis inhibited IRP2 Ser-157 phosphorylation. Humans have two Cdc14 homologues, Cdc14A and Cdc14B, that differ in cellular localization and function. Cdc14B localizes to interphase nucleoli and the mitotic spindle (50, 43, 51) while Cdc14A localizes to interphase centrosomes and to the cytoplasm of interphase and mitotic cells (42, 43, 33, 52). Thus, it is likely that IRP2 and Cdc14A interact in the cytoplasm during mitotic exit.
IRP2 RNA-binding Activity Is Reduced during G2/M—The reduction in RNA-binding activity of IRP2, but not IRP1, during G2/M coincided with Ser-157 phosphorylation and increased ferritin synthesis. Ferritin mRNAs were not significantly altered during the cell cycle, indicating that the increase in ferritin synthesis during G2/M was due to translational regulation by IRP2. Although these data suggest that Ser-157 phosphorylation inhibits the binding of IRP2 to RNA, we have been unable to show that in vitro phosphorylated IRP2 has reduced RNA-binding activity.4 The inconsistency between our in vivo and in vitro results is not apparent, but may be due to one or more missing factors in the in vitro assay. Ser-157 phosphorylation in vivo may recruit one or more proteins that block the interaction between IRP2 and RNA or may be part of a hierarchical phosphorylation cascade. It is interesting to note that Ser-157 is located in the 73-amino acid domain that was proposed to be required for iron-dependent IRP2 degradation (19). This model, however, has been challenged by several groups (53–55). Due to the location of Ser-157, we predicted that it would not have a role in the iron-dependent IRP2 degradation, and as expected, the degradation rates of WT and S157A FLAG-IRP2 were similar. Therefore, it is possible that the 73-amino acid domain serves as a binding site for a protein whose interaction with IRP2 is dependent on the prior phosphorylation of Ser-157 by Cdk1.
Relevance of Ferritin and TfR1 Regulation during the Cell Cycle—What is the significance of IRP2 Ser-157 phosphorylation during mitosis? We propose that increased ferritin synthesis during G2/M is a protective mechanism to prevent iron-induced DNA damage. Mitosis is a vulnerable phase of the cell cycle due to the global repression of transcription (56) and the inability to repair damaged DNA (57). In addition, the accumulation of ferritin during G2/M may protect cells from the forthcoming iron influx during mitotic exit. It is well known that transferrin-dependent iron uptake ceases during early mitosis but resumes during late anaphase and cytokinesis (58–60). IRP2 Ser-157 phosphorylation is possibly a preemptive iron-independent mechanism to up-regulate ferritin during mitosis, which may function dually to protect cells from iron-toxicity and to store iron for use during the next round of cell division.
We also show that TfR1 mRNA is cell cycle-regulated. Several studies have found that increased TfR1 cell surface expression confers a growth advantage to both malignant and nonmalignant cells (61–64). The mechanisms controlling TfR1 expression during cellular proliferation are not entirely elucidated but may include transcriptional regulation by the c-myc oncogene (65) or post-transcriptional regulation by phosphorylated IRP2 as described in our study. We observe a slight reduction in TfR1 steady-state protein levels during G2/M but no decrease in TfR1 mRNA levels. During mitosis, TfR1 is sequestered in endosomes due to reduced endosomal recycling to the cell surface (58–60). Whether endosomally sequestered TfR1 is degraded during mitosis is unknown. The reduction in TfR1 protein and the sequestration of TfR1 in endosomes during mitosis may protect mitotic cells by limiting iron uptake.
As cells exited mitosis, IRP2 RNA-binding activity was restored and TfR1 mRNA levels were increased. We cannot determine whether the increase in TfR1 mRNA during late G2/M was due to mRNA stabilization by dephosphorylated IRP2 or due to increased transcription, which is restored at the end of mitosis (56). Our data suggest that the coordinate regulation of ferritin and TfR1 expression during mitotic exit and G1/S increase the availability of iron for growth during G1 phase and for DNA synthesis during S phase. This idea is consistent with many studies showing that iron is required for cell cycle progression (66) and that ferritin expression can influence cellular proliferation. Sequestration of intracellular iron by overexpression of ferritin-H (67) or mitochondrial ferritin (68) suppresses cellular proliferation. Conversely, ferritin repression stimulates short term cellular growth by increasing the labile iron pool (69). These studies demonstrate that intracellular iron regulation, which is primarily facilitated by the IRPs, is essential for cellular proliferation. Our study identifies IRP2 Ser-157 phosphorylation as a molecular mechanism to regulate ferritin and TfR expression during the cell cycle, which may potentially modulate cellular proliferation. Although we have focused on ferritin and TfR, it is likely that other IRE-mRNAs are regulated by IRP2 during the cell cycle.
Regulation of Cdc14A mRNA during the Cell Cycle—What is the functional significance of the Cdc14A and IRP2 interaction? The dephosphorylation of Ser-157 by Cdc14A at the M/G1 transition may restore IRP2 RNA-binding activity during G1, which would increase intracellular iron levels by reducing ferritin and increasing TfR1 expression. It is tempting to speculate that a feedback mechanism may exist between IRP2 and Cdc14A. For example, IRP2 binding to Cdc14A mRNA during interphase would function to stabilize Cdc14A mRNA, whereas the inhibition of IRP2 RNA-binding during G2/M would destabilize Cdc14A mRNA at a time when excess Cdc14A activity may prematurely inactivate Cdk1. It is not clear if Cdc14A activity or IRE-variant protein levels are regulated during the mammalian cell cycle. One study showed that Cdc14A phosphatase activity increased slightly during S phase and late mitosis while protein levels of the non-IRE variant remained relatively constant (43). Due to the lack of IRE-variant specific Cdc14A antibody, we could not measure Cdc14A protein levels. However, our qRT-PCR results showed that both Cdc14A IRE and non-IRE mRNAs were increased during S phase. Consequently, we are unable to conclude that IRP2 is solely responsible for the regulation of the Cdc14A IRE variant during the cell cycle. It is possible, however, that both variants are similarly regulated, but by different mechanisms.
It is interesting to note that Cdc14A IRE mRNA was shown to be regulated by iron deficiency but not excess (14). DFO treatment increased Cdc14A IRE mRNA levels in HEK293 and MCF7 cells but not in HeLa or K562 cells (14). Thus, if the Cdc14A IRE is functional, its regulation is not iron-dependent and is cell type-specific. We find that Cdc14A IRE and non-IRE mRNA levels are increased 2-fold in HeLa cells during S phase. It is therefore possible that the DFO-mediated increase in Cdc14A IRE mRNA in HEK293 and MCF7 cells (14) is due to DFO-induced S phase arrest (8). Whether IRPs regulate Cdc14A IRE mRNA during the cell cycle has yet to be determined.
Supplementary Material
Acknowledgments
We are grateful to Dr. Bob Schackmann for performing the Edman phosphate release sequencing, Dr. Wayne Green for assistance with flow cytometry, and Dr. Chad Nelson for performing mass spectrometry. We thank Dr. Jiri Lukas for the Myc-Cdc14A plasmid (Institute of Cancer Biology, Danish Cancer Society). We thank Drs. Don Ayer and Matthew Movsesian for critical reading of the manuscript and helpful comments.
This work was supported, in whole or in part, by National Institutes of Health Grant R01GM045201 (to E. A. L.). DNA sequencing and the syntheses of peptide and DNA oligonucleotides were performed by Core facilities supported by National Institutes of Health Cancer Center Support Grant 2P30CA42014. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental text and Figs. S1–S3.
Footnotes
The abbreviations used are: Cdk2, cyclin-dependent kinase 2; Cdk1, cyclin-dependent kinase 1; IRP1, iron regulatory protein 1; IRP2, iron regulatory protein 2; IRE, iron-responsive element; TfR1, transferrin receptor 1; HIF-2α, hypoxia-inducible factor 2α; Cdc14A, cell division cycle 14A; PMA, phorbol 12-myristate 13-acetate; DFO, deferoxamine mesylate; FAC, ferric ammonium citrate; CI, catalytically inactive; WT, wild type; PBS, phosphate-buffered saline; Bis-Tris, 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol; RT, reverse transcription; qRT, quantitative real-time RT.
M. L. Wallander, S. J. Romney, and E. A. Leibold, unpublished observation.
References
- 1.Hentze, M. W., Muckenthaler, M. U., and Andrews, N. C. (2004) Cell 117285 –297 [DOI] [PubMed] [Google Scholar]
- 2.Dunn, L. L., Rahmanto, Y. S., and Richardson, D. R. (2007) Trends Cell Biol. 1793 –100 [DOI] [PubMed] [Google Scholar]
- 3.Nyholm, S., Mann, G. J., Johansson, A. G., Bergeron, R. J., Graslund, A., and Thelander, L. (1993) J. Biol. Chem. 26826200 –26205 [PubMed] [Google Scholar]
- 4.Kulp, K. S., Green, S. L., and Vulliet, P. R. (1996) Exp. Cell Res. 22960 –68 [DOI] [PubMed] [Google Scholar]
- 5.Gao, J., and Richardson, D. R. (2001) Blood 98842 –850 [DOI] [PubMed] [Google Scholar]
- 6.Fu, D., and Richardson, D. R. (2007) Blood 110752 –761 [DOI] [PubMed] [Google Scholar]
- 7.Buss, J. L., Torti, F. M., and Torti, S. V. (2003) Curr. Med. Chem. 101021 –1034 [DOI] [PubMed] [Google Scholar]
- 8.Yu, Y., Wong, J., Lovejoy, D. B., Kalinowski, D. S., and Richardson, D. R. (2006) Clin. Cancer Res. 126876 –6883 [DOI] [PubMed] [Google Scholar]
- 9.Rouault, T. A. (2006) Nat. Chem. Biol. 2406 –414 [DOI] [PubMed] [Google Scholar]
- 10.Wallander, M. L., Leibold, E. A., and Eisenstein, R. S. (2006) Biochim. Biophys. Acta-Mol. Cell Res. 1763668 –689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gray, N. K., Pantopoulos, K., Dandekar, T., Ackrell, B. A. C., and Hentze, M. W. (1996) Proc. Natl. Acad. Sci. U. S. A. 934925 –4930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cox, T. C., Bawden, M. J., Martin, A., and May, B. K. (1991) EMBO J. 101891 –1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanchez, M., Galy, B., Muckenthaler, M. U., and Hentze, M. W. (2007) Nat. Struct. Mol. Biol. 14420 –426 [DOI] [PubMed] [Google Scholar]
- 14.Sanchez, M., Galy, B., Dandekar, T., Bengert, P., Vainshtein, Y., Stolte, J., Muckenthaler, M. U., and Hentze, M. W. (2006) J. Biol. Chem. 28122865 –22874 [DOI] [PubMed] [Google Scholar]
- 15.Cmejla, R., Petrak, J., and Cmejlova, J. (2006) Biochem. Biophys. Res. Commun. 341158 –166 [DOI] [PubMed] [Google Scholar]
- 16.Haile, D. J., Rouault, T. A., Tang, C. K., Chin, J., Harford, J. B., and Klausner, R. D. (1992) Proc. Natl. Acad. Sci. U. S. A. 897536 –7540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Phillips, J. D., Guo, B., Yu, Y., Brown, F. M., and Leibold, E. A. (1996) Biochemistry 3515704 –15714 [DOI] [PubMed] [Google Scholar]
- 18.Guo, B., Yu, Y., and Leibold, E. A. (1994) J. Biol. Chem. 26924252 –24260 [PubMed] [Google Scholar]
- 19.Iwai, K., Klausner, R. D., and Rouault, T. A. (1995) EMBO J. 145350 –5357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanson, E. S., and Leibold, E. A. (1998) J. Biol. Chem. 2737588 –7593 [DOI] [PubMed] [Google Scholar]
- 21.Hanson, E. S., Foot, L. M., and Leibold, E. A. (1999) J. Biol. Chem. 2745047 –5052 [DOI] [PubMed] [Google Scholar]
- 22.Schneider, B. D., and Leibold, E. A. (2003) Blood 1023404 –3411 [DOI] [PubMed] [Google Scholar]
- 23.Meyron-Holtz, E. G., Ghosh, M. C., and Rouault, T. A. (2004) Science 3062087 –2090 [DOI] [PubMed] [Google Scholar]
- 24.LaVaute, T., Smith, S., Cooperman, S., Iwai, K., Land, W., Meyron-Holtz, E., Drake, S. K., Miller, G., Abu-Asab, M., Tsokos, M., Switzer, R., 3rd, Grinberg, A., Love, P., Tresser, N., and Rouault, T. A. (2001) Nat. Genet. 27209 –214 [DOI] [PubMed] [Google Scholar]
- 25.Meyron-Holtz, E. G., Ghosh, M. C., Iwai, K., LaVaute, T., Brazzolotto, X., Berger, U. V., Land, W., Ollivierre-Wilson, H., Grinberg, A., Love, P., and Rouault, T. A. (2004) EMBO J. 23386 –395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cooperman, S. S., Meyron-Holtz, E. G., Olivierre-Wilson, H., Ghosh, M. C., McConnell, J. P., and Rouault, T. A. (2005) Blood 1061084 –1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Galy, B., Ferring, D., Minana, B., Bell, O., Janser, H. G., Muckenthaler, M., Schumann, K., and Hentze, M. W. (2005) Blood 1062580 –2589 [DOI] [PubMed] [Google Scholar]
- 28.Eisenstein, R. S., Tuazon, P. T., Schalinske, K. L., Anderson, S. A., and Traugh, J. A. (1993) J. Biol. Chem. 26827363 –27370 [PubMed] [Google Scholar]
- 29.Schalinske, K. L., and Eisenstein, R. S. (1996) J. Biol. Chem. 2717168 –7176 [DOI] [PubMed] [Google Scholar]
- 30.Pitula, J. S., Deck, K. M., Clarke, S. L., Anderson, S. A., Vasanthakumar, A., and Eisenstein, R. S. (2004) Proc. Natl. Acad. Sci. U. S. A. 10110907 –10912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clarke, S. L., Vasanthakumar, A., Anderson, S. A., Pondarre, C., Koh, C. M., Deck, K. M., Pitula, J. S., Epstein, C. J., Fleming, M. D., and Eisenstein, R. S. (2006) EMBO J. 25544 –553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown, N. M., Anderson, S. A., Steffen, D. W., Carpenter, T. B., Kennedy, M. C., Walden, W. E., and Eisenstein, R. S. (1998) Proc. Natl. Acad. Sci. U. S. A. 9515235 –15240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mailand, N., Lukas, C., Kaiser, B. K., Jackson, P. K., Bartek, J., and Lukas, J. (2002) Nat. Cell Biol. 4318 –322 [DOI] [PubMed] [Google Scholar]
- 34.Van der Geer, P. (1993) in Protein Phosphorylation: a Practical Approach (Hardie, D. G., ed) pp.31 –59, Oxford University Press, Oxford
- 35.MacDonald, J. A., Mackey, A. J., Pearson, W. R., and Haystead, T. A. J. (2002) Mol. Cell Proteomics 1314 –322 [DOI] [PubMed] [Google Scholar]
- 36.Leibold, E. A., and Munro, H. N. (1988) Proc. Natl. Acad. Sci. U. S. A. 852171 –2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mackey, A. J., Haystead, T. A. J., and Pearson, W. R. (2003) Nucleic Acids Res. 313859 –3861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malumbres, M., and Barbacid, M. (2005) Trends Biochem. Sci. 30630 –641 [DOI] [PubMed] [Google Scholar]
- 39.Goto, H., Tomono, Y., Ajiro, K., Kosako, H., Fujita, M., Sakurai, M., Okawa, K., Iwamatsu, A., Okigaki, T., Takahashi, T., and Inagaki, M. (1999) J. Biol. Chem. 27425543 –25549 [DOI] [PubMed] [Google Scholar]
- 40.Visintin, R., Craig, K., Hwang, E. S., Prinz, S., Tyers, M., and Amon, A. (1998) Mol. Cell 2709 –718 [DOI] [PubMed] [Google Scholar]
- 41.Shou, W., Seol, J. H., Shevchenko, A., Baskerville, C., Moazed, D., Chen, Z. W., Jang, J., Charbonneau, H., and Deshaies, R. J. (1999) Cell 97233 –244 [DOI] [PubMed] [Google Scholar]
- 42.Bembenek, J., and Yu, H. (2001) J. Biol. Chem. 27648237 –48242 [DOI] [PubMed] [Google Scholar]
- 43.Kaiser, B. K., Zimmerman, Z. A., Charbonneau, H., and Jackson, P. K. (2002) Mol. Biol. Cell 132289 –2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lanzetti, L., Margaria, V., Melander, F., Virgili, L., Lee, M. H., Bartek, J., and Jensen, S. (2007) J. Biol. Chem. 28215258 –15270 [DOI] [PubMed] [Google Scholar]
- 45.Vazquez-Novelle, M. D., Esteban, V., Bueno, A., and Sacristan, M. P. (2005) J. Biol. Chem. 28029144 –29150 [DOI] [PubMed] [Google Scholar]
- 46.Paulsen, M. T., Starks, A. M., Derheimer, F. A., Hanasoge, S., Li, L., Dixon, J. E., and Ljungman, M. (2006) Mol. Cancer 525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu, Y., Kovacevic, Z., and Richardson, D. R. (2007) Cell Cycle 61982 –1994 [DOI] [PubMed] [Google Scholar]
- 48.Nigg, E. A. (2001) Nat. Rev. Mol. Cell Biol. 221 –32 [DOI] [PubMed] [Google Scholar]
- 49.Blethrow, J. D., Glavy, J. S., Morgan, D. O., and Shokat, K. M. (2008) Proc. Natl. Acad. Sci. U. S. A. 1051442 –1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li, L., Ernsting, B. R., Wishart, M. J., Lohse, D. L., and Dixon, J. E. (1997) J. Biol. Chem. 27229403 –29406 [DOI] [PubMed] [Google Scholar]
- 51.Cho, H. P., Liu, Y., Gomez, M., Dunlap, J., Tyers, M., and Wang, Y. (2005) Mol. Cell Biol. 254541 –4551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yuan, K., Hu, H., Guo, Z., Fu, G., Shaw, A. P., Hu, R., and Yao, X. (2007) J. Biol. Chem. 28227414 –27423 [DOI] [PubMed] [Google Scholar]
- 53.Bourdon, E., Kang, D. K., Ghosh, M. C., Drake, S. K., Wey, J., Levine, R. L., and Rouault, T. A. (2003) Blood Cells Mol. Dis. 31247 –255 [DOI] [PubMed] [Google Scholar]
- 54.Hanson, E. S., Rawlins, M. L., and Leibold, E. A. (2003) J. Biol. Chem. 27840337 –40342 [DOI] [PubMed] [Google Scholar]
- 55.Wang, J., Chen, G., Muckenthaler, M., Galy, B., Hentze, M. W., and Pantopoulos, K. (2004) Mol. Cell Biol. 24954 –965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gottesfeld, J. M., and Forbes, D. J. (1997) Trends Biochem. Sci. 22197 –202 [DOI] [PubMed] [Google Scholar]
- 57.Rieder, C. L., and Maiato, H. (2004) Dev. Cell 7637 –651 [DOI] [PubMed] [Google Scholar]
- 58.Warren, G., Davoust, J., and Cockcroft, A. (1984) EMBO J. 32217 –2225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schweitzer, J. K., Burke, E. E., Goodson, H. V., and D'Souza-Schorey, C. (2005) J. Biol. Chem. 28041628 –41635 [DOI] [PubMed] [Google Scholar]
- 60.Boucrot, E., and Kirchhausen, T. (2007) Proc. Natl. Acad. Sci. U. S. A. 1047939 –7944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Larrick, J. W., and Cresswell, P. (1979) J. Supramol. Struct. 11579 –586 [DOI] [PubMed] [Google Scholar]
- 62.Galbraith, R. M., and Galbraith, G. M. (1981) Immunology 44703 –710 [PMC free article] [PubMed] [Google Scholar]
- 63.Frazier, J. L., Caskey, J. H., Yoffe, M., and Seligman, P. A. (1982) J. Clin. Invest. 69853 –865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chitambar, C. R., Massey, E. J., and Seligman, P. A. (1983) J. Clin. Invest. 721314 –1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.O'Donnell, K. A., Yu, D., Zeller, K. I., Kim, J.-w., Racke, F., Thomas-Tikhonenko, A., and Dang, C. V. (2006) Mol. Cell Biol. 262373 –2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Le, N. T. V., and Richardson, D. R. (2002) Biochim. Biophys. Acta Rev. Cancer 160331 –46 [DOI] [PubMed] [Google Scholar]
- 67.Cozzi, A., Corsi, B., Levi, S., Santambrogio, P., Albertini, A., and Arosio, P. (2000) J. Biol. Chem. 27525122 –25129 [DOI] [PubMed] [Google Scholar]
- 68.Nie, G., Chen, G., Sheftel, A. D., Pantopoulos, K., and Ponka, P. (2006) Blood 1082428 –2434 [DOI] [PubMed] [Google Scholar]
- 69.Kakhlon, O., Gruenbaum, Y., and Cabantchik, Z. I. (2001) Blood 972863 –2871 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.