

Abstract
Lysyl oxidase (LOX) is a potent chemokine inducing the migration of varied cell types. Here we demonstrate that inhibition of LOX activity by β-aminopropionitrile (BAPN) in cultured rat aortic smooth muscle cells (SMCs) reduced the chemotactic response and sensitivity of these cells toward LOX and toward PDGF-BB. The chemotactic activity of PDGF-BB was significantly enhanced in the presence of a non-chemotactic concentration of LOX. We considered the possibility that extracellular LOX may oxidize cell surface proteins, including the PDGF receptor-β (PDGFR-β), to affect PDGF-BB-induced chemotaxis. Plasma membranes purified from control SMC contained oxidized PDGFR-β. The oxidation of this receptor and other membrane proteins was largely prevented in cells preincubated with BAPN. Addition of purified LOX to these cells restored the profile of oxidized proteins toward that of control cells. The high affinity and capacity for the binding of PDGF-BB by cells containing oxidized PDGFR-β was diminished by ∼2-fold when compared with cells in which oxidation by LOX was prevented by BAPN. Phosphorylated members of the PDGFR-β-dependent signal transduction pathway, including PDGFR-β, SHP2, AKT1, and ERK1/ERK2 (p44/42 MAPK), turned over faster in BAPN-treated than in control SMCs. LOX knock-out mouse embryonic fibroblasts mirrored the effect obtained with SMCs treated with BAPN. These novel findings suggest that LOX activity is essential to generate optimal chemotactic sensitivity of cells to chemoattractants by oxidizing specific cell surface proteins, such as PDGFR-β.
Lysyl oxidase
(LOX)3 catalyzes the
oxidation of specific lysine residues within extracellular elastin and
collagen thus generating residues of α-aminoadipic-δ-semialdehyde
within these proteins (1).
These peptidyl aldehydes can then undergo condensation with vicinal
α-aminoadipic-δ-semialdehyde or unmodified lysine residues to form
inter- and intrapeptide covalent cross-linkages that stabilize these fibrous
proteins. In addition to the non-ionic aldehyde product, the LOX-catalyzed
reaction acting on protonated lysine produces stoichiometric amounts of
hydrogen peroxide and ammonium, as shown in Reaction
1.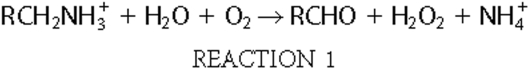
LOX is synthesized as an N-glycosylated preproprotein, which is secreted as the 50-kDa proprotein, proLOX. proLOX is activated by proteolysis to release the C-terminal moiety of the proprotein as the 32-kDa non-glycosylated functional catalyst (LOX) and the glycosylated propeptide in the extracellular space (2). Deletion of the LOX gene does not alter embryonic development of the knock-out mouse but is perinatal lethal due to major defects in cardiovascular tissue and the respiratory system (3, 4). In recent years, four lysyl oxidase-like proteins (LOXL), LOXL-1, -2, -3, and -4, have been identified in mammalian cells (5). LOXL1 activity has recently been implicated in elastin homeostasis (6). The spectrum of substrates and biological functions of LOXL-2, -3, and -4 remain to be established.
Basic research on LOX began with the demonstration of the oxidation of peptidyl lysine in an elastin substrate by a crude aortic extract (7). Subsequent investigations operated on the assumption that the function of this enzyme was restricted to its oxidation of its extracellular elastin and collagen substrates. Thus, it was of considerable interest that purified LOX was found to readily oxidize lysine within a variety of basic globular proteins (pI ≥ 8.0) in vitro, suggesting that its substrate specificity in vivo might be broader than initially thought (8). Nevertheless, the limited view of LOX function pertained until the striking observation was made that LOX acted as a suppressor of ras-induced carcinogenesis (9, 10). Subsequently, LOX has been implicated in a number of novel biological functions, including the regulation of the promoter activity of collagen type III (11), the control of cell adhesion and growth (12), the metastatic phenotype of certain tumors in adult animals (13, 14), and gene regulation (15). Of additional interest is the finding that the mature extracellular catalyst can be found within the nuclei of fibrogenic vascular smooth muscle cells and fibroblasts (16, 17).
We have previously reported that LOX purified from bovine aorta actively induces the migration of peripheral blood monocytes (18) and vascular smooth muscle cells (SMCs) (19). Moreover, this chemotactic activity of LOX was fully inhibitable by β-aminopropionitrile (BAPN), which inactivates catalysis by LOX, or by the presence of catalase in the chemotactic chamber assays (19). The conclusions were drawn, therefore, that the enzymatic activity of LOX and the hydrogen peroxide product of its enzyme activity underlie the chemotactic response. Consistent with the principle that chemokines initiate cell migration by binding to cell membrane receptors, these studies also noted that the chemotactic response of SMCs to LOX did not reflect the interaction of this enzyme with soluble forms of its extracellular substrates secreted into the medium, but more likely required the direct interaction of this enzyme with the cell membrane (19). The chemotactic response of vascular SMCs to LOX may prove to be relevant to certain pathologies. For example, normally quiescent vascular SMCs actively migrate from the arterial media to developing atherosclerotic lesions in the sub-endothelial region of diseased arteries, which, in turn, may relate to the observation that total LOX enzyme activity is significantly increased in the arterial lesions of atherosclerotic rabbits (20). A more tangible relationship between LOX-induced migration and pathology has recently been demonstrated. Kirschmann and colleagues (13) have reported that the malignant invasion of breast cancer cells is induced by LOX and noted that this chemotactic response is inhibited by BAPN and by the presence of catalase, supporting the catalysis- and peroxide-dependent mechanism of LOX-induced chemotaxis reported earlier. LOX has also been implicated in the migration of malignant dermal cells in squamous cell carcinoma (21) and in the directed movement of neurons (22).
Despite the number of well documented cellular and biological processes dependent on LOX activity, the relevant molecular mechanisms, including the identification of cell protein substrates that might be oxidized by LOX in these instances, has yet to be accomplished. We have approached this issue in part by studying the role of LOX in chemotaxis, a basic cellular response underlying several of the biological functions in which LOX is involved. Specifically, we have focused on the effect of LOX activity on chemotaxis induced by LOX and by PDGF-BB in rat aorta SMCs. The results reveal that oxidation of specific plasma membrane proteins by LOX is required for optimal chemotactic responses of SMCs. This newly documented priming effect of LOX on chemotaxis is clearly distinguishable from the acute chemotactic effect of this enzyme previously described (19).
EXPERIMENTAL PROCEDURES
Materials—Rat recombinant, protein carrier-free PDGF-BB (R&D Systems), 125I-Monoiodo Bolton-Hunter reagent (Amersham Biosciences Corporation), rabbit anti-dinitrophenol antibody, fluorescein isothiocyanate-conjugated goat anti-rabbit antibody, and rabbit anti-actin antibody (Invitrogen), oxy-blot protein oxidation detection kit (Chemicon International), rabbit polyclonal anti-PDGFR-β (Santa Cruz Biotechnology Inc.), rabbit polyclonal antibodies against prosphoproteins of the PDGFR-β signal transduction pathway described in the text (Cell Signaling Technology), 30% hydrogen peroxide, glucose oxidase, and catalase (Sigma-Aldrich), and OptiPrep (Axis-Shield) were purchased from the indicated companies. The sources of other reagents are indicated in the text.
Cell Culture Preparation—Neonatal rat aorta SMCs were isolated and expanded as described (23). Cells were seeded at a density of 7.5 × 104 cell/ml in 2 ml of medium per well of a 6-well plate. Freshly isolated cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% bovine calf serum, 1 mm sodium pyruvate (Invitrogen), 0.1 mm non-essential amino acids (Invitrogen), and 1% penicillin/streptomycin (Invitrogen). Cell counting was carried out by hemocytometry in triplicate cultures using the trypan blue exclusion method. Wild-type and lox-/- embryos from heterozygous crosses were used to derive mouse embryonic fibroblasts (MEFs). E14.5 embryos were dissociated with sterile scissors, trypsinized, and transferred to 60-mm tissue culture dishes. After 2-3 days, cells were trypsinized and expanded as described (4). Cells of the embryo rat aorta SMC line A7r5 (American Type Culture Collection) were cultured and expanded as described above for SMCs. Lysyl oxidase was purified from bovine aorta (24).
Chemotaxis Assays—SMCs, MEFs, and A7r5 cells were grown from seeding to 80-90% confluence in the absence (control) or in the presence of 100 μm BAPN. The cell layers were rinsed three times with Hanks' balanced salt solution. In some experiments, cells were further incubated with pure LOX (5 units/ml) for 30 min in a medium designed for optimal LOX activity and cell viability containing 5.5 mm glucose, 4.2 mm NaHCO3, 0.1 mm CaCl2, 0.8 mm MgSO4 in PBS (pH 7.8). SMCs and A7r5 were starved for 24 h and MEFs for 6 h in Dulbecco's modified Eagle's medium serum-free medium containing 0.2% BSA with or without 100 μm BAPN, and washed three times in Hanks' balanced salt solution. Cell layers were then harvested by incubation at 37 °C for 15 min in a medium containing 2 mm EDTA/Hanks' balanced salt solution, 25 mm HEPES (pH 7.4), and 0.05% TrypZean, a recombinant bovine trypsin (Sigma-Aldrich), and then collected by centrifugation at 1500 rpm for 5 min. This harvesting procedure did not result in the degradation of PDGFR-β as judged by the expected apparent molecular masses of 180-185 kDa determined by Western blots using specific antibody (not shown). Sedimented cells were resuspended in an appropriate volume of quenching medium composed of serum-free Dulbecco's modified Eagle's medium, 5% BSA, 2 mm CaCl2, and 2 mm MgCl2, counted using Trypan blue to assess viability (>90%), and diluted to 1.0 × 106 cells/ml in serum-free Dulbecco's modified Eagle's medium containing 0.2% BSA. Chemotaxis assays, performed in 96-well, 8-μm MultiScreen MIC plates (Millipore), were initiated immediately after cells were harvested, counted, and diluted. Plates and reagents were brought to room temperature (25 °C). Serum-free medium (150 μl) containing or lacking the designated chemoattractants was added to the wells of the feeder tray (lower plate). The cell migration plate (upper plate) was placed on the feeder tray, and 100 μl of serum-free medium containing 5 × 104 cells was added to each well. The mounted plate was covered and incubated for 1-3 h at 37 °C in a 5% CO2 incubator. The cells and media from the migration plate were gently removed and discarded, and the migration plate was rinsed once with Hanks' balanced salt solution and placed onto a new feeder tray containing 150 μl of the cell detachment solution containing 0.25% TrypZean, 1 mm EDTA in Dubecco's PBS without Ca2+ or Mg2+, and incubated for 30 min at 37 °C. During this time, the migration plate was gently lifted from and then reassembled onto the feeder tray to allow complete dislodging of the cells from the underside of the membrane. Finally, 100 μl taken from each well of the feeder tray was transferred to the corresponding wells of a new 96-well plate. Cell content in the wells was determined in a Tecan Infinite M200 microplate reader using the CyQuant NF cell proliferation kit assay (Invitrogen).
Oxy-immunofluorescence Microscopy—Cells were grown in the wells of the Nunc Lab-Tek II CC2 Chamber Slide System to ∼80% confluence under the conditions as described herein, including the starvation in serum-free medium and washing steps. Cells were fixed in PBS containing 3% formaldehyde for 20 min at room temperature, washed three times with PBS, incubated for 10 min at room temperature in PBS containing 10 mm ammonium chloride to quench the excess of formaldehyde, and then washed twice with PBS. Carbonyl residues generated by the oxidative deamination of lysyl residues in cell surface proteins of cell monolayers were covalently derivatized with 1 ml/well of 1.5 mm dinitrophenylhydrazine (DNPH) in 0.5 m trifluoroacetic acid for 15 min at room temperature with gentle shaking. Cells not derivatized with DNPH were incubated in 0.5 m trifluoroacetic acid for 15 min at room temperature in the absence of DNPH. The DNPH solution was aspirated and 0.5 m Tris base (1 ml/well) was added to neutralize the residual trifluoroacetic acid. The fact that neither anti-core histone antibody nor rhodamine-phalloidin were able to bind to their target proteins when fixed cells were derivatized but not permeabilized (not shown) indicated that the acidic treatment of the fixed cell monolayer, under the conditions described above, did not result in cell permeabilization. Wells were rinsed two times with PBS, incubated for 2 h at room temperature in blocking buffer (PBS/3% BSA). The resulting dinitrophenyl adducts were reacted with anti-dinitrophenol antibodies (anti-DNP) in PBS/3% BSA buffer for 2 h at 4 °C. Wells were rinsed three times for 5 min each with PBS and then incubated with a 1/500 dilution of fluorescein isothiocyanate-labeled secondary antibody in PBS/3% BSA buffer for 1 h at 4 °C. Fluorescent images were captured in a Nikon Eclipse E400 equipped with ocular lens CFIUW 10×/25 and objective lenses 40×/0.65 Ph2 DL and 20×/0.40 Ph1 DL, using a Spot RT KE digital camera (Diagnostic Instruments, Inc.) driven by the Spot imaging software V 4.6.
Plasma Membrane Purification—Cultured cells were grown in 150 mm plates, rinsed and then harvested by gently scraping each plate in 5 ml of lysis buffer (10 mm NaCl, 20 mm Tris-HCl, pH 7.5), containing 1 μl/ml medium of protease inhibitor mixture for use with mammalian cell extracts (Sigma) and 0.5 mm phenylmethylsulfonyl fluoride (PMSF). The resulting cell suspension was left on ice for 15 min to allow swelling and then disrupted by 20 strokes in a 12 ml Dounce homogenizer. The homogenate was subjected to zonal rate centrifugation twice for 5 min at 700 × g to sediment the nuclear fraction and cell debris. The post-nuclear supernatant (S700 × g) was either concentrated by ultracentrifugation at 150,000 × g for 1 h or loaded directly onto a 6-40% OptiPrep (Iodixanol) gradient. When S700 × g was concentrated by ultracentrifugation, the supernatant (S150K × g) consisted of the cytoplasmic fraction while the pellet (P150K × g) was the total membrane fraction of the cells depleted of nuclear membranes. This P150K × g was then gently resuspended in 500 μl of lysis buffer and loaded onto a 6-40% OptiPrep gradient composed of 150 mm NaCl, Tricine-NaOH (pH 7.4), and 1 mm EDTA. A linear gradient was formed by overlaying 18 fractions (211 μl) of 40.0%, 38.0%, 36.0%, 34.0%, 32.0%, 30.0%, 28.0%, 26.0%, 24.0%, 22.0%, 20.0%, 18.0%, 16.0%, 14.0%, 12.0%, 10.0%, 8.0%, and 6.0% in a 4.3-ml tube of the SW 60Ti rotor. Equilibrium sedimentation of the sub-cellular membrane fraction P150K × g was achieved by centrifugation at 200,000 × g for 130 min at 4 °C. Twenty fractions (215 μl) from the top of the tube were collected in new tubes. The refractive index of each fraction (50 μl) was determined using a digital display refractometer, and the corresponding density, ρ, was calculated from ρ = (η × A) - B, where η is the refractive index and the constant values used, A = 3.344 and B = 3.458, are for an OptiPrep gradient in NaCl/Tricine solution. To identify the actual sedimentation density of the PM fraction in the gradient, SMC surface proteins were biotinylated as follows: rinsed cell monolayers in 150-mm plates were incubated in 5 ml of biotinylation buffer (1 mg/ml biotin (Pierce), 10 mm triethanolamine (pH 8.3), and 150 mm NaCl), and incubated for 25-30 min at 4 °C to prevent endocytosis. Plates were rinsed three times with PBS, and the excess biotin was quenched by incubation in 5 ml of quenching buffer (100 mm glycine (pH 8.3) in PBS) for an additional 15 min at 4 °C. Surface biotinylated cells were then subjected to lysis, zonal rate centrifugation, and equilibrium sedimentation in an OptiPrep gradient as described above. Western blot analysis using horseradish peroxidase-conjugated streptavidin (Invitrogen) to probe gradient fractions revealed that plasma membranes sedimented at a density of 1.065 ± 0.021 g/ml corresponding to fractions 6 (8% Opti-Prep) to 10 (16% OptiPrep) withdrawn from the top of the tube. Alternatively, plasma membranes were recovered after equilibrium sedimentation in the 6-18% interface of a stepwise gradient consisting of 6%, 18%, and 30% OptiPrep layers. Ten 150-mm plates were processed to obtain 100-150 μg of purified plasma membrane, free of antigenic signals toward anti-Sec61β (ER marker), anti-syntaxin 6 (Golgi marker), and anti-histone H1 (nucleus marker) (not shown). Detection of oxidized proteins in solution was done using an oxy-blot protein oxidation detection kit (Chemicon International) following the manufacturer's instructions.
Enzyme Assays and Immunoprecipitation—Lysyl oxidase and glucose oxidase activities were determined fluorometrically by monitoring the production of H2O2 through the oxidation of Amplex Red (Invitrogen) as described for lysyl oxidase activity (25) with modification. The substrate used for lysyl oxidase was a synthetic lysine:tyrosine (4:1) heteropolymer present at 5 μg/ml in the assay mixture. Glucose oxidase activity was measured in PBS containing 5 mm glucose. One unit of activity of either enzyme is the amount of enzyme required to produce 50 pmol of H2O2/min. The kinetics of fluorescence change was monitored using a TECAN Infinite M200 microplate reader. PDGFR-β was immunoprecipitated with a rabbit polyclonal antibody against rat PDGFR-β (Santa Cruz Biotechnology) using a protein A/G-agarose immunoprecipitation kit (KPL) following the manufacturer's instructions.
Iodination of PDGF-BB and Binding of 125I-PDGF-BB to Cell Monolayers—PDGF-BB was iodinated using the Bolton-Hunter reagent (26). Briefly, 50 μg of freeze-dried, recombinant rat, carrier-free PDGF-BB (R&D Systems) was resuspended in 50 μl of 100 mm sodium borate, pH 8.5, and added to 125I-Monoiodo Bolton-Hunter reagent (1000 μCi) that had been dried under nitrogen and incubated for 15 min at 4 °C. 200 μl of quenching solution (0.1 m sodium borate, pH 8.5, and 0.2 m glycine) was added and the mixture was incubated for an additional 10 min at 4 °C followed by the addition of 50 μl of 100 mm sodium borate containing 5 mg/ml BSA. Two 5-μl aliquots of the mixture were used to assess the efficiency of iodination, and the remaining 295 μl was loaded onto a D-Salt polyacrylamide desalting column (6-kDa cut-off, Pierce) that had been equilibrated in 100 mm acetic acid containing 1 mg/ml BSA. Fractions of 500 μl were collected and 125I-PDGF-BB eluted in the first peak of radioactivity (fractions 4th to 6th) with a specific radioactivity of 25,000 cpm/ng. Binding of 125I-PDGF-BB was measured in monolayers of cells grown in 24-well cluster culture plates. Cells were seeded on the plates at a density of 5 × 104 cells/well, grown to near confluence, starved in serum-free medium for 48 h, washed three times with cold binding buffer (25 mm Hepes, pH 7.4, 125 mm NaCl, 5 mm MgSO4, 5 mm KCl, and 1 mm CaCl2, containing 2 mg/ml BSA) and incubated in 1 ml of binding buffer for 30 min at 4 °C. The binding buffer was aspirated, and 1 ml of fresh binding buffer containing the indicated concentrations of 125I-PDGF-BB at a specific radioactivity of 88,000 cpm/ng was added and further incubated at 4 °C for 4 h. After washing four times with 1 ml of binding buffer at 4 °C, cells were solubilized with 1 ml of 1% Triton X-100, 10% glycerol, 25 mm Hepes, pH 7.5, 1 mg/ml BSA. Radioactivity was measured in a gamma counter. Nonspecific binding was determined in the presence of unlabeled PDGF-BB at 500 ng/ml. Specific binding was obtained by subtracting the nonspecific binding from the total binding. The dissociation constant (Kd) and the maximum binding capacity (Bmax) for 125I-PDGF-BB were determined by least-squares, non-linear regression fitting of the hyperbolic function for ligand binding to one site saturation. Cell numbers were determined in triplicate parallel cultures by trypsinization and counting the detached cells in a hemocytometer.
PDGF-BB Signal Transduction Pathway—Activation of SHP2, Akt, and ERK1/ERK2 (p44/42 MAPK), assessed as downstream components of PDGFR-β in response to PDGF-BB binding, was assayed in cell monolayers by monitoring their phosphorylated state using specific anti-phosphoprotein antibodies. Cells grown on 6-well plates were incubated for 24 h (SMC and A7r5) or for 6 h (MEF) in serum-free medium and then stimulated with PDGF-BB (25 ng/ml) for the indicated times at 37 °C in a CO2 incubator. The reaction was stopped by aspirating the media, and cells were immediately lysed with 200 μl/well extraction buffer containing 20 mm Tris, pH 7.5, 5 mm EGTA, 150 mm NaCl, 1% Nonidet P-40, 0.1 mm Na3VO4, 1 mm NaF, 10 mm sodium β-glycerophosphate, 0.5 mm phenylmethylsulfonyl fluoride, 1 μl/ml protease inhibitor mixture (Sigma). Cell extracts were clarified by centrifugation at 10,000 × g for 10 min at 4 °C, and the supernatants were saved. Protein concentrations were determined by the bicinchoninic acid method (Pierce) using BSA as the standard. To assess phosphorylation of SHP2, Akt, and ERK1/ERK2 (p44/42 MAPK) and PDGFR-β, total protein extracts (30 μg/lane) were separated by 4.0-20.0% gradient SDS-PAGE. Proteins were transferred to polyvinylidene difluoride membranes in 25 mm Tris, 192 mm glycine, 20% methanol using a Mini Trans-Blot blotter (Bio-Rad) at 30 V overnight and under continuous cooling at 4 °C. Blots were blocked in PBS buffer containing 0.1% Tween 20 and 5% w/v nonfat dry milk for 1 h and then incubated overnight with gentle agitation at 4 °C with a mixture of the anti-phosphoprotein antibodies, anti-P-SHP2, anti-P-Akt, anti-P-ERK1/ERK2(p44/42 MAPK), and anti-P-PDGFR-β, each diluted 1/1000 in PBS buffer containing 0.1% Tween 20 and 5% w/v BSA. Membranes were washed three times for 5 min each with PBS/T buffer and incubated for 1 h with horseradish peroxidase-conjugated, goat anti-rabbit-IgG antibody (1:2000) in PBS buffer containing 0.1% Tween 20 and 5% w/v BSA. After washing four times for 10 min each with PBS/T, membranes were developed with a chemiluminescence substrate for detection of horseradish peroxidase (Pierce), and images were captured in a Chemi Doc XRS imager (Bio-Rad) using the Quantity One v 4.6 imaging software.
Real-time PCR—Total RNA was extracted from the cells in 4 m guanidinium thiocyanate as described previously (27). Genomic DNA was removed by incubation with RNase-free DNase I (New England BioLabs, M0303S) in the presence of RNase inhibitor. The RNA was annealed with random hexamer primers, and first strand synthesis was carried out with MuLV reverse transcriptase. Negative controls were performed without reverse transcriptase. Real-time PCR was done on an Applied Biosystems 7300 Real-Time PCR system using ABI TaqMan gene expression assays for rat elastin (RN01499782_m1), rat lysyl oxidase (RN00566984_m1), and the eukaryotic 18 S rRNA endogenous control (4308329). The cycling parameters were 50 °C for 10 min, 95 °C for 2 min, 40 cycles of 95 °C for 15 s, and 60 °C for 1 min. Results were calculated by applying the ΔΔCT method, using 18 S rRNA as the endogenous control.
Analysis of LOX Secretion—Secreted LOX protein and activity were analyzed in samples (2 ml) of conditioned medium withdrawn from SMC cultures at different time points. Samples were concentrated 10-fold by ultrafiltration at 4 °C in Centricon Ultracel, a 10-kDa molecular mass cut-off device (Millipore, Amicon Ultra). LOX activity was assayed as described above using lysine:tyrosine (4:1) heteropolymer as a substrate. Secreted LOX protein was visualized by Western blots using the antipeptide antibody against the sequence 293DEFSHYDLLDASTQRR308 in rat mature LOX.
RESULTS
Chemotaxis Is Decreased by Inhibition of LOX in SMC—It has been previously shown that LOX promotes chemotactic migration in monocytes (18), vascular SMCs (19), and fibroblasts (28). It has also been documented that the production of hydrogen peroxide accompanying the oxidation of unknown substrate(s) by LOX appeared to be essential for the chemotactic response of cells to this enzyme (19). Preincubation of SMCs in culture with BAPN, an irreversible inhibitor of lysyl oxidase activity, does not significantly alter cell morphology, attachment, or growth (data not shown) whereas it clearly inhibits LOX catalytic activity by acting as a suicide substrate (29). However, when preincubated with BAPN for 5 days in culture followed by removal of BAPN by thorough washing of the cell layers with PBS, the chemotactic response of these cells to LOX (Fig. 1, bar 9 versus bar 10) or to PDGF (Fig. 1, bar 3 versus bar 4), an active chemokine for vascular SMCs (30), was significantly reduced. As previously reported (19), the chemotactic migration of these cells to LOX was prevented if 100 μm BAPN was added to the feeder tray together with LOX (bar 9 versus bar 11, Fig. 1). In contrast, PDGF-induced chemotaxis in SMC was insensitive to the presence of 100 μm BAPN in the feeder tray during the actual migration assay (Fig. 1, bar 3 versus bar 5).
FIGURE 1.

Chemotactic response of SMC and MEF. Rat aorta SMCs (SMC) grown in the absence (control cells) or in the presence of 100 μ m BAPN (BAPN-treated cells) were subjected to chemotaxis analysis as described under “Experimental Procedures.” The following compounds were tested as chemoattractants: PDGF-BB (20 ng/ml), glucose oxidase (2 units; GO), LOX (2 units), and H2O2 (12 μìM). i The inhibitory effect on LOX-induced chemotaxis by BAPN and catalase (2 units, Cat), added with LOX in the feeder tray (bar 9 versus bars 11 and 13) confirmed the requirement for LOX activity in this process (19). RFU stands for relative fluorescent units. Compounds applied to the feeder tray are indicated in the abscissa. The error bars are standard deviations of the mean values obtained from three independent experiments (n = 12).
The induction of chemotaxis by a variety of chemokines such as PDGF requires interaction of the chemokine with its receptor at the cell surface leading to activation of chemotaxis-promoting signal transduction pathways (31). The molecular basis of LOX-dependent chemotaxis is not known other than the demonstrated requirement for its catalytic activity and the consequent production of its hydrogen peroxide product (19). If hydrogen peroxide produced by LOX acting on endogenous substrates was necessary and sufficient to induce chemotaxis, it would be expected that the production of hydrogen peroxide by another enzyme, such as glucose oxidase (32), would elicit a similar chemotactic effect. Addition of 2 units (i.e. 100 pmol of H2O2/min) of glucose oxidase in the feeder tray did not induce a significant chemotactic response of SMCs (Fig. 1, bar 7 versus bar 1) as did occur in the presence of 2 units of LOX (Fig. 1, bar 9 versus bar 1). Moreover, the addition to the chemotaxis chamber of a calibrated quantity of hydrogen peroxide, equivalent to that expected to result from the optimal chemotactically active amount of LOX, induced minimal cell motility in the absence of added LOX (Fig. 1, bar 15 versus bar 1). Higher concentrations of hydrogen peroxide did not stimulate but did inhibit the chemotactic response (not shown). Thus, the production of hydrogen peroxide by LOX is not sufficient to elicit the full chemotactic response of cells to LOX. The A7r5 immortalized rat vascular SMC line displayed a similar chemotactic response to the conditions and effectors shown for the neonatal rat SMC seen in Fig. 1 (not shown). Taken together, these results point to the conclusion that the chemotactic response to exogenous LOX, or to PDGF, was inhibited if the enzyme activity of endogenous LOX was inhibited by the presence of BAPN during culturing of the cells prior to the chemotaxis assays. Thus, our working hypothesis is that the oxidation of specific cell proteins by endogenous LOX “primes” the cell for optimum chemotactic responses to exogenous LOX or to other chemotactic agents, the latter is illustrated here by the use of PDGF.
The Magnitude and Sensitivity of SMC Chemotaxis Are Modulated by the Catalytic Activity of LOX—To further characterize this LOX-dependent effect, we compared the concentration dependence of the chemotactic responses of SMC to LOX and to PDGF-BB. As shown (Fig. 2), LOX (Fig. 2A), and PDGF-BB (Fig. 2B) each induced chemotaxis in these cells at optimal concentrations of 20 nm (LOX) and 10 ng/ml (PDGF-BB). In contrast, the maximal responses in cells pretreated with BAPN occurred at 62 nm LOX and 30 ng/ml PDGF-BB. Moreover, the maximal chemotactic response in BAPN-pretreated cells was reduced when compared with the cells, which had not been pretreated with BAPN. The effect was prominent in the case of PDGF-BB where a 44% reduction was seen at the peaks of these responses (Fig. 2B). Thus, it appears that inhibition of LOX activity during culture of these cells decreases their sensitivity and maximum chemotactic response to LOX and to PDGF-BB, further suggesting a “priming” role for endogenous LOX in the development of chemotactic responses. Consistent with this possibility, the chemotactic response to PDGF-BB is significantly increased (1.7-fold) when determined in the presence of a concentration of added LOX (2.5 nm), which is insufficient to induce chemotaxis when present as the only added chemokine (Fig. 2C).
FIGURE 2.

Chemotactic response of SMC is controlled by catalytically functional lysyl oxidase. Chemotaxis driven by LOX (A), PDGF-BB (B), and PDGF-BB in the presence of 2.5 nm LOX (C) in control (filled circles) and BAPN-treated SMC (empty circles) were assayed as in Fig. 1, using compounds in the range of concentrations indicated in the plots. Error bars represent the standard deviations from the means derived from three independent experiments (n = 12).
Oxidative States of Cell Surface Proteins—Clearly, the early events in chemotaxis occur at the cell surface. The inhibition of chemotaxis by the inhibition of LOX activity suggested that the oxidized state of cell surface proteins might contribute importantly to chemokine effectiveness. In preparation for the testing of this possibility, we developed a procedure to visualize the oxidative state of cell surface proteins in SMC. Thus, monolayers of SMC were grown on microscope slide chambers, rinsed with buffer, fixed with formaldehyde, reacted with DNPH in acidic medium, and then sequentially incubated with anti-DNP, and fluorescently labeled second antibody. Control (Fig. 3A) and BAPN-treated (Fig. 3B) SMC that were not derivatized with DNPH showed weak background fluorescence likely due to minimal cross-reactivity or nonspecific absorption of anti-DNP antibody on cell surface proteins. Derivatization of cells with DNPH strongly increased the fluorescent signal in control cells (Fig. 3C) but not in BAPN-treated cells (Fig. 3D). These results are consistent with the presence of oxidized, protein-linked carbonyl residues at the cell surface. Appropriate controls indicated that the process of fixation and derivatization of the cell monolayers did not result in permeabilization of the cell membrane toward the antibodies used for detection (see “Experimental Procedures”). It is important to note possible limitations of this oxy-immunofluorescence method. Specifically, it does not resolve oxidized plasma membrane proteins from oxidized extracellular matrix proteins, nor does it discriminate between integral and peripherally associated plasma membrane proteins. It is also possible that some intracellular proteins may react to give a signal, overlapping the signal from cell surface proteins. This could be particularly variable depending on the cell type under investigation. Moreover, the dynamic range for the signal intensity response to the oxidized state of cell surface proteins has not yet been established. In view of these possibilities, we further investigated the oxidized state of proteins that remain associated with the plasma membrane after its purification following a standard biochemical procedure.
FIGURE 3.

Oxidized state of cell surface proteins. Oxidized cell surface proteins in SMC were visualized after derivatization with DNPH using anti-DNP antibody and fluorescein isothiocyanate-labeled secondary antibody (green) employing the oxy-immunofluorescent microscopy method described under “Experimental Procedures.” Non-derivatized control (A) and BAPN-treated (B) cell monolayers. Derivatized control (C) and BAPN-treated (D) cell monolayers. The green fluorescence images produced by fluorescein isothiocyanate-conjugated secondary antibody (left side) and the blue signal images from 4′,6-diamidino-2-phenylindole staining of the cell nuclei (right side) of the same field were merged (center). The merged images indicate the lack of detection of oxidized proteins in nuclei, as expected for the non-permeabilized cells used in this method. Scale bars are at the right-hand corners. Images are representative of 15 fields observed by scanning the entire slide area.
PDGF Receptor-β Undergoes LOX-dependent Oxidation in SMC Plasma Membranes—Plasma membranes from control and BAPN-treated SMC were purified, and the oxidized state of the membrane-associated proteins was analyzed by the oxy-Western blot procedure. Several oxidized proteins with molecular masses of 190 kDa, 100-120 kDa, 74 kDa, 61 kDa, and 44 kDa were detected in the plasma membrane extract from control cells (Fig. 4A, lane 1). The oxidized state of these proteins was significantly reduced in BAPN-treated cells (Fig. 4A, lane 2), consistent with the conclusion that a peptidyl lysyl oxidase was the target of BAPN. Plasma membrane purified from cells that had been pretreated with BAPN and then incubated in culture with purified LOX (5 units/ml for 30 min) after BAPN was thoroughly eliminated, largely regained the pattern of oxidized proteins observed in control cells (Fig. 4A, lane 3). Cell monolayers subjected to a similar acute treatment with LOX recovered 60% (19,477 ± 1,132 relative fluorescent units) of the chemotactic response to PDGF-BB observed in control cells (32,415 ± 1,308 relative fluorescent units). Thus, BAPN inhibits the oxidation of membrane proteins and chemotaxis during cell culture, but after its removal and subsequent incubation of the cells with exogenous LOX, the oxidation of membrane proteins, and a partial chemotactic response, are regained because the prior oxidation of these proteins by endogenous LOX, and the chemotactic response of the cells to PDGF-BB (Fig. 1, lanes 3 and 4), were prevented by BAPN. Moreover, the result in Fig. 4A demonstrates that the oxidative state of cell surface proteins is preserved throughout isolation and purification of the cell membranes described above. Thus, carbonyl groups generated on lysine side chains either by endogenous or exogenous LOX appear to be stable. An additional oxidized band of 32 kDa corresponding to LOX, identified by anti-LOX antibodies, was detected in this sample (not shown). This could reflect the presence of the LTQ cofactor of LOX, which bears two carbonyl groups (33), and, possibly, oxidized lysine residues within the enzyme. The presence of LOX in the purified membrane fraction points toward the association of LOX with the plasma membrane that is sufficiently stable to have survived the isolation procedure possibly reflecting an enzyme-protein substrate complex. An additional oxidized cell surface protein was identified as the PDGF receptor-β by reaction with specific antibody (Fig. 4B, lane 1). The oxidation of this receptor is significantly reduced in BAPN-treated cells (Fig. 4B, lane 2). In vitro LOX activity assays revealed that 2 units of purified bovine aorta LOX readily oxidized plasma membrane proteins (10 μg) at a rate of 29.5 pmol of H2O2/min but did not oxidize cytoplasmic proteins (10 μg) or nuclei-free, sub-cellular membrane proteins (10 μg) further supporting the conclusion that proteins at the plasma membrane are oxidizable substrates of LOX.
FIGURE 4.

Oxidation of PDFGR-β is largely prevented by inhibition of LOX. A, plasma membranes from SMCs that were subjected to different treatments were purified, resolved by electrophoresis in 13.5% SDS-polyacrylamide gel electrophoresis, and analyzed by the oxy-Western blot as described under “Experimental Procedures.” Purified plasma membranes (30 μg) from control cells (lane 1), from BAPN-treated cells (lane 2), and from BAPN-treated cells subsequently incubated with pure LOX (5 units/ml culture medium) (lane 3). B, purified plasma membranes (50 μg) from control (lane 1) and from BAPN-treated cells (lane 2) were dispersed in radioimmune precipitation assay buffer and immunoprecipitated with antibody against PDFGR-β. Each immune pellet was derivatized with DNPH and subjected to oxy-blot analysis. Blots are representative of the results from three independent experiments.
Inhibition of LOX Activity Influences Binding Affinity of PDGF Receptor-β for PDGF-BB—The reduction in the chemotactic response of SMC to PDGF in cells pretreated with BAPN (Fig. 2B) could be related to BAPN-inhibitable changes in the binding properties of the PDGF-BB receptor. This possibility was assessed by measuring key parameters for the binding of PDGF-BB to monolayers of SMC and A7r5 cells. Indeed, the magnitudes of the binding dissociation constants and total binding values for PDGF-BB in control SMCs (Kd = 82.1 ± 16.2 pm; Bmax = 36,440 ± 756 molecules/cell) decreased in cells pretreated with BAPN (Kd = 137.4 ± 23.0 pm; Bmax = 23,428 ± 671 molecules/cell; Fig. 5A). The PDGF-BB binding parameters in A7r5 cells were similarly altered by BAPN pretreatment. The affinity for PDGF-BB and total binding properties of the control A7R5 cells (Kd = 63.8 ± 11.7 pm; Bmax = 37,134 ± 689 molecules/cell) decreased in the BAPN-pretreated cells (Kd = 156.6 ± 15.2 pm; Bmax = 32,022 ± 519), as shown in Fig. 5B. These results provide additional evidence indicating that the expression of LOX activity during cell culture results in the optimal binding affinity and maximal binding capacity of PDGFR-β.
FIGURE 5.

Binding parameters of PDGF-BB to SMC and A7r5 cells. Binding of PDGF-BB to SMC and A7r5 cells in culture was done using 125I-PDGF-BB as described under “Experimental Procedures.” The binding of PDGF-BB was measured over a wide concentration range of the ligand (10-3500 pm) in control (filled circles) and BAPN-treated (open circles) cells. Bmax and Kd values are shown under the corresponding plot. Error bars are the standard deviations of the mean values of eight determinations (n = 8).
LOX Activity Decreases the Turnover of the PDGF Receptor-β Signal Transduction Pathway—In view of the effects seen on the PDGF receptor, the functional states of components of the signal transduction pathway activated by this receptor in response to pretreatment of the cells with BAPN were investigated. PDGFR-β tyrosine kinase is activated upon binding of PDGF-BB, generating several autophosphorylated, phosphotyrosine residues that act as docking sites for a number of proteins containing a Src homology 2 domain, including Src, phospholipase C-1, phosphatidylinositol 3′-kinase, Shp2 protein-tyrosine phosphatase, and several other proteins critically involved in signal transduction pathways controlling cell division, growth, and chemotaxis (34). Because Shp2 (35), Akt (36, 37), and ERK1 and ERK2 (p44/42 MAPK) (38) become phosphorylated in response to PDGFR-β activation, the engagement of these proteins in the PDGF signal transduction pathway can be monitored by immunoblots using specific anti-phosphoprotein antibodies. PDGFR-β, Shp2, Akt, and ERK1/ERK2 (p44/42 MAPK) become phosphorylated within 5 min of the addition of PDGF-BB to an SMC monolayer (Fig. 6A). In control cells, the maximal phosphorylation of PDGFR-β seen at 15 min is followed by a slow decay within 90 min. The decay of phosphorylated PDGFR-β is more abrupt in cells pretreated with BAPN with complete loss of signal beyond 15 min. A similar pattern was observed for the phosphorylation of Shp2, which is relatively stable throughout the time course in control cells but decays significantly in BAPN-treated cells after 30 min. The phosphorylated state of Akt and ERK1/ERK2 (p44/42 MAPK) also decayed faster in BAPN-treated cells than in control cells (Fig. 6A).
FIGURE 6.

PDFGR-β signal transduction pathway is sensitive to LOX activity. Analysis of the PDFGR-β signal transduction pathway by monitoring the phosphorylated state of several intermediates was done as described under “Experimental Procedures.” A, Western blot of total SMC extract, at different times after the addition of PDGF-BB, probed with the anti-phosphoprotein antibodies against the proteins indicated on the right-hand side column. B, is a membrane from experiment A that was re-probed with antibodies against PDGFR-β. The graphs on the right side show the densitometry plots of the Western blot bands.
The actual levels of PDGFR-β in SMC were assessed as a function of the time after addition of PDGF-BB. The detected quantity of PDGFR-β was reduced by 44% within 30 min in control SMC, whereas a reduction of 70% was detected in BAPN-treated cells at 30 min (Fig. 6B). These results further support the correlation between LOX activity and the endurance of PDGFR-β after binding of PDGF-BB. The enhanced rate of decrease in the steady-state levels of PDGFR-β likely reflects a higher rate of protein turnover resulting from endocytosis and subsequent lysosomal degradation.
Chemotaxis Is Decreased by Inactivation of LOX Gene—To address the concern that BAPN may have effects other than inhibiting LOX activity, the chemotactic response of wild type (lox+/+) and LOX knock-out (lox-/-) mouse embryonic fibroblasts (MEFs) toward LOX and PDGF-BB were compared in control MEF and in MEF that were pretreated with BAPN or with purified LOX. LOX-induced chemotaxis in lox+/+ MEF (Fig. 7, bar 3 versus bar 1) was reduced by ∼62.5% in lox-/- MEF (Fig. 7, bar 5 versus bar 2). BAPN treatment inhibited LOX-induced chemotaxis by 81.0% in lox+/+ MEF (Fig. 7, bar 3 versus bar 4) but had no effect on lox-/- MEF (Fig. 7, bar 5 versus bar 6). Treatment of lox-/- MEF monolayers with purified LOX for 15 and 30 min before the chemotaxis assay progressively increased their chemotactic response toward LOX (Fig. 7, bars 7 and 8 versus bar 5). Similarly PDGF-BB-induced chemotaxis in lox+/+ MEF (Fig. 7, bar 9 versus bar 1) was lower by ∼86.0% in lox-/- MEF (Fig. 7, bar 11 versus bar 2). Pretreatment of the cells with BAPN inhibited PDGF-BB-induced chemotaxis by 82.0% in lox+/+ MEF (Fig. 7, bar 10 versus bar 9) but had no effect on the negligible chemotactic response of lox-/- MEF (Fig. 7, bar 12 versus bar 11).
FIGURE 7.

Chemotaxis is decreased by inactivation of the LOX gene. Wild type (lox+/+) and LOX knock-out (lox-/-) MEF were grown in the absence (control cells), or in the presence of 100 μm BAPN (BAPN-treated cells). LOX-treated refers to lox-/- cells that were treated with pure LOX (5 units/ml culture medium) for 15 min and 30 min (indicated on top of the bars 7, 8, 13, and 14). After the indicated treatment cells were subjected to chemotaxis analysis as described under “Experimental Procedures.” PDGF-BB (20 ng/ml) and LOX (2 units) were tested as chemoattractants. Additions to the feeder tray are indicated in the abscissa. RFU stands for relative fluorescent units. The error bars are standard deviations of the mean values obtained from three independent experiments (n = 12).
Pretreatment of lox-/- MEF with pure LOX also increased chemotactic response to PDGF in a time-dependent manner (Fig. 7, lanes 13 and 14 versus 11). These results further support the hypothesis that LOX activity is required for optimal chemotactic response. In the context of similar studies in SMC presented in Fig. 1, we conclude that cells not expressing endogenous LOX fail to respond to LOX when it is added as the chemoattractant, because cell surface proteins in those cells had not been previously oxidized during cell culture due to the absence of LOX in the lox-/- cells. Thus, the chemotaxis priming effect that we propose as being due to endogenous LOX did not and cannot occur in the lox-/- cells. Nevertheless, priming effects were observed upon addition of purified LOX to lox-/- MEF (Fig. 7, bars 7, 8, 13, and 14).
The Magnitude and Sensitivity of MEF to Chemokines Are Modulated by LOX—Because the chemotactic response of lox-/- MEF to 20 ng/ml PDGF-BB is significantly less than that of lox+/+ MEF (Fig. 7), it was relevant to compare their response to several concentrations of this chemoattractant. As shown (Fig. 8), lox+/+ MEF responded maximally at 16 ng/ml PDGF-BB (Fig. 8, filled circles), whereas the maximal response seen with the lox-/- cells occurred at ∼28 ng/ml (Fig. 8, empty circles). The magnitude of the maximal response of the lox-/- cells was slightly less than that of the wild-type cells. The reduced chemotactic sensitivity of lox-/- MEF toward PDGF-BB resembles that of BAPN-treated SMC toward this chemokine (Fig. 2B). Thus, the lack of LOX enzyme activity, due either to inhibition by BAPN (Fig. 2B) or by inactivation of the LOX gene (Fig. 8) decreases the chemotactic sensitivity of cells toward PDGF-BB.
FIGURE 8.

The magnitude and sensitivity of MEF chemotaxis are modulated by LOX chemotaxis driven by PDGF-BB in lox+/+ (filled circles) and lox-/- MEF (open circles) were assayed as in Fig. 1 for SMCs, using PDGF-BB in the range of concentrations indicated in the abscissa. Error bars represent the standard deviations from the means derived from three independent experiments (n = 12).
Inactivation of the LOX Gene Influences the Binding Affinity of PDGF Receptor-β for PDGF-BB—The reduction in the chemotactic sensitivity toward PDGF-BB suggested that a change in the binding properties of its receptor might be involved. As shown (Fig. 9A), the affinity and maximal binding capacity of lox+/+ MEF for PDGF-BB (Kd = 55.3 ± 12.5 pm; Bmax = 3,550 ± 137 molecules/cell) were both reduced in lox-/- MEF (Kd = 196.1 ± 23.4 pm; 2,307 ± 115). Pretreatment of lox+/+ MEF with BAPN (Fig. 9B) also reduced the affinity and maximal binding capacity (Kd = 83.8 ± 10.5 pm; 2,197 ± 137 molecules/cell), similar to the values observed for lox-/- MEF (Fig. 9A). BAPN-treated lox-/- MEF (Kd = 156.3 ± 11.7 pm; Bmax = 2,197 ± 126 molecules/cells) showed affinity and maximal binding similar to control lox-/- MEF (Kd = 196.1 ± 23.4 pm; Bmax = 2,307 ± 115 molecules/cells) (Fig. 9C). These results provide additional evidence suggesting that BAPN does not affect the chemotactic response beyond its inhibitory effect on LOX activity.
FIGURE 9.

Inactivation of the LOX gene influences binding affinity of PDGFR-β for PDGF-BB. Binding of PDGF-BB to MEF in culture was done using 125I-PDGF-BB as described under “Experimental Procedures.” The binding of PDGF-BB was measured over a wide concentration range of the ligand (10-3500 pm). Bmax and Kd values are shown under the corresponding plot. Error bars are the standard deviations of the mean values of eight determinations (n = 8).
Inactivation of the LOX Gene Increases the Turnover of Phosphorylated Intermediates of the PDGF Receptor-β Signal Transduction Pathway—The PDGFR-β signal transduction pathway was analyzed in lox+/+ and lox-/- MEF as described above for SMC (Fig. 6A). The autophosphorylation of PDGFR-β was not detected in MEFs (Fig. 10A), possibly due to a lower level of PDGFR-β expressed in these cells (<3,700 molecules/cell; Fig. 9A) when compared with SMCs (<37,000 molecules/cell; Fig. 5A). Nevertheless, the degree of phosphorylation of Shp2, Akt, and ERK1/ERK2 (p44/42 MAPK) decayed faster in lox-/- MEF than in lox+/+ MEF, thus correlating with the results obtained with BAPN on SMC (Fig. 6A) and supporting the idea that the effects of BAPN on chemotaxis are due to the inhibition of LOX activity. Moreover, these results strongly support a causal connection between LOX activity and the longevity of the signal transduction pathway triggered by PDGF-BB. The levels of PDGFR-β in lox+/+ MEF and lox-/- MEF were also assessed as a function of the time after addition of PDGF-BB. The levels of PDGFR-β declined by 36% in lox+/+ cells and by 65% in lox-/- cell after 30 min of activation by PDGF-BB (Fig. 10B). This faster degradation of the activated PDGFR-β correlates with the reduced chemotactic response of these cells (Fig. 7, bar 5 versus bar 3).
FIGURE 10.

Inactivation of the LOX gene increases the turnover of the PDGF receptor-β signal transduction pathway. Analysis of the PDFGR-β signal transduction pathway by monitoring the phosphorylated state of several intermediates was done as described under “Experimental Procedures.” A, Western blot of total MEF extract, at different times after the addition of PDGF-BB, probed with the anti-phosphoprotein antibodies against the proteins indicated on the right-hand side column. B, is the membrane from experiment A that was re-probed with antibodies against PDGFR-β. The graphs on the right side show the densitometry plots of the Western blot bands.
Timeline for LOX Activity and Expression, Protein Oxidation, and Chemotactic Response—The demonstration that pretreatment with LOX restores protein oxidation in the plasma membrane (Fig. 4) and chemotaxis sensitivity of cells genetically deficient in LOX (Fig. 7) prompted us to analyze the timeline of events correlating LOX activity and expression with plasma membrane protein oxidation and chemotactic response. LOX mRNA, LOX secretion, and LOX activity were at their lowest levels while cells were expanding in culture (days 1-3, Fig. 11). As the cells approached confluence (∼90%), by day 4, LOX secretion and activity increased by ∼3-fold, whereas LOX mRNA showed only a minor increase. At cell confluence, LOX activity and secretion approximated 80% of their maximal values. Although the level of LOX secretion was nearly maximal after day 7, LOX mRNA increased ∼3-fold from days 7-14. Because the cells are exposed to their own secreted LOX activity during this time in culture, plasma membrane proteins, with molecular masses approximating 180 kDa, became progressively oxidized (Fig. 11B). The time-line for protein oxidation shown in Fig. 11B was paralleled by an equivalent increase in the chemotactic response of the cells. Thus, cells rapidly growing (1-3 days) produced a low level of LOX, a low degree of oxidation of their plasma membrane proteins, and a low chemotactic response. As LOX secretion increased the cells reached stationary growth (day 4 and beyond), and the oxidation of cell surface proteins and their chemotactic response were augmented. Due to this progressive exposure of cells to secreted LOX, the priming effect of LOX is maximal when cells have reached the stationary phase and beyond. Apparently, cells in the growth phase are not primed for the optimal chemotactic response, because secretion of LOX is very low at that time. The observed delayed secretion of LOX in cultured SMC (Fig. 11) was also reported in rat fetal lung cells (39).
FIGURE 11.

Timeline for LOX activity/expression, protein oxidation, and chemotactic response. A, graph representing LOX secretion (open circles), LOX activity (filled circles), and LOX mRNA (black bars) as a function of the days in culture of SMC. Elastin mRNA (gray bars) was also analyzed as known positive control (54). Secreted LOX plot (open circles) was generated from the densitometry analysis of the Western blot signals shown in the abscissa. The percentage of confluence as a function of time in culture is also indicated in the abscissa. B, oxy-Western blot analysis of purified plasma membrane (10 μg/well) resolved in 4.0-20.0% gradient SDS-polyacrylamide gel electrophoresis and the corresponding protein staining, at different times of cells in culture. C, chemotactic response of cultured cells to LOX (black bars) and to PDGF-BB (gray bars) as a function of time in culture. Densitometry analysis (filled circles) from the region around 180 kDa indicated by the dotted line in the oxy-blot graph in B.
The question arises as to how this chronic exposure of cells to LOX activity over a period of 5-6 days, resulting in a gradual increment in chemotactic response (Fig. 11C), compares with the acute effect of exogenously added LOX capable of restoring the chemotactic response to LOX and PDGF-BB by ∼70 and 50%, respectively, in lox-/- cells (Fig. 7). A reasonable approximation can be drawn from the data in Fig. 11A. It can be assumed that cells in culture are exposed to an average of 0.02 unit/ml of LOX activity for 6 days, i.e. 0.02 unit × 144 h = 2.9 unit hours. Applying this criterion to the data obtained in the acute exposure of cells to 5 units/ml exogenously added LOX for 30 min yields a value of 2.5 unit hours, quite similar to the value of 2.9 unit hours estimated for the condition relying only upon the effect of endogenous LOX. Accordingly, the acute LOX pretreatment condition (Fig. 7) closely reconstitutes the condition prevailing under chronic exposure to endogenous LOX (Figs. 1 and 2).
DISCUSSION
The present study has explored biological events that appear to underlie the unusual ability of lysyl oxidase to induce chemotaxis in vascular SMCs. The data support our previous findings that catalytic expression by LOX is critical for this cellular response to this enzyme (19) and provide new insight into possible cellular site(s) where oxidative deamination of lysine residues by this enzyme occurs as an important precedent to cell migration not only in response to LOX but also in response to PDGF-BB.
The use of BAPN as an irreversible inhibitor of LOX activity has been an important criterion for the role of LOX catalysis not only in this report but in others, as well, which also note the relationship between chemotaxis and the catalytic function of LOX with various cell types (22, 40-43). In the present study, BAPN has been applied at a concentration of 100 μm, which previously has been shown to selectively inhibit LOX among a group of other amine oxidases (44). It is important to reiterate that BAPN has been used for over five decades to study connective tissue homeostasis and to block LOX activity in the test tube, in cell culture, and in animal studies. This aminonitrile inhibits lysyl oxidase activity by a catalysis-dependent suicidal inactivation mechanism: the initial step of oxidation of the β-amino group of BAPN (H2NCH2CH2CN) generates an intermediate that remains covalently bound to the active site, thus irreversibly inactivating the enzyme (29).
The key role of the catalytic expression of LOX-dependent stimulation or induction of chemotaxis was supported here by the parallel studies with lox-/- cells and by the demonstration that BAPN-inhibitable oxidation of cell membrane proteins occurs in SMCs in conjunction with decreases in key chemotactic parameters.
In the present study, we observed that glucose oxidase producing hydrogen peroxide at an equivalent rate to that produced by LOX did not elicit chemotaxis in SMC (Fig. 1, bars 7 and 8 versus bars 1 and 2), strongly suggesting that hydrogen peroxide by itself is insufficient to trigger the chemotactic response observed with LOX. It is not known if hydrogen peroxide is involved in the chronic priming effect taking place when cells in culture are naturally exposed to endogenous LOX.
The present study has also shown that the chemotactic response of lox-/- cells toward LOX was reduced when compared with the wild-type cells, supporting the conclusion that the effect of BAPN is primarily related to its inhibition of LOX activity rather than that of LOX-like enzymes. BAPN-treated cells required approximately a 2-3-fold greater concentration of exogenous LOX or of PDGF-BB to reach the maximum chemotactic response in each case, which, in turn, was reduced in magnitude in comparison to that of the control cells not pretreated with BAPN. The enhancement of the PDGF-BB-mediated chemotaxis by LOX (Fig. 2C) provides additional evidence for the involvement of LOX activity in the preparation of cells for the optimum response to chemotactic agents other than LOX itself.
The hypothesis that LOX activity has a “priming” effect required for optimal chemotactic response was reinforced by the observation that the low chemotactic response of lox-/- cells was markedly enhanced by acute exposure to LOX activity (Fig. 7), mimicking the condition existing in normal cultured cells (Fig. 11). Most interestingly, the oxy-immunofluorescence and Western blotting analyses for oxidized cell membrane proteins revealed that specific SMC surface proteins were oxidized in a BAPN-inhibitable manner, consistent with the participation of endogenous LOX in these effects.
At least five BAPN-sensitive oxidized proteins with apparent molecular masses of 44-190 kDa (Fig. 4A, lanes 1 and 3) were found, included among which was the PDGF receptor-β. A significant increase in Kd and reduction in Bmax for the binding of PDGF-BB to BAPN-treated SMC and A7r5 monolayers appears to be a consequence of the chronic blockage of LOX activity by the presence of BAPN during cell culture. Overall, the collected results support the hypothesis that the reduced chemotactic response toward PDGF-BB observed in BAPN-treated cells reflects the inhibition of peptidyl lysine oxidation in this receptor by endogenous LOX. It is of interest in this regard that the extracellular domain of PDGFR-β contains nine lysine residues some of which occur in sequence segments that have features that have been shown to favor LOX catalysis in model peptides (44). For example, the sequence 158TLHEKKV164 within this receptor has a net positive charge and contains a glutamate N-terminal to the first lysine residue. The kcat/Km value for the... EK... sequence within a polyglycine 11-mer peptide is several times higher than that lacking a glutamate in this position (45). Moreover, the grouping of positively charged residues in this receptor is consistent with the earlier demonstration that proteins with a net cationic charge (pI > 8.O) are optimal substrates for the expression of LOX activity (8). The precise site(s) of oxidation of this receptor by LOX remain to be established.
In addition to the oxidation of the receptor, the increased rate of decay of phosphorylated intermediates in the signal transduction pathway triggered upon activation of PDGFR-β in BAPN-treated SMC and in lox-/- MEF cells; points to a protective role of LOX in these downstream components of the chemotactic response, which are critically involved in cell migration and proliferation (34). Thus the data support the conclusion that the longevity of phosphorylated intermediates in the PDGFR-β signal-transduction pathway as well as that of the activated receptor at the cell surface are both diminished by inhibition of LOX activity during cell culture.
Recycling of activated receptors is usually detected as a delay in their clearance after activation. Although activated PDGFR-β does not normally recycle, its intracellular trafficking may lead to recycling in specific cellular backgrounds, such as in T-cell phosphatase -/- fibroblasts (46). The present data reveal that PRGFR-β is cleared 2-3-fold faster in BAPN-pretreated SMC and in lox-/- MEF than in control SMC and wild-type MEF. Moreover, the initial level of resting PRGFR-β (at zero time) is very similar in control and BAPN-pretreated SMC and in wild-type and lox-/- MEF cells, suggesting that BAPN pretreatment or the lack of LOX gene did not alter the rates of synthesis/degradation (turnover) to unbalance its resting, steady-state level. Thus, we conclude that the oxidative state of activated PRGFR-β is an important factor in the determination of its lifetime span.
Although the molecular basis of the apparent role of H2O2 in chemotactic responses to exogenous LOX is not yet evident, it is relevant to note that the established mechanism of chemotaxis by PDGF-β involves the activation of NADPH oxidase at the cytosolic surface of the plasma membrane (47). The H2O2 product of NADPH oxidase activity is considered to act as a second messenger activating downstream signal transduction molecules by acting on protein tyrosine phosphatases (48), leading to the chemotactic response to PDGF. It is likely that the molar quantity of H2O2 produced as NADPH is oxidized by this enzyme would significantly exceed that which would be expected to result from the oxidation of a few lysine residues by LOX. Nevertheless, H2O2 produced by LOX in intimate contact with a protein substrate at the cell surface might escape dilution by the extracellular milieu and diffuse into the cell with possible local consequences to signal transduction component(s) required for chemotaxis. Such a mechanism would be consistent with the lack of a chemotactic effect of the calibrated amounts of H2O2 added to cells in the present study. Understanding the molecular role of H2O2 in the observed chemotactic response toward LOX may reveal additional mechanisms by which this second messenger is critically involved in chemotaxis. It is known that polyamines, such as spermine, spermidine, and putrescine affect cell migration and proliferation. Amine oxidase enzymes can generate cytotoxic products, for example, in addition to hydrogen peroxide, various aldehyde products. Thus, the products of semicarbazide-sensitive amine oxidase activity could also be involved in the LOX-mediated modulation of chemotactic response reported here. Semicarbazide-sensitive amine oxidase, a 100-kDa protein localized in outer membranes of vascular smooth muscles and endothelia, could be one of the plasma membrane proteins oxidized by LOX (Fig. 4A). The identification of all cell surface proteins oxidized by LOX will help understanding the impact on cellular responses of this new, beneficial, enzyme-catalyzed, post-translational modification taking place in the extracellular milieu.
The fact that cellular proteins are susceptible to non-enzymatic oxidation in vivo has been known for many years (49). The bulk of cellular protein oxidation is produced by chemically induced covalent modification of several amino acid residues either directly by reactive oxygen species or indirectly by reaction with secondary by-products of oxidative stress (50). The chemical oxidation of intracellular proteins (51) is generally conceived of as a triggering mechanism for protein turnover by proteasomal degradation (52). In contrast to enzyme-mediated oxidation, non-enzymatic, chemical oxidation of proteins is neither specific for amino acid residue(s) within the protein substrate nor is it subject to biochemical regulation per se. Rather, the process is balanced by the redox state maintained by cellular antioxidant systems (53). Conversely, the concepts of specificity and regulation are fully applicable to the enzymatic oxidation of proteins. The findings presented here that the oxidation of cell surface proteins, such as PDGFR-β, by LOX favorably modulates the chemotactic response of the cell points to a previously unrecognized aspect of protein oxidation with important functional significance.
Acknowledgments
We gratefully acknowledge the expert review of this manuscript by Drs. Barbara Smith and Barbara Schreiber from Boston University School of Medicine.
This work was supported, in whole or in part, by National Institutes of Health Grant HL13262-31 (to H. K. and K. R.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: LOX, lysyl oxidase; LOXL, LOX-like; A7r5, embryonic rat aorta SMC line; AKT1, v-akt murine thymoma viral oncogene homolog 1; BAPN, β-aminoproprionitrile; DNP, dinitrophenol; DNPH, dinitrophenylhydrazine; ERK1/ERK2, extracellular signal-regulated kinase; MEF, mouse embryonic fibroblasts; SMC, neonatal rat aorta smooth muscle cells; PDGFR-β, platelet-derived growth factor β; SHP2, SRC homology 2 domain-containing protein-tyrosine phosphatase; PBS, phosphate-buffered saline; BSA, bovine serum albumin; Tricine, N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine; MAPK, mitogen-activated protein kinase.
References
- 1.Lucero, H. A., and Kagan, H. M. (2006) Cell Mol. Life Sci. 632304 -2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Panchenko, M. V., Stetler-Stevenson, W. G., Trubetskoy, O. V., Gacheru, S. N., and Kagan, H. M. (1996) J. Biol. Chem. 2717113 -7119 [DOI] [PubMed] [Google Scholar]
- 3.Mäki, J. M., Räsänen, J., Tikkanen, H., Sormunen, R., Mäkikallio K., Kivirikko, K. I., and Soininen, R. (2002) Circulation 1062503 -2509 [DOI] [PubMed] [Google Scholar]
- 4.Mäki, J. M., Sormunen, R., Lippo, S., Kaarteenaho-Wiik, R., Soininen, R., and Myllyharju, J. (2005) Am. J. Pathol. 167927 -936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Csiszar, K. (2001) Prog. Nucleic Acids Res. Mol. Biol. 701 -32 [DOI] [PubMed] [Google Scholar]
- 6.Liu, X., Zhao, Y., Gao, J., Pawlyk, B., Starcher, B., Spencer, J. A., Yanagisawa, H., Zuo, J., and Li, T. (2004) Nat. Genet. 36178 -182 [DOI] [PubMed] [Google Scholar]
- 7.Pinnell, S. R., and Martin, G. R. (1968) Proc. Natl. Acad. Sci. U. S. A. 61 708-716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kagan, H. M., Williams, M. A., Williamson, P. R., and Anderson, J. M. (1984) J. Biol. Chem. 25911203 -11207 [PubMed] [Google Scholar]
- 9.Kenyon, K., Contente, S., Trackman, P. C., Tang, J., Kagan, H. M., and Friedman, R. M. (1991) Science 253 802. [DOI] [PubMed] [Google Scholar]
- 10.Jeay, S., Pianetti, S., Kagan, H. M., and Sonenshein, G. E. (2003) Mol. Cell Biol. 72251 -2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giampuzzi, M., Botti, G., Di Luca, M., Arata, L., Ghiggeri, G., Gusmano, R., Ravazzolo, R., and Di Donato, A. (2000) J. Biol. Chem. 27536341 -36349 [DOI] [PubMed] [Google Scholar]
- 12.Giampuzzi, M., Oleggini, R., and Di Donato, A. (2003) Biochim. Biophys. Acta 1647239 -244 [DOI] [PubMed] [Google Scholar]
- 13.Kirschmann, D. A., Seftor, E. A., Fong, S. F. T., Nieva, D. R. C., Sullivan, C. M., Edwards, E. M., Sommer, P., Csiszar, K., and Hendrix, M. J. C. (2002) Cancer Res. 624478 -4483 [PubMed] [Google Scholar]
- 14.Erler, J. T., Bennewithm, K. L., Nicolau, M., Dornhöfer, N., Kong, C., Le, Q. T., Chi, J. T., Jeffrey, S. S., and Giaccia, A. J. (2006) Nature 4401222 -1226 [DOI] [PubMed] [Google Scholar]
- 15.Payne, S. L., Hendrix, M. J., and Kirschmann, D. A. (2007) J. Cell Biochem. 1011338 -1354 [DOI] [PubMed] [Google Scholar]
- 16.Li, W., Nellaiappan, K., Strassmaier, T., Graham, L., Thomas, K. M., and Kagan, H. M. (1997) Proc. Natl. Acad. Sci. U. S. A. 9412817 -12822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nellaiappan, K., Risitano, A., Liu, G., Nicklas, G., and Kagan, H. M. (2000) J. Cell Biochem. 79 576-582 [DOI] [PubMed] [Google Scholar]
- 18.Lazarus, H. M., Cruikshank, W. W., Narasimhan, N., Kagan, H. M., and Center, D. M. (1995) Matrix Biol. 14 727-731 [DOI] [PubMed] [Google Scholar]
- 19.Li, W., Liu, G., Chou, I. N., and Kagan, H. M. (2000) J. Cell Biochem. 78550 -557 [PubMed] [Google Scholar]
- 20.Kagan, H. M., Raghavan, J., and Hollander, W. (1981) Arteriosclerosis 1287 -291 [DOI] [PubMed] [Google Scholar]
- 21.Bouez, C., Reynaud, C., Noblesse, E., Thépot, A., Gleyzal, C., Kanitakis, J., Perrier, E., Damour, O., and Sommer, P. (2006) Clin. Cancer Res. 121463 -1469 [DOI] [PubMed] [Google Scholar]
- 22.Laczko, R., Szauter, K. M., Jansen, M. K., Hollosi, P., Muranyi, M., Molnar, J., Fong, K. S., Hinek, A., and Csiszar, K. (2007) Neuropathol. Appl. Neurobiol. 33 631-643 [DOI] [PubMed] [Google Scholar]
- 23.Schreiber, B. M., Martin B. M., Hollander, W., and Franzblau, C. (1988) Atherosclerosis 69 69-79 [DOI] [PubMed] [Google Scholar]
- 24.Kagan, H. M., and Cai, P. (1995) Methods Enzymol. 258122 -132 [DOI] [PubMed] [Google Scholar]
- 25.Palamakumbura, A. H., and Trackman, P. C. (2002) Anal. Biochem. 300245 -251 [DOI] [PubMed] [Google Scholar]
- 26.Bolton, A. E., and Hunter, W. M. (1973) Biochem. J. 133529 -539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolfe, L. B., Rich, C. B., Goud, H. D., Terpstra, A. J., Bashir, M., Rosenbloo, J., Sonnenshein, G. E., and Foster, J. A. (1993) J. Biol. Chem. 26812418 -12436 [PubMed] [Google Scholar]
- 28.Nelson, J. M., Diegelmann, R. F., and Cohen, I. K. (1988) Proc. Soc. Exp. Biol. Med. 188346 -352 [DOI] [PubMed] [Google Scholar]
- 29.Tang, S. S., Trackman, P. C., and Kagan, H. M. (1983) J. Biol. Chem. 2584331 -4338 [PubMed] [Google Scholar]
- 30.Shimokado, K., and Higaki, M. (1997) Ann. N. Y. Acad. Sci. 811130 -133 [DOI] [PubMed] [Google Scholar]
- 31.Rönnstrand, L., and Heldin, C.-H. (2001) Int. J. Cancer 91757 -762 [DOI] [PubMed] [Google Scholar]
- 32.Leskovac, V., Trivić, S., Wohlfahrt, G., Kandrac, J., and Pericin, D. (2005) Int. J. Biochem. Cell Biol. 37731 -750 [DOI] [PubMed] [Google Scholar]
- 33.Wang, S. X., Mure, M., Medzihradszky, K. F., Burlingame, A. L., Brown, D. E., Dooley, D. M., Smith, A. J., Kagan, H. M., and Klinman, J. P. (1996) Science 2731078 -1084 [DOI] [PubMed] [Google Scholar]
- 34.Heldin, C. H., Ostman, A., and Ronnstrand, L. (1998) Biochim. Biophys. Acta 1378F79 -F113 [DOI] [PubMed] [Google Scholar]
- 35.Vogel, W., and Ullrich, A. (1996) Cell Growth & Differ. 71589 -1597 [PubMed] [Google Scholar]
- 36.Klippel, A., Kavanaugh, W. M., Pot, D., and Williams, L. T. (1997) Mol. Cell Biol. 17 338-344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Franke, T. F., Kaplan, D. R., Cantley, L. C., and Toker, A. (1997) Science 275665 -668 [DOI] [PubMed] [Google Scholar]
- 38.Bardwell, A. J., Flatauer, L. J., Matsukuma, K., Thorner, J., and Bardwell, L. (2001) J. Biol. Chem. 27610374 -10386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thomassin, L., Werneck, C. C., Broekelmann, T. J., Gleyzal, C., Hornstra, I. K., Mecham, R. P., and Sommer, P. (2005) J. Biol. Chem. 28042848 -42855 [DOI] [PubMed] [Google Scholar]
- 40.Payne, S. L., Fogelgren, B., Hess, A. R., Seftor, E. A., Wiley, E. L., Fong, S. F., Csiszar, K., Hendrix, M. J., and Kirschmann D. A. (2005) Cancer Res. 6511429 -11436 [DOI] [PubMed] [Google Scholar]
- 41.Farjanel, J., Sève, S., Borel, A., Sommer, P., and Hulmes, D. J. S. (2005) Osteoarthritis and Cartilage 13120 -128 [DOI] [PubMed] [Google Scholar]
- 42.Bulut, T., Bilsel, Y., Yanar, H., Yamaner, S., Balik, E., Solakoglu, S., and Koser, M. (2004) J. Invest. Surg. 17211 -219 [DOI] [PubMed] [Google Scholar]
- 43.Gilad, G. M., and Gilad, V. H. (2001) Eur. J. Pharmacol. 43069 -72 [DOI] [PubMed] [Google Scholar]
- 44.Tang, S. S., Chichester, C. O., and Kagan, H. M. (1989) Connect. Tissue Res. 19 93-103 [DOI] [PubMed] [Google Scholar]
- 45.Nagan, N., and Kagan, H. M. (1994) J. Biol. Chem. 26922366 -22371 [PubMed] [Google Scholar]
- 46.Karlsson, S., Kowanetz, K., Sandin, A., Persson, C., Ostman, A., Heldin, C. H., and Hellberg, C. (2006) Mol. Biol. Cell 174846 -4855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sundaresan, M., Yu, Z. X., Ferrans, V. J., Irani, K., and Finkel, T. (1995) Science 270296 -299 [DOI] [PubMed] [Google Scholar]
- 48.Veal, E. A., Day, A. M., and Morgan, B. A. (2007) Mol. Cell. 261 -14 [DOI] [PubMed] [Google Scholar]
- 49.Stadtman, E. R. (2006) Free Radic. Res. 401250 -1258 [DOI] [PubMed] [Google Scholar]
- 50.Shacter, E. (2000) Drug Metab. Rev. 32307 -326 [DOI] [PubMed] [Google Scholar]
- 51.Dean, R. T., Fu, S., Stocker, R., and Davies, M. J. (1997) Biochem. J. 324 1-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davies, K. J. (2001) Biochimie (Paris) 83301 -310 [DOI] [PubMed] [Google Scholar]
- 53.Genestra, M. (2007) Cell. Signal. 191807 -1819 [DOI] [PubMed] [Google Scholar]
- 54.Toselli, P., Faris, B., Sassoon, D., Jackson, B. A, and Franzblau, C. (1992) Matrix 12 321-332 [DOI] [PubMed] [Google Scholar]