

Summary
Mutations in the lamin A/C gene cause the rare genetic disorder Hutchinson-Gilford progeria syndrome (HGPS). The prevalent mutation results in the production of a mutant lamin A protein with an internal 50 amino acid deletion which causes a cellular aging phenotype characterized by growth defects, limited replicative lifespan and nuclear membrane abnormalities. The relevance of these findings to normal human aging is however unclear. In this study we demonstrate that increased levels of wild-type lamin A in normal human cells result in decreased replicative lifespan and nuclear membrane abnormalities that lead to apoptotic cell death and senescence in a manner that is strongly reminiscent of the phenotype shown by HGPS cells. In contrast to the accelerated aging defects observed in HGPS cells, the progeroid phenotype resulting from increased expression of wild-type lamin A can be rescued by over-expression of ZMPSTE24, the metalloproteinase responsible for the removal of the farnesylated carboxyl terminal region of lamin A. Furthermore, farnesyltransferase inhibitors also serve to reverse the progeroid phenotype resulting from increased lamin A expression. Significantly, cells expressing elevated levels of lamin A display abnormal lamin A localization and similar alterations in the nuclear distribution of lamin A are also observed in cells from old-age individuals. These data demonstrate that the metabolism of wild-type lamin A is delicately poised and even in the absence of disease-linked mutations small perturbations in this system are sufficient to cause prominent nuclear defects and result in a progeroid phenotype.
Introduction
Hutchinson Gilford Progeria Syndrome (HGPS) is a rare autosomal dominant genetic disease that is observed in about 1 in 4 million children (Ackerman & Gilbert-Barness, 2002; Pollex & Hegele, 2004). Affected infants appear normal at birth but begin to display growth retardation, bone abnormalities, osteoporosis, baldness and scleradermatous skin between 6 to 12 months of age. HGPS children typically die between the ages of 13 and 16 years from myocardial infarction or stroke caused by progressive arterial occlusive disease. HGPS is caused by mutations in the lamin A/C gene (De Sandre-Giovannoli et al., 2003; Eriksson et al. 2003). Lamin A is synthesized as a precursor molecule (prelamin A), which undergoes several processing steps to produce a mature lamin A polypeptide. The carboxyl terminus of prelamin A contains a CAAX-box motif which is subject to farnesylation and has been suggested to play an important role in the localization of this protein to the periphery of the inner nuclear membrane. Upon farnesylation, the last three amino acids are removed by proteolytic cleavage and the C-terminal cysteine is methylated. A second proteolytic cleavage is subsequently carried out by the ZMPSTE24 metalloproteinase, which removes the last 15 amino acids containing the farnesylated residue to generate mature lamin A. The most prevalent mutation in HGPS patients is a de-novo heterozygous C to T transition in exon 11 of the lamin A/C gene, which results in the production of a cryptic splice site. Translation of mutant lamin A mRNA produces a lamin A protein with a 50 amino acid deletion in the region that contains the ZMPSTE24 cleavage site. This results in the production of a mutant protein, termed progerin, which retains the farnesylated tail (De Sandre-Giovannoli et al., 2003; Eriksson et al., 2003). Cells from HGPS patients display severe growth defects and abnormal nuclear morphology. Since HGPS is an autosomal dominant disease, the genetics suggest that the mutated protein acts in a dominant negative fashion and acquires novel pathological properties that are distinct from the wild-type lamin A protein. However, it remains unresolved whether normal lamin A metabolism plays a central role in cellular longevity. Here we show that ectopic expression of wild-type lamin A as well as expression of progerin in normal human cells results in defective replicative lifespan and nuclear membrane blebbing that leads to apoptotic cell death and senescence. Moreover, we demonstrate that the progeroid phenotype resulting from increased expression of wild-type lamin A can be reverted by over-expression of ZMPSTE24 or farnesyltransferase inhibitors. We further show that cells expressing elevated levels of lamin A display an abnormal pattern of lamin A localization that resembles that observed in HGPS cells and cells from old-age individuals. Taken together our data suggest that lamin A metabolism may play an important role in human aging.
Results
To examine the role of the normal lamin A metabolism in cellular aging, we studied the proliferative capacity and nuclear morphology of normal diploid fibroblasts with that of matched cells expressing progerin or elevated levels of wild-type lamin A. Cell lines were transduced with recombinant lentiviruses expressing mouse flag-(pre)lamin A or flag-progerin and the green fluorescence protein (GFP) under the control of the human mismatch repair protein 2 (MSH2) gene promoter (Figure 1A). In parallel, cells were also transduced with a control lentivirus expressing GFP. Two independent cell lines from each transduction were then subjected to fluorescence activated cell sorting (FACS) to select cells expressing GFP. Western blot analysis shows that the cell lines express the expected flag-tagged proteins (Figure 1B). Interestingly, expression of progerin leads to down regulation of endogenous lamin A (Figure 1B), a phenomenon observed in certain HGPS fibroblasts (De Sandre-Giovannoli et al., 2003; Eriksson et al., 2003). The significance of this observation however remains to be determined. The growth property of each cell line was then monitored throughout their replicative lifespan. Cells expressing progerin display a marked defect in growth, which closely resembles the growth defect observed in cells from HGPS patients (Figure 1, panel C, n=4; p<0.0001 for a two-way comparison of cell growth between progerin expressing fibroblasts and control fibroblasts from passages 5 to 13; panel D). This result demonstrates that cells expressing flag-tagged progerin have growth kinetics similar to HGPS cells. Surprisingly, cells expressing elevated levels of wild-type lamin A also exhibit a slow-growth phenotype. For the first few passages these cells display slightly better growth rates than cells expressing progerin (Figure 1 C, n= 4; p < 0.05 for a two-way comparison of cell growth between cells expressing progerin and cell expressing increased wild-type lamin A levels from passages 6 to 8). At later passages however, the growth rate of cells expressing elevated levels of wild-type lamin A is not significantly different from cells expressing progerin (Figure 1 C; n=4; p > 0.05 for a two-way comparison of cell growth between cells expressing progerin and cell expressing increased wild-type lamin A levels from passages 9 to 13). These results suggest that elevated levels of wild-type lamin A induce a growth defect qualitatively similar to that of progerin-expressing cells after passage 8. To make sure that the lamin A pathway was not overwhelmed by massive over expression of the flag tagged protein, we measured the relative levels of lamin A in both control and flag-lamin A expressing fibroblasts by western blot analysis (Figure 1 E). We estimate that the levels of lamin A are ~ 10 % (fibroblast line # 1) to ~15 % (fibroblast line #2) higher in the two flag-lamin A expressing fibroblast lines when compared to the vector control fibroblasts. To assure that expression of ectopic wild-type lamin A was not limited to a few cells, we examined lamin A levels in control cells and cells overexpressing wild-type lamin A at the single cell level by indirect confocal microscopy. This analysis shows that there are only small differences in overall lamin A expression in the transduced cells and cell-to-cell variations in lamin A levels are comparable to that of control cells (supplementary Figure S1). Thus, these data demonstrate that small increases in the lamin A levels are sufficient to cause a growth defect that is similar to that observed in cells expressing progerin albeit with slightly delayed kinetics.
Figure 1.
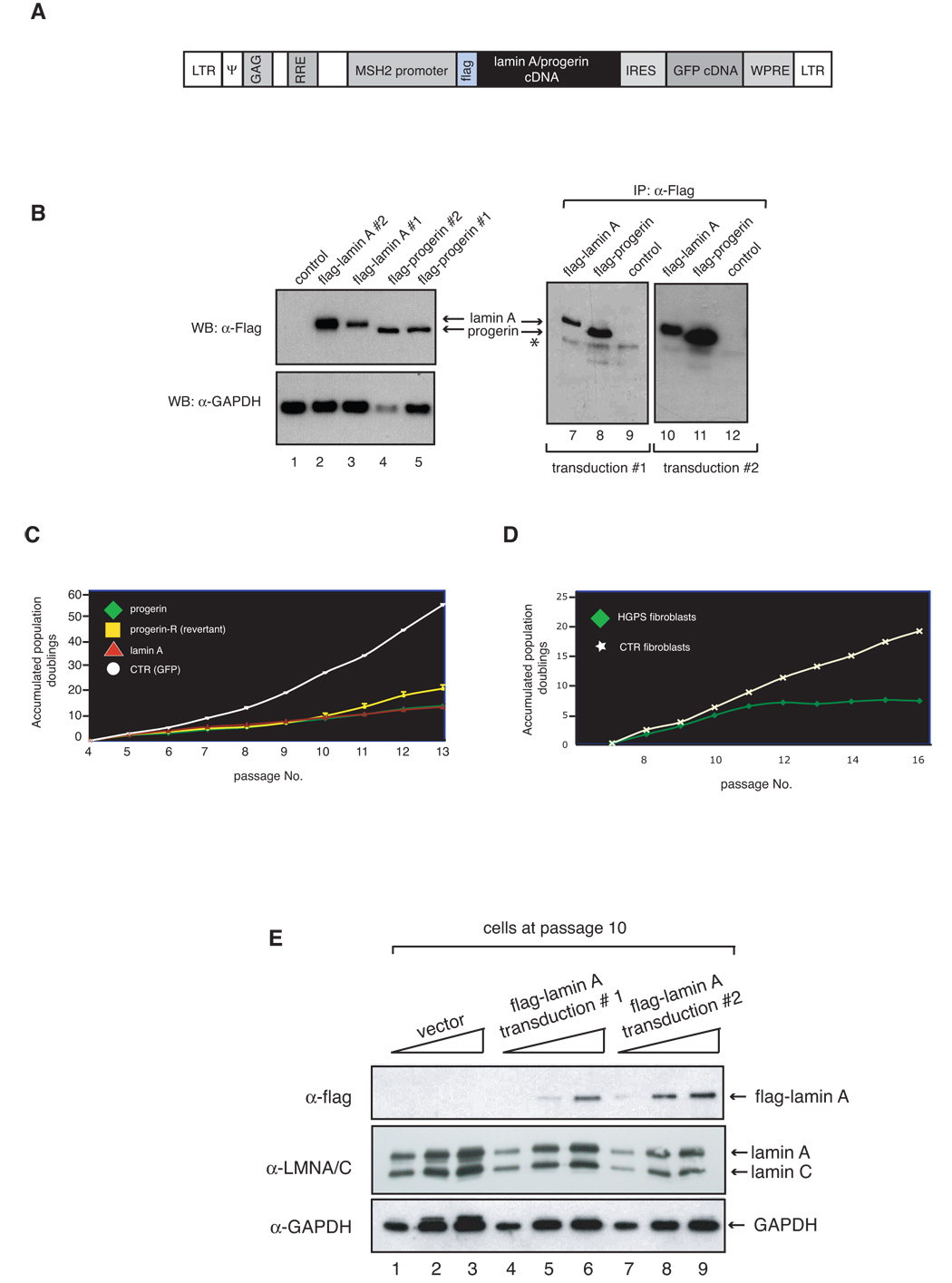
Elevated levels of wild-type lamin A in normal human fibroblasts results in decreased cell growth. A. Map of the lentiviral vector used to generate the human primary fibroblast cell lines expressing wild-type flag-lamin A and flag-progerin. LTR= Long Terminal Repeats; ψ= encapsidation signal; GAG gene; RRE= Rev response element; IRES= internal ribosome entry sequence; WPRE= woodchuck hepatitis virus regulatory element. B. Cell lysates were prepared from fibroblasts transduced with lentiviruses expressing flag-lamin A (lanes 2, 3, 7 and 10), Flag-progerin (lanes 4, 5, 8 and 11), or GFP-only (lanes 1, 9 and 12), were either analyzed by SDS-PAGE, and immunoblotted with anti-flag and anti-GAPDH antibody (lanes 1–5) or subjected to anti-flag immunoprecipitation and analyzed by western blotting with flag antibody (lanes 7–12). 15 µl and 300 µl of cell lysates were used in western blot analyses and immunoprecipitation reactions, respectively. Asterisk refers to signal by IgG heavy chains. C. Growth curves of stably transduced fibroblasts are shown as accumulated population doublings. D. Growth curves of HGPS fibroblasts (AG11498) and normal fibroblasts (GM00038). Another HGPS (HGADFN003) and normal (GM00316) fibroblast lines showed a growth pattern similar to AG11498 and GM00038, respectively. E. Lamin A levels in flag-lamin A expressing cells demonstrate a small increase in lamin A when compared to control cells. Lamin A levels were measured in total protein extracts (lanes 1, 4 and 7:10 µg, lanes 2, 5 and 8: µg 20 and lanes 3, 6 and 9: 30 µg) prepared from fibroblast lines transduced with vector lentivirus or lentiviruses expressing flag-lamin A at passage 10 by western blot analysis. The levels of flag-lamin A and total lamin A were quantified and normalized to lamin C and GAPDH by Imagequant software as described in Materials and Methods.
Intriguingly, we observed that while the two independently generated progerin-expressing cell lines behave in a similar manner during the first few passages, one cell line began to grow better after passage 10. To determine whether the increase in growth was associated with reduced progerin expression we prepared cell extracts from each cell line at passage 12 and examined the expression of the flag-tagged proteins by western blot analysis (Figure 2A). This analysis indicates that the progerin-transduced fibroblasts which show increased growth rate (termed progerin-revertant) do not express any detectable amounts of progerin. In contrast, all other transduced fibroblast lines expressed the expected flag-tagged proteins. This finding strongly supports the hypothesis that the growth defect in cells expressing progerin is functionally linked to expression of the mutant protein and is not due to a transduction artifact. The reason for the loss of expression of progerin in the revertant cell line is unclear at this time but may be the result of promoter silencing as this cell line also failed to express the green fluorescent protein (GFP) marker at passage 12 (data not shown). Moreover, semi quantitative analysis of western blots data indicates that progerin levels are slightly increasing over passages in the cell line that continues to ectopically express progerin (Figure 2B). Collectively, these results demonstrate that human fibroblasts expressing progerin or increased levels of wild-type lamin A display a limited replicative lifespan when compared to the control cell line indicating that the defect in proliferation caused by progerin can be recapitulated by increased expression of wild-type lamin A.
Figure 2.
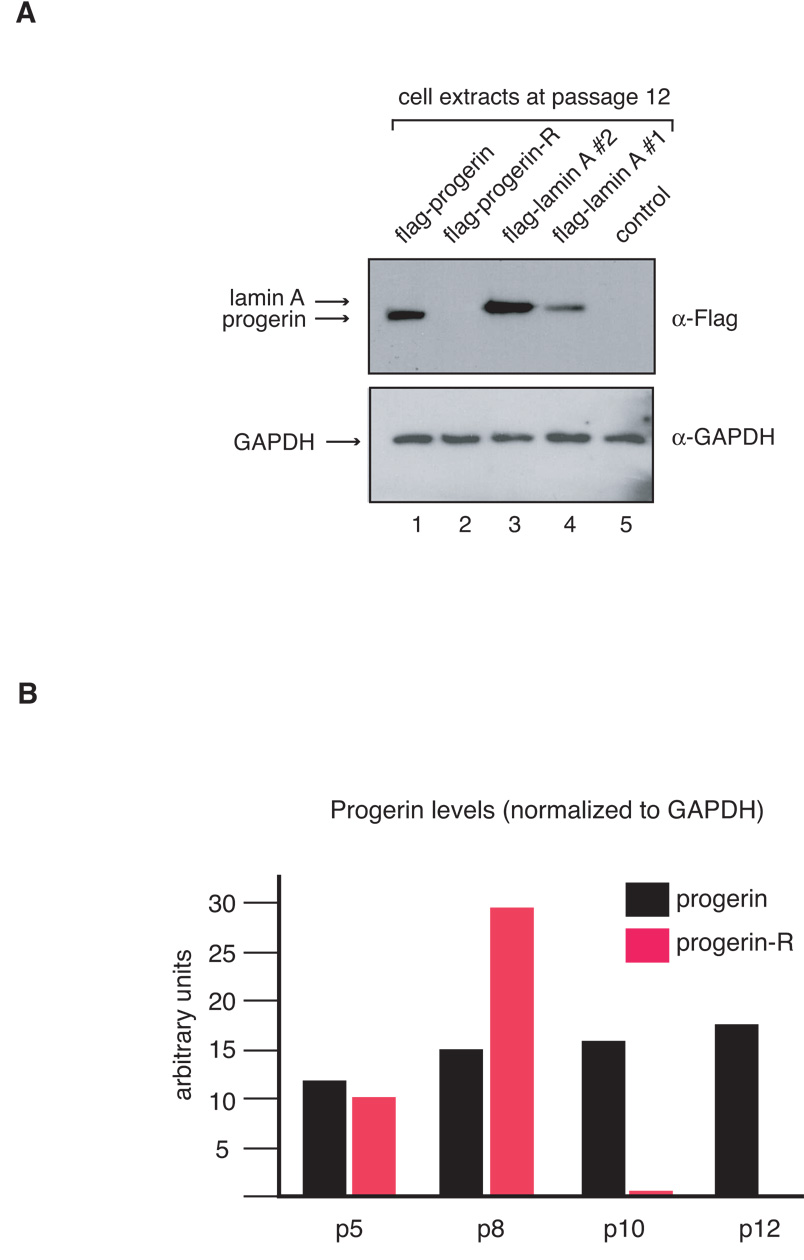
A Western blot analysis of extracts prepared from transduced fibroblasts at passage 12 shows loss of progerin expression in one of the cell lines (upper panel: flag antibody; lower panel: GAPDH antibody, as a control for sample loading). B. Progerin levels display a small passage-dependent increase. Progerin levels were quantified and normalized to lamin C using Imagequant software.
HGPS cells display a high frequency of nuclear membrane abnormalities, which include nuclear membrane lobulation and blebbing (Figure 3, panel A; Goldman et al., 2004). To test whether expression of progerin or elevated levels of flag-lamin A in normal human fibroblasts leads to similar phenotypic changes, the transduced cell lines were stained with DAPI and examined microscopically. Cells expressing progerin show a significant number of cells with lobulated nuclei (Figure 3). However the number of cells with nuclear abnormalities did not increase with the passage number, suggesting that the cells with blebs are either dying or fail to undergo cell division. In contrast, the progerin-revertant cell line, which initially shows an elevated number of cells with nuclear membrane abnormalities, displayed a sharp decrease in cells with nuclear blebs after passage 11 when the progerin expression was no longer detected. This demonstrates that alterations in nuclear morphology require sustained expression of progerin which is reversible on progerin shutdown. Interestingly, cells expressing flag-lamin A also show a dramatic increase in the number of cells with nuclear blebs, indicating that bleb formation is not an exclusive consequence of progerin expression. However, unlike cells expressing progerin, cell lines expressing flag-lamin A display a passage-dependent increase in the number of cells with nuclear membrane abnormalities. It is important to note that blebs were also occasionally seen in wild-type cells (Figure 3, Panel A, bottom left panel), but whether this event is linked to alteration in lamin A expression remains to be determined. In summary, these results demonstrate that increased expression of lamin A leads to growth defects and alterations in nuclear morphology similar to that observed in cells from HGPS patients. These observations were originally made using the epitope tagged mouse lamin A protein. To ensure that these results were not a consequence of either the presence of the flag epitope or due to species-specific differences between human and mouse lamin A, we carried out similar experiments using the flag-tagged and untagged human lamin A. In all cases similar results were obtained, indicating that increased levels of either human or mouse lamin A are sufficient to cause growth retardation and nuclear morphology abnormalities in human fibroblasts (supplemental Figure S2).
Figure 3.
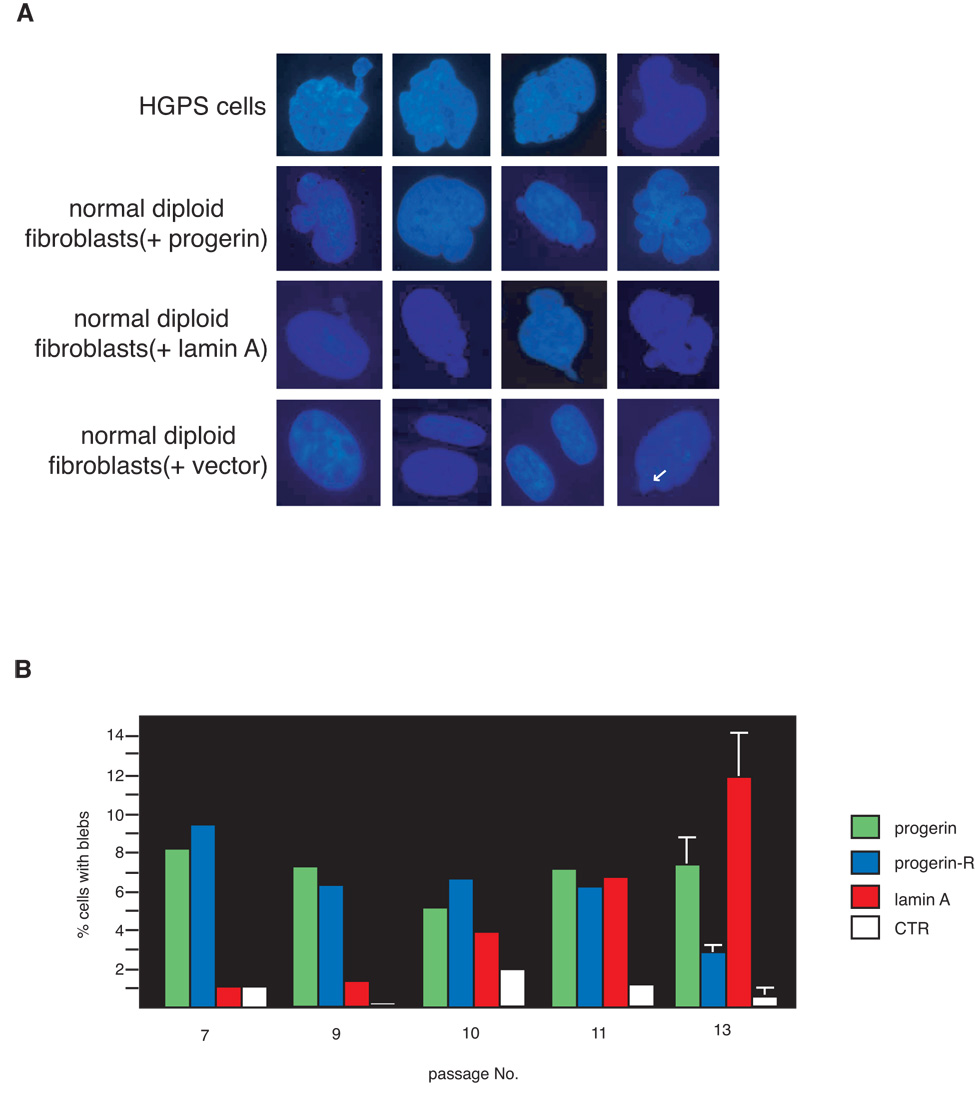
Increased levels of wild-type lamin A in primary human fibroblasts are sufficient to induce the formation of nuclear blebs. A. Fluorescence microscopy of DAPI stained cells show typical nuclei of HGPS fibroblasts and normal diploid fibroblasts expressing either flag-progerin, wild-type flag-lamin A, or GFP. White arrow in the bottom right panel shows nuclear blebs that are occasionally seen in normal fibroblasts. B. Quantitation of the cells with one or more nuclear blebs from passages 7 to 13. For each experiment, 300 cells were scored by two independent observers for nuclear blebs as described in Materials and Methods. Data shown for cells at passages 7 through 11 represent averages of two measurements per cell line while data for passage 13 represent mean +/− standard deviation of 3 measurements in two independent cell lines for each group.
To identify the processes responsible for the proliferative defect observed in fibroblasts expressing flag-lamin A and progerin, we counted the numbers of senescent and apoptotic cells at each passage. Cell senescence was examined by staining the cells for the presence of senescence-associated β-galactosidase (SA-βGal), a marker that is expressed by senescent, but not by pre-senescent and quiescent fibroblasts, or terminally differentiated cells (Dimri & Campisi, 1994). These analyses demonstrate that cells expressing progerin display a progressive, passage-dependent increase in the percentage of senescent cells over a time frame which spanned passages 5 to 10 compared to control cells (Figure 4, panel A, n=4; p=0.0062 for a two-way comparison between control and progerin expressing cells), which levels off after passage 10. Importantly, senescence seems to be directly linked to expression of progerin, since the loss of progerin expression observed in the revertant cell line is associated with a significant decrease in the number of senescent cells. A similar but slightly delayed trend was observed in the cells expressing flag-lamin A; while the early passages display a level of cell senescence comparable to control cells, there was a substantial increase in senescent cells after passage 7, where the percentage of senescent cells reached levels similar to the late passage (passage 13) cells expressing progerin.
Figure 4.
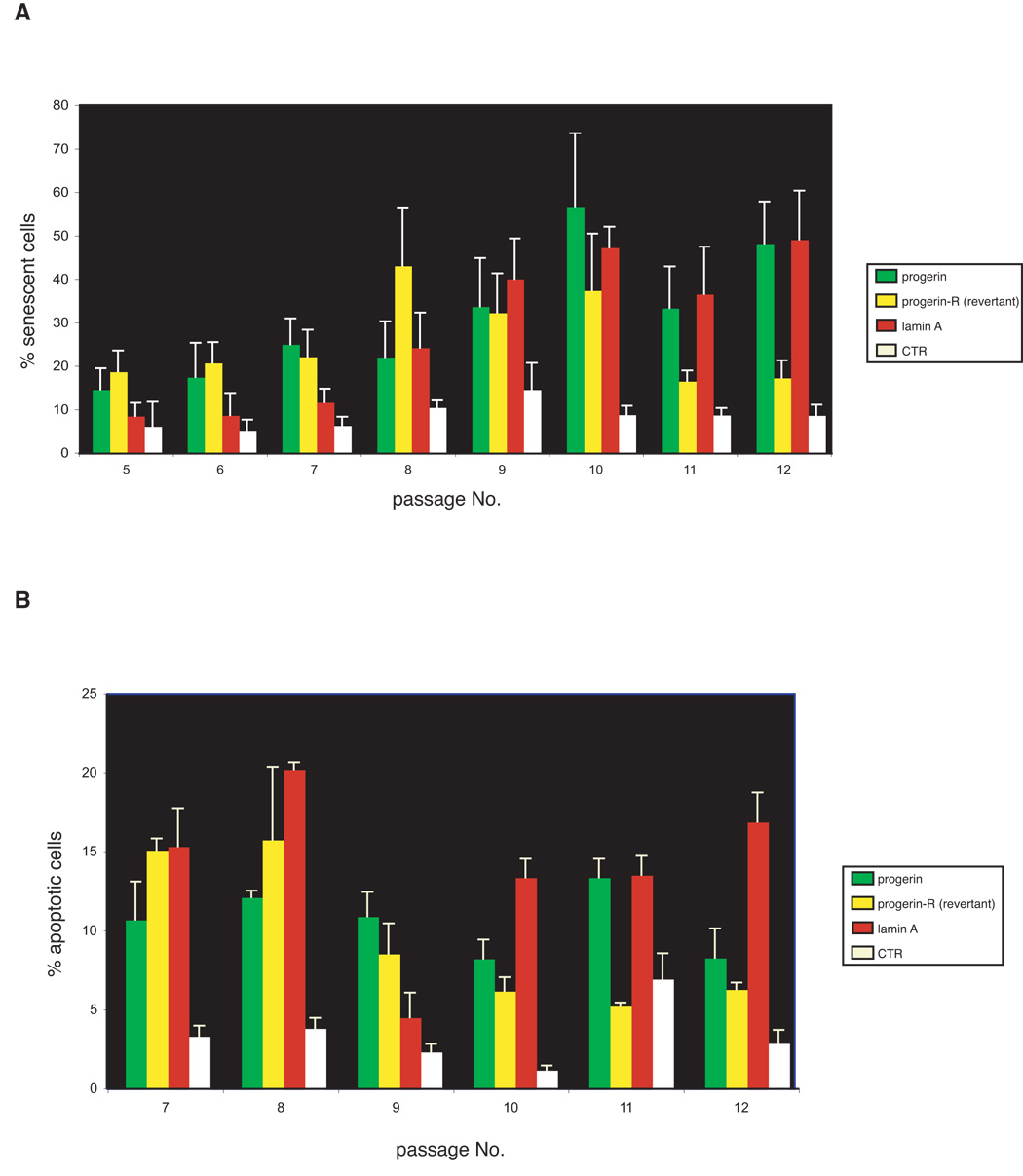
Cell senescence and apoptosis contribute to the cellular aging phenotype of cells expressing elevated levels of wild-type lamin A. A. The percentage of senescent cells at passage 5 through 12 was determined by staining for senescence-associated β-galactosidase as described in Materials and Methods. B. The percentage of apoptotic cells at passage 7 through 12 was determined by Tunnel assays as described in the supplementary Materials and Methods. Data shown represent mean +/− standard deviation of three independent measurements.
To determine whether abnormalities in the morphology of the nuclear membrane in cells expressing wild-type lamin A or HGPS cells was linked to senescence, we examined the cells with blebs for the presence of senescence-associated β-galactosidase (SA-βGal). This analysis reveals that approximately 80% of cells overexpressing wild-type lamin A showing nuclear membrane blebs are senescent, while 50% of HGPS cells with blebs are senescent (supplementary Figure S3). Thus, although there is a significant correlation between nuclear membrane abnormalities and cellular senescence in both cell lines, the relative difference in the number of cells with nuclear blebs that are senescent between cells expressing wild-type lamin A and HGPS cells may underscore a divergence in the pathways that lead to the progeroid phenotype.
To determine whether cell death also contributes to the poor growth of cells expressing progerin or elevated level of lamin A, we detected and counted apoptotic cells between passage 7 and 12 using the TUNEL assay. Both cell lines display elevated levels of apoptotic cells, which are significantly higher than those of control cells (Figure 4, panel B, n=4; p< 0.0001 for a two-way comparison between control cells and either cells with elevated levels of lamin A or progerin expressing cells between passage 7 and 12); however, unlike senescent cells, the percentage of apoptotic cells is high at early passages and remains fairly constant at each passage in both lines expressing progerin and wild-type flag lamin A, with the exception of flag-lamin A expressing cells at passage 9, which showed levels of apoptosis comparable to those of control cells. Notably, the levels of apoptotic cells in the progerin-revertant cell culture declines after passage 9, which correlates with the loss of progerin expression in this cell line. Collectively, these results indicate that both cell death and senescence contribute to limiting the proliferative capacity of cells expressing either progerin or elevated levels of lamin A.
Recent studies have shown that treatment with farnesyl transferase inhibitors (FTIs) can reverse the nuclear morphology defects in HGPS cells and Hela cells transiently expressing progerin (Capell et al., 2005; Glynn & Glover, 2005; Mallampalli et al., 2005; Toth et al., 2005; Yang et al., 2005). These results suggest that accumulation of farnesylated progerin is, at least in part, responsible for the formation of nuclear blebs. This set of experiments did not however investigate whether FTIs are also capable of reversing the growth defects associated with progerin expression. Since elevated levels of lamin A induce growth defects and abnormal nuclear architecture similar to that observed in cells expressing progerin, we tested the effect of FTI on the cellular defects observed both in cells expressing progerin as well as those with elevated lamin A levels. For this purpose, control cells (GFP-vector) and cells expressing flag-lamin A or progerin at passage 13 were split into two aliquots and incubated in media containing the FTI L-744832 or vehicle only (DMSO). Fourteen days of continuous treatment with FTI resulted in improved cell growth and a reduction in the number of nuclear blebs in progerin-expressing cells (Figure 5, Panel A; n=3; p=0.071 for a two-way comparison of growth between cell pairs treated with DMSO and FTI; Panel B; n=3; p=0.093 for a two-way comparison of nuclear blebs between cell-pairs treated with DMSO and FTI). This indicates that prevention of the accumulation of farnesylated progerin can partially revert the growth defects and morphological alterations of the nuclear membrane of progerin-expressing cells. Importantly, in cells over-expressing lamin A, a statistically significant increase in growth and a significant decrease in the number of cells with nuclear membrane blebs is observed upon FTI treatment (Figure 5, Panel A; n=3; p <0.0001 for a two-way comparison in growth between cell-pairs treated with DMSO and FTI; Panel B; n=3; p =0.005 for a two-way comparison of nuclear blebs between cell-pairs treated with DMSO and FTI). In contrast, FTI treatment does not alter growth rates or nuclear morphology in normal (control) or progerin-revertant cells (Figure 5, Panel A, n=3; p=0.628 (progerin-R) and p=0.384 (control) for a two-way comparison of growth between cell-pairs treated with DMSO and FTI; Panel B, p =0.102 (progerin-R) and p=0.177 (control) for a two-way comparison of nuclear membrane blebs between cell-pairs treated with DMSO and FTI). These data therefore demonstrate that a two week treatment with FTIs does not have an overt toxic effect on growth or nuclear morphology in human cells but can rescue the growth defect and nuclear membrane abnormalities exhibited by progerin and wild-type flag-lamin A expressing cells.
Figure 5.
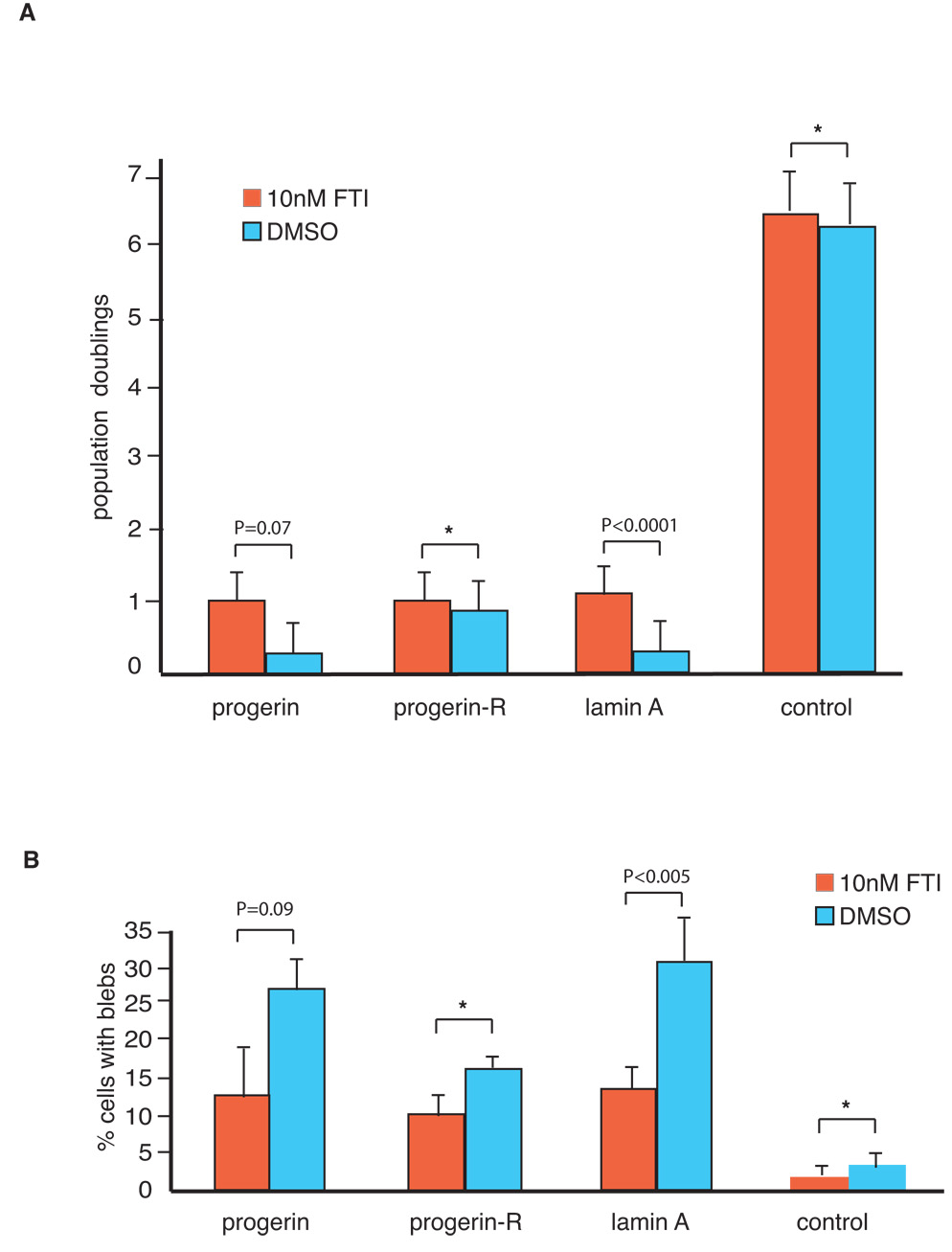
Treatment with farnesyl transferase inhibitors improves growth and reduces nuclear blebs in fibroblasts expressing increased levels of wild-type lamin A. Fibroblasts expressing flag-lamin A, flag-progerin and GFP (control) at passage 13 were incubated in media containing the 10 nM farnesyl transferase inhibitor (FTI) L-744832 or in media containing the vehicle only (DMSO) for 14 days. Media was changed every four days. Cells were then analyzed for growth (A) and nuclear morphology (B) as described in Materials and Methods. Data shown represent mean +/− standard deviation of three independent measurements. Statistical significance between treatment groups was determined by a two-tailed student’s t-test. Asterisks: the data does not reach statistical significance.
ZMPSTE24, a metalloprotease involved in the lamin A maturation process, is not predicted to affect progerin metabolism, as the cleavage site is absent in progerin. However, elevated levels of ZMPSTE24 are predicted to reduce the toxicity caused by over expression of wild-type lamin A if increased synthesis or accumulation of farnesylated prelamin A intermediates play a causal role in the senescent phenotype observed in the cells over-expressing lamin A. To test this idea we determined whether the growth defects and nuclear abnormalities of fibroblasts over-expressing wild-type lamin A could be rescued by increasing the amount of ZMPSTE24 in the cell. Specifically, GFP control, flag-lamin A and flag-progerin expressing fibroblasts at passage 13 were transduced with either a lentivirus expressing epitope tagged ZMPSTE24 or GFP and their growth rates and nuclear morphology were examined 2 weeks after transduction. This analysis shows that over-expression of ZMPSTE24 improves the growth properties and leads to a significant decrease in the number of nuclear blebs in cells expressing elevated levels of wild-type lamin A (Figure 6, Panel A; n=3; p <0.0001 for a two-way comparison of growth between cell-pairs transduced with the control virus and the ZMPSTE24-expressing virus; Panel B; n=3; p<0.0001 for a two-way comparison of number of cells with nuclear blebs between cell-pairs transduced with the control virus and the ZMPSTE24-expressing virus). In contrast, cell growth and nuclear blebs in cells expressing progerin, which lacks the ZMPSTE24 cleavage site, were not affected by ZMPSTE24 over-expression (Figure 6, Panel A; n=3; p =0.157 for a two-way comparison of growth between cell-pairs transduced with control virus and ZMPSTE24-expressing virus; panel B; n=3; p=0.148 for a two-way comparison of number of cells with nuclear blebs between cell-pairs transduced with the control virus and the ZMPSTE24-expressing virus). These data are consistent with the hypothesis that elevated levels of ZMPSTE24, which may serve to increase the rate of prelamin A maturation, can rescue the growth defects and nuclear morphology abnormalities resulting from lamin A over-expression.
Figure 6.
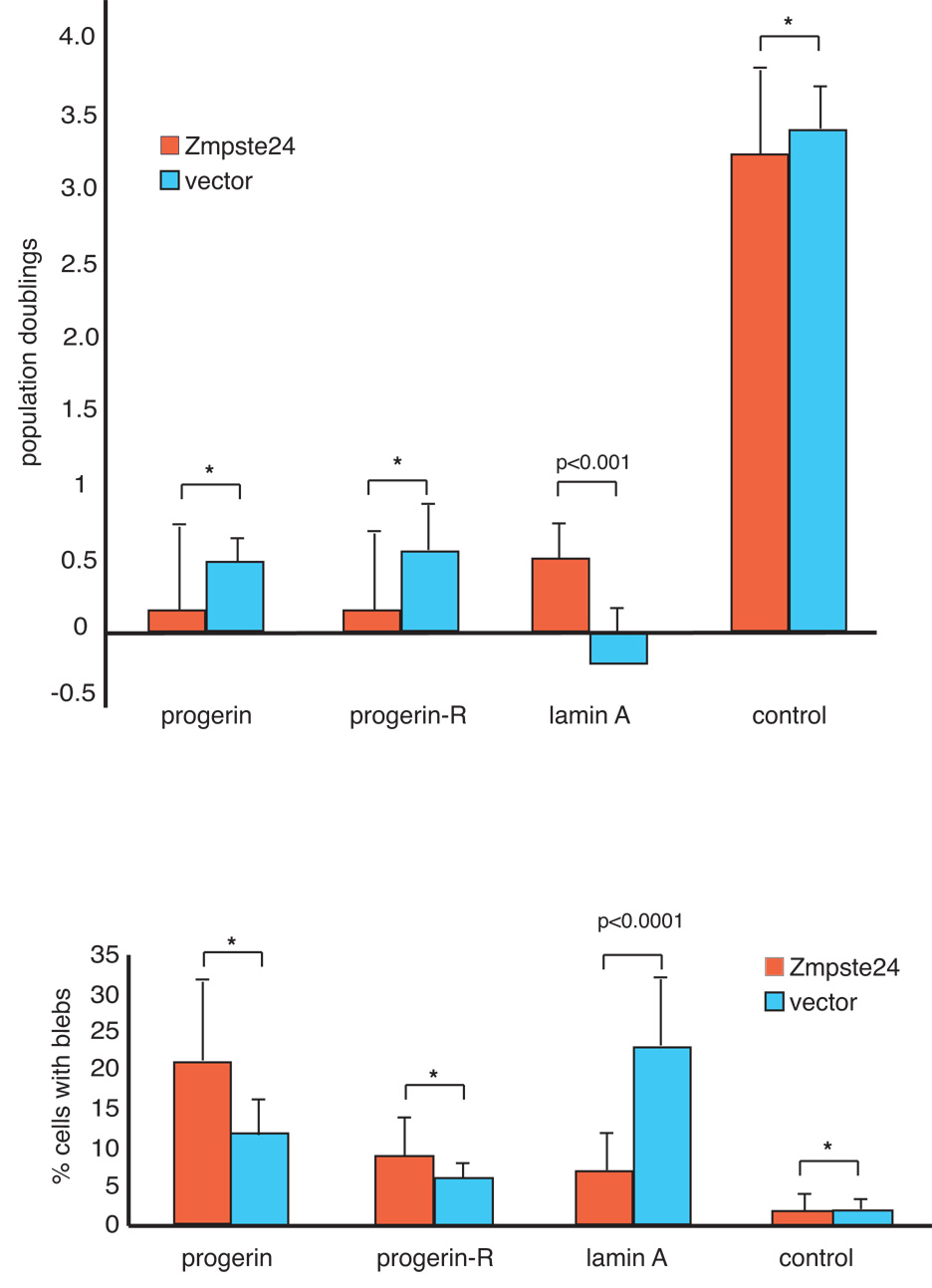
Over-expression of ZMPSTE24 improves growth and reduces nuclear blebs in fibroblasts with elevated levels of wild-type lamin A. Fibroblasts expressing flag-lamin A, progerin and GFP (control) were transduced at passage 13 with lentiviruses expressing ZMPSTE24 or vector control. Two weeks after transduction, the cells were analyzed for cell growth (A) and nuclear morphology (B) as described in supplementary Materials and Methods. Data shown represent the mean +/− standard deviation of three independent measurements. Statistical significance between treatment groups was determined by a two-tailed student’s t-test. Asterisk denotes p values greater than 0.05.
To study the stoichiometry of prelamin A to mature lamin A in normal and flag-lamin A expressing cells we carried out western blot analysis of extracts prepared from cells grown in normal media or media containing either 0.1 µM or 1 µM FTI for 48 hours using lamin A/C and flag antibodies, and an antibody against prelamin A. This analysis shows that both normal and lamin A over-expressing cells treated with FTI accumulate prelamin A (Figure 7A, lanes 2, 3 and 5, 6). However, we did not observe any detectable level of prelamin A in cells expressing ectopic lamin A (Figure 7A lane 1), even when we used an antibody raised against prelamin A (data not shown). As the western blot analysis may not be sufficiently sensitive to detect small increases in prelamin A intermediates, we follow the biosynthesis and maturation of lamin A by metabolically labeling cells for 17 hours with 35S-methionine. The levels of lamin A and its biosynthetic precursors were analyzed by SDS-polyacrylamide gel electrophoresis and fluorography of immunoprecipitated proteins. The results of these experiments indicate that cells expressing either untagged or flag-tagged lamin A display a 1.5 to 2.0 fold increase in the steady-state levels of prelamin A intermediates compared to control cells (Figure 7B). Since this analysis provides an average fold-increase for all intermediates, depending on the nature of the toxic product, the specific level of increase could be more significant. This analysis also indicates that the rate of biosynthesis of mature lamin A in cells expressing wild-type lamin A is not significantly different from that of control cells.
Figure 7.
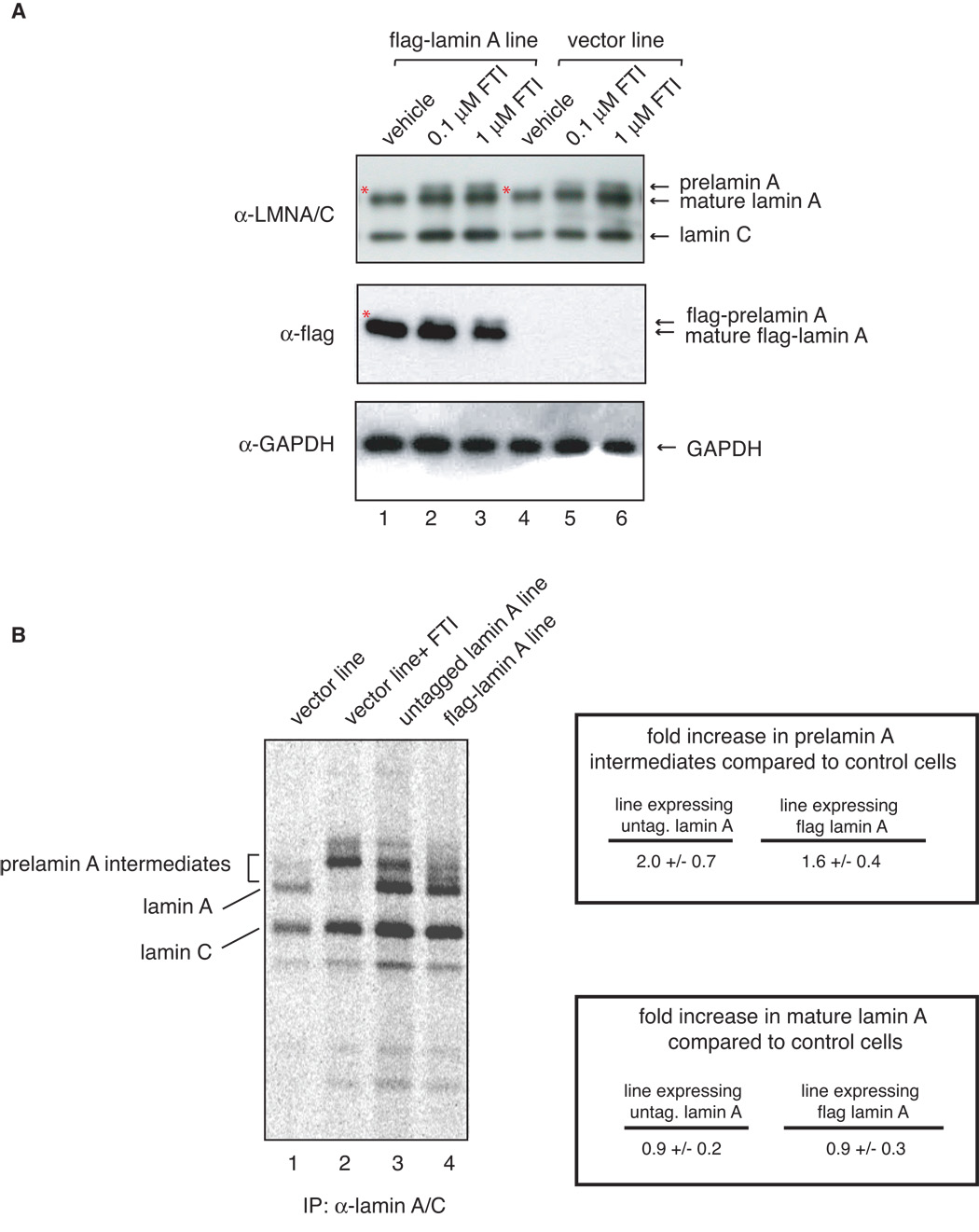
A There is no detectable accumulation of prelamin A in untreated flag-lamin A expressing fibroblasts. Fibroblasts expressing flag-lamin A and control at passage 10 were treated with vehicle only (lanes 1 and 4), and 0.1 µM (lanes 2 and 5) or 1 µM (lanes 3 and 6) farnesyl transferase inhibitor L-744832 for 48 hours, collected and analyzed by western blot analysis with lamin A/C (top panel), flag (middle panel) and GAPDH (lower panel) antibodies. To the best of resolution we do not see prelamin A accumulation in vector and flag-lamin A expressing fibroblasts treated with vehicle, even at lighter exposure (data not shown). However, Lamin A and flag-lamin A accumulate in FTI-treated control and flag-lamin A expressing fibroblasts. Asterisks show absence of prelamin A and flag-prelamin A in vehicle-treated cells. B. Immunoprecipitation analysis of prelamin A intermediates in metabolically labeled cells. Control fibroblasts, which were either untreated (lane 1) or treated with FTI (lane 2), and fibroblasts expressing either untagged (lane 3) or flag-tagged (lane 4) human wild type lamin A and were subjected to long term metabolic labeling and analyzed by fluorography as described in Materials and Methods. Biosynthesis of lamin A precursors and mature lamin A was visualized by phosphorimager and quantitated. Amounts of prelamin A intermediates and mature lamin A in each sample were normalized to lamin C and are shown as fold increase over that of control cells.
To investigate whether lamin A overexpression influences the cellular distribution of lamin A, we performed immunofluorescence analysis in control cells and cells expressing elevated levels of either flag-lamin A or untagged lamin A. In control fibroblasts lamin A shows uniform nuclear rim and diffuse nucleoplasmic staining. However, the distribution of lamin A in cells expressing elevated levels of lamin A is significantly altered when compared to control cells (Figure 8). Specifically, cells expressing elevated levels of lamin A display lamin A aggregates at the nuclear periphery (rim) and atypical lamin A “folds-like” structures (Figure 8A and C), which penetrate the nucleoplasmic area (supplemental Figure S4). Both patterns are rarely observed in passage-matched control cells, but are a prominent feature of progeria cells (Figure 8 and data not shown). To assess whether these alterations in the pattern of lamin A accumulation were characteristics of normal cellular aging, we performed immunofluorescence analyses of human diploid fibroblasts isolated from young (17 to 23 years of age) and old-age (90 and 91 years of age) individuals. Interestingly, cells from old-age individuals but not cells from young individuals display uneven distribution of lamin A along the nuclear rim and lamin A folds that are similar to those observed in cells expressing elevated levels of lamin A (Figure 8). Since these alterations are rarely observed in fibroblasts from young individuals, these data suggest that abnormal accumulation of lamin A may represent a phenotypic change relevant to cellular aging.
Figure 8.
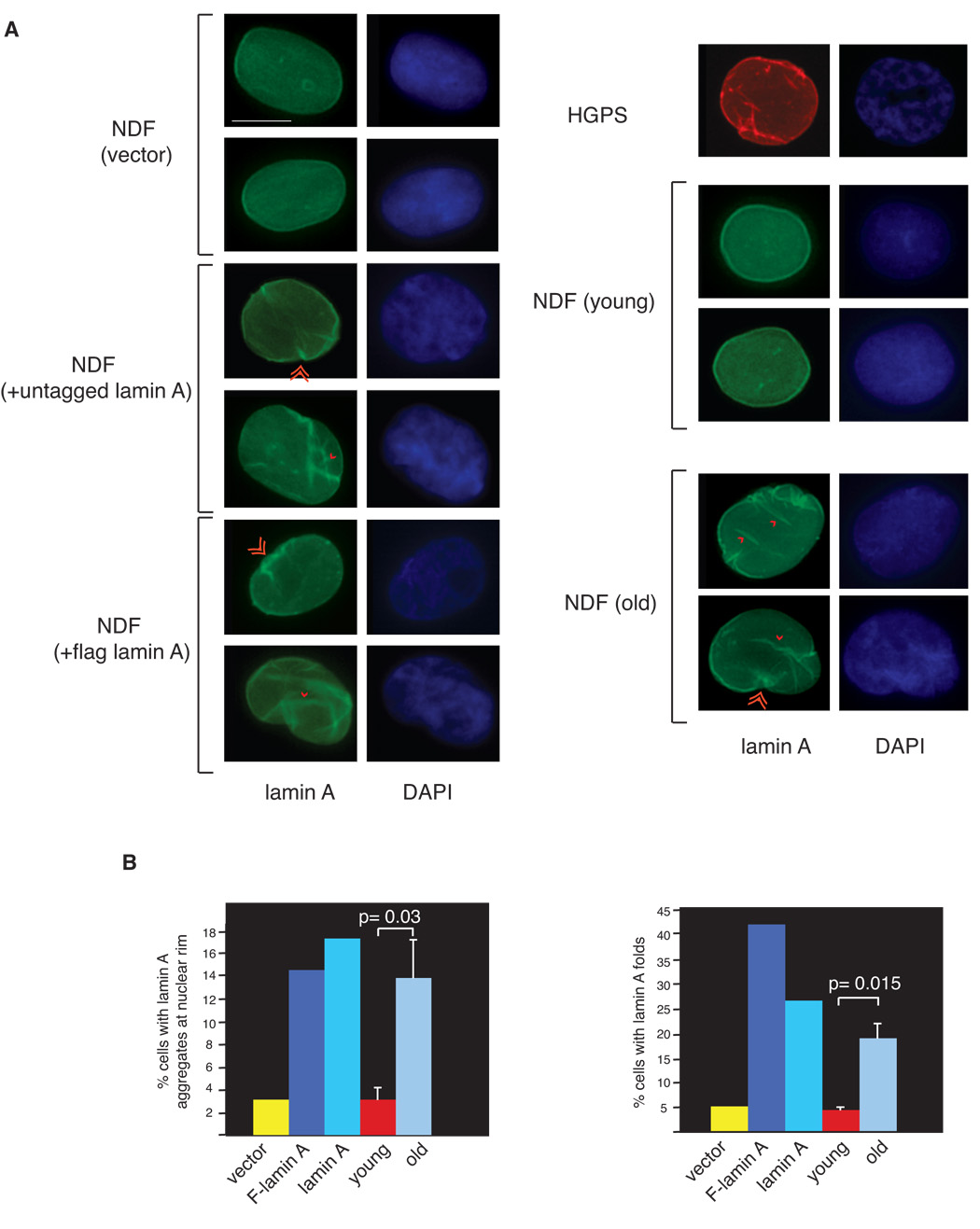
A Abnormal pattern of lamin A localization in cells expressing elevated levels of wild-type lamin A resembles that of cells from old-age individuals. Immunofluorescence microscopy on primary diploid fibroblasts transduced with control lentivirus and lentiviruses expressing either lamin A or Flag-lamin A, primary fibroblasts from young (30 y) and old (87 y) healthy individuals and fibroblasts from a HGPS patient. DAPI, 4′,6′-diamidino-2-phenylindole. Single and double arrowheads show lamin A aggregates at nuclear rim and lamin A folds, respectively. Bar: 10 µm B. Quantitation of percentage of cells (n= 300) showing either lamin A aggregates at the nuclear rim (right panel) or lamin A folds (left panel). Data for young and old age fibroblasts represent mean +/− standard deviation of analyses performed using 3 distinct cell lines from young and old healthy individuals. Statistical significance of the differences between young and old individuals was determined by a two-tailed student’s t-test.
Discussion
In this study we demonstrate that ectopic expression of wild-type lamin A in normal human cells leads to reduced replicative lifespan and induces nuclear membrane abnormalities that result in apoptotic cell death and senescence. Importantly, our data show that these phenotypes can be rescued by over-expression of ZMPSTE24, the metalloproteinase responsible for the removal of the farnesylated carboxyl terminal region of lamin A or by treatment with a farnesyltransferase inhibitor. These findings suggest that toxicity is likely related to alterations in the prelamin A metabolism. The processing of prelamin A into mature lamin A is a multistep process and our initial analysis suggests that small increases in the steady-state levels of one or more lamin A precursors are responsible for the observed phenotype. The fact that either FTI treatment or ZMPSTE23 overxpression rescue both growth and nuclear membrane abnormalities strongly suggests that the toxic prelamin A intermediate is farnesylated. The precise identification of this toxic molecule requires further biochemical analyses and is an important goal of our future studies.
As prelamin A is processed into mature lamin A by farnesylation and proteolytic cleavage in normal cells, our results are consistent with the hypothesis that toxicity is a function of two parameters: the number of prelamin A molecules produced and the length of time a prelamin A intermediates are present in a cell [toxicity α (number of prelamin A molecules) (processing time of farnesylated prelamin A intermediates)]. In HGPS cells, the synthesis of progerin leads to the formation of a stable farnesylated protein that is constitutively embedded in the nuclear membrane (number of prelamin A molecule = normal; processing time ~ large) (Figure 9, ii). Thus toxicity is caused by the presence of a permanently farnesylated protein in the nuclear membrane. In contrast, elevated expression of wild-type prelamin A leads to an increase in the number of prelamin A molecules that are funneled into this pathway (Figure 9, iii). Thus, toxicity in this event does not appear to be caused by an increase in the processing time of prelamin A intermediates, but rather to an increase in the number of prelamin A molecules synthesized at a given time (processing time = normal; number of prelamin A molecules = elevated). This functional relationship predicts that over expression of ZMPSTE24 would be beneficial in cells over expressing wild-type lamin A, as it would serve to reduce the time that prelamin A precursors are present in the nuclear membrane (Figure 9, iv)). This latter prediction is borne out by our experiments.
Figure 9.
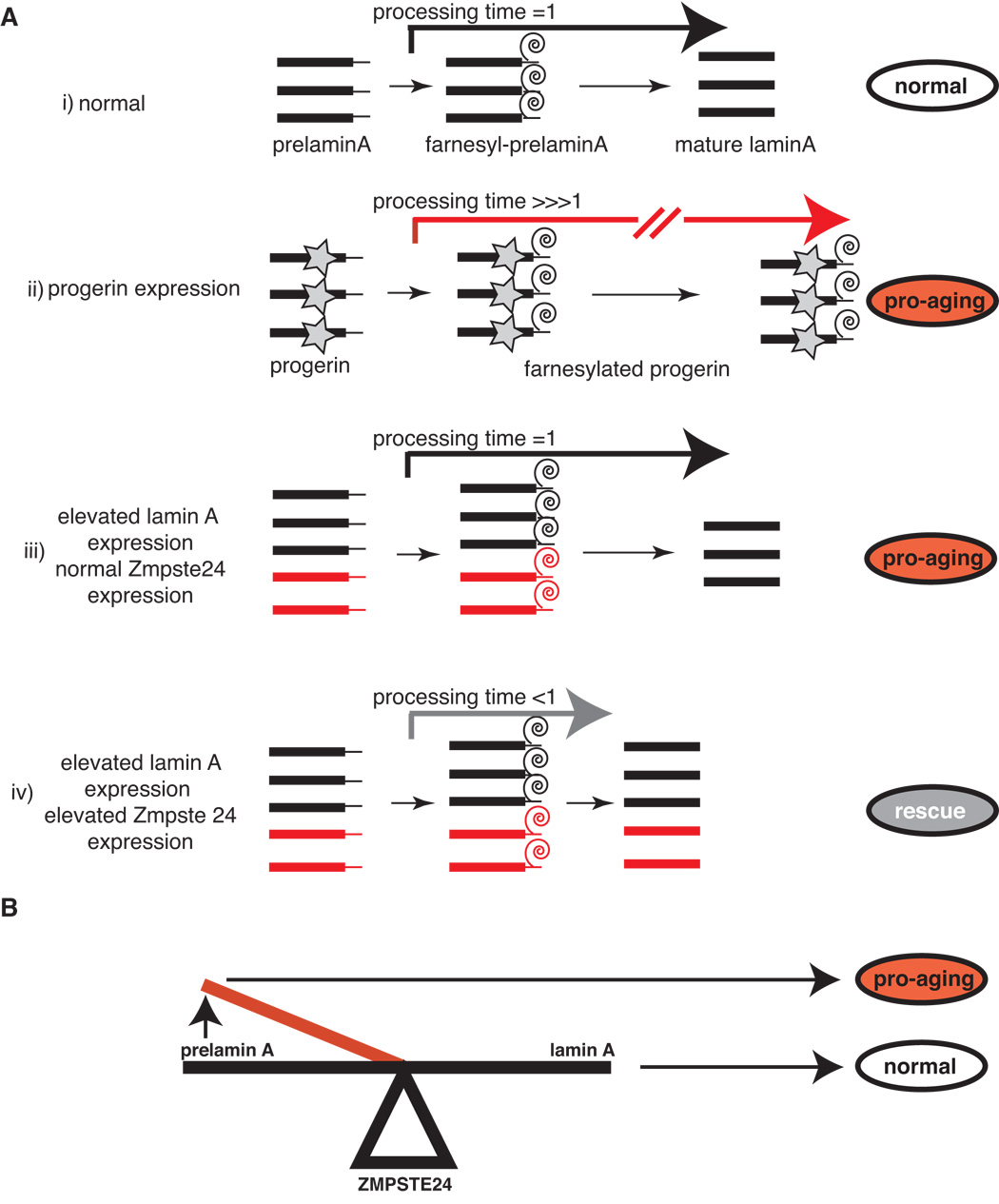
A. Hypothetical model for the toxicity of progerin and elevated levels of wild-type lamin A is shown. Prelamin A processing time in normal cells has been set arbitrarily as 1. Abnormal processes/steps are shown in red. i): maturation pathway of prelamin A in normal cells; ii): progeria cells display an exceptionally high processing time as the farnesyl group on mutant lamin A cannot be cleaved by ZMPSTE24. This conditions lead to cellular aging that can be partially reversed by decreasing the number of farnesylated molecules in the cell by treatment with FTIs iii): Elevated levels of wild-type lamin A expression do not change the prelamin A processing time as the synthesis of mature lamin is not affected, but increase the overall number of prelamin A intermediates at a given time. This conditions lead to cellular aging. iv): over-expression of ZMPSTE24 reduces the prelamin A processing time and rescues the cellular aging phenotype in cells with elevated levels of wild-type lamin A expression. B. Lamin A metabolism is precisely poised such that small increases in the steady-state levels of one or more prelamin A intermediates caused by increased lamin A expression or decreased ZMPSTE24 activity can propel the cell into a cellular aging phenotype.
A recent study has shown that a small amount of progerin is produced in normal cells by infrequent use of the cryptic splice site on exon 11 (Scaffidi & Misteli; 2006) and has suggested that this rare splicing event in lamin A RNA can result in age-associated nuclear defects in cells from older individuals who do not carry the HGPS mutation. We did not observe any detectable level of progerin protein in cells overexpressing wild-type lamin A even at late passages by western blot analysis (Figure 1 and Figure 2). Furthermore, RT-PCR analysis did not reveal any difference in the amount of progerin RNA present in control and wild-type lamin A overexpressing fibroblasts, as wells as fibroblasts from young and old-age individuals (supplementary Figure S5). Thus, these results demonstrate that the observed phenotypes including defective proliferation, nuclear bleb formation, irregular lamin A localization and cell death and senescence, are induced by wild-type lamin A rather than aberrant accumulation of progerin.
Both wild-type and mutant lamin A can lead to cellular aging. However, it is significant that the aging phenotype exhibited by over-expression of lamin A and progerin have slightly different kinetics. For example, a significant increase in the percentage of senescent cells is observed at passage 7 in lamin A over-expressing cells compared to passage 5 in progerin expressing cells (Figure 4, Panel A). Moreover, nuclear membrane blebs are observed only after nine passages in cells expressing elevated lamin A levels, while progerin expressing cells have a more rapid (within hours) effect on nuclear morphology (Figure 3, Panel B; Goldman et al., 2004). Lastly, while more than 80% of the cells expressing elevated levels of wild-type lamin A with nuclear membrane blebs are senescent, cell senescence is observed in only 50% of progerin expressing cells with nuclear blebbing (supplementary Figure S3). These observations suggest that there may be qualitative differences between expression of progerin and elevated levels of lamin A. This delayed kinetics in the manifestation of the aging phenotype exhibited by lamin A over-expression may be the reason why transient over expression of lamin A did not lead to a defective phenotype in previous studies (Capell et al., 2005; Glynn & Glover, 2005; Mallampalli et al., 2005).
Expression of the disease-linked progerin induces dramatic changes in the pattern of lamin A localization, which are likely to contribute to the onset of HGPS. Our study demonstrates that elevated levels of lamin A induce similar changes in the pattern of lamin A localization, including the formation of lamin A aggregates at the nuclear periphery and lamin A folds-like structures. Interestingly, a similar pattern of lamin A localization is observed in cells from old-age individuals but rarely seen in cells from young individuals, suggesting that alterations in the nuclear distribution of lamin A are relevant cellular changes that occur during the aging process.
Collectively, these data suggest that the lamin A metabolism is a finely poised system where minimal alterations in the pathway that result in the synthesis of elevated levels of lamin A production or reduced ZMPSTE24 activity can propel the system into an aging phenotype (Figure 8, Panel B). Consistent with this model mutations in Zmstpe24 have been shown to lead to growth retardation, skeletal abnormalities and cutaneous atrophy (Agarwal et al., 2003; Navarro et al., 2005) and ZMPSTE24 knock out mice display a progeroid phenotype (Bergo et al., 2002; Pendas et al., 2002). Thus, our data show that even in the absence of disease-linked genetic mutations, minor perturbations in the expression of lamin A can cause striking nuclear defects and profoundly affect the course of cellular aging. Importantly, these results demonstrate that the lamin A metabolism may not be limited to its role in the development of an accelerated aging phenotype in rare syndromes but may be a more general contributor to normal cellular aging.
Experimental Procedures
Cell culture
Primary dermal fibroblast cell lines from HGPS patients (AG11498 and HGADFN003), healthy newborn (GM00038 and GM00316), young-age (AG06234, 17y; AG11747, 22y; AG07804, 23y) and old-age (AG06291, 90y; AG12788, 91y; Ag07725, 91y) individuals were obtained from the Coriell Cell Repository. Cells were grown in DMEM supplemented with 15% fetal bovine serum, 2mM L-glutamine, 100U/ml penicillin and 100µg/ml streptomycin at 37°C in 5% CO2, and 3% O2. Cells seeded at 1.4 × 105 per 100 mm diameter dish were passaged when cultures reached 85% confluency. Cell growth was measured by calculation of accumulated population doublings using the formula (log H − log S)/ log2, where log H is the logarithm of the number of cells harvested and log S is the logarithm of the number of cells seeded on the first day of each passage, as described in Bridger & Kill (2004).
Construction of lamin A, progerin and ZMPSTE24 vectors and generation of stably transduced cell lines
Mouse lamin A cDNA was purchased from ATCC (MGC-19217) and cloned into the pCR4-topo vector by PCR using the following primers: 5’ -TCCATAGFFAGACCCCGTCACAG-3’/ 5’-GAATTCTTACATGATGCTGCAGTTCTG- 3’ Sequence accuracy was verified by DNA sequencing. Progerin cDNA (lamin A HGPS mutant, G608G(C → T transition at position 1824 of exon 11 of the lamin A/C gene) was generated by introducing the HGPS-specific 150 nucleotide deletion in the wild-type lamin A cDNA using a two step PCR-based protocol with the following primers: PCR-1, 5’-TCCATAGFFAGACCCCGTCACAG-3’/ 5’-TCTGGGCTCCCGCTCCA-3’, PCR-2, 5’-TCCATAGFFAGACCCCGTCACAG-3’/ 5’-GATTACATGATGCTGCAGTTCTGGGA GCTCTGGGCTCCCGC-3’. The PCR product was then cloned into the pCR4-TopoTA vector (Invitrogen) and the accuracy of the sequence was verified by DNA sequencing. The lamin A and progerin cDNAs were subcloned into the Nde1-EcoR1 sites of the pVL1393-Flag vector (Comai et al., 1994). Flag-tagged wild-type and progerin lamin A cDNAs were then subcloned into the BamHI/Xba I sites of the lentiviral transfer vector pkey204 MSH2-IRES-GFP (kind gift of Drs. Richard Fishel and Kristine Yoder, Kimmel Cancer Institute, Thomas Jefferson University) to generate pkey-Flag-mLMNA and pkey-mProgerin. Recombinant lentiviruses were generated as previously described Li et al. (2004). Lentiviruses used for the expression of flag-tagged and untagged human lamin A were obtained by subcloning the human lamin A cDNA (ATCC No. 7517636) into the lentiviral transfer vector pkey204 MSH2-IRES-GFP to generate pkey-Flag-hLMNA and pkey-hLMNA, using an approach similar to that outlined above. For lentiviral infection, transfected 293T cell cultures were trypsinized, seeded onto 100 mm plates, and incubated at 37° C for 24 hours. The supernatant containing viral particles was collected, filtered and equal volumes of each viral supernatant were added to normal diploid human fibroblast cultures that were approximately 40% confluent. After 6 hours incubation at 37° C, the supernatant was removed, the cells were washed twice with phosphate saline buffer (PBS) and incubated in Dulbecco’s modified Essential medium containing 10% serum at 37° C. Transduced cells expressing the green fluorescent protein (GFP) were selected by fluorescence activated cell sorting (FACS). The expression of flag-tagged proteins was then analyzed by immunoblotting with anti-flag antibodies (Sigma). Each lentivirus was transduced in two independent fibroblast cell lines and each cell line was grown in duplicate. The human ZMPSTE24 cDNA was purchased from ATCC (No. MGC-33086) and cloned into the pCR4-TOPO vector using a PCR-based approach with the following primers: 5’-CATATGGGGATGTGGGCATC-3’/ 5’-TCTAGATCAGTGTTGCTTCATAGTTTTCAAAGC-3’. The ZMPSTE24 cDNA was subsequently subcloned into the Nde1-Pst1 sites of the pVL1393-Flag vector (Comai et al.,1994) and the DNA fragment containing the flag epitope and the ZMPSTE24 cDNA was then subcloned into the BamHI/Xba I sites of the lentiviral transfer vector pkey204 MSH2-IRES-GFP to generate pkey-ZMPSTE24. The sequences of the intermediate and final construct were verified by DNA sequencing. Lentivirus production and cell transduction were carried out as described above.
Senescence-associated β-galactosidase assay
SA-βGal activity was determined as described in Dimri & Campisi (1994). Briefly, cells were washed in PBS, fixed for 3–5 min at room temperature in 3% formaldehyde, washed, and incubated for 12–16 hours at 37°C with freshly made staining solution containing 1mg/ml 5-bromo-4-chloro-3-indolyl β-D-galactoside (X-Gal), 40 mM citric acid/sodium phosphate, pH 6.0, 5mM potassium ferrocyanide, 5mM potassium ferricyanide, 150 mM NaCl, 2mM MgCl2. The percentage of SA β-gal cells in each cell lines was determined by scoring positive cells by microscopic examination of 150 to 500 cells from at least three independent samples for each cell line. The scorer was blind to the identity of the sample.
Analysis of cell death
Apoptotic cells were detected using the Terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end-labeling (TUNEL) assay according to the manufacturer's instructions (Roche). Briefly, cells were seeded on a chambered slide each time the cells were passaged, allowed to adhere, and fixed for 1 hour in 4% paraformaldehyde in PBS. Cells were then permeabilized in 0.1% triton X-100/0.1% sodium citrate in PBS 2 minutes on ice. Cells were washed in PBS and a labeling mix containing fluorescein dUTP and terminal deoxynucleotidyltransferase was added to the cells and incubated for1 h at 37°C in a dark humidified atmosphere. The labeled cells were then washed in PBS and Converter-AP was added to the cells and incubated 30 min at 37°C. Subsequently the cells were again washed in PBS and the substrate solution was added to the cells and incubated for 30 min at room temperature in the dark. The percentage of apoptotic cells in at least three independent samples was determined by scoring 300 to 600 cells from at least three independent samples from each cell line under 100x microscopic magnification.
Nuclear Morphology Analysis
Nuclear membrane alterations were analyzed by fluorescence microscopy of DAPI-stained cells. 2.3×103 cells were seeded in chambered slides and were analyzed at each passage. Cells were washed in PBS and fixed using 4% paraformaldehyde for 5 min at room temperature. Fixed cells were then washed again in PBS and incubated in a permeabilization solution (0.1% Triton X-100, 0.1% Sodium citrate) for 5 minutes on ice. Cells were then washed with PBS and DAPI (1µg/ml in PBS) was added for 1 minute in the dark. Subsequently the cells were washed in a permeabilization solution followed by PBS to remove the detergent. Each slide was then treated with Antifade reagent and allowed to dry in the dark. Nuclear morphology was analyzed with a fluorescence microscope at 200, 400, and 1000x magnification. Nuclei with blebs were considered as those with one or more lobulations resulting in misshaped nuclei. At each passage, two independent observers scored 300 cells for each cell line. For the comparison between cells grown in the presence or absence of FTI and between cells with and without ZMPSTE24 over-expression, 300 cells were counted in three plates by two independent observers, who were blinded to the identity of the samples.
Farnesyl Transferase Inhibitor treatment
Stably transduced cells at passage 12 were seeded (1×104 cells) on 6 well plates for growth rates and chamber slides for nuclear morphology. Cells were grown in DMEM supplemented with either 10 nM farnesyltransferase inhibitor L-744832 (Biomol, Plymouth Meeting, PA) or DMSO and 15% fetal bovine serum, 2mM L-glutamine, 100 U/ml penicillin and 100ug/ml streptomycin at 37°C in 5% CO2, and 3% O2. Media was changed every 4 days. After 14 days in culture, cell proliferation rates were determined by counting the cells in a hemocytometer under an inverted microscope at each passage. Counts were calculated and plotted as described above. Nuclear morphology was examined by fluorescent microscopy of DAPI-stained cells.
Western blot analysis
Human fibroblasts were washed twice in PBS, collected, and lysed in SDS Sample Buffer at 95°C for 5 minutes. Cell extracts were resolved by SDS-PAGE and transferred to PVDF membranes. Blots were probed with anti-prelamin A (goat polyclonal; Santa Cruz, sc-6214), anti-GAPDH (goat polyclonal; Santa Cruz, sc-20357), anti-Lamin A/C (rabbit polyclonal; Santa Cruz, sc-20681), anti-lamin A (rabbit polyclonal; Novus) anti-Flag (mouse monoclonal, Sigma, F-3165) or anti-ZMPSTE24 (Abgent). Prelamin A antisera was a generous gift of Dr. Stephen Young (UCLA). Immunoreactive bands were detected with the appropriate horseradish peroxidase-conjugated secondary antibodies (Pierce) and visualized by enhanced chemiluminescence (Amersham). For the determination of the relative levels of lamin A in control and lamin A over-expressing fibroblasts, 10, 20 and 30 µg of total proteins were subjected to western blot analysis using lamin A/C, flag and GAPDH antibodies. The band intensities were measured by densitometry analysis using Gene Tool software (Syngene Inc., USA) and the increment of lamin A in the lamin A-overexpressing cells was calculated as the lamin A/GAPDH ratio over lamin C/GAPDH ratio relative to the endogenous protein. The same approach was used to measure the relative levels of progerin.
Immunofluorescence protocols and confocal microscopy
For immunofluorescence staining, cells were fixed with 4% paraformaldehyde in PBS (pH 7.2) and permeabilized with 0.5% Triton X-100 in PBS. After 30 min incubation in blocking buffer (10% FCS in PBS), cells were incubated with primary antibodies at 4°C overnight. Cells were then washed three times with PBS and incubated with FITC- or Texas Red-conjugated secondary antibodies for 2 hrs at room temperature. The cells were then washed with PBS, counterstained with 0.05 µg/ml 4′, 6’-diamidino-2-phenylindole (DAPI) in PBS and mounted in Immunon™ mounting (Shandon, PA). The samples were analyzed using Nikon fluorescence microscope with 100X and 64X objective lenses. Image acquisition and analyses were carried out using Metamorph software (Molecular Devices). For confocal microscopy analysis, images were captured using a Zeiss LSM510 confocal microscope. Exposure times were kept constant for each fluorescence channel within each experiment and antibody used. 300 randomly selected cells were scored to determine the percentage of cells with lamin A aggregates at the nuclear rim and cells with lamin A folds.
Metabolic labeling of cells
Fibroblasts were radiolabeled for 16 hours with [35S]methionine (50 µCi/ml; specific activity, 1175 Ci/mmol). After labeling, the cells were harvested, sonicated in RIPA buffer (50mM Tris pH 8.0, 150 mM NaCl, 1% NP40, 0.5% deoxycholate, 0.1% SDS) and equal counts equivalent of each extract were subjected to immunoprecipitation using antibodies against lamin A/C. Immunoprecipitated products were separated by SDS-polyacrylamide gel electrophoresis and gels were soaked in EN3HANCE. Dried gels were exposed to phosphoimager screens and data were analyzed using Imagequant software (Molecular Dynamics) and expressed as ratios of prelamin A intermediates or mature lamin A over lamin C. To account for potential differences in inputs between samples, an aliquot of each extract was resolved on a separate SDS-polyacrylamide gel and dry gels were exposed to phosphoimager and analyzed using ImageQuant software (Molecular Dynamics).
RT-PCR analysis
RNA from each cell line was extracted and purified using the RNeasy kit (Quiagen) according to the manufacturer’s instructions. For each sample, 3µg of RNA were transcribed using the first strand cDNA synthesis kit from Amersham Biosciences for 1h at 37°C, after 10 min denaturation at 65°C. Primers for specific detection of Δ150 LMNA were located across the aberrant splice junction and in exon 12 (HGPS2F: 5’-AGCGGCTCAGGAGCCCAG---AG-3’; hLAex12R: 5’- CAGGGAAAAGGAAGGGAGGAG-3’). Primers used to amplify all LMNA transcripts were located across the splice junction of exon 11 and 12 and in exon 12 (hLAex11/12F: 5’-CAGCCCCCGAACCCAG---AG-3’; hLAex12R: 5’-CAGGGAAAAGGAAGGGAGGAG-3’). Primers for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were used for normalization (GAPDH-F: 5’-TGAAGGTCGGAGTCAACGGATTTGGT-3’; GAPDH-R: 5’-CCATGTGGGCCATGAGGTCCACCAC-3’). PCR reactions were carried out using 41 amplification cycles. PCR products were separated on 2% agarose gels and stained with Etidium Bromide.
Statistical analysis
Data were evaluated by Least Squares Means, non-parametric Kruskal-Wallis test, two-tailed student’s t-test, and p values were calculated using the statistical programs SAS and PROC MIXED.
Supplementary Material
Figure S1. Overall levels of lamin A in 20 control cells and 20 cells expressing ectopic wild-type lamin A. Lamin A was visualized by indirect immunofluorescence using lamin A specific antibody and FITC-conjugated secondary antibodies, and pixel intensity per cell was determined using Metamorph software and plotted as arbitrary units.
Figure S2. A. Growth curves of fibroblasts stably transduced with lentiviruses expressing the indicated proteins are shown as accumulated population doublings. B. Quantitation of cells with one or more nuclear blebs. For each experiment, 300 cells were stained with DAPI and scored by two independent observers. Data shown represent mean +/− standard deviation of 3 measurements in two independent cell lines for each group.
Figure S3. The percentage of fibroblasts with nuclear blebs that are senescent was determined by simultaneously staining cells with DAPI and for senescence-associated β-galactosidase as described in the Materials and Methods.
Figure S4. Lamin A folds penetrate the nucleoplasmic space. Confocal microscopy was performed on fibroblasts transduced with lentiviruses expressing either flag-lamin A or lamin A, fibroblasts from old-age individual and fibroblasts from a progeria patient. Images collected from two focal planes passing through the cell nucleus are shown. Bar: 10 µm
Figure S5. RT-PCR analysis of lamin A splicing in control fibroblasts, fibroblasts overexpressing wild-type lamin A, HGPS fibroblasts and fibroblasts from 3 young and 3 old-age individuals. Schematic representation of LMNA exons 9–12. The HGPS-specific splice site (vertical line with asterisk) and the position of the different primer sets used for RT-PCR are indicated in the diagram. The detection of wild-type and progerin mRNA using the indicated set of primers was carried out as described in Materials and Methods. RT-PCR analysis with primers that detect GAPDH mRNA was used as control.
Aknowledgments
We thank members of the Comai and Reddy labs for valuable suggestions during the course of this study. This investigation was supported by the Zumberge Innovation Fund and a grant from the Progeria Research Foundation awarded to L. C., and was conducted in a facility constructed with support from Research Facilities Improvement Program Grant Number C06 RR014514-01, C06 RR10600-01 and C06 CA62528 from the National Center for Research Resources, National Institutes of Health. J. C. is supported by an NIH Predoctoral Fellowship (F31 GM076861).
References
- Ackerman J, Gilbert-Barness E. Hutchinson-Gilford progeria syndrome: a pathologic study. Pediatr Pathol Mol Med. 2002;21:1–13. doi: 10.1080/pdp.21.1.1.13. [DOI] [PubMed] [Google Scholar]
- Agarwal AK, Fryns JP, Auchus RJ, Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet. 2003;12:1995–2001. doi: 10.1093/hmg/ddg213. [DOI] [PubMed] [Google Scholar]
- Bergo MO, Gavino B, Ross J, Schmidt WK, Hong C, Kendall LV, Mohr A, Meta M, Genant H, Jiang Y, Wisner ER, Van Bruggen N, Carano RA, Michaelis S, Griffey SM, Young SG. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc Natl Acad Sci U S A. 2002;99:13049–13054. doi: 10.1073/pnas.192460799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridger JM, Kill IR. Aging of Hutchinson-Gilford progeria syndrome fibroblasts is characterised by hyperproliferation and increased apoptosis. Exp Gerontol. 2004;39:717–724. doi: 10.1016/j.exger.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Capell BC, Erdos MR, Madigan JP, Fiordalisi JJ, Varga R, Conneely KN, Gordon LB, Der CJ, Cox AD, Collins FS. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2005;102:12879–12884. doi: 10.1073/pnas.0506001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comai L, Zomerdijk JC, Beckmann H, Zhou S, Admon A, Tjian R. Reconstitution of transcription factor SL1: exclusive binding of TBP by SL1 or TFIID subunits. Science. 1994;266:1966–1972. doi: 10.1126/science.7801123. [DOI] [PubMed] [Google Scholar]
- De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, Levy N. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003;300:2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- Dimri GP, Campisi J. Molecular and cell biology of replicative senescence. Cold Spring Harb Symp Quant Biol. 1994;59:67–73. doi: 10.1101/sqb.1994.059.01.010. [DOI] [PubMed] [Google Scholar]
- Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glynn MW, Glover TW. Incomplete processing of mutant lamin A in Hutchinson-Gilford progeria leads to nuclear abnormalities, which are reversed by farnesyltransferase inhibition. Hum Mol Genet. 2005;14:2959–2969. doi: 10.1093/hmg/ddi326. [DOI] [PubMed] [Google Scholar]
- Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, Collins FS. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2004;101:8963–8968. doi: 10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Navarro S, Kasahara N, Comai L. Identification and biochemical characterization of a Werner's syndrome protein complex with Ku70/80 and poly(ADP-ribose) polymerase-1. J Biol Chem. 2004;279:13659–13667. doi: 10.1074/jbc.M311606200. [DOI] [PubMed] [Google Scholar]
- Mallampalli MP, Huyer G, Bendale P, Gelb MH, Michaelis S. Inhibiting farnesylation reverses the nuclear morphology defect in a HeLa cell model for Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2005;102:14416–14421. doi: 10.1073/pnas.0503712102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro CL, Cadinanos J, De Sandre-Giovannoli A, Bernard R, Courrier S, Boccaccio I, Boyer A, Kleijer WJ, Wagner A, Giuliano F, Beemer FA, Freije JM, Cau P, Hennekam RC, Lopez-Otin C, Badens C, Levy N. Loss of ZMPSTE24 (FACE-1) causes autosomal recessive restrictive dermopathy and accumulation of Lamin A precursors. Hum Mol Genet. 2005;14:1503–1513. doi: 10.1093/hmg/ddi159. [DOI] [PubMed] [Google Scholar]
- Pendas AM, Zhou Z, Cadinanos J, Freije JM, Wang J, Hultenby K, Astudillo A, Wernerson A, Rodriguez F, Tryggvason K, Lopez-Otin C. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet. 2002;31:94–99. doi: 10.1038/ng871. [DOI] [PubMed] [Google Scholar]
- Pollex RL, Hegele RA. Hutchinson-Gilford progeria syndrome. Clin Genet. 2004;66:375–381. doi: 10.1111/j.1399-0004.2004.00315.x. [DOI] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312:1059–1063. doi: 10.1126/science.1127168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth JI, Yang SH, Qiao X, Beigneux AP, Gelb MH, Moulson CL, Miner JH, Young SG, Fong LG. Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc Natl Acad Sci U S A. 2005;102:12873–12878. doi: 10.1073/pnas.0505767102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Bergo MO, Toth JI, Qiao X, Hu Y, Sandoval S, Meta M, Bendale P, Gelb MH, Young SG, Fong LG. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. Proc Natl Acad Sci U S A. 2005;102:10291–10296. doi: 10.1073/pnas.0504641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Overall levels of lamin A in 20 control cells and 20 cells expressing ectopic wild-type lamin A. Lamin A was visualized by indirect immunofluorescence using lamin A specific antibody and FITC-conjugated secondary antibodies, and pixel intensity per cell was determined using Metamorph software and plotted as arbitrary units.
Figure S2. A. Growth curves of fibroblasts stably transduced with lentiviruses expressing the indicated proteins are shown as accumulated population doublings. B. Quantitation of cells with one or more nuclear blebs. For each experiment, 300 cells were stained with DAPI and scored by two independent observers. Data shown represent mean +/− standard deviation of 3 measurements in two independent cell lines for each group.
Figure S3. The percentage of fibroblasts with nuclear blebs that are senescent was determined by simultaneously staining cells with DAPI and for senescence-associated β-galactosidase as described in the Materials and Methods.
Figure S4. Lamin A folds penetrate the nucleoplasmic space. Confocal microscopy was performed on fibroblasts transduced with lentiviruses expressing either flag-lamin A or lamin A, fibroblasts from old-age individual and fibroblasts from a progeria patient. Images collected from two focal planes passing through the cell nucleus are shown. Bar: 10 µm
Figure S5. RT-PCR analysis of lamin A splicing in control fibroblasts, fibroblasts overexpressing wild-type lamin A, HGPS fibroblasts and fibroblasts from 3 young and 3 old-age individuals. Schematic representation of LMNA exons 9–12. The HGPS-specific splice site (vertical line with asterisk) and the position of the different primer sets used for RT-PCR are indicated in the diagram. The detection of wild-type and progerin mRNA using the indicated set of primers was carried out as described in Materials and Methods. RT-PCR analysis with primers that detect GAPDH mRNA was used as control.