

Abstract
Sprouty (SPRY) proteins modulate receptor-tyrosine kinase signaling and, thereby, regulate cell migration and proliferation. Here, we have examined the role of endogenous human SPRY2 (hSPRY2) in the regulation of cellular apoptosis. Small inhibitory RNA-mediated silencing of hSPRY2 abolished the anti-apoptotic action of serum in adrenal cortex adenocarcinoma (SW13) cells. Silencing of hSPRY2 decreased serum- or epidermal growth factor (EGF)-elicited activation of AKT and ERK1/2 and reduced the levels of EGF receptor. Silencing of hSPRY2 also inhibited serum-induced activation of p90RSK and decreased phosphorylation of pro-apoptotic protein BAD (BCL2-antagonist of cell death) by p90RSK. Inhibiting both the ERK1/2 and AKT pathways abolished the ability of serum to protect against apoptosis, mimicking the effects of silencing hSPRY2. Serum transactivated the EGF receptor (EGFR), and inhibition of the EGFR by a neutralizing antibody attenuated the anti-apoptotic actions of serum. Consistent with the role of EGFR and perhaps other growth factor receptors in the anti-apoptotic actions of serum, the tyrosine kinase binding domain of c-Cbl (Cbl-TKB) protected against down-regulation of the growth factor receptors such as EGFR and preserved the anti-apoptotic actions of serum when hSpry2 was silenced. Additionally, silencing of Spry2 in c-Cbl null cells did not alter the ability of serum to promote cell survival. Moreover, reintroduction of wild type hSPRY2, but not its mutants that do not bind c-Cbl or CIN85 into SW13 cells after endogenous hSPRY2 had been silenced, restored the anti-apoptotic actions of serum. Overall, we conclude that endogenous hSPRY2-mediated regulation of apoptosis requires c-Cbl and is manifested by the ability of hSPRY2 to sequester c-Cbl and thereby augment signaling via growth factor receptors.
The Sprouty (SPRY)2 family of proteins has emerged as an important modulator of receptor-tyrosine kinase signaling, and this function of SPRY proteins has been conserved throughout evolution. Drosophila SPRY was the first member of this family to be identified and has been shown to regulate tracheal branching in response to fibroblast growth factor (1). Studies that followed demonstrated that Drosophila SPRY also inhibited the actions of EGF (2). The four mammalian SPRY isoforms (SPRY1–4) have also been shown to modulate growth factor-mediated actions (for reviews, see Refs. 3 and 4). The loss of mouse SPRY2 increases lung branching morphogenesis (5), whereas mouse SPRY4 inhibits angiogenesis (6) and causes pulmonary hypoplasia (7). SPRY2 and SPRY1 decrease uteretic branching and kidney development (8, 9), demonstrating that the SPRY proteins play a profound role in regulating tubular morphogenesis. SPRY proteins also play a role in the development of other organs such as the brain and limbs (10–12).
At the cellular level, overexpression of SPRY1 (13, 14), SPRY2 (15–18), and SPRY4 (6) inhibit migration and proliferation of a variety of cell types in response to serum and growth factors. Stimulation of cells with EGF results in the translocation of the human SPRY2 (hSPRY2) from the vicinity of microtubules to membrane ruffles (15, 19), and the abrogation of translocation of hSPRY2 to membrane ruffles obliterates the ability of the protein to inhibit cell migration and proliferation (15). We have previously shown that hSPRY2, in part, mediates its anti-migratory actions by increasing the amount of soluble protein-tyrosine phosphatase 1B (20) and decreases growth factor-mediated activation of Rac1 (21). The ability of hSPRY2 to decrease Rac1 activation also contributes to its anti-migratory, but not the anti-proliferative actions (21). We have also demonstrated that hSPRY2 increases phosphatase and tensin homologue deleted on chromosome 10 (PTEN), and the anti-proliferative actions of hSPRY2 require PTEN (18). Although a large number of reports have used overexpressed SPRY proteins to study their functions, relatively few studies have examined the role of endogenous SPRY proteins in modulating cellular events. In this context, studies with SPRY2 knock-out mice have shown no obvious phenotypes except impaired hearing due to altered cytoarchitecture of the organ of Corti (22) and enteric neuronal hyperplasia and esophageal achalasia (23). However, the role of endogenous SPRY proteins in regulating events at the cellular level remain largely unknown.
Importantly, although Sprouty proteins have predominantly been thought of as inhibitors of receptor-tyrosine kinases, they can also positively regulate growth factor actions. Thus, previous studies reported that hSPRY2 can augment the activation of ERK1/2 in response to EGF by decreasing c-Cbl-mediated degradation of the epidermal growth factor receptor (EGFR) (Refs. 24–26; for reviews, also see Refs. 27 and 28). Likewise, by increasing EGFR signaling, hSPRY2 has been shown to facilitate the differentiation of PC12 cells into the neuronal phenotype (26).
In this report, by silencing the endogenous hSPRY2 in SW13 cells and primary mouse embryonic fibroblasts, we show that endogenous hSPRY2 is necessary for the anti-apoptotic actions of serum. The knockdown of endogenous hSPRY2 decreased the amount of EGFR and downstream signaling via the AKT and ERK pathways in response to both EGF and serum. Furthermore, silencing of endogenous hSPRY2 decreased serum-mediated activation of RSK, the immediate downstream kinase of ERK1/2, and phosphorylation of its target, proapoptotic protein, BAD. The anti-apoptotic action of serum was mediated by both the ERK1/2 and AKT pathways and was mimicked by the combination of the MEK and phosphatidylinositol 3-kinase inhibitors. Serum transactivated the EGFR, and inhibition of the EGFR attenuated the anti-apoptotic actions of serum. Consistent with a role for the EGFR and perhaps other growth factor receptors in the actions of serum, the tyrosine kinase binding domain of c-Cbl (Cbl-TKB) protected against EGFR down-regulation and preserved the anti-apoptotic actions of serum when hSPRY2 was silenced. Additionally, in c-Cbl null cells, the ability of endogenous Spry2 to modulate the anti-apoptotic actions of serum was obliterated. Moreover, transduction of TAT-HA-hSPRY2, but not the mutant (Y55F) that does not bind Cbl into cells in which endogenous hSPRY2 had been silenced by siRNA, rescued the anti-apoptotic actions of serum. Thus, this is the first demonstration that endogenous hSPRY2, by modulating the function of c-Cbl, regulates cellular apoptosis.
EXPERIMENTAL PROCEDURES
Cells and Culture Conditions
Adrenal cortex adenocarcinoma (SW13) cells obtained from American Type Culture Collection (ATCC) were maintained in Leibovitz’s (L15) medium supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, and penicillin/streptomycin in a humidified 37 °C incubator in the absence of CO2. The Spry2wt/loxP mice (C57/B6) in which the entire coding sequence (exon 2) was floxed were generated through inGenious Targeting Labs (Stonybrook) using the construct shown in supplemental Fig. S1. Primary mouse embryonic fibroblasts from heterozygous Spry2wt/loxP mice were isolated and cultured as described previously (18). Forty eight hours before experimentation, the Spry2wt/loxP cells were infected with retrovirus to express either GFP or Cre-recombinase-GFP fusion protein, and Spry2 expression in these cells was monitored by Western analyses to confirm the Spry2+/+ and Spry2+/− phenotypes. Wild type and c-Cbl−/− mouse embryonic fibroblasts (MEFs) were obtained from Dr. Hamid Band (Northwestern University, Chicago, IL) and cultured in minimal essential medium, 10% FBS, 2 mM penicillin/streptomycin, 1× nonessential amino acids, and 0.5 mM β-mercaptoethanol.
Constructs and Purification of Proteins
Wild type TAT-HA-hSPRY2 was constructed as described previously (15). TAT-HA-hSPRY2-Y55F, CT-3A (P304A, P307A, R309A), and Y55F-CT-3A mutants were generated by site-directed mutagenesis under the following conditions; one cycle of 95 °C for 3 min followed by 16 cycles of 95 °C 1 min, 52 °C 1 min, and 68 °C 8 min followed by 1 cycle of 68 °C for 60 min in a Robocycler PCR machine using Pfu Turbo DNA polymerase (Stratagene). The following primers were used: Y55F sense, 5′ AAC ACC AAT GAG TTT ACA GAG GGG CCT ACT GTC GTC-3′, and antisense, 5′-AGG CCC CTC TGT AAA CTC ATT GGT GTT TCG GAT GGC T-3′; CT-3A sense, 5′-TGC AAA GTT GCC ACT GTC GCC CCT GCT AAC TTT GAA AAA CC-3′, and antisense, 5′-GGT TTT TCA AAG TTA GCA GGG GCG ACA GTG GCA ACT TTG CA-3′. The wild type, various mutant forms of TAT-HA-hSPRY2, and TAT-HA-GFP were purified and used as described previously (15). FLAG-tagged hSPRY3 and Cbl-TKB plasmids were generous gift from Dr. Graeme R. Guy, Institute of Molecular and Cell Biology, Singapore.
Western Analyses
Cell lysates were prepared in the SDS sample buffer under reducing conditions, and proteins were separated by SDS-PAGE. Proteins were transferred to a nitrocellulose membrane for 1 h at 100 V in the cold. Membranes were blocked either in Tris-buffered saline containing 5% (w/v) nonfat dry milk with 0.1% Tween 20 or in phosphate-buffered saline containing 3% (w/v) nonfat dry milk. The following primary antibodies were used: affinity-purified anti-SPRY2 antibody (Rockland, Gilbertsville, PA and Sigma); anti-SPRY1 and SPRY4 antibodies (generous gifts from Dr. Graeme R. Guy, Institute of Molecular and Cell Biology, Singapore); anti-HA (HA.11 monoclonal antibody, Covance Research Products, Berkeley, CA); anti-Akt (polyclonal antibody), anti-phospho-Akt (Thr-380, polyclonal and Ser-473, 4E2 monoclonal), anti-phospho-ERK1/2 (CT), anti-phospho-BAD (Ser-112, polyclonal), anti-BAD (polyclonal) (Cell Signaling, Beverly, MA); monoclonal anti-actin antibody (MP Biomedicals, Aurora, OH); anti-Erk1/2 antibody (Upstate Biotechnology, Lake Placid, NY); anti-RSK and anti-SPRY3 antibodies (Santa Cruz Biotechnology, Santa Cruz, CA).
hSPRY2 Knockdown Using siRNA
SW13 cells were plated at 50–70% confluency in 35-mm dishes. hSPRY2 was knocked down as described in the transit TKO (Mirus, MI) protocol. Briefly, 6 μl of transit TKO reagent was mixed thoroughly with 200 μl Opti-MEM and incubated for 12 min at room temperature. To this 2.4 μl of 10 μM hSPRY2 siRNA or mutant hSPRY2 siRNA was added and incubated at room temperature for another 12 min before dropwise addition to the dishes containing 1 ml of regular media. Cells were incubated for 48 h before experimentation. The 27-ribonucleotide-long hSPRY2-specific siRNA (5′-GAU CAG AUC AGA GCC AUC CGA AAC ACC-3′ (sense)/5′-GGU GUU UCG GAU GGC UCU GAU CUG AUC-3′ (antisense)), mutant hSPRY2 siRNA (5′-GGA GUU UCG CAU GGC UAU GAU CUG CUC-3′ (sense)/5′-GAG CAG AUC AUA GCC AUG CGA AAC UCC-3′ (anti-sense)), and scrambled hSPRY2 siRNA (5′-AGU CGA ACU GAA CCG UAC CTT-3′ (sense)/5′-GGU ACG GUU CAG UUC GAC UTT-3′ (antisense)) were synthesized commercially (Integrated DNA Technologies). These sequences do not share any homology with other hSPRY isoforms.
Cellular Apoptosis
Sham, hSPRY2 siRNA, or mutant hSPRY2 siRNA-transfected SW13 cells were plated on a 24-well plate (50,000 cells/well). Cells were serum-starved for 36 h and then exposed to 10% fetal bovine serum for 15 min. When indicated, the MEK inhibitor (UO126, 20 μM) or phosphoinositide 3-kinase inhibitor LY294002 (50 μM) was added 15 min before serum treatment. Cells were then treated with or without 20 ng/ml TNF-α plus 25 μg/ml cycloheximide (TNFα/CHX) to induce apoptosis for 1h at 37 °C in a humidified incubator. Thereafter, DNA fragmentation was measured using the cell death detection kit (Roche Applied Science) as described previously (29, 30). Absorbance at 405 nm was normalized with the protein concentrations from the corresponding parallel wells in the same experiment. The validity of the DNA fragmentation method was also confirmed by monitoring the cleavage of PARP as an index of apoptosis using the anti-cleaved and total PARP antibodies (Cell Signaling).
Activation of c-Cbl
SW13 cells treated with mutant siRNA or siRNA against hSPRY2 were serum-starved and then challenged with EGF (50 nM) or serum (10%). Ten minutes later the cells were lysed in phosphate-buffered saline, pH 7.4, containing 2% nonaethylene glycol monododecyl ether (C12E9), 0.1% SDS, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM sodium orthovanadate, 50 mM sodium fluoride, 5 mM sodium pyrophosphate, 50 mM β-glycerophosphate, and protease inhibitors. The c-Cbl was immunoprecipitated using anti-c-Cbl antibody (BD Transduction Laboratories). Western blots of the immunoprecipitated c-Cbl were performed with anti-Tyr(P)-99 antibody (Santa Cruz Biotechnology).
RESULTS
Silencing of Endogenous hSPRY2 Attenuates the Anti-apoptotic Actions of Serum
Because we have previously shown that hSPRY2 decreases cell proliferation (15, 17, 18), we reasoned that hSPRY2 may regulate cellular apoptosis. To investigate the role of endogenous hSPRY2 in regulation of cellular apoptosis, we utilized small inhibitory RNA specific against hSPRY2 (hSPRY2 siRNA) to decrease endogenous expression of hSPRY2 in SW13 cells. As controls, cells were either sham-transfected or transfected with equimolar amounts of mutant siRNA (harboring three mutations in the hSPRY2 siRNA sequence). As compared with the controls (sham or mutant siRNAs), transfection with hSPRY2 siRNA markedly (>80%) decreased the endogenous levels of hSPRY2 (Fig. 1A). We did not observe any gross morphological changes in the cells after knocking down endogenous hSPRY2 amount (not shown). Moreover, as shown in supplemental Fig. S2A, silencing of hSPRY2 in SW13 cells did not alter the amounts of endogenous hSPRY1 and hSPRY4. Although our antibody detected the overexpressed hSPRY3 in SW13 cells, hSPRY3 was not detectable in the lysates of SW13 cells (supplemental Fig. S2B).
FIGURE 1. Silencing of endogenous hSPRY2 decreases the anti-apoptotic actions of serum.
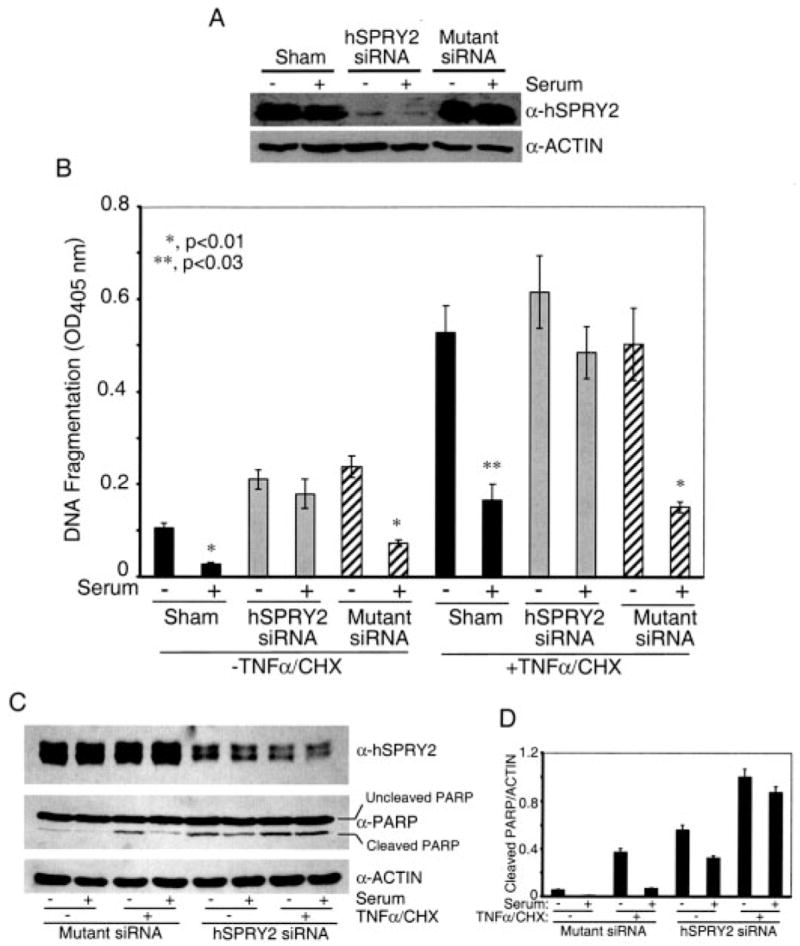
Panel A, SW13 cells were transfected with sham and 20 nM each hSPRY2 siRNA or mutant hSPRY2 siRNA (Mutant siRNA) and 48 h later plated on 35-mm dishes. After 36 h of serum deprivation, cells were lysed in Laemmli buffer, and proteins were separated on 10% polyacrylamide gels. The amount of SPRY2 was detected by immunoblotting to assess the efficiency of silencing by the siRNA. Actin was used to assess equal loading of samples. Panel B, cells treated as described in panel A were plated after 48 h of transfection onto 24-well plates. After 36 h of serum deprivation, the cells were treated with or without serum for 15 min. Vehicle or a mixture of TNF-α (20 ng/ml) and CHX (25 μg/ml) were added to the cells and incubated for 1 h before measuring DNA fragmentation as described under “Experimental Procedures.” Data are the mean ± S.D. of A405 nm per 50,000 cells (n = 6). The significance of differences was assessed by Student’s unpaired t test. Panel C, same as panel B, except that cells were lysed in Laemmli sample buffer, and proteins were separated by SDS-PAGE followed by immunoblotting with cleaved and total PARP antibodies. Panel D, quantification of the cleaved PARP normalized to endogenous actin from three independent blots similar to panel C.
In apoptosis assays, serum deprivation (without TNF) induced some degree of apoptosis that was reversed by serum addition when endogenous hSPRY2 levels were not manipulated, but this action of serum was obliterated when hSPRY2 was silenced (Fig. 1B). Similarly, the anti-apoptotic actions of serum in the presence of TNF-α were also markedly attenuated when hSPRY2 was silenced (Fig. 1B). Furthermore, identical observations were also made using cleaved PARP as a monitor of cellular apoptosis (Figs. 1, C and D), confirming the validity of the DNA fragmentation method used in Fig. 1B. Notably, although silencing of hSPRY2 decreased the anti-apoptotic actions of serum, EGF- and serum-induced migration of SW13 cells was either enhanced or not affected, respectively.3
Endogenous hSPRY2 Regulates Serum and EGF-elicited Activation of ERK1/2 and AKT
Because the ERK1/2 and AKT pathways contribute toward cell survival, next we monitored the ability of serum to activate these enzymes when hSPRY2 was silenced. As shown in Fig. 2, panels A–C, siRNA-mediated knockdown of hSPRY2 markedly attenuated the ability of serum to stimulate AKT and ERK1/2. Because hSPRY2 has previously been shown to modulate the signaling via growth factor receptors (25, 26, 31–34) and because serum mediates its action via activation of multiple receptors including growth factor receptors, we investigated whether silencing of endogenous hSPRY2 altered the ability of EGF to activate the ERK1/2 and AKT pathways. Similar to our findings with serum, EGF-elicited activation of ERK1/2 and AKT was also markedly attenuated when hSPRY2 was silenced (Fig. 2, D and E). Moreover, the amount of EGFR was also decreased when hSPRY2 was silenced (Fig. 2, D and E). Interestingly, the transduction of Y55F mutant of TAT-HA-hSPRY2, which is not phosphorylated on Tyr-55 by growth factors (31, 33) and which acts as a “dominant negative” form of hSPRY2 (35), also decreased serum- and EGF- mediated activation of ERK1/2 and AKT, respectively, and decreased the amount of EGFR in SW13 cells (supplemental Fig. S3). Previous studies have shown that Tyr-55 phosphorylation of hSPRY2 provides a docking site for the ubiquitin ligase c-Cbl and sequesters c-Cbl so that ubiquitylation and degradation of growth factor receptors such as the EGFR is diminished (25, 33, 34, 36). Silencing of hSPRY2 or transduction of the dominant negative Y55F mutant of hSPRY2 would produce an opposite effect and decrease the amount of EGFR. Thus, the decrease in EGFR when hSPRY2 is silenced may be explained by the ability of hSPRY2 to bind and sequester c-Cbl and would, therefore, explain the attenuation of ERK1/2 and AKT activation by EGF when hSPRY2 is silenced (discussed further below).
FIGURE 2. Silencing of endogenous hSPRY2 decreases AKT and ERK1/2 activation.

Panel A, after transfection of SW13cells with 20nM each hSPRY2 siRNA and mutant siRNA or sham, cells were grown in serum-free medium for 36h and then exposed to fetal bovine serum for 15 min. Total cell lysates (50 μg of protein) were analyzed by immunoblotting for total AKT, phospho (p)-Thr-308 AKT, total (t) ERK1/2, and phospho-ERK1/2. The anti-SPRY2 blot shows the efficiency of knockdown. Panels B and C, quantification of the ratio of phospho-AKT/total AKT and phospho-ERK1/2/total ERK1/2, respectively, from three independent blots similar to those in panel A. Statistical significance was assessed by Student’s unpaired t test (n = 3). Panel D, same as panel A except that the cells were stimulated with EGF (50 nM) for the indicated times, and cell lysates were immunoblotted for phospho-AKT Thr-308, phospho-ERK1/2, and EGFR. Total AKT and ERK1/2 were used as loading controls. Anti-hSPRY2 blot shows the efficiency of knockdown. Panel E, the bar graphs show the quantification of phospho-AKT/total AKT, phospho-ERK1/2/total ERK1/2, and EGFR/total AKT from three similar experiments to those in panel D. Statistical significance was assessed by Student’s unpaired t test (n = 3).
Silencing of hSPRY2 Decreases the Activation of p90RSK and Phosphorylation of Its Pro-apoptotic Protein Substrate BAD
Cellular apoptosis is modulated by a number of regulatory proteins, including BAD (37, 38). Because we observed decreased ERK1/2 activation when hSPRY2 was knocked down, we reasoned that proteins downstream of ERK1/2 might be responsible for the increased apoptosis. Because the pro-apoptotic protein BAD is regulated by phosphorylation on Ser-112 by p90RSK (24), the immediate downstream kinase of ERK1/2, we examined the activation of p90RSK by monitoring its phosphorylation on Thr-573 by ERK1/2 (39) as well as the phosphorylation of BAD on Ser-112 by p90RSK (24). Sham, hSPRY2 siRNA, or mutant siRNA-transfected SW13 cells that had been serum-starved for 36 h were stimulated with serum for 15 min, and cell lysates were immunoblotted for phospho-Thr-573 p90RSK and phospho-Ser-112 BAD. As shown in Fig. 3, serum augmented the phosphorylation of p90RSK and BAD in sham and mutant siRNA-transfected cells. However, when hSPRY2 was knocked down, the ability of serum to activate p90RSK and phosphorylate BAD on Ser-112, the p90RSK site, was abrogated. Notably, the amounts of total p90RSK and BAD were not affected by silencing hSPRY2 expression (Fig. 3A). Quantification of phospho-BAD and phospho-RSK is shown in panels B and C of Fig. 3, respectively. Thus, consistent with the increased apoptosis when hSPRY2 was silenced, the phosphorylation and activation of p90RSK and subsequent phosphorylation of pro-apoptotic BAD on Ser-112 were decreased. These findings are consistent with, and explain the decreased anti-apoptotic action of serum when endogenous hSPRY2 is depleted.
FIGURE 3. Silencing of endogenous hSPRY2 decreases p90RSK activation and phosphorylation of BAD.
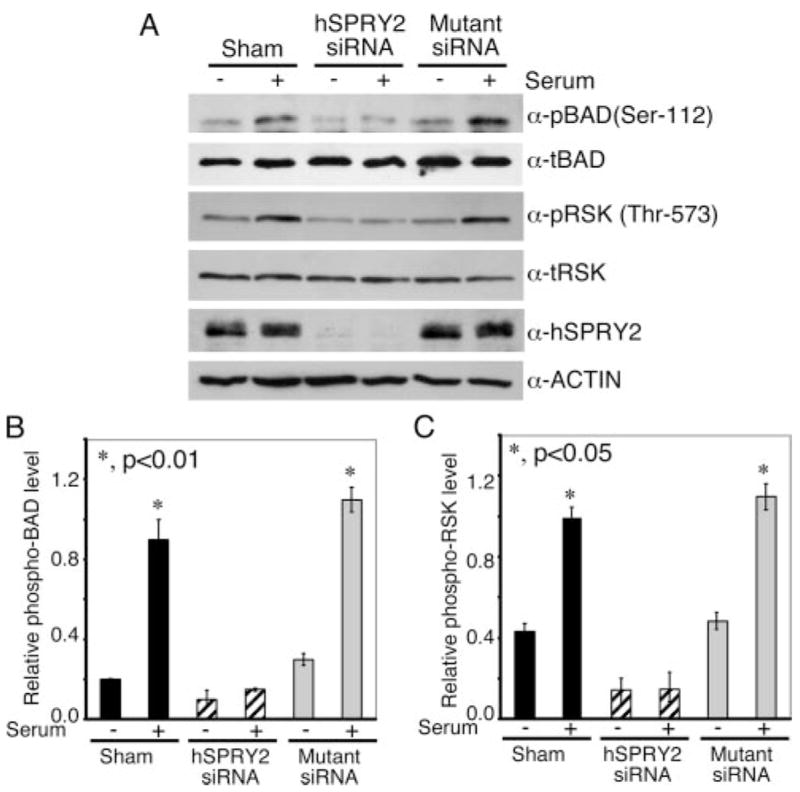
Panel A, sham and 20 nM each hSPRY2 siRNA or mutant siRNA-transfected SW13 cells were serum-starved for 36 h and subsequently stimulated with 10% FBS for 15 min. Cells were lysed in SDS sample buffer and immunoblotted using phospho (p)-Ser-112 BAD and phospho (p)-Thr-573 RSK antibodies. Total BAD, RSK, and actin immunoblots show equal loading of the proteins. Anti-hSPRY2 blot shows the efficiency of hSPRY2 knock down. Representative blots from three independent experiments are shown. t-, total. Panels Band C show the quantification of the ratio of phospho-BAD/total BAD and phospho-RSK/total RSK, respectively, from three independent blots. Statistical significance was assessed by Student’s unpaired t test (n = 3).
Contribution of ERK1/2 and AKT in Anti-apoptotic Actions of Serum
The data from Figs. 2 and 3 suggested that the increased cellular apoptosis when hSPRY2 is silenced is likely associated with the decreased activation of ERK1/2 and AKT that play a crucial role in cell survival. Therefore, using inhibitors of upstream kinases in the ERK1/2 and AKT pathways, we investigated the role of these two pathways in the anti-apoptotic actions of serum. For this purpose, after serum starvation, cells were treated with UO126 (MEK inhibitor) or LY294002 (phosphatidylinositol 3-kinase inhibitor) for 15 min before testing the anti-apoptotic actions of serum on TNFα/CHX-induced cellular apoptosis. As observed before (Fig. 1B), serum markedly decreased TNFα/CHX-induced apoptosis in mutant siRNA-treated cells (Fig. 4A). When present individually, both the MEK (U0126) and the phosphatidylinositol 3-kinase (LY294002) inhibitors further increased cellular apoptosis, and the ability of serum to reverse this was attenuated but not completely obliterated (Fig. 4A). However, when U0126 and LY294002 were added together, the anti-apoptotic action of serum in control (mutant siRNA-treated cells) was completely obliterated (Fig. 4A). Similar observations were also made in sham-treated controls (not shown). In contrast, when hSPRY2 was silenced, neither U0126 nor LY294002 further enhanced apoptosis, and the ability of serum to rescue cells was abolished whether the inhibitors were present or not (Fig. 4A). These data show that in control SW13 cells, ERK1/2 and AKT pathways both contribute to the anti-apoptotic actions of serum and that when both pathways are inhibited, the ability of serum to protect against apoptosis is overcome. Moreover, the data in Fig. 4A show that silencing of hSPRY2, which results in the inhibition of both ERK1/2 and AKT signaling, is mimicked by the combined effects of U0126 and LY294002. Therefore, when hSPRY2 expression is decreased by siRNA, neither of the two inhibitors shows any further increase in apoptosis. The data in Fig. 4B show that in control (sham and mutant siRNA-treated) cells, U0126 inhibited ERK1/2 and p90RSK activation by serum to the same extent as that observed in cells in which hSPRY2 expression had been silenced. However, as expected, U0126 did not alter activation and phosphorylation of AKT on S473 in the control cells (Fig. 4B).
FIGURE 4. The ERK1/2 and AKT pathways contribute toward abolishing the anti-apoptotic actions of serum when hSPRY2 is silenced.
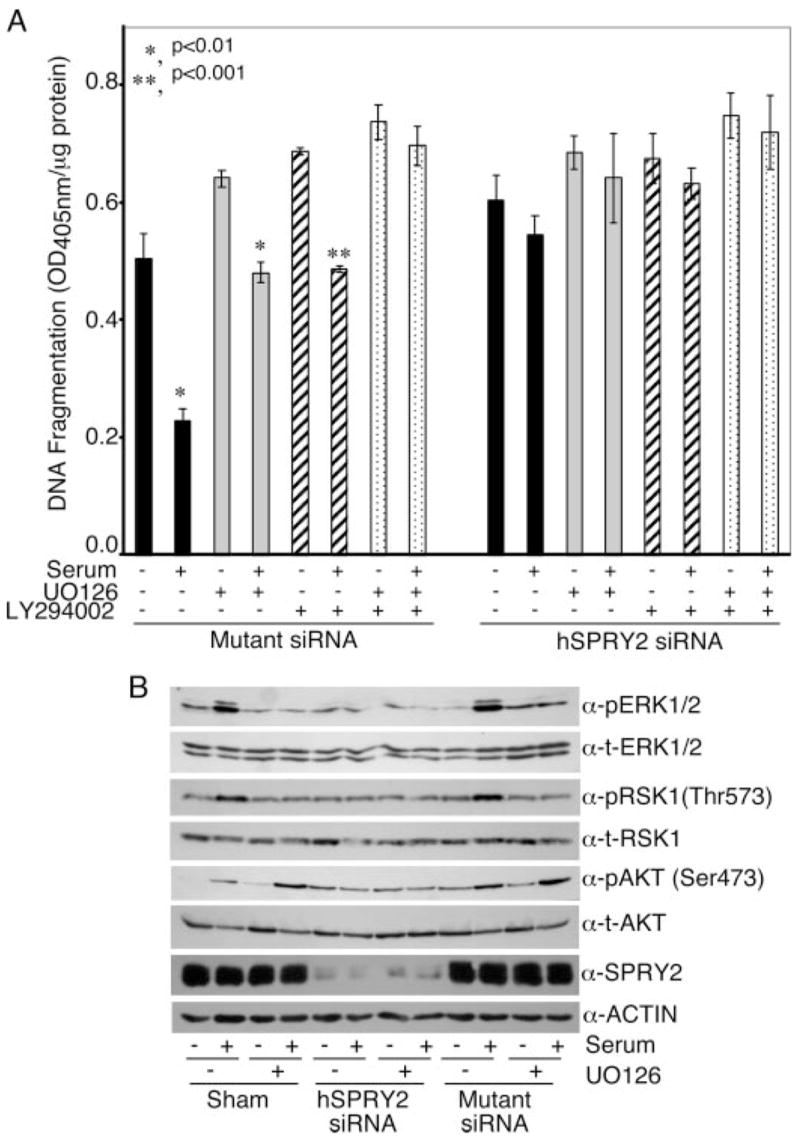
Panel A is same as panel B in Fig. 1, except that the cells were preincubated with the ERK1/2 inhibitor UO126 (20 μM) or the phosphatidylinositol 3-kinase inhibitor LY294002 (50 μM) for 15 min before the addition of serum for another 15 min. DNA fragmentation was measured as described under “Experimental Procedures.” Data are the mean ± S.E. of A405 nm per 50,000 cells (n = 6). p <0.01 (*) and p <0.001 (**) using Student’s unpaired t test. Panel B, cells were treated as described in panel A. After serum stimulation, cells were lysed in SDS sample buffer and immunoblotted for phospho (p)-ERK1/2, phospho-Ser-473AKT, and phospho-Thr-573 RSK. Total (t) ERK1/2, AKT, and RSK show equal loading of the proteins.
Serum Mediates Its Anti-apoptotic Actions via Activation of Growth Factor Receptors Such as the EGFR
As alluded to above, serum mediates its cellular actions via activation of numerous receptors, including growth factor receptors. The growth factor receptors may be directly activated by various growth factors present in the serum or indirectly activated (transactivated) by agonists such as lysophosphatidic acid that are key components of serum (40, 41). Therefore, we next examined the contribution of the growth factor receptors, such as the EGFR, in mediating the anti-apoptotic actions of serum. As shown in Fig. 5A, incubation of SW13 cells with an EGFR antibody (Ab528b) that competes with EGFR ligands for binding to the extracellular region of the receptor, partially blocked the anti-apoptotic actions of serum. Moreover, as shown in Fig. 5B, serum enhanced the autophosphorylation of the EGFR on Tyr-1173, and this activation of the EGFR was abolished by preincubation of cells with the EGFR neutralizing antibody (Ab528b); panel C (Fig. 5) shows the quantification of the pEGFR as a ratio of total EGFR. These data show that the EGFR, in part, contributes toward the anti-apoptotic actions of serum. This example with the EGFR would also suggest that other growth factor receptors such as platelet-derived growth factor receptor may also mediate the actions of serum.
FIGURE 5. EGFR activation contributes toward the anti-apoptotic actions of serum.
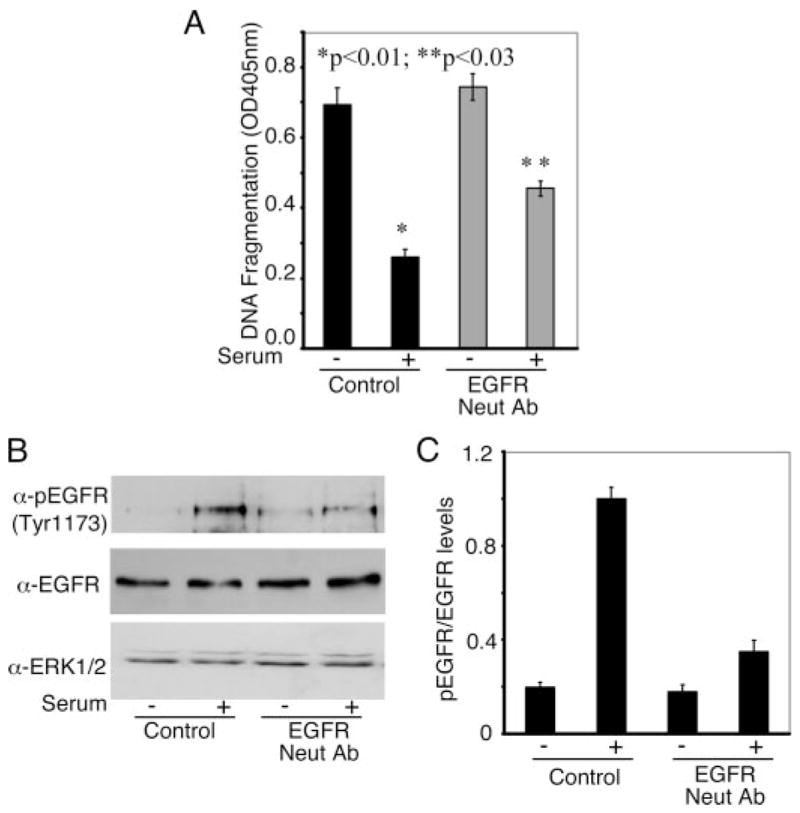
Panel A, SW13 cells were serum-starved overnight followed by one wash with phosphate-buffered saline, pH 6.5. Thereafter cells were incubated in the serum-free medium for another 2 h followed by treatment with 15 μg/ml EGFR neutralizing antibody (Ab528B; Neut Ab) for 1 h. Cells were treated with or without 10% FBS for 15 min followed by treatment with or without TNF-α in the presence of cycloheximide. DNA fragmentation assay was performed as described under “Experimental Procedures.” Data are the mean ± S.E. of A405 nm per 50,000 cells (n = 6). Statistical significance was assessed using Student’s unpaired t test. Panel B, cells from parallel plates in the experiments shown in panel A were lysed in Laemmli buffer and immunoblotted for phospho-EGFR Tyr-1173 antibody. Total EGFR and ERK1/2 were used as loading control. Panel C, quantification of the levels of phospho-EGFR normalized to the total EGFR from three independent experiments similar to panel B.
The Tyrosine Kinase Binding Domain of c-Cbl (c-Cbl-TKB) Restores the Anti-apoptotic Actions of Serum When hSPRY2 Is Silenced
Because growth factor receptors such as the EGFR mediate the anti-apoptotic actions of serum and because the amount of EGFR is decreased when hSPRY2 is silenced, it would appear that the diminution of the biological actions of serum and signaling in the absence of hSPRY2 may be related to the number of EGFR and other growth factor receptors on cells. As mentioned above, after their activation, phosphotyrosine residues on growth factor receptors such as the EGFR and platelet-derived growth factor receptor serve as docking sites for the tyrosine kinase binding (TKB) domain of c-Cbl which facilitates their ubiquitylation by c-Cbl and targeting toward degradation (42–44). Activation of growth factor receptors also results in phosphorylation of Tyr-55 on hSPRY2, and this serves as an alternate binding site for the TKB domain of c-Cbl (31, 33). Therefore, we reasoned that in the absence of hSPRY2, more c-Cbl would be available to bind activated growth factors and target them toward degradation. However, the expression of a truncated, dominant negative form of c-Cbl (c-Cbl-TKB) that contains the TKB domain of c-Cbl should compete with endogenous c-Cbl, protect the growth factor receptors from degradation, and restore the anti-apoptotic actions of serum even when hSPRY2 is silenced. As shown in Fig. 6A, overexpression of c-Cbl-TKB did not alter the ability of serum to protect control (mutant siRNA transfected) cells from apoptosis. As shown before, hSPRY2-specific siRNA decreased hSPRY2 content, decreased the amount of EGFR (Fig. 6B), and antagonized the anti-apoptotic actions of serum (Fig. 6A). However, expression of c-Cbl-TKB restored the ability of serum to protect cells from apoptosis even when hSPRY2 was silenced (Fig. 6A). Moreover, expression of c-Cbl-TKB maintained the amount of EGFR at control levels when hSPRY2 was silenced. These data (Figs. 6, A and B) together with those in Fig. 5 support the notion that serum mediates its actions by activating growth factor receptors and that silencing of hSPRY2, by decreasing the amount of growth factor receptors, diminishes the anti-apoptotic actions of serum.
FIGURE 6. Role of c-Cbl in regulation of apoptosis by endogenous Sprouty 2.
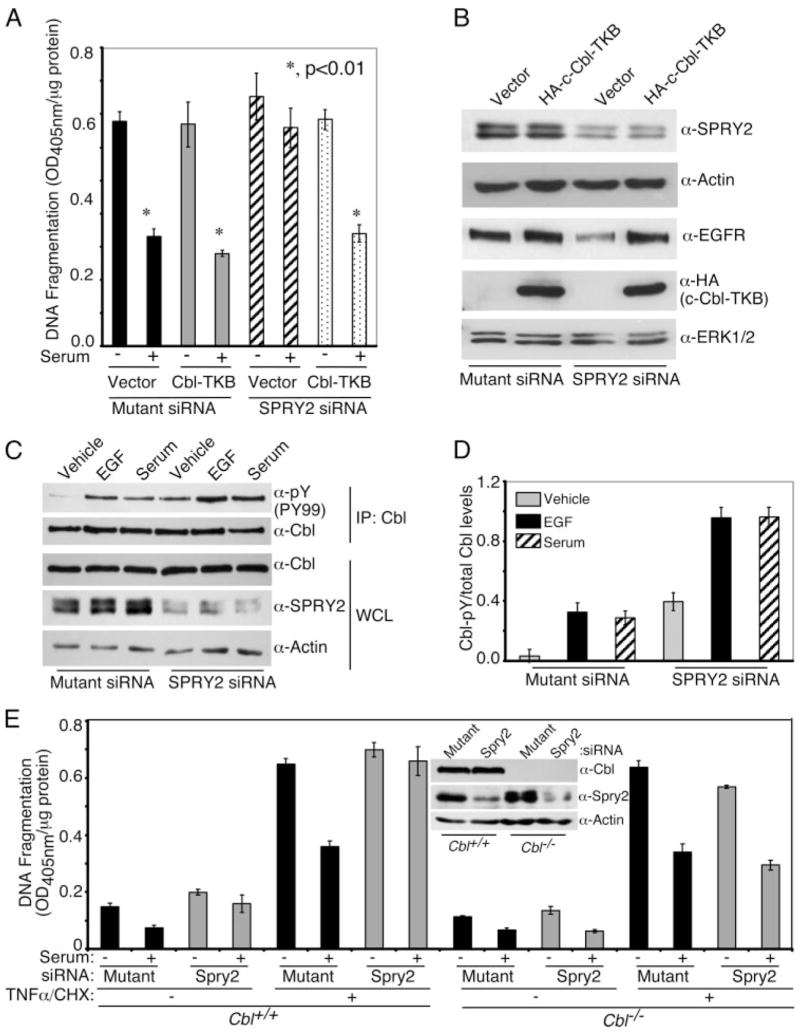
Panel A, SW13 cells were plated at 50% confluency on 35-mm dishes and were transfected with 20 nM each hSPRY2 siRNA or mutant siRNA or sham. Twenty-four hours later, cells were transfected with vector alone or vector expressing HA-c-Cbl-TKB as indicated. After 24 h, cells were re-plated on 24-well plates and serum-starved for 36 h. DNA fragmentation assay was performed as described before. Statistical significance was assessed using Student’s unpaired t test (n = 6). Panel B, cells from panel A before the DNA fragmentation assay were lysed in Laemmli buffer, and proteins were immunoblotted using anti-SPRY2, anti-HA (HA-c-Cbl-TKB) and anti-EGFR antibodies. Total ERK and actin were used as loading controls. Panel C, SW13 cells were transfected with mutant siRNA or SPRY2 siRNA. After 48 h cells were serum-starved overnight followed by an additional serum-free medium change 2 h before stimulating the cells with 50 nM EGF, 10% FBS, or vehicle for 10 min. Cells were lysed, and Cbl was immunoprecipitated. Immunoprecipitates (IP) were resolved on SDS-PAGE, and the membrane was probed with phosphotyrosine (Tyr(P)-99 (PY99)) antibody. The membrane was further reprobed with Cbl antibody. An SPRY2 blot in the whole cell lysate (WCL) show the knockdown. Actin was used for loading control. Panel D, quantification of tyrosine-phosphorylated Cbl normalized to the total Cbl in the immunoprecipitates from three independent blots similar to panel C. Panel E, Cbl−/− and Cbl+/+ MEFs were transfected with mutant siRNA or SPRY2 siRNA. Forty-eight hours after transfection, cells were serum-starved overnight followed by stimulation with 10% FBS for 10 min. Cells were then treated with or without TNFα/CHX for another hour, and DNA fragmentation was monitored. The inset shows the SPRY2 knock down in Cbl−/−and Cbl+/+ cells. Actin was used as the loading control.
hSPRY2 Regulates the Activation of c-Cbl, and Endogenous c-Cbl Is Required for hSPRY2-mediated Regulation of Cell Survival
To determine if hSPRY2 regulates c-Cbl activation, which is dependent on binding to growth factor receptors and tyrosine phosphorylation of c-Cbl (43, 45), the experiments shown in Fig. 6C were performed. EGF and serum resulted in increased tyrosine phosphorylation of c-Cbl both in control (mutant siRNA) and hSPRY2 siRNA-transfected cells. However, in the absence of hSPRY2, the amount of tyrosine-phosphorylated c-Cbl was greater than in control cells (Fig. 6C); Fig. 6D shows quantification of tyrosine-phosphorylated c-Cbl as a ratio of the total c-Cbl in the immunoprecipitates. Notably, the total amount of c-Cbl in whole cell lysates is not altered by silencing of hSPRY2 (Fig. 6C). Overall, these data are consistent with the notion that silencing of endogenous hSPRY2 permits more c-Cbl to be recruited to receptor-tyrosine kinases, resulting in more c-Cbl being activated for the ubiquitylation and ultimately degradation of EGFR and perhaps other growth factor receptors.
Further evidence for a role of c-Cbl in mediating the modulatory actions of Spry2 on serum-induced cell survival was examined in MEFs from mice in which c-Cbl was genetically ablated (Fig. 6E). Hence, as observed in SW13 cells, in wild type MEFs, silencing of endogenous Spry2 obliterated the anti-apoptotic actions of serum. However, in c-Cbl null MEFs, silencing of hSPRY2 did not alter the ability of serum to protect cells from apoptosis (Fig. 6E). These data clearly show that c-Cbl mediates the ability of Spry2 to regulate the anti-apoptotic actions of serum.
Wild Type hSPRY2, but Not Its Mutants That Do Not Bind c-Cbl or CIN85, Rescues the Anti-apoptotic Actions of Serum after Silencing Endogenous hSPRY2
The data presented thus far demonstrate that c-Cbl is required for the modulatory actions of hSPRY2 on cell survival and also suggest that endogenous hSPRY2, by binding c-Cbl, protects cell surface growth factor receptors from degradation to mediate the anti-apoptotic actions of serum. To further confirm this notion, we used mutant or hSPRY2 siRNA-treated SW13 cells and transduced them with wild type TAT-HA-hSPRY2 or its mutants that either do not bind c-Cbl (Y55F TAT-HA-SPRY2) (31, 33) or do not bind Cbl interacting protein p85 (CIN85/endophilins) (46) (P304A, R307A, R309A; TAT-HA-hSPRY2 CT-3A) or a mutant hSPRY2 in which both the c-Cbl and CIN85 binding sites are mutated (TAT-HA-hSPRY2 Y55F CT-3A) (Fig. 7A). Control cells were transduced with TAT-HA-GFP. CIN85 clusters c-Cbl for efficient EGFR internalization and degradation (47) and the disruption of either the c-Cbl or CIN85 binding to hSPRY2 attenuates the ability of hSPRY2 to decrease EGFR degradation (46). Therefore, we reasoned that if hSPRY2 facilitated the anti-apoptotic actions of serum by sequestering c-Cbl and protecting growth factor degradation, then the mutants of hSPRY2 that do not bind c-Cbl or CIN85 should not rescue the anti-apoptotic actions of serum when endogenous hSPRY2 was silenced. Previously, we have shown that hSPRY2 and GFP, when tagged with an 11 amino acid human immunodeficiency virus TAT, sequence can be transduced into a variety of different cells with ≥99% efficiency (15, 17, 18, 20, 21). Thus, 24 h after treatment with hSPRY2-specific siRNA or mutant siRNA, SW13 cells were split and plated on a 24-well plate (50,000 cells/well). After serum deprivation (36 h) in the presence of 20 μg/ml each of TAT-HA-tagged proteins, cells were stimulated with or without serum for 15 min followed by treatment with TNFα/CHX for another 1 h. In mutant siRNA-transfected cells (i.e. controls), the ability of serum to protect against apoptosis was not altered by transduction of any of the hSPRY2 forms, except Y55F hSPRY2, which acted as a dominant negative and obliterated the anti-apoptotic actions of serum (Fig. 7B). As shown before, when hSPRY2 was silenced, in TAT-HA-GFP-transduced cells the ability of serum to protect cells from apoptosis was obliterated (Fig. 7B). On the other hand, the transduction of TAT-HA-hSPRY2 into SW13 cells in which endogenous hSPRY2 had been silenced rescued the anti-apoptotic actions of serum, demonstrating a critical role for hSPRY2 in regulating cellular apoptosis (Fig. 7B). Consistent with a role for hSPRY2 in sequestering c-Cbl, transduction of the Y55F hSPRY2 did not restore the anti-apoptotic actions of serum (Fig. 7B). Likewise, the mutant forms of hSPRY2 with compromised CIN85 binding (TAT-HA-hSPRY2 CT-3A) or that do not bind both c-Cbl and CIN85 (TAT-HA-hSPRY2 Y55F CT-3A) did not restore the anti-apoptotic actions of serum (Fig. 7B). That hSPRY2 was effectively knocked down by the hSPRY2-specific siRNA is shown in Fig. 7C, and the successful transduction of TAT-HA-GFP or wild type and mutant forms of TAT-HA-hSPRY2 is shown in Fig. 7D.
FIGURE 7. Reintroduction of wt-hSPRY2, but not its mutant forms that do not bind c-Cbl or CIN85, restores the anti-apoptotic actions of serum after silencing of endogenous hSPRY2.

Panel A, schematic representation of the wt and different mutants of TAT-HA-hSPRY2 used. Panel B, hSPRY2 siRNA or mutant siRNA (20 nM each)-transfected SW13 cells (50,000 cells/well) were serum-starved for 36 h in the presence of 20 μg/ml each TAT-HA tagged GFP, WT SPRY2 (WT), Y55F SPRY2 (Y55F), PXXPPR to AXXAPA SPRY2 (CT-3A), or Y55F-CT-3A-hSPRY2 (Y55F-CT-3A) as illustrated in Panel A. Cells were stimulated with 10% FBS in the presence of TAT proteins, and TNFα/CHX were then added and incubated for 1 h. DNA fragmentation was then monitored as described under “Experimental Procedures.” Statistical significance was assessed by Student’s unpaired t test (n = 6). Panel C, lysates of cells after transfection with mutant or hSPRY2 siRNA were analyzed by immunoblotting for SPRY2 to determine the efficiency of endogenous SPRY2 silencing. Actin was used as a loading control. Panel D, cell lysates from parallel plates similar to those in panel B were immunoblotted to monitor the presence of HA-tagged TAT proteins.
As an alternate approach to decrease endogenous Spry2 content and to demonstrate the generality of our findings that endogenous Spry2 regulates the anti-apoptotic action of serum, we used primary mouse embryonic fibroblasts (PMEFs) from Spry2wt/loxP mice. These cells were treated with retrovirus to express either GFP (control) or Cre-recombinase to decrease the amount of Spry2 expression. As shown in Fig. 8, Cre-recombinase effectively decreased the amount of Spry2 expression in the PMEFs from Spry2wt/loxP mice (Fig. 8A). Moreover, as observed in SW13 cells, the ability of serum to protect PMEFs from apoptosis was abolished when Spry2 expression was decreased (Fig. 8B). Furthermore, the transduction of TAT-HA-hSPRY2 restored the anti-apoptotic actions of serum in the Cre-recombinase-treated PMEFs (Fig. 8B). Notably, the transduction of TAT-HA-hSPRY2 in control cells did not significantly alter the anti-apoptotic actions of serum. The successful transduction of TAT-HA-hSPRY2 and TAT-HA-GFP is shown in Fig. 8C. These data demonstrate that Sprouty2 facilitates the anti-apoptotic actions of serum in primary (PMEFs) cells and cells lines (SW13). Moreover, the regulation of apoptosis by Sprouty2 is preserved in human- and mouse-derived cells.
FIGURE 8. Role of Spry2 in the anti-apoptotic actions of serum in PMEFs.

Spry2wt/loxP PMEFs were infected with retrovirus to express either GFP or Cre-recombinase GFP. After 48 h cells were lysed in Laemmli sample buffer, and proteins were immunoblotted for Spry2 to determine efficiency of knock down (panel A). Total ERK was used to assess equal loading of proteins. Panel B, cells treated as described in panel A were re-plated on 24-well plates and serum-starved for 36 h in the presence of 10 μg/ml TAT-HA-GFP or TAT-HA-SPRY2. Cells were stimulated with 10% FBS in the presence of TAT-proteins for 15 min. TNFα/CHX were then added and incubated for 1 h. DNA fragmentation was then monitored as described under “Experimental Procedures.” Statistical significance was assessed by Student’s unpaired t test (n = 6). Panel C, cells from parallel plates similar to those in panels B and E were lysed, and samples were immunoblotted to monitor the presence of HA-tagged TAT-proteins.
DISCUSSION
In this report we have investigated the role of endogenous hSPRY2 in the regulation of cell survival. Although significant amounts of other hSPRY isoforms (hSPRY4 and hSPRY1) are also expressed in SW13 cells (supplemental Fig. S2), it is the specific knock down of hSPRY2 that abolishes the anti-apoptotic actions of serum. This suggests that there is not much redundancy among the expressed hSPRY isoforms and that the functions of each isoform may be selective and specific. In this context a recent report (48) has shown that although all four isoforms of mammalian Sprouty proteins interact with caveolin-1, they differ in their cooperation with caveolin-1 in regulating signaling from specific growth factors.
An important finding reported here is that endogenous hSPRY2 plays a crucial, permissive role in the anti-apoptotic actions of serum. Thus, the depletion of endogenous SPRY2 in SW13 cells or PMEFs by siRNA or genetic manipulations, respectively, abolished the anti-apoptotic actions of serum. We also show that serum mediates its actions via activation of growth factor receptors, such as the EGFR, and the inhibition of the EGFR attenuates both the transactivation of the EGFR and the anti-apoptotic actions of serum. Concerning the downstream signaling processes, silencing of hSPRY2 markedly attenuates the activation of the ERK1/2 and AKT pathways in response to both serum and growth factors such as EGF with a concomitant decrease in activation of p90RSK and phosphorylation of the proapoptotic protein BAD. Notably, in SW13 cells both the AKT and ERK1/2 pathways appear to contribute to the anti-apoptotic action of serum, and inhibition of either pathway partly attenuated the ability of serum to protect against apoptosis. However, when both the AKT and ERK1/2 pathways were inhibited, the ability of serum to protect against apoptosis was obliterated.
Mechanistically, the attenuation of serum-elicited downstream signaling after silencing of hSPRY2 is related to the amount of growth factor receptors such as EGFR. Thus, it is now well established that hSPRY2 provides an alternate docking site for c-Cbl on Tyr-55 after this residue is phosphorylated (31, 33). The sequestration of c-Cbl in this manner decreases ubiquitylation and degradation of growth factor receptors, such as the EGFR. Indeed, when hSPRY2 was silenced, the amount of EGFR was decreased (Fig. 2D), and because serum mediates its actions via activation of EGFR (Fig. 5) and possibly other growth factor receptors, the downstream signaling is also diminished. Consistent with a role for c-Cbl in mediating the permissive actions of hSPRY2 in the anti-apoptotic actions of serum, the expression of the dominant negative c-Cbl (Cbl-TKB) in cells when hSPRY2 was silenced mimicked the actions of hSPRY2 and permitted serum to protect against apoptosis. Moreover, Cbl-TKB also restored the amount of EGFR (Fig. 6A). A role for c-Cbl in mediating the actions of Spry2 is clearly shown by the experiments using wild type and c-Cbl null MEFs. Thus, ablation of c-Cbl expression obliterated the ability of Spry2 to modulate cell survival in response to serum (Fig. 6E). That the hSPRY2 regulates c-Cbl activity and function is shown by our findings that when hSPRY2 was silenced, the activation of c-Cbl by both EGF and serum was markedly enhanced (Figs. 6, C and D).
Further evidence for the involvement of c-Cbl in the modulation of the anti-apoptotic actions of hSPRY2 is provided by our experiments with the Y55F mutant of hSPRY2. Hence, the Y55F mutant of hSPRY2 that does not bind c-Cbl (31, 33) but acts as a dominant negative form of hSPRY2 mimicked the actions of silencing hSPRY2. Thus, the Y55F hSPRY2 decreased AKT and ERK1/2 activation by EGF as well as serum (supplemental Fig. S3) and also obliterated the anti-apoptotic actions of serum (Fig. 7B). Additionally, Y55F hSPRY2 also decreased the amount of EGFR (supplemental Fig. S3, D and E). Moreover, when hSPRY2 was silenced, TAT-HA-hSPRY2, but not TAT-HA-hSPRY2 Y55F mutant, was able to rescue the anti-apoptotic actions of serum. Together these data demonstrate that endogenous hSPRY2, by modulating the amount of growth factor receptors (via c-Cbl), plays an important role in regulating cell survival in response to serum.
The C-terminal proline-rich region of hSPRY2 (304PXX-PPR309) has been shown to bind CIN85, and the mutation of this region or the c-Cbl binding Tyr-55 attenuates the ability of hSPRY2 to inhibit internalization and degradation of growth factor receptors (31, 46). In keeping with the findings of Haglund et al. (46) that both the CIN85 and c-Cbl binding on hSPRY2 are necessary to protect growth factor receptor degradation, our data also show that mutation of either the proline-rich region (hSPRY2 CT-3A) or the c-Cbl binding site (hSPRY2 Y55F) abrogates the ability of hSPRY2 to restore the anti-apoptotic actions of serum. Thus, although the c-Cbl binding site (Tyr-55) is intact in the hSPRY2 CT-3A mutant, unlike the wt-hSPRY2, this mutant cannot restore the anti-apoptotic actions of serum. These findings lend further credence to the contention that hSPRY2, by sequestering c-Cbl, reduces growth factor receptor degradation and permits serum to activate downstream signaling that protects cells from apoptosis.
Interestingly, in the context of the CT-3A mutations, the Y55F mutation also loses its ability to act as a dominant negative, suggesting that the C-terminal proline-rich region is necessary for the dominant-negative actions of Y55F mutant of hSPRY2. Recently, in studies involving the overexpression of hSPRY2 and fibroblast growth factor, the C-terminal proline-rich region of hSPRY2 has also shown to be the site of Grb2 interaction (49). By binding Grb2, this proline-rich region has been shown to attenuate the activation of ERK1/2 by growth factors, such as fibroblast growth factor (49). Notably in our studies, silencing of hSPRY2 diminishes the activation of ERK1/2 as well as AKT and not augment them. Thus, it would appear that the hSPRY2/Grb2 interactions that would inhibit ERK1/2 activation do not play a role in the functions of endogenous hSPRY2 studied in this report. In this respect, recently the N-terminal region comprising amino acid 50–60 of hSPRY2 has also been shown to bind protein phosphatase 2A and dephosphorylate hSPRY2 on two serine residues and expose the C-terminal proline-rich region (50). Interestingly, although both protein phosphatase 2A (PP2A) and c-Cbl compete for binding the N-terminal region encompassed by amino acid 50–60, there are distinct cellular pools of hSPRY2 that either bind PP2A or c-Cbl (50). Our evidence, especially the experiments with Cbl-TKB and c-Cbl null MEFs, suggest a central role for c-Cbl in preserving the amount of growth factor receptors and in reinstating the ability of serum to protect cells from apoptosis when endogenous hSPRY2 is depleted. Thus, it would appear that the pool of hSPRY2 that is accessible for c-Cbl interaction is the predominant pool of hSPRY2 that regulates the ability of serum to induce cell survival.
In conclusion, our data show that serum-mediated anti-apoptotic actions in SW13 cells involve the activation of receptor-tyrosine kinases such as the EGFR. Silencing of endogenous hSPRY2 by decreasing the amount of growth factor receptors attenuates the activation of the pro-survival downstream signaling processes such as ERK1/2, AKT, and RSK. As determined by genetic ablation, c-Cbl is required to mediate the regulatory actions of hSPRY2 on cellular apoptosis. Endogenous hSPRY2, by sequestering c-Cbl, protects growth factor receptors such as EGFR from degradation and permits serum to protect cells from apoptosis. The mutation of either the c-Cbl or CIN85 binding region on hSPRY2 abrogates its ability to restore the anti-apoptotic function of serum, confirming the role of c-Cbl/CIN85/hSPRY2 interactions in this process. In this context this is the first report demonstrating a novel role for hSPRY2 in regulating cell survival.
Supplementary Material
Acknowledgments
We thank Drs. Graeme R. Guy and Chee Wai Fong, Institute of Molecular and Cell Biology, Singapore for the gift of anti-Spry1 and Spry4 antibodies as well as the Cbl-TKB and hSPRY3 vectors. We are greatful to Dr. Hamid Band, North Western University for the Cbl−/− MEFs. We are also grateful to Dr. Adriano Marchese, Loyola University Chicago for helpful discussions of our work.
Footnotes
This research was supported by National Institutes of Health Grant GM073181 (to T. B. P.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
The abbreviations used are: SPRY2, Sprouty 2; hSPRY2, human SPRY2; SW13 cells, adrenal cortex adenocarcinoma cells; EGF, epidermal growth factor; EGFR, EGF receptor; siRNA, small interfering RNA; HA, hemagglutinin; ERK1/2, extracellular signal regulated kinase 1/2; TNF-α, tumor necrosis factor-α; CHX, cycloheximide; p90RSK, p90 ribosomal S6 kinase; BAD, BCL2-antagonist of cell death; MEK, mitogen-activated protein kinase/extracellular signal-regulated kinase kinase; TKB domain, tyrosine kinase binding domain; FBS, fetal bovine serum; GFP, green fluorescent protein; MEF, mouse embryonic fibroblast; PMEF, primary MEF; PARP, poly(ADP-ribose) polymerase; wt, wild type.
F. Edwin and T. B. Patel, unpublished observations.
References
- 1.Hacohen N, Kramer S, Sutherland D, Hiromi Y, Krasnow MA. Cell. 1998;92:253–263. doi: 10.1016/s0092-8674(00)80919-8. [DOI] [PubMed] [Google Scholar]
- 2.Reich A, Sapir A, Shilo B. Development. 1999;126:4139–4147. doi: 10.1242/dev.126.18.4139. [DOI] [PubMed] [Google Scholar]
- 3.Mason JM, Morrison DJ, Albert Basson M, Licht JD. Trends Cell Biol. 2006;16:45–54. doi: 10.1016/j.tcb.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 4.Kim HJ, Bar-Sagi D. Nat Rev Mol Cell Biol. 2004;5:441–450. doi: 10.1038/nrm1400. [DOI] [PubMed] [Google Scholar]
- 5.Tefft JD, Lee M, Smith S, Leinwand M, Zhao J, Bringas P, Jr, Crowe DL, Warburton D. Curr Biol. 1999;9:219–222. doi: 10.1016/s0960-9822(99)80094-3. [DOI] [PubMed] [Google Scholar]
- 6.Lee SH, Schloss DJ, Jarvis L, Krasnow MA, Swain JL. J Biol Chem. 2001;276:4128–4133. doi: 10.1074/jbc.M006922200. [DOI] [PubMed] [Google Scholar]
- 7.Perl AK, Hokuto I, Impagnatiello MA, Christofori G, Whitsett JA. Dev Biol. 2003;258:154–168. doi: 10.1016/s0012-1606(03)00106-4. [DOI] [PubMed] [Google Scholar]
- 8.Chi L, Zhang S, Lin Y, Prunskaite-Hyyrylainen R, Vuolteenaho R, Itaranta P, Vainio S. Development. 2004;131:3345–3356. doi: 10.1242/dev.01200. [DOI] [PubMed] [Google Scholar]
- 9.Gross I, Morrison DJ, Hyink DP, Georgas K, English MA, Mericskay M, Hosono S, Sassoon D, Wilson PD, Little M, Licht JD. J Biol Chem. 2003;278:41420–41430. doi: 10.1074/jbc.M306425200. [DOI] [PubMed] [Google Scholar]
- 10.Furthauer M, Reifers F, Brand M, Thisse B, Thisse C. Development. 2001;128:2175–2186. doi: 10.1242/dev.128.12.2175. [DOI] [PubMed] [Google Scholar]
- 11.Minowada G, Jarvis LA, Chi CL, Neubuser A, Sun X, Hacohen N, Krasnow MA, Martin GR. Development. 1999;126:4465–4475. doi: 10.1242/dev.126.20.4465. [DOI] [PubMed] [Google Scholar]
- 12.Chambers D, Mason I. Mech Dev. 2000;91:361–364. doi: 10.1016/s0925-4773(99)00288-9. [DOI] [PubMed] [Google Scholar]
- 13.Impagnatiello MA, Weitzer S, Gannon G, Compagni A, Cotten M, Christofori G. J Cell Biol. 2001;152:1087–1098. doi: 10.1083/jcb.152.5.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gross I, Bassit B, Benezra M, Licht JD. J Biol Chem. 2001;276:46460–46468. doi: 10.1074/jbc.M108234200. [DOI] [PubMed] [Google Scholar]
- 15.Yigzaw Y, Cartin L, Pierre S, Scholich K, Patel TB. J Biol Chem. 2001;276:22742–22747. doi: 10.1074/jbc.M100123200. [DOI] [PubMed] [Google Scholar]
- 16.Lee CC, Putnam AJ, Miranti CK, Gustafson M, Wang LM, Vande Woude GF, Gao CF. Oncogene. 2004;23:5193–5202. doi: 10.1038/sj.onc.1207646. [DOI] [PubMed] [Google Scholar]
- 17.Zhang C, Chaturvedi D, Jaggar L, Magnuson D, Lee JM, Patel TB. Arterioscler Thromb Vasc Biol. 2005;25:533–538. doi: 10.1161/01.ATV.0000155461.50450.5a. [DOI] [PubMed] [Google Scholar]
- 18.Edwin F, Singh R, Endersby R, Baker SJ, Patel TB. J Biol Chem. 2006;281:4816–4822. doi: 10.1074/jbc.M508300200. [DOI] [PubMed] [Google Scholar]
- 19.Lim J, Wong ES, Ong SH, Yusoff P, Low BC, Guy GR. J Biol Chem. 2000;275:32837–32845. doi: 10.1074/jbc.M002156200. [DOI] [PubMed] [Google Scholar]
- 20.Yigzaw Y, Poppleton HM, Sreejayan N, Hassid A, Patel TB. J Biol Chem. 2003;278:284–288. doi: 10.1074/jbc.M210359200. [DOI] [PubMed] [Google Scholar]
- 21.Poppleton HM, Edwin F, Jaggar L, Ray R, Johnson LR, Patel TB. Biochem Biophys Res Commun. 2004;323:98–103. doi: 10.1016/j.bbrc.2004.08.070. [DOI] [PubMed] [Google Scholar]
- 22.Shim K, Minowada G, Coling DE, Martin GR. Dev Cell. 2005;8:553–564. doi: 10.1016/j.devcel.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 23.Taketomi T, Yoshiga D, Taniguchi K, Kobayashi T, Nonami A, Kato R, Sasaki M, Sasaki A, Ishibashi H, Moriyama M, Nakamura K, Nishimura J, Yoshimura A. Nat Neurosci. 2005;8:855–857. doi: 10.1038/nn1485. [DOI] [PubMed] [Google Scholar]
- 24.Fang X, Yu S, Eder A, Mao M, Bast RC, Jr, Boyd D, Mills GB. Oncogene. 1999;18:6635–6640. doi: 10.1038/sj.onc.1203076. [DOI] [PubMed] [Google Scholar]
- 25.Egan JE, Hall AB, Yatsula BA, Bar-Sagi D. Proc Natl Acad Sci U S A. 2002;99:6041–6046. doi: 10.1073/pnas.052090899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wong ES, Fong CW, Lim J, Yusoff P, Low BC, Langdon WY, Guy GR. EMBO J. 2002;21:4796–4808. doi: 10.1093/emboj/cdf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christofori G. Nat Cell Biol. 2003;5:377–379. doi: 10.1038/ncb0503-377. [DOI] [PubMed] [Google Scholar]
- 28.Li X, Wheldon L, Heath JK. Biochem Soc Trans. 2003;31:1445–1446. doi: 10.1042/bst0311445. [DOI] [PubMed] [Google Scholar]
- 29.Deng W, Poppleton H, Yasuda S, Makarova N, Shinozuka Y, Wang DA, Johnson LR, Patel TB, Tigyi G. J Biol Chem. 2004;279:47871–47880. doi: 10.1074/jbc.M405443200. [DOI] [PubMed] [Google Scholar]
- 30.Chaturvedi D, Poppleton HM, Stringfield T, Barbier A, Patel TB. Mol Cell Biol. 2006;26:4586–4600. doi: 10.1128/MCB.01422-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fong CW, Leong HF, Wong ES, Lim J, Yusoff P, Guy GR. J Biol Chem. 2003;278:33456–33464. doi: 10.1074/jbc.M301317200. [DOI] [PubMed] [Google Scholar]
- 32.Hall AB, Jura N, DaSilva J, Jang YJ, Gong D, Bar-Sagi D. Curr Biol. 2003;13:308–314. doi: 10.1016/s0960-9822(03)00086-1. [DOI] [PubMed] [Google Scholar]
- 33.Mason JM, Morrison DJ, Bassit B, Dimri M, Band H, Licht JD, Gross I. Mol Biol Cell. 2004;15:2176–2188. doi: 10.1091/mbc.E03-07-0503. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Rubin C, Litvak V, Medvedovsky H, Zwang Y, Lev S, Yarden Y. Curr Biol. 2003;13:297–307. doi: 10.1016/s0960-9822(03)00053-8. [DOI] [PubMed] [Google Scholar]
- 35.Sasaki A, Taketomi T, Wakioka T, Kato R, Yoshimura A. J Biol Chem. 2001;276:36804–36808. doi: 10.1074/jbc.C100386200. [DOI] [PubMed] [Google Scholar]
- 36.Wong ES, Lim J, Low BC, Chen Q, Guy GR. J Biol Chem. 2001;276:5866–5875. doi: 10.1074/jbc.M006945200. [DOI] [PubMed] [Google Scholar]
- 37.Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 38.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 39.Richards SA, Fu J, Romanelli A, Shimamura A, Blenis J. Curr Biol. 1999;9:810–820. doi: 10.1016/s0960-9822(99)80364-9. [DOI] [PubMed] [Google Scholar]
- 40.Moolenaar WH. Trends Cell Biol. 1994;4:213–219. doi: 10.1016/0962-8924(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 41.Tigyi G, Miledi R. J Biol Chem. 1992;267:21360–21367. [PubMed] [Google Scholar]
- 42.Waterman H, Levkowitz G, Alroy I, Yarden Y. J Biol Chem. 1999;274:22151–22154. doi: 10.1074/jbc.274.32.22151. [DOI] [PubMed] [Google Scholar]
- 43.Levkowitz G, Waterman H, Ettenberg SA, Katz M, Tsygankov AY, Alroy I, Lavi S, Iwai K, Reiss Y, Ciechanover A, Lipkowitz S, Yarden Y. Mol Cell. 1999;4:1029–1040. doi: 10.1016/s1097-2765(00)80231-2. [DOI] [PubMed] [Google Scholar]
- 44.Grovdal LM, Stang E, Sorkin A, Madshus IH. Exp Cell Res. 2004;300:388–395. doi: 10.1016/j.yexcr.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 45.Kassenbrock CK, Anderson SM. J Biol Chem. 2004;279:28017–28027. doi: 10.1074/jbc.M404114200. [DOI] [PubMed] [Google Scholar]
- 46.Haglund K, Schmidt MH, Wong ES, Guy GR, Dikic I. EMBO Rep. 2005;6:635–641. doi: 10.1038/sj.embor.7400453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kowanetz K, Szymkiewicz I, Haglund K, Kowanetz M, Husnjak K, Taylor JD, Soubeyran P, Engstrom U, Ladbury JE, Dikic I. J Biol Chem. 2003;278:39735–39746. doi: 10.1074/jbc.M304541200. [DOI] [PubMed] [Google Scholar]
- 48.Cabrita MA, Jaggi F, Widjaja SP, Christofori G. J Biol Chem. 2006;281:29201–29212. doi: 10.1074/jbc.M603921200. [DOI] [PubMed] [Google Scholar]
- 49.Lao DH, Chandramouli S, Yusoff P, Fong CW, Saw TY, Tai LP, Yu CY, Leong HF, Guy GR. J Biol Chem. 2006;281:29993–30000. doi: 10.1074/jbc.M604044200. [DOI] [PubMed] [Google Scholar]
- 50.Lao DH, Yusoff P, Chandramouli S, Philp RJ, Fong CW, Jackson RA, Saw TY, Yu CY, Guy GR. J Biol Chem. 2007;282:9117–9126. doi: 10.1074/jbc.M607563200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.