

Abstract
Trefoil factor 3 (intestinal trefoil factor) is a cytoprotective factor in the gut. Here, we compared the effect of trefoil factor 3 with tumor necrosis factor-α on (1) activation of nuclear factor-κB in intestinal epithelial cells; (2) expression of Twist protein (a molecule essential for down-regulation of nuclear factor-κB activity in vivo); and (3) production of interleukine-8. We showed that Twist protein is constitutively expressed in intestinal epithelial cells. Tumor necrosis factor-α induced persistent degradation of Twist protein in intestinal epithlial cells via a signal pathway linked to proteasome, which was associated with prolonged activation of nuclear factor-κB. In contrast to tumor necrosis factor, trefoil factor 3 triggered transient activation of nuclear factor-κB and prolonged up-regulation of Twist protein in intestinal epithelial cells via an ERK kinase-mediated pathway. Unlike tumor necrosis factor-α, transient activation of NF-κB by trefoil factor 3 is not associated with induction of IL-8 in cells. To examine the role of Twist protein in intestinal epithelial cells, we silenced the Twist expression by siRNA. Our data showed that trefoil factor 3 induced interleukine-8 production after silencing Twist in intestinal epithelial cells. Together, these observations indicated that (1) trefoil factor 3 triggers a diverse signal from tumor necrosis factor-α on the activation of nuclear factor-κB and its associated molecules in intestinal epithelial cells; and (2) trefoil factor 3-induced Twist protein plays an important role in the modulation of inflammatory cytokine production in intestinal epithelial cells.
Keywords: Intestinal Epithelial Cells, Trefoil factor 3, Signal Transduction, NF-κB
Keywords: TFF, trefoil factor; GI, gastrointestinal; TFF3, trefoil factor 3; NF-κB, nuclear factor-κB; IL-8, interleukin-8; TNF, tumor necrosis factor-α
INTRODUCTION
NF-κB is a transcription factor involved in important biological processes including inflammation and apoptosis (23, 55). Many extracellular stimuli induce NF-κB activation. The physiological roles of NF-κB are both cell type- and stimulus-dependent (23, 55). Previous investigations have shown that activation of NF-κB by extracellular stimuli such as tumor necrosis factor α (TNF) is predominantly mediated by a distinctive signal pathway linked to degradation of IκBα. Activated NF-κB complex is translocated into the nucleus and binds to the promoter regions of its targeted genes (23).
In recent times, a growing body of evidence suggests the existence of autoregulatory loops for a net negative regulation of NF-κB functions in mammalian cells (3, 4, 6, 10, 38, 43, 44). For example, activation of NF-κB results not only in up-regulation of genes involved in inflammation and cell survival but also in synthesizing/re-synthesizing NF-κB-dependent, negative regulators of NF-κB signaling, such as IκBα, Bcl-3, A20, nitric oxide and prostaglandin E2 (6, 10, 25, 26, 29, 39, 43, 44). Newly synthesized IκBα and Bcl-3 are translocated into the nucleus and serve to terminate NF-κB action (3, 6), whereas A20, nitric oxide and prostaglandin E2 are responsible for the blocking of NF-κB pathway (12, 13, 28). Overexpression of IκBα results in cardioprotection in trauma (7). Mice lacking A20 suffer from severe systemic inflammation (28, 43). Taken together, the negative regulatory loop of NF-κB ensures transient generation of intracellular signaling to prevent uncontrolled-NF-κB activation. It is critical to limit inflammatory injury by terminating and blocking proinflammatory cytokine-induced NF-κB activity in vivo.
Twist protein is a basic helix-loop-helix transcription factor. In mammals, Twist contributes to the morphogenesis of cranial neural tube and participates in the regulation of muscular differentiation (9). Previously, Sosic et al. have demonstrated that (1) Twist protein interacts with transcription factor NF-κB; (2) Twist is essential for the down-regulation of NF-κB activity in vivo; and (3) NF-κB regulates Twist gene expression (43). Thus, Twist protein is a NF-κB associated protein. It plays an important role in the regulation of the negative regulatory loop of NF-κB pathway and the attenuation of inflammatory responses (42). Recently, Twist has been shown to be present in mouse mammary epithelial cell lines (18), suggesting that it may be involved in the regulation of physiological or phathophysiological processes in epithelial tissues.
The trefoil factor (TFF) family is a group of extracellular peptides that were originally found in the gastrointestinal (GI) tract (37, 51, 53). These peptides contain distinctive cystine-rich “trefoil” domains and are resistant to proteolytic degradation (16, 37, 51). Three mammalian TFFs have been identified, namely, pS2 (TFF1), SP (TFF2), and intestinal trefoil factor (ITF or TFF3). TFFs are expressed in several tissues of the body but most pronouncedly in the GI tract. They are usually associated with the mucous layer in the GI tract. Under normal circumstances TFF1 and TFF2 are expressed in the human stomach, whereas TFF3 is expressed in the small and large intestine. The expression of TFF has been demonstrated to be up-regulated in GI tract ulcers (2). In contrast to TGF-alpha and EGF, TFFs are expressed rapidly in response to injury. It has been shown that intestinal epithelium is the targeted tissue of TFFs (17).
TFF3 is predominantly expressed in mucous epithelia. A major source for TFF3 is goblet cells of the small and large intestine. Maximal TFF3 expression in the GI tract was observed in the distal portions of the ileum and the colon (31). Secretion of TFF3 is evoked by certain neurotransmitters and inflammatory mediators (33). Among TFFs, the physiological functions of TFF3 have been well characterized. Previous studies demonstrated that TFF3 enhances restitution in intestinal epithelial cells and sustains mucosa integrity (35). We showed that TFF3 protects epithelial cells against reactive oxygen species induced-injury (45). In addition, we found that TFF3 also induces activation of NF-κB in intestinal epithelial cells and demonstrated that TFF3 prevents apoptosis of intestinal epithelial cells via NF-κB pathway (8). Pretreatment of the GI mucosa with TFF3 protects epithelium against various injuries (5, 24, 45). However, it is still unknown whether TFF3 modulates inflammatory responses in intestinal epithelial cells. In the present study, we examined (1) how TFF3 activates NF-κB in intestinal epithelial cells; (2) whether Twist protein is expressed in intestinal epithelial cells and regulated by TFF3 and TNF; and (3) whether Twist protein plays a role in modulation of expression of NF-κB-targeted proinflammatory cytokines such as IL-8 in intestinal epithelial cells.
MATERIALS AND METHODS
Reagents.
All cell culture media were obtained from GIBCO Invitrogen Corp. (Carlsbad, CA, USA). Polyclonal antibodies against Twist (SC-15393), IκBα (SC-371), p50 (SC-114X), and p65 (SC-109X) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-phosphotyrosine mAb (clone 4G10) was purchased from Upstate Biotechnology (Lake Placid, NY, USA). HRP-conjugated goat anti-rabbit IgG (Catalog #111-035-144) and anti-mouse IgG (Catalog #115-035-062) were obtained from Jackson Immuno Research Laboratories, Inc (West Grove, PA, USA). mAb against GAPDH was purchased from Abcam, Inc (Cambridge, MA, USA). Chemicals and molecular biology reagents were purchased from Sigma Chemical Company (St. Louis, MO, USA). Reagents for electrophoresis and Western blotting were supplied by Bio-Rad Laboratories (Hercules, CA, USA). Precasted 4–20% iGels were purchased from Gradipore Ltd (Frenchs Forest, Australia). Quanti Kine R Human IL-8 Immunoassay Kit was purchased from R and D Systems (Minneapolis, MN, USA). Protease inhibitor cocktail tablets were supplied by Roche Molecular Biochemicals (Indianapolis, IN, USA). All tissue culture plasticwares were supplied by Costar (Cambridge, MA, USA). U126 and PD98059 (selective inhibitors for ERK kinase) and MG132 (a potent proteasome inhibitor) were obtained from Biomol (Plymouth Meeting, PA, USA). ECL System and [γ-32P]-ATP were supplied by Amersham Pharmacia Biotech (Piscataway, NJ, USA).
Preparation of Rat Recombinant TFF3 in Yeast.
Rat TFF3 was expressed from Pichia pastoris using a method modified from our previous protocol (8). The recombinant protein was purified with a FPLC (AKTA FPLC, Amersham Pharmacia Biotech). The purified TFF3 peptide was visualized as a single band with the silver staining technique. Its biological activity was assessed by the cell migration assay using IEC-18 cells. The peptide induced approximate three-fold increase in cell migration.
Cell Culture.
HT-29 and IEC-18 cells were purchased from American Type Culture Collection (Rockville, MD, USA) and cultured in a water-saturated atmosphere with 5% CO2 at 37°C. HT-29 cells (passages 20–35 after receipt from ATCC) were maintained in Dulbecco’s Modified Eagle’s Minimum Essential Medium (DMEM) containing 100 unit/ml penicillin, 100 μg/ml streptomycin, and 10% heat-inactivated fetal bovine serum (FBS). IEC-18 cells were maintained in DMEM supplemented with 10% heat-inactivated FBS, insulin, 1% nonessential amino acids, 100 unit/ml penicillin, and 100 μg/ml of streptomycin.
Western blotting (47).
Cells were lysed in a buffer containing 2 mM Tris-Cl (pH 7.6), 30 mM NaCl, 1 mM EDTA, 1 mM DTT, 1 mM PMSF, complete protease inhibitor cocktail (1 tablet/10ml), and 1% Notidet-40. Total cell lysate was processed through sonication at 4°C and followed by centrifugation at 10000 X g for 10 minutes at 4°C. Supernatants were mixed with an equal volume of 2 X Laemmli’s buffer and boiled for 5 minutes. Thirty micrograms of protein were resolved on 4–20% SDS-PAGE gel along with molecular weight standards. The proteins were then transferred onto a nitrocellulose membrane as described before (57). The membranes containing sample proteins were used for immunodetection of Twist protein. Briefly, blots preincubated with PBS containing 5% skim milk were reacted with primary antibody against Twist protein (1:500) for 1 hour at room temperature. After incubation, the blot was washed four times with PBS containing 0.05% Tween 20 (PBS-T), and then incubated with PBS-T containing 1:10000 diluted HRP-conjugated goat anti-rabbit IgG for 1 hour at room temperature. After additional washing with PBS-T, immune complexes on the blot were visualized using the ECL system. Blots were stripped and reprobed with mAb against GAPDH (1:10000) following a standard procedure (45).
Immunoprecipitation:
Lysate containing 0.5 mg of proteins was treated with 1 μg of rabbit anti-IκBα antibodies in 800 μl of lysate buffer at 4°C overnight, and the immune complexes were precipitated with protein A/G-Sepharose beads. The beads were thoroughly washed, resuspended in SDS sample buffer, and boiled for 5 min. After boiling, the proteins were resolved on 4–20% SDS-PAGE gel, electrotransferred to a nitrocellulose membrane, and probed with mouse anti-phosphotyrosine monoclonal antibody (1:1000). The blot was then treated with HRP-conjugated goat anti-mouse IgG and finally detected by ECL reagent.
Preparation of Nuclear Extracts from Cells (8).
After removing medium, cells (5 x 106) were washed with cold PBS containing 0.5 mM DTT, directly treated with 500 μl of buffer A containing 10 mM Hepes (pH7.9), 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 0.1% NP-40, scraped off Petri dishes, and incubated for 15 minutes at 4°C. The nuclei were collected by a brief centrifugation using a microcentrifuge. The supernatant was removed. The nuclear pellet was washed with buffer A, resuspended in 150 μl of buffer B containing 20 mM Hepes (pH7.9)/1.5 mM MgCl2/0.5 mM DTT/0.42 M NaCl/0.2mM EDTA/25% glycerol/0.5mM PMSF, and incubated for 15 minutes on ice. Following centrifugation at 14,000 x g for 10 minutes at 4°C, the supernatant (nuclear protein fraction) was diluted in buffer C containing 20mM Hepes (pH7.9)/50 mM KCl/0.5 mM DTT/0.2 mMEDTA/0.5 mM PMSF to 300 μl (5x106 cells) and stored at −80°C. Protein concentration was measured by the Bradford’s method.
Electrophoretic Mobility Shift Assay (EMSA) and Supershift Assay.
NF-κB DNA-binding activity was determined by EMSA as described before (8). Briefly, the NF-κB consensus oligonucleotide was labeled by [γ -32P]ATP (3,000 Ci/mmmol, 10 mCi/ml) with T4 polynucleotide kinase. Nuclear extracts (5 μg) were added to 10 μl of gel shift binding buffer (40% glycerol, 10 mM Tris.HCl, pH7.5, 50 mM NaCl, 0.5 mM EDTA, 1 mM MgCl2, 0.5 mM DTT, 0.01 mg/ml PolydIdC). 1 μl of 32P-labeled oligonucleotide probe was added and the mixture was incubated at room temperature for 20 min. Electrophoresis was done on a 6% polyacrymide gel with 0.5 X TBE buffer. The gel was dried and analyzed using a phosphorimaging system. Supershift experiments were performed as follows. Nuclear extracts were incubated with 32P-labeled oligonucleotide (1 μl) at room temperature for 20 min. 1 μg of antibody against p50, p65, p52, c-Rel, or RelB subunit of NF-κB was subsequently added, samples were incubated at room temperature for 15 min, and electrophoresis was performed. To confirm the specificity of NF-κB DNA binding activity, a competitive experiment was done by adding 1 μl (1750 pmol/ml) of unlabelled oligonucleotide to the reaction mixture.
ELISA assay for quantitation IL-8 in conditioned medium.
Cells were harvested by trypsinization and seeded at 5 X 104 cells/well in 96-well tissue culture plates. Cells were further incubated for 2 days to reach confluence. The medium of confluent monolayers was changed to serum deprived-medium, and cells were cultured for overnight. Medium was then changed to fresh serum deprived-medium containing different stimulators and incubated for indicated times. Medium was collected and IL-8 was detected using Quantikine human IL-8 immunoassay kit (R and D). The protocol provided by the manufacturer was followed. Standard curves were generated for IL-8 using the standard provided in the kit and the concentration of IL-8 in the cell supernatant was determined by interpreting from the appropriate standard curve.
Immunohistochemical staining:
Rat intestinal tissues were sectioned with a cryostat system, fixed with acetone for 10 min at −20 °C, and air-dried. Slides were rinsed with PBS and incubated with PBS containing 1% BSA for 10 min. Next, slides were incubated with rabbit pAb against Twist (1:500) for 30 min, washed, and incubated with biotinylated goat anti-rabbit IgG for 30 min. After washing, slides were incubated with FITC-labeled streptavidin for 30 min, washed, and covered with FluorSave reagent (Calbiochem, San Diego, CA). The staining was performed at room temperature. Finally, slides were visualized under a fluorescence microscope.
siRNA-mediated Twist gene silencing.
The siRNA duplexes targeting the human Twist mRNA (Gene Bank Accession No: NM_000474) were designed with the most efficient siRNA-2-duplexes design protocol and synthesized by Qiagen (Valencia, CA). The targeting sequences of double-stranded siRNA are: TGG GAT CAA ACT GGC CTG CAA and TAA GAA CAC CTT TAG AAA TAA. These sequences were submitted to BLAST search to ensure that only the TWIST gene was targeted by the TWIST siRNA, and control sequences (Non-silencing labeled Control siRNA) were not homologous to any known genes. HT29 cells were transfected using RNAi Human/Mouse Control Kit (Qiagen, Valencia, CA). In brief, HT29 cells were seeded onto 6-well plates (4 x 104 cells/well) 24 hrs before siRNA treatment. For transfection, siRNA (1.9 μg/well) was transfected with the RNAiFect reagent (Qiagen, Valencia, CA), according to the manufacture’s instruction. Twenty-four hours after transfection, culture medium was changed. Cells were used for designed experiments on the fourth day after transfection.
Statistics.
Data were expressed as means + s.e.m. Analysis of variance and one-way analysis of variance (ANOVA) followed by Fisher’s protected least significant difference post-hoc test were used to assess the significant of differences; P< 0.05 was considered significant.
RESULTS
In the present study, we used intestinal epithelial cell lines including HT-29 and IEC-18 lines. The cell lines for studying the mechanisms of TFF3 action in intestinal epithelia are selected based on the following rationales: (1) Epithelial cell lines from both small and large intestine are selected, since TFF3 is expressed in both. (2) The cell lines selected are well-characterized, and consist of a homogeneous cell population, and are easily maintained in culture. (3) The cells to be used should express many characteristics of the normal intestinal epithelium. (4) The cells to be used should respond to rat TFF3 stimulation in vitro. IEC-18 is a non-transformed rat small intestinal epithelial cell line derived from undifferentiated crypt epithelial cells. HT-29 cell line originates from human colon carcinomas and shows many characteristic features of intestinal epithelium. Both IEC-18 and HT-29 cell lines have previously been used for TFF3 study by various investigators and respond to rat TFF3 stimulation (24, 36, 47). Therefore, we selected them for our studies.
1. TFF3 activates NF-κB in intestinal epithelial cells in a manner different from TNF.
Our recent investigation suggests that TFF3 activates NF-κB in intestinal epithelial cells (8). Here, we further examined the kinetics of TFF3-induced NF-κB activation. HT-29 cells were treated with TFF3 (2.5 μM) for 0.5, 1, 2, 4, 8, and 24 hours respectively. At the end of the treatment, nuclear proteins were extracted and EMSA assay was performed with 32P-labelled NF-κB consensus probe. As shown in Fig. 1A, a low basal level of NF-κB activity was detected in resting HT-29 cells. NF-κB binding activity was induced within 30 min after treatment and rapidly fell thereafter, reaching control levels by 60 min. The specificity of DNA/NF-κB binding was confirmed with a 100-fold excess of unlabeled κB-oligonucleotide (data not shown). Incubation of nuclear extracts with anti-p65 antibody resulted in the abrogation in NF-κB/DNA complex, whereas anti-p50 antibody had no effect (Fig. 1A). In addition, the NF-κB/DNA complex was not reduced by antibodies against p52, c-Rel, or RelB (data not shown). Together, the data suggests that NF-κB activated by TFF3 in human intestinal epithelial cells was composed of p65 homodimer. In contrast, TNF induced strong NF-κB activation in HT-29 cells and the effect persisted for 24 hours (Fig. 1B). Supershift assay revealed that TNF-activated NF-κB in intestinal epithelial cells contained p50/p65 heterodimers (Fig. 1B).
Fig. 1. Effect of TFF3 and TNF on activation of NF-κB in human intestinal epithelial cells.
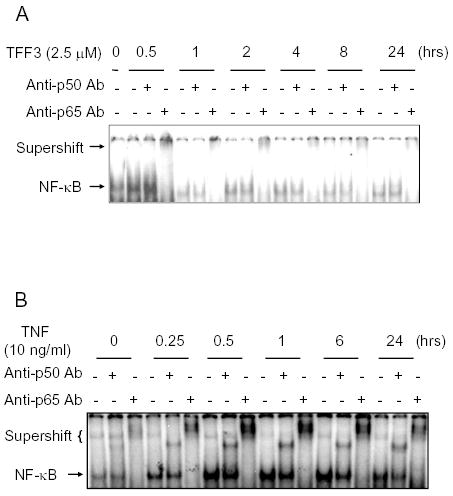
A. Time course of TFF3 effect on activation of NF-κB in human intestinal epithelial cells. Nuclear extracts from HT-29 cells either unstimulated or stimulated for indicated times with TFF3 (2.5 μM) were subjected to EMSA/supershift assay as described under Material and Methods. Data are representative of 3 separate experiments.
B. Time course of TNF effect on activation of NF-κB in human intestinal epithelial cells. Nuclear extracts from HT-29 cells either unstimulated or stimulated for indicated times with TNF (10 ng/ml) were subjected to EMSA/supershift assay as described under Material and Methods. Data are representative of 3 separate experiments.
Extracellular stimulus activates NF-κB mainly through inducing either rapid degradation of IκBα protein following phosphorylation of Ser-32 and Ser-36 of IκBα by IκB kinase or tyrosine phosphorylation of IκBα protein without proteolytic degradation of IκBα (21, 23). To dissect the process through which TFF3 activates NF-κB in intestinal epithelium, we investigated whether TFF3 induces degradation of IκBα. For this, HT-29 cells were subjected to treatment with TFF3 (2.5 μM), total cellular proteins were isolated, and Western blot was performed with rabbit anti-IκBα polyclonal antibody. Strikingly, treatment of HT-29 cells with TFF3 resulted in partial degradation of IκBα within 30 min. IκBα was actively re-synthesized 2 hours after the treatment and increased thereafter (Fig. 2A). The data suggested phosphorylation of Ser-32 and Ser-36 of IκBα following TFF3 stimulation. To further explore whether TFF3 also induces tyrosine phosphorylation of IκBα protein, whole cell lysates from untreated and TFF3-treated HT-29 cells were immunoprecipitated either with preimmune sera or with anti-IκBα antibody followed by Western blot with anti-phosphotyrosine antibody. In the positive control, we subjected cells to treatment with pervanadate (100 μM) for 30 min, lysate, immunoprecipitation, and Western blotting. As shown in Fig. 2B, IκBα immunoprecipitated from pervanadate-treated cells was tyrosine-phosphorylated. In contrast, tyrosine phosphorylated IκBα was not detected in TFF3-treated cells.
Fig. 2. Effect of TFF3 on modulation of IκBα in intestinal epithelial cells.

A. Stimulation with TFF3 results in partial degradation of IκBα. Cell lysates (30 μg) from HT-29 cells either unstimulated or stimulated for indicated times were subjected to SDS-PAGE gel electrophoresis, transferred to membranes. The membranes were subjected to Western blotting analysis with anti-IκBα pAb as described under Material and Methods. Top panel: Representative autoradiographs of an immunoblot. Bottom panel: Densitometric analysis of immunoblot data. *P<0.05 versus 0 hr. Data expressed as means + s.e.m. of three determinations.
B. Stimulation with TFF3 does not result in tyrosine phosphorylation of IκBα. Cell lysates (400 μg) from HT-29 cells either unstimulated or stimulated for indicated times with 2.5 μM TFF3 were processed for immunoprecipitation with anti-IκBα antibody. Resulting immunocomplexes were subjected to SDS-PAGE gel electrophoresis and transferred to membranes. The membranes were subjected to Western blotting by using anti-PY mAb as described under Material and Methods. Data are representative of 3 separate experiments. Van: pervanadate.
To compare the effect of TFF3 and TNF on the degradation of IκBα, HT-29 cells were also treated cells with TNF followed by detection of IκBα protein with Western blot. We confirmed that IκBα protein was extensively degraded 30 min after TNF administration and subsequently resynthesized (data not shown). Together, the data suggested that TFF3 induces partial degradation of IκBα but without induction of its tyrosine phosphorylation in intestinal epithelial cells. The effect is associated with transient activation of p65/p65 homodimer NF-κB. In contrast to TFF3, TNF induced more degradation of IκBα and prolonged activation of p50/p65 heterodimer NF-κB in intestinal epithelial cells.
2. TFF3 does not induce IL-8 production in intestinal epithelial cells.
Persistent activation of NF-κB in intestinal epithelial cells results in up-regulation of several NF-κB targeted proinflammatory cytokines such as IL-8. However, whether transient activation of NF-κB induces inflammatory mediators is not clear. As we have shown that TFF3 induces transient activation of NF-κB in intestinal epithelial cells, we further examined the ability of TFF3 to induce the release of IL-8 by intestinal epithelial cells. For this we subjected HT-29 cells to stimulation with TFF3 (2.5 μM) or TNF (10 ng/ml) for 1 hour. We then removed the medium, washed the cells with PBS, added fresh standard medium, and chased for 23 hours. Thereafter, we measured IL-8 in culture medium with ELISA assay. As shown in Fig. 3, TNF strongly induced IL-8 release in HT-29 cells, whereas TFF-3 showed no effect. The data indicated that transient activation of NF-κB by TFF3 did not result in IL-8 production in intestinal epithelial cells.
Fig. 3. Modulation of IL-8 production by TNF and TFF3 in intestinal epithelial cells.

HT-29 cells were treated with TNF (10 ng/ml) and TFF3 (2.5 μM) for 24 hrs respectively. Conditioned medium from untreated or treated cells were subjected to measurement of IL-8 with ELISA assay as described under Material and Methods. Results are the means + s.e.m. of three independent experiments. **, significantly different from the control at P<0.01.
3. Twist is constitutively expressed in intestinal epithelial cells.
Twist plays an important role in the attenuation of inflammation in multiple tissues such as muscles and skin (43). It blocks NF-κB regulated gene expression (43). To determine whether intestinal epithelium expresses Twist, total cellular protein extracted from intestinal epithelial cells including HT-29 and IEC-18 cell lines were used for Western blot analysis. Rat skeletal muscle tissue was used as the positive control. As shown in Fig. 4A, Twist protein was present in both HT-29 and IEC-18 cells. Furthermore, the localization of Twist protein in rat small intestinal tissue was visualized by immunofluorescence microscopy using anti-Twist polyclonal antibody. As shown in Fig. 4B, cytosol of intestinal epithelial cells and some cells in the lamina propria stained with anti-Twist pAb. In contrast, intestinal epithelium did not stain with control pre-immune serum (Fig. 4C). The data suggests that Twist protein is present in intestinal epithelial cells in vivo.
Fig. 4. Expression of Twist in intestinal epithelium.

A. Twist is constitutively expressed in intestinal epithelial cells in vitro. Total cellular proteins were isolated from rat hindlimb muscles, IEC-18 and HT-29 cells respectively. Thirty micrograms of protein were subjected to SDS-PAGE gel electrophoresis, transferred to membranes, and analyzed by Western blotting using anti-twist Ab (1:500 dilution).
B and C. Localization of Twist protein in the rat small intestinal tissue. Cryostat sections from the small intestine of a normal rat were stained with immunofluorescence using antibody against Twist (B) or rabbit pre-immune serum (C). Sections were observed by immunofluorescence microscopy. Original magnification: X100
4. Effect of TNF on the expression of Twist protein in intestinal epithelial cells.
Recently, Sosic et al. demonstrated that Twist plays an important role in the attenuation of inflammation in multiple tissues such as muscles and skin (43). To investigate whether inflammatory mediators such as TNF modulate the expression of Twist in intestinal epithelial cells, IEC-18 cells were stimulated with TNF (10 ng/ml) for up to 24 hours. Cells were then lysed and processed for Western blot. Time course analysis revealed that levels of Twist protein in IEC-18 cells dropped within 30 min of treatment with TNF and persisted in low levels for up to 24 hours (Fig. 5A). Under similar conditions, TNF also induced decrease of Twist protein in HT-29 cells (Fig. 5B). These data suggests that TNF induces the degradation of Twist in intestinal epithelial cells.
Fig. 5. Time course of down-regulation of Twist protein in intestinal epithelial cells by treatment with TNF.

HT-29 (panel A) and IEC-18 (panel B) cells were subjected to stimulation with TNF (10 ng/ml) for different time points and lysis thereafter. Total protein lysates (30 μg) were subjected to SDS-PAGE gel electrophoresis and transferred to membranes. The blots were subjected to Western blotting using anti-twist Ab. The membranes were stripped and immunoblotted with anti-GAPDH mAb (housekeeping gene). Twist protein expression at each time point was normalized to that of GAPDH expression by using densitometry, and normalized content was compared with that at time 0. Top panel: Representative autoradiographs of an immunoblot. Bottom panel: Densitometric analysis of immunoblot data. **P<0.005 versus 0 hr. ***P<0.001 versus 0 hr. Data expressed as means + s.e.m. of three determinations.
Previous studies have shown that modulation of various signaling proteins by TNF requires 26S proteasome (19, 20). To investigate whether proteasome plays a role in TNF-induced degradation of Twist, HT-29 cells were pretreated with MG132 (50 μM, a selective proteasome inhibitor) for 60 minutes, followed by stimulation with TNF for 30 min. As shown in Fig. 6, pretreatment of cells with MG132 resulted in blocking the effect of TNF on the expression of Twist protein. Epoxomicin (32), another selective proteasome inhibitor, also blocked the TNF effect. This result suggests that TNF-triggered degradation of Twist in intestinal epithelial cells is mediated by proteasome.
Fig. 6. Role of proteasome on TNF-induced degradation of Twist protein in intestinal epithelial cells.

Selective proteasome inhibitors block the effect of TNF on reduction of Twist protein in intestinal epithelial cells. Cell lysates (30 μg) from HT-29 cells untreated or treated with indicated stimulators were subjected to SDS-PAGE gel electrophoresis and transferred to membranes. The membranes were subjected to Western blotting and densitometric analysis as in Fig. 5. Results are the means + s.e.m. of three independent experiments. *, significantly different from control at P<0.05. MG132 (MG, 50 μM) and epoxomicin (Epx, 100 nM) are selective proteasome inhibitors.
5. Effect of TFF3 on the expression of Twist protein in intestinal epithelial cells.
TFF3 is an important GI peptide. It protects intestinal epithelial cells against injuries (35). Previous investigations have demonstrated that TFF3 is up-regulated in intestinal inflammatory conditions such as inflammatory bowel disease (2). However, the role of TFF3 in inflammation remains unclear. We first investigated whether TFF3 regulates Twist expression. We treated intestinal epithelial cells with TFF3 (2.5 μM) at different time points and isolated total cellular protein for Western blot analysis. As shown in Fig. 7, treatment with TFF3 resulted in up-regulation of Twist protein in HT-29 cells within 30 min. The effect persisted for 24 hours after the treatment.
Fig. 7. Effect of TFF3 on the expression of Twist protein in intestinal epithelial cells.
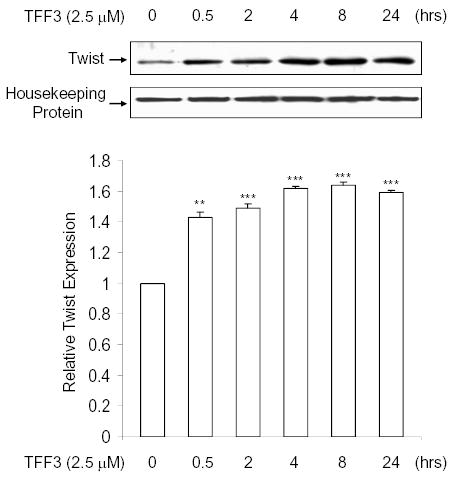
Cell lysates (30 μg) from HT-29 cells untreated or treated for indicated times with TFF3 (2.5 μM) were subjected to SDS-PAGE gel electrophoresis and transferred to membranes. The immunoblotting was performed as in Fig. 5. Top panel: Representative autoradiographs of an immunoblot. Bottom panel: Densitometric analysis of immunoblot data. **P<0.005 versus 0 hr. ***P<0.001 versus 0 hr. Data expressed as means + s.e.m. of three determinations.
It has been shown that TFF3 activates ERK kinase in intestinal epithelial cells (49). To examine the signal mechanisms by which TFF3 up-regulates the expression of Twist protein, HT-29 cells were pretreated with U126 (10 μM, a potent ERK kinase inhibitor) for 60 minutes and followed by treatment with TFF3 (2.5 μM) for 4 and 24 hrs respectively. As shown in Fig. 8, TFF3 increased the expression of Twist protein in intestinal epithelial cells. Pretreatment of cells with U126 resulted in blocking the effect of TFF3 on expression of Twist protein. In addition, we obtained similar results by using PD98059, another potent ERK inhibitor (data not shown). Together, these data suggest that ERK kinase mediates TFF3 action.
Fig. 8. U126, a potent ERK inhibitor, blocks TFF3 effect on reduction of Twist protein in intestinal epithelial cells.

Cell lysates (30 μg) from IEC-18 cells untreated or treated with indicated stimulators were subjected to SDS-PAGE gel electrophoresis and transferred to membranes. The membranes were subjected to Western blotting and densitometric analysis as in Fig. 5. Results are the means + s.e.m. of three independent experiments. *, significantly different from untreated at P<0.05.
6. Twist plays a role in blocking IL-8 production in TFF3-stimulated intestinal epithelial cells.
To eliminate Twist protein from HT-29 cells, we silenced twist expression using siRNA strategy. As demonstrated in Fig. 9A, treatment with siRNA for twist resulted in attenuation of Twist protein expression in intestinal epithelial cells within 5 days. To examine whether Twist protein modulates IL-8 expression in TFF3-stimulated cells, HT-29 cells were transfected with Twist siRNA for 4 days and treated with TFF3 (2.5 μM) for 18 hrs. Conditioned-medium was then collected for IL-8 assay. As shown in Fig. 9B, HT-29 cells constitutively secrete IL-8 protein. The basal level of IL-8 in cells with silenced Twist was not changed compared to the control group, suggesting that constitutive IL-8 expression in HT-29 cells is not modulated by Twist protein. In contrast, TFF3 markedly induced IL-8 secretion in HT-29 cells with silenced Twist, indicating endogenous Twist protein plays a role in balancing IL-8 expression during transient NF-κB activation in intestinal epithelial cells by TFF3.
Fig. 9. RNAi reduces twist expression, which results in induction of IL-8 expression in TFF3-stimulated intestinal epithelial cells.

A. Reduction of endogenous Twist gene expression using siRNA. HT-29 cells were transfected with siRNA against Twist as described under Material and Methods. Five days later, cell lysates were immunoblotted for Twist and GAPDH respectively. Data are representative of 3 separate experiments. siTwist, siRNA for Twist gene; siNS, non-specific siRNA.
B. Reduction of Twist expression results in induction of IL-8 in TFF3-treated intestinal epithelial cells. HT-29 cells were treated with siTwist, siNS or no siRNA and, 4 days later, stimulated with TFF3 (2.5 μM) for 18 hrs. Conditioned medium from untreated or treated cells were subjected to measurement of IL-8 with ELISA assay as described under Material and Methods. Results are the means + s.e.m. of three independent experiments. *, significantly different from the control at P<0.05.
DISCUSSIONS
A growing body of evidence suggests that transiently activated NF-κB in non-inflammatory states and persistently activated NF-κB during inflammation play different pathophysiological roles in vivo. During the initial phase of inflammation, proinflammatory cytokines and mediators induce prolonged NF-κB activation in various inflammatory cells and endothelium. The activated NF-κB further up-regulates the xpression of a number of proinflammatory molecules such as endothelial cell adhesion molecules and macrophage inflammatory protein-2, which triggers neutrophilic infiltration and tissue injury (1, 27, 34). Thus, persistent NF-κB activation during the early phase of inflammation amplifies inflammatory response in vivo.
In contrast, transient activation of NF-κB prior to inflammatory stimulation results in the anti-inflammatory response. For example, several investigators have found that transient activation of NF-κB is required for the heart to tolerate ischemia/reperfusion-induced myocardial stunning and myocardial infarction (41, 54, 56). Pretreatment of rats with LMW-HA induces hepatoprotection against inflammatory insults via transient activation of NF-κB (52). In the intestine, preconditioning with lipopolysaccharide results in transient activation of NF-κB and induces protective mechanisms against intestinal dysfunction (40).
In the present study, we demonstrated that TFF3-induced NF-κB activation is a transient event. The transient activation of NF-κB by TFF3 is followed by induction of Twist protein, a NF-κB associated negative regulatory molecule for NF-κB pathway. Previous studies have shown that Twist interacts with NF-κB (43). It represses NF-κB activity and attenuates the inflammatory response via suppression of NF-κB activity (43). We showed that silencing expression of Twist in TFF3-treated cell results in the induction of IL-8, a NF-κB regulated proinflammatory cytokine. Thus, TFF3-induced transient activating NF-κB is associated with strengthening the negative regulatory loop of NF-κB, which inhibits pro-inflammatory cytokine expression in intestinal epithelial cells and protects against inflammation of the GI mucosa.
Intestinal epithelial cells play a unique role in the GI inflammation through its ability to release proinflammatory cytokines such as IL-1β and IL-8. Previously, we and others have shown that inflammatory mediators induce NF-κB activation in the intestine (14, 15, 22, 48). The activated NF-κB further up-regulates the expression of a number of proinflammatory molecules such as endothelial cell adhesion molecules and macrophage inflammatory protein-2, which result in neutrophilic infiltration and mucosal injury (1, 27, 34). NF-κB is controlled by a negative control loop. However, it is not clear whether the loop is regulated during the inflammation. Here, we reported for the first time that Twist protein, a novel molecule in the loop, is present in intestinal epithelial cells in vivo. TNF induces markedly degradation of Twist protein in intestinal epithelial cells by a proteasome-dependent mechanism. The effect is associated with TNF-induced NF-κB activity in the cells, which suggests that (1) cytokines regulate the expression of molecules in the loop; and (2) reduction of Twist protein by TNF may result in maintaining NF-κB activity. Persistent activation of NF-κB causes prolonged expression of genes including both induction of and activation by NF-κB.
Previous studies have shown that TFF3 is expressed in goblet cells and secreted onto intestinal lumen in normal circumstance (37, 51, 53). It targets intestinal epithelial cells. In contrast to TNF, TFF3 induces up-regulation of Twist in intestinal epithelial cells. Thus, TFF3 probably plays an important role in maintaining Twist protein in intestinal epithelial cells, which may contribute to down-regulation of inflammation in vivo.
The physiological function of Twist in intestinal epithelial cells is not clear. Previously, Twist has been found to play an important role in negative regulation of NF-κB in vivo (43). Thus, we hypothesize that endogenous Twist in intestinal epithelial cells is involved in controlling proinflammatory cytokine expression during inflammation. This hypothesis is further supported by our observation that TFF3 induces IL-8 production after elimination of Twist protein from intestinal epithelial cells. Furthermore, since TNF induces degradation of Twist in intestinal epithelial cells, which is associated with prolonged NF-κB activation, to prevent cytokine-induced down-regulation of Twist protein may be a novel strategy for blocking inflammation in the GI tract.
In addition to participation in negative regulation of NF-κB signal pathway, induction of Twist has been shown to play an essential role in the prevention of cells from undergoing apoptosis (30). Interestingly, we and others showed that TFF3 protects intestinal epithelial cells against apoptosis (8, 50). Taken together, TFF3-induced Twist protein may mediate anti-apoptotic effect of TFF3 in intestinal epithelial cells.
Previously, we and others have demonstrated that intestinal epithelial cells are the target of TFF3 (51). TFF3 binds to intestinal epithelial cells (11, 46, 47). In response to TFF3 stimulation, intestinal epithelial cells release nitric oxide and prostaglandins (45, 47). TFF3 activates several intracellular molecules such as NF-κB and ERK in intestinal epithelial cells (8, 49). In the present study, we found that TFF3 enhances Twist protein level in intestinal epithelial cells. We further showed that the selective inhibitor of ERK kinase attenuates the effect of TFF3 on up-regulation of Twist protein, suggesting that TFF3 activates a distinctive signal pathway involved in the modulation of Twist protein. These observations indicate that multiple intracellular regulatory molecules including ERK, IκBα, Twist, and NF-κB complex may participate to mediate the effect of TFF3. Dissection of the distinctive signal pathway linking these molecules is the subject of our ongoing research.
In summary, we found that TFF3 activates intestinal epithelial NF-κB in a mechanism distinctive from TNF. We demonstrated for the first time that intestinal epithelial cells constitutively express Twist protein, a novel negative regulator for NF-κB pathway. We showed that TNF, which induces prolonged NF-κB activation, induces degradation of Twist protein in intestinal epithelial cells. The TNF effect is mediated by the proteasome activity. In contrast, TFF3, which activates NF-κB in a transient event, up-regulates Twist protein in intestinal epithelial cells. The effect of TFF3 is mediated by endogenous ERK activity. In addition, we showed that Twist protein plays an important role in silencing IL-8 production in NF-κB activated intestinal epithelial cells. Further understanding of these mechanisms will provide new insights into the controlling process involved in activation of NF-κB in inflammation, and may lead to develop a new pharmaceutical to block GI inflammation.
Acknowledgments
This work was supported in part by the Grant R01DK064240 (to X.-D.T.) from National Institutes of Health, a grant from Crohn’s and Colitis Foundation of America (to X.-D.T.), and Eloise and Warren Batts Investigator Award (to X.-D.T.). We thank Yu Lu for technical assistance. Additionally, we thank Adrienne G. Woodworth and Leah M. Edmonson for preparation of the manuscript. Y.-Q. Z. is a visiting scholar from China Medical University (Shenyang, P. R. China, 110001).
References
- 1.Ajuebor MN, Swain MG. Role of chemokines and chemokine receptors in the gastrointestinal tract. Immunology. 2002;105:137–143. doi: 10.1046/j.1365-2567.2002.01309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alison MR, Chinery R, Poulsom R, Ashwood P, Longcroft JM, Wright NA. Experimental ulceration leads to sequential expression of spasmolytic polypeptide, intestinal trefoil factor, epidermal growth factor and transforming growth factor alpha mRNAs in rat stomach. J Pathol. 1995;175:405–414. doi: 10.1002/path.1711750408. [DOI] [PubMed] [Google Scholar]
- 3.Arenzana-Seisdedos F, Thompson J, Rodriguez MS, Bachelerie F, Thomas D, Hay RT. Inducible nuclear expression of newly synthesized I kappa B alpha negatively regulates DNA-binding and transcriptional activities of NF- kappa B. Mol Cell Biol. 1995;15:2689–2696. doi: 10.1128/mcb.15.5.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science. 1995;270:286–290. doi: 10.1126/science.270.5234.286. [DOI] [PubMed] [Google Scholar]
- 5.Babyatsky MW, deBeaumont M, Thim L, Podolsky DK. Oral trefoil peptides protect against ethanol- and indomethacin-induced gastric injury in rats. Gastroenterology. 1996;110:489–497. doi: 10.1053/gast.1996.v110.pm8566596. [DOI] [PubMed] [Google Scholar]
- 6.Brasier AR, Lu M, Hai T, Lu Y, Boldogh I. NF-kappa B-inducible BCL-3 expression is an autoregulatory loop controlling nuclear p50/NF-kappa B1 residence. J Biol Chem. 2001;276:32080–32093. doi: 10.1074/jbc.M102949200. [DOI] [PubMed] [Google Scholar]
- 7.Carlson DL, White DJ, Maass DL, Nguyen RC, Giroir B, Horton JW. Ikappa B overexpression in cardiomyocytes prevents NF-kappa B translocation and provides cardioprotection in trauma. Am J Physiol. 2003;284:H804–H814. doi: 10.1152/ajpheart.00394.2001. [DOI] [PubMed] [Google Scholar]
- 8.Chen YH, Lu Y, De Plaen IG, Wang LY, Tan XD. Transcription factor NF-kappaB signals antianoikic function of trefoil factor 3 on intestinal epithelial cells [In Process Citation] Biochem Biophys Res Commun. 2000;274:576–582. doi: 10.1006/bbrc.2000.3176. [DOI] [PubMed] [Google Scholar]
- 9.Chen ZF, Behringer RR. Twist is required in head mesenchyme for cranial neural tube morphogenesis. Genes Dev. 1995;9:686–699. doi: 10.1101/gad.9.6.686. [DOI] [PubMed] [Google Scholar]
- 10.Chiao PJ, Miyamoto S, Verma IM. Autoregulation of I kappa B alpha activity. Proc Natl Acad Sci U S A. 1994;91:28–32. doi: 10.1073/pnas.91.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chinery R, Cox HM. Immunoprecipitation and characterization of a binding protein specific for the peptide, intestinal trefoil factor. Peptides. 1995;16:749–755. doi: 10.1016/0196-9781(95)00045-l. [DOI] [PubMed] [Google Scholar]
- 12.Conte E, Bonaiuto C, Nesci C, Crimi N, Vancheri C, Messina A. Nuclear factor-kappaB activation in human monocytes stimulated with lipopolysaccharide is inhibited by fibroblast conditioned medium and exogenous PGE2. FEBS Lett. 1997;400:315–318. doi: 10.1016/s0014-5793(96)01409-3. [DOI] [PubMed] [Google Scholar]
- 13.D’Acquisto F, Sautebin L, Iuvone T, Di Rosa M, Carnuccio R. Prostaglandins prevent inducible nitric oxide synthase protein expression by inhibiting nuclear factor-kappaB activation in J774 macrophages. FEBS Lett. 1998;440:76–80. doi: 10.1016/s0014-5793(98)01407-0. [DOI] [PubMed] [Google Scholar]
- 14.De Plaen IG, Tan XD, Chang H, Qu XW, Liu QP, Hsueh W. Intestinal NF-kappaB is activated, mainly as p50 homodimers, by platelet-activating factor. Biochim Biophys Acta. 1998;1392:185–192. doi: 10.1016/s0005-2760(98)00024-1. [DOI] [PubMed] [Google Scholar]
- 15.De Plaen IG, Tan XD, Chang H, Wang L, Remick DG, Hsueh W. Lipopolysaccharide activates nuclear factor kappaB in rat intestine: role of endogenous platelet-activating factor and tumour necrosis factor. Br J Pharmacol. 2000;129:307–314. doi: 10.1038/sj.bjp.0703055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dignass A, Lynch-Devaney K, Kindon H, Thim L, Podolsky DK. Trefoil peptides promote epithelial migration through a transforming growth factor beta-independent pathway. J Clin Invest. 1994;94:376–383. doi: 10.1172/JCI117332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoffmann W, Jagla W, Wiede A. Molecular medicine of TFF-peptides: from gut to brain. Histol Histopathol. 2001;16:319–334. doi: 10.14670/HH-16.319. [DOI] [PubMed] [Google Scholar]
- 18.Howe LR, Watanabe O, Leonard J, Brown AM. Twist is up-regulated in response to Wnt1 and inhibits mouse mammary cell differentiation. Cancer Res. 2003;63:1906–1913. [PubMed] [Google Scholar]
- 19.Hu X. Proteolytic signaling by TNFalpha: caspase activation and IkappaB degradation. Cytokine. 2003;21:286–294. doi: 10.1016/s1043-4666(03)00107-8. [DOI] [PubMed] [Google Scholar]
- 20.Hu X, Bryington M, Fisher AB, Liang X, Zhang X, Cui D, Datta I, Zuckerman KS. Ubiquitin/proteasome-dependent degradation of D-type cyclins is linked to tumor necrosis factor-induced cell cycle arrest. J Biol Chem. 2002;277:16528–16537. doi: 10.1074/jbc.M109929200. [DOI] [PubMed] [Google Scholar]
- 21.Imbert V, Rupec RA, Livolsi A, Pahl HL, Traenckner EB, Mueller-Dieckmann C, Farahifar D, Rossi B, Auberger P, Baeuerle PA, Peyron JF. Tyrosine phosphorylation of I kappa B-alpha activates NF-kappa B without proteolytic degradation of I kappa B-alpha. Cell. 1996;86:787–798. doi: 10.1016/s0092-8674(00)80153-1. [DOI] [PubMed] [Google Scholar]
- 22.Jobin C, Panja A, Hellerbrand C, Iimuro Y, Didonato J, Brenner DA, Sartor RB. Inhibition of proinflammatory molecule production by adenovirus- mediated expression of a nuclear factor kappaB super-repressor in human intestinal epithelial cells. J Immunol. 1998;160:410–418. [PubMed] [Google Scholar]
- 23.Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 24.Kindon H, Pothoulakis C, Thim L, Lynch-Devaney K, Podolsky DK. Trefoil peptide protection of intestinal epithelial barrier function: cooperative interaction with mucin glycoprotein. Gastroenterology. 1995;109:516–523. doi: 10.1016/0016-5085(95)90340-2. [DOI] [PubMed] [Google Scholar]
- 25.Laherty CD, Hu HM, Opipari AW, Wang F, Dixit VM. The Epstein-Barr virus LMP1 gene product induces A20 zinc finger protein expression by activating nuclear factor kappa B. J Biol Chem. 1992;267:24157–24160. [PubMed] [Google Scholar]
- 26.Laherty CD, Perkins ND, Dixit VM. Human T cell leukemia virus type I Tax and phorbol 12-myristate 13- acetate induce expression of the A20 zinc finger protein by distinct mechanisms involving nuclear factor kappa B. J Biol Chem. 1993;268:5032–5039. [PubMed] [Google Scholar]
- 27.Laskowski I, Pratschke J, Wilhelm MJ, Gasser M, Tilney NL. Molecular and cellular events associated with ischemia/reperfusion injury. Ann Transplant. 2000;5:29–35. [PubMed] [Google Scholar]
- 28.Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000:2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lowenstein CJ, Alley EW, Raval P, Snowman AM, Snyder SH, Russell SW, Murphy WJ. Macrophage nitric oxide synthase gene: two upstream regions mediate induction by interferon gamma and lipopolysaccharide. Proc Natl Acad Sci U S A. 1993;90:9730–9734. doi: 10.1073/pnas.90.20.9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maestro R, Dei Tos AP, Hamamori Y, Krasnokutsky S, Sartorelli V, Kedes L, Doglioni C, Beach DH, Hannon GJ. Twist is a potential oncogene that inhibits apoptosis. Genes Dev. 1999;13:2207–2217. doi: 10.1101/gad.13.17.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsuoka Y, Pascall JC, Brown KD. Quantitative analysis reveals differential expression of mucin (MUC2) and intestinal trefoil factor mRNAs along the longitudinal axis of rat intestine. Biochim Biophys Acta. 1999;1489:336–344. doi: 10.1016/s0167-4781(99)00186-4. [DOI] [PubMed] [Google Scholar]
- 32.Meng L, Mohan R, Kwok BH, Elofsson M, Sin N, Crews CM. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo anti-inflammatory activity. Proc Natl Acad Sci U S A. 1999;96:10403–10408. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moro F, Levenez F, Durual S, Plaisancie P, Thim L, Giraud AS, Cuber J. Secretion of the trefoil factor TFF3 from the isolated vascularly perfused rat colon. Regul Pept. 2001;101:35–41. doi: 10.1016/s0167-0115(01)00257-9. [DOI] [PubMed] [Google Scholar]
- 34.Olson TS, Ley K. Chemokines and chemokine receptors in leukocyte trafficking. Am J Physiol Regul Integr Comp Physiol. 2002;283:R7–28. doi: 10.1152/ajpregu.00738.2001. [DOI] [PubMed] [Google Scholar]
- 35.Podolsky DK. Mechanisms of regulatory peptide action in the gastrointestinal tract: trefoil peptides. J Gastroenterol. 2000;35 (Suppl 12):69–74. [PubMed] [Google Scholar]
- 36.Sands BE, Ogata H, Lynch-Devaney K, deBeaumont M, Ezzell RM, Podolsky DK. Molecular cloning of the rat intestinal trefoil factor gene. Characterization of an intestinal goblet cell-associated promoter. J Biol Chem. 1995;270:9353–9361. doi: 10.1074/jbc.270.16.9353. [DOI] [PubMed] [Google Scholar]
- 37.Sands BE, Podolsky DK. The trefoil peptide family. Annu Rev Physiol. 1996;58:253–273. doi: 10.1146/annurev.ph.58.030196.001345. [DOI] [PubMed] [Google Scholar]
- 38.Scheinman RI, Cogswell PC, Lofquist AK, Baldwin AS., Jr Role of transcriptional activation of I kappa B alpha in mediation of immunosuppression by glucocorticoids. Science. 1995:283–286. doi: 10.1126/science.270.5234.283. [DOI] [PubMed] [Google Scholar]
- 39.Schmedtje JF, Jr, Ji YS, Liu WL, DuBois RN, Runge MS. Hypoxia induces cyclooxygenase-2 via the NF-kappaB p65 transcription factor in human vascular endothelial cells. J Biol Chem. 1997;272:601–608. doi: 10.1074/jbc.272.1.601. [DOI] [PubMed] [Google Scholar]
- 40.Schwarz NT, Engel B, Eskandari MK, Kalff JC, Grandis JR, Bauer AJ. Lipopolysaccharide preconditioning and cross-tolerance: the induction of protective mechanisms for rat intestinal ileus. Gastroenterology. 2002;123:586–598. doi: 10.1053/gast.2002.34777. [DOI] [PubMed] [Google Scholar]
- 41.Shinmura K, Xuan YT, Tang XL, Kodani E, Han H, Zhu Y, Bolli R. Inducible nitric oxide synthase modulates cyclooxygenase-2 activity in the heart of conscious rabbits during the late phase of ischemic preconditioning. Circ Res. 2002;90:602–608. doi: 10.1161/01.res.0000012202.52809.40. [DOI] [PubMed] [Google Scholar]
- 42.Sosic D, Olson EN. A new twist on twist--modulation of the NF-kappa B pathway. Cell Cycle. 2003;2:76–78. [PubMed] [Google Scholar]
- 43.Sosic D, Richardson JA, Yu K, Ornitz DM, Olson EN. Twist regulates cytokine gene expression through a negative feedback loop that represses NF-kappaB activity. Cell. 2003;112:169–180. doi: 10.1016/s0092-8674(03)00002-3. [DOI] [PubMed] [Google Scholar]
- 44.Sun SC, Ganchi PA, Ballard DW, Greene WC. NF-kappa B controls expression of inhibitor I kappa B alpha: evidence for an inducible autoregulatory pathway. Science. 1993;259:1912–1915. doi: 10.1126/science.8096091. [DOI] [PubMed] [Google Scholar]
- 45.Tan XD, Chen Y, Liu Q, Gonzalez-Crussi F, Liu X. Prostanoids mediate the protective effect of trefoil factor 3 in oxidant-induced intestinal epithelial cell injury: role of cyclooxygenase-2. J Cell Sci. 2000;113:2149–2155. doi: 10.1242/jcs.113.12.2149. [DOI] [PubMed] [Google Scholar]
- 46.Tan XD, Hsueh W, Chang H, Wei KR, Gonzalez-Crussi F. Characterization of a putative receptor for intestinal trefoil factor in rat small intestine: identification by in situ binding and ligand blotting. Biochem Biophys Res Commun. 1997;237:673–677. doi: 10.1006/bbrc.1997.7144. [DOI] [PubMed] [Google Scholar]
- 47.Tan XD, Liu QP, Hsueh W, Chen YH, Chang H, Gonzalez-Crussi F. Intestinal trefoil factor binds to intestinal epithelial cells and induces nitric oxide production: priming and enhancing effects of mucin. Biochem J. 1999;338:745–751. [PMC free article] [PubMed] [Google Scholar]
- 48.Tan XD, Sun X, Gonzalez-Crussi FX, Gonzalez-Crussi F, Hsueh W. PAF and TNF increase the precursor of NF-kappa B p50 mRNA in mouse intestine: quantitative analysis by competitive PCR. Biochim Biophys Acta. 1994;1215:157–162. doi: 10.1016/0005-2760(94)90105-8. [DOI] [PubMed] [Google Scholar]
- 49.Taupin D, Wu DC, Jeon WK, Devaney K, Wang TC, Podolsky DK. The trefoil gene family are coordinately expressed immediate-early genes: EGF receptor- and MAP kinase-dependent interregulation. J Clin Invest. 1999;103:R31–R38. doi: 10.1172/JCI3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Taupin DR, Kinoshita K, Podolsky DK. Intestinal trefoil factor confers colonic epithelial resistance to apoptosis. Proc Natl Acad Sci U S A. 2000;97:799–804. doi: 10.1073/pnas.97.2.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thim L. Trefoil peptides: from structure to function. Cell Mol Life Sci. 1997;53:888–903. doi: 10.1007/s000180050108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wolf D, Schumann J, Koerber K, Kiemer AK, Vollmar AM, Sass G, Papadopoulos T, Bang R, Klein SD, Brune B, Tiegs G. Low-molecular-weight hyaluronic acid induces nuclear factor-kappaB- dependent resistance against tumor necrosis factor alpha-mediated liver injury in mice. Hepatology. 2001;34:535–547. doi: 10.1053/jhep.2001.27218. [DOI] [PubMed] [Google Scholar]
- 53.Wright NA, Hoffmann W, Otto WR, Rio MC, Thim L. Rolling in the clover: trefoil factor family (TFF)-domain peptides, cell migration and cancer. FEBS Lett. 1997;408:121–123. doi: 10.1016/s0014-5793(97)00424-9. [DOI] [PubMed] [Google Scholar]
- 54.Xuan YT, Tang XL, Banerjee S, Takano H, Li RC, Han H, Qiu Y, Li JJ, Bolli R. Nuclear factor-kappaB plays an essential role in the late phase of ischemic preconditioning in conscious rabbits. Circ Res. 1999;84:1095–1109. doi: 10.1161/01.res.84.9.1095. [DOI] [PubMed] [Google Scholar]
- 55.Yamamoto Y, Gaynor RB. Role of the NF-kappaB pathway in the pathogenesis of human disease states. Curr Mol Med. 2001;1:287–296. doi: 10.2174/1566524013363816. [DOI] [PubMed] [Google Scholar]
- 56.Zhao TC, Taher MM, Valerie KC, Kukreja RC. p38 Triggers late preconditioning elicited by anisomycin in heart: involvement of NF-kappaB and iNOS. Circ Res. 2001;89:915–922. doi: 10.1161/hh2201.099452. [DOI] [PubMed] [Google Scholar]
- 57.Zhu YQ, Lu Y, Tan XD. Monochloramine induces reorganization of nuclear speckles and phosphorylation of SRp30 in human colonic epithelial cells: role of protein kinase C. Am J Physiol Cell Physiol. 2003;285:C1294–C1303. doi: 10.1152/ajpcell.00090.2003. [DOI] [PubMed] [Google Scholar]