

Abstract
BAR domains are proteins that sense and sculpt curved membranes in cells, furnishing a relatively well-studied example of mechanisms employed in cellular morphogenesis. We report a computational study of membrane bending by BAR domains at four levels of resolution, described by 1), all-atom molecular dynamics; 2), residue-based coarse-graining (resolving single amino acids and lipid molecules); 3), shape-based coarse-graining (resolving overall protein and membrane shapes); and 4), a continuum elastic membrane model. Membrane sculpting performed by BAR domains collectively is observed in agreement with experiments. Different arrangements of BAR domains on the membrane surface are found to lead to distinct membrane curvatures and bending dynamics.
INTRODUCTION
The morphology of living cells features a large variety of membrane shapes, forming barriers, organelles, and compartments, which are sculpted actively by the cellular machinery to various levels of curvature (1–3). Generation of membrane curvature is an essential step in cellular morphogenesis, typically achieved with the help of various proteins that employ different mechanisms to perform this task (4–13). In turn, the membrane curvature can affect structure and function of the associated proteins (see, e.g., (14–17)). One relatively well-studied family of membrane-sculpting proteins are the BAR domains, which are found in many organisms and drive the formation of tubular and vesicular membrane structures in a variety of cellular processes, including fission of synaptic vesicles, endocytosis, and apoptosis (18,19). In the in vitro experiments, BAR domains bind to liposomes and convert low-curvature spheres to high-curvature tubules (20). The structures of several types of BAR domains have been resolved (21–29). They all form dimers with a high density of positively charged residues on one side of their surface. Recent molecular dynamics (MD) simulations (30,31) have provided a dynamic picture with atomistic details of how a single BAR-domain induces a local curvature on a negatively charged membrane through electrostatic interactions and matching of the protein shape by the membrane.
Despite its vital role for cellular structure and function, cellular membrane morphogenesis is only starting to be understood. This is due to the heterogeneity of the process, where the constituent lipids exist in partially disordered phases and participating proteins are distributed nonuniformly, restricting the application of many experimental imaging methods. However, MD simulations (32–34) can offer a description of the process with single-atom resolution, as shown here for the case of BAR domains, and in Chandler et al. (13) for integral membrane proteins of bacterial photosynthetic chromatophores. Unfortunately, with current computing capabilities, molecular dynamics is limited to system sizes and timescales that are too small and too short for many morphogenesis events. Thus, many aspects of membrane morphogenesis in general, and action of BAR domains in particular, remain largely unknown. Here, we report a computational study of single and multiple amphiphysin BAR domains interacting with negatively charged membranes. To overcome the limitations of atomistic MD simulations, we utilize coarse-grained modeling and a multiscale approach at four levels of resolution, which allows us to reach timescales up to several microseconds.
Many questions remain unanswered about how multiple BAR domains bend membranes, such as why the radii of curvature of BAR domain-induced tubes and vesicles fall in a wide range of values, while the intrinsic radius of curvature of the protein itself is ∼110 Å (for amphiphysin BAR domain). In principle, multiple BAR domains could act either individually or in concert to bend membranes. The latter possibility has been favored in the literature since cryo-electron microscopy images of BAR domain-induced tubules allow one to distinguish striations (20,21), presumed to be rows of BAR domains. Recent work by Frost et al. (29) has shown for the first time that BAR domains indeed align in rows, or spirals, on the surface of the membrane tubules, as seen in Fig. 1. The arrangement shown in Fig. 1 is formed by F-BAR domains that polymerize and aggregate in a spiral to tubulate the membrane; other members of the BAR domain family, such as amphiphysin BAR domains, form similar arrangements (20,29). Thus, the experimental evidence suggests that BAR domains act concertedly in close, well-ordered formation, forming longitudinal lines around the tubules. Clearly, differences in the formation lead to the observed variability in the size of tubules, and one may wonder how lateral arrangements of BAR domains in parallel rows determine the tubule size. To investigate this, we set up two models for line formation (Fig. 2), one with laterally staggered lines, and the other with nonstaggered lines. Results of our simulations show a clear difference between curvatures induced by the two formations on a membrane, with the staggered arrangement producing a higher curvature than the nonstaggered one (∼300 vs. 1000 Å). An individual BAR domain in our simulations produces a local membrane curvature with radius in the range 100–500 Å. The dynamics of membrane bending is also strongly influenced by the specific BAR domain arrangement. A number of simulations were performed for each arrangement studied, providing a sampling that elucidates the variations in individual bending events.
FIGURE 1.

Membrane tube formed concertedly by BAR domains. Shown is a cryo-electron microscopy view of F-BAR domains that sculpt a tubular membrane of ∼670 Å diameter. A single F-BAR domain is marked by the solid oval. The image is reproduced from Frost et al. (29).
FIGURE 2.

Arrangements of BAR domains studied. The systems studied by simulations are periodic; the simulation box is highlighted as a solid square, and a few periodic images are shown in dimmed color; boundaries of periodic cells are marked by dotted lines. (A) Nonstaggered one-row arrangement. (B) Staggered two-row arrangement.
The multiscale approach employed acts at four levels of resolution: all-atom, single-residue, single-protein, and continuum. Coarse-grained (CG) MD techniques for biomolecules are currently under active development, but are usually focused on applications to specific types of biological systems, such as membranes (35–47), or to specific proteins (48–50), although recent studies have investigated peptide- and protein-membrane systems or large number of different proteins (17,51–63). At an even coarser level of description, continuum models have been used to describe the elastic properties, remodeling, and fusion of membranes (see, e.g., (64–68)), including those being bent by BAR domains (69). Our four-level description is based on these models and on our own coarse-graining approaches, which resulted recently in the development of a residue-based CG model (51,57) and a shape-based CG model (52,53). The latter was originally developed for proteins only; here we describe an extension of this model to lipids.
The atomistic description is provided through all-atom MD simulations with the CHARMM force field (70). The next level of coarseness is furnished by MD with residue-based CG (51,57), which describes single residues by just a few CG beads (e.g., an amino acid is represented by two beads, one for the backbone and another for the side chain). The shape-based CG model (52,53) uses CG beads to represent protein segments, whose dynamics is described using MD simulations as well. For all three levels, MD simulations are performed using NAMD (71). The fourth level of our description involves a continuum elastic membrane model. The models, at each level, are parameterized based on properties from the higher-resolution models, as well as from experimental data. Results from computations across all four levels are consistent with experimental observations, whenever available, as well as with previous all-atom simulations (30,31).
METHODS
BAR domain-membrane systems have been simulated using four levels of description (Fig. 3): all-atom molecular dynamics (MD), residue-based CG (RBCG) MD (57) with ∼10 atoms being represented by a single CG bead, shape-based CG (SBCG) MD (52) with ∼150 atoms per CG bead, and a continuum elastic membrane model. MD simulations were performed using NAMD (71). Analysis and visualization were carried out with VMD (72).
FIGURE 3.

Residue-based coarse-grained (RBCG) and shape-based coarse-grained (SBCG) models. (A) Overlap of all-atom and RBCG models for a DOPC lipid and AWLFV peptide. RBCG uses ∼10 atoms per CG bead; an amino acid is represented by one bead for the backbone and another one for the side chain. (B) SBCG model of a protein segment, with ∼150 atoms per CG bead. Each CG bead has the same color as the all-atom domain represented by the bead. (C) Side- and top-views of the SBCG model of a small DOPC membrane patch. Each SBCG molecule corresponds to 2.2 lipids on average and consists of two beads, one representing the lipid heads (cyan) and the other the lipid tails (white). (D) DOPC membrane represented by all-atom, RBCG, and SBCG models. (E–G) BAR domain viewed from top and side, in all-atom (E), RBCG (F), and SBCG (G) representations. The BAR domain is a homodimer; the monomers are shown in purple and green. In the SBCG model, the two monomers are connected by bonds (orange) to preserve the integrity of the dimer.
Systems with one or six BAR domains were simulated. In the case of six BAR domains, two arrangements of proteins were studied: a single row of six BAR domains and two parallel rows of three BAR domains. Periodic boundary conditions were assumed, resulting in the arrangements illustrated in Fig. 2. Both arrangements involve parallel, longitudinal rows of BAR domains, but one formation features laterally aligned rows (Fig. 2 A) and the other one exhibits lateral staggering of the rows (Fig. 2 B). These staggered and nonstaggered arrangements were simulated using RBCG and SBCG models. We will employ the nomenclature one-row arrangement and two-row arrangement for systems shown in Fig. 2, A and B, (e.g., refer to the respective simulations as 6BAR-1row-RB and 6BAR-2row-RB).
The spacing between rows of BAR domains (70–100 Å) is similar to that inferred from cryo-electron micrographs of tubules formed by amphiphysin BAR domains (50–100 Å between those observed in (20,21)). The pitch of the spiral in Fig. 1 is ∼60 Å, but varies significantly from case to case (29). Also, Fig. 1 shows F-BAR domains, which are different from amphiphysin BAR domains investigated here (F-BAR domains are larger and produce shallower curvature).
All-atom simulations
We performed an all-atom simulation of a single BAR domain on a patch of DOPC/DOPS membrane. This simulation served mainly for the parameterization of the CG models. The coordinates of Drosophila melanogaster amphiphysin BAR domain were obtained from the Protein Data Bank (PDB code 1URU (21)). The missing 35 residues near the N-terminus were modeled according to the structure suggested in Gallop et al. (25), namely, as a short helix and a flexible link, using the MOLEFACTURE plug-in of VMD (72). The base system of 72 DOPC lipids (obtained from http://persweb.wabash.edu/facstaff/fellers/ (73)) was replicated once along one dimension, fully hydrated, and equilibrated for 15 ns. Then, 30% of the PC headgroups were changed to PS headgroups randomly and uniformly. The DOPC/DOPS system was equilibrated for 10 ns.
The equilibrated membrane patch was replicated to yield a patch with initial dimensions of 460 × 98 Å2. A BAR domain was placed onto the surface of the membrane with its N-terminal helices partially buried between the lipid headgroups. The TIP3P water model (74) was used to solvate the system and Na+ ions were subsequently added to neutralize the net charge, resulting in a system of 671,331 atoms with dimensions 440 × 97 × 155 Å3. The system was equilibrated for 0.5 ns with the atoms of BAR domain being harmonically constrained to their initial positions (the constraints' spring constant was 1 kcal/(mol Å2)), and then simulated for 25 ns without any constraints (simulation 1BAR-AA).
The CHARMM (70,75) force field was used. Parameters for the charged PS headgroup were generated by adding a carboxylate group to the phosphatidylethanolamine headgroup in CHARM27 (75). Periodic boundary conditions were applied, and simulations assumed the NpT ensemble (temperature 310 K and pressure 1 atm). A Langevin thermostat with a damping coefficient of 0.5 ps−1 maintained temperature; pressure was maintained via a Langevin-piston barostat with a piston period and damping time of 2 ps each. Short-range nonbonded interactions were cut off smoothly between 10 and 12 Å. The PME algorithm was used to compute long-range electrostatic interactions. The implementation of these algorithms in NAMD is described in Phillips et al. (71). Simulations of membrane patches were performed with an integration time step of 1 fs; a 2 fs time step was employed in simulation 1BAR-AA (see below).
Residue-based CG simulations
The residue-based CG (RBCG) method has been extensively applied to study lipid-protein systems, namely, high-density lipoproteins (51,57–59), and, thus, appears to be suitable for simulations of membrane sculpting by proteins. The method was originally developed to extend Marrink's CG lipid model (37,40,46) to proteins (51). A correspondence of ∼10 atoms per bead characterizes the level of coarse-graining (Fig. 3 A). A DOPC lipid is represented by 14 CG beads: one for the choline group, one for the phosphate group, two for each of the glycerol groups, and 10 to represent the two hydrocarbon tails. Four water molecules are represented by a single CG bead; an ion together with its first hydration shell (six water molecules) is represented by one CG bead; each amino acid is represented by two CG beads, one for the backbone and one for the side chain (glycine is represented by a single backbone CG bead). CG beads are assigned effective interaction potentials, reproducing hydrophobic/hydrophilic properties, and time evolution is described using classical MD. The RBCG model and parameters used in this work are the same as those in the literature (57,58), except for a few features specified in the following.
Originally, RBCG was parameterized to maintain only secondary structure of a protein and has been successful in describing the protein component of HDL, which is a chain of α-helices, but it was not designed to maintain tertiary structure of proteins. When RBCG was applied to simulate a BAR domain, its tertiary structure was not preserved, because it is determined by interactions such as hydrogen bonds, which are missing in the RBCG description. To overcome this limitation, we added potential energy terms for harmonic bonds connecting the protein beads that otherwise are not bonded. The flexibility of the protein's tertiary structure observed in the all-atom simulation of the BAR domain was matched in the RBCG model, using a set of extra bonds and tuning their strength (implemented through a NAMD (71) feature for adding extra bonded interactions). The constraints, or extra bonds, were set between the beads representing the protein backbone. The main purpose was to maintain the intrinsic bending of α-helices in the protein and the distance between the α-helices. Extra bonds were established if two backbone beads were in the same α-helix and on the same side of the helix, within a distance of 4–20 Å from each other, or, if the two backbone beads were at adjacent surfaces of two α-helices, with distances between the beads ranging from 4–15 Å. The full list of these extra bonds is provided in Data S1 in the Supplementary Material. The strength was the same for all constraints, K = 5 kcal/(mol Å2), and the bond lengths were equal to the distances between the beads as found in the crystal structure. As a result, the protein was not heavily constrained, yet the tertiary structure (the crescent shape) was maintained.
The choice of constraint strength is illustrated in Fig. 4. Several quantities, such as the characteristic distances and angles shown in Fig. 4 A as well as Cα-RMSD (root mean-square deviation), are chosen to monitor the dynamics of the overall protein structure. The quantities chosen are: LCOM, the distance between the centers of mass (COM) of the two monomers; LETE, the end-to-end dimer distance between the COMs for clusters of atoms at the ends of two monomers, the clusters being composed of residues 149–177 (we define the COM of this cluster for one monomer as the end point); Lmonomer, the end-to-end distance for one monomer between its end point and the COM of a cluster composed of residues 82–87, 90–95, and 230–235; θdimer, the dimer opening angle formed by the end point of one monomer, COM of the cluster of residues 37–125 and 199–242 from both monomers, and the end point of the other monomer; θmonomer, the monomer opening angle between the end point, COM of residues 190–206, and COM of residues 199–242 in the same monomer. These quantities were monitored in the all-atom simulation of BAR domain on a patch of membrane (simulation 1BAR-AA), and in equivalent RBCG simulations with different strength of the constraints. The values were measured after the first 20 ns of simulations, to allow an emerging structure to stabilize, and were averaged over the subsequent 5 ns. The averages with the associated error bars are shown in Fig. 4, B–G (the values for Lmonomer and θmonomer were additionally averaged over the quantities observed for the two monomers). The results show that the choice of the spring constant K = 5 kcal/(mol Å2) provides the weakest set of constraints that maintains the dimer shape well. Simulations with weaker, or no constraints, result in a significant change of the structure. Stronger constraints make the protein stiffer than in the all-atom simulation, as can be seen, e.g., from the RMSD values (Fig. 4 B).
FIGURE 4.

Constraints are necessary to maintain the tertiary structure and interdomain arrangement for the RBCG model of BAR domain. (A) A BAR domain dimer is shown in top and side view. Several distances and angles are chosen that characterize the overall structure of the protein. These are the distance between the centers of mass of the two monomers (LCOM), end-to-end distance for the whole dimer (LETE), end-to-end distance for one monomer (Lmonomer), and the opening angles for the dimer and for the monomer (θdimer and θmonomer). Averages of these values, as well as of Cα-RMSD for the dimer, are shown in panels B–G for, from left to right, the all-atom simulation (1st bar), and RBCG simulations with K = 5 kcal/(mol Å2) (2nd bar), K = 25 kcal/(mol Å2) (3rd bar), K = 0.5 kcal/(mol Å2) (4th bar), and K = 0 (5th bar). Restraints with K = 5 kcal/mol Å2 provide the best agreement between the all-atom and RBCG simulations, and, thus, was used further on.
All-atom simulations of a BAR domain, both present and previous ones (30,31), indicate that the interaction that drives membrane bending is of electrostatic nature. In the previous RBCG simulations (37,51,57), electrostatics never played an important role, and the use of a relative dielectric constant ɛ = 20 was satisfactory. However, our attempts to obtain membrane curvature in RBCG simulations of the BAR domain with ɛ = 20 failed. Varying ɛ, we found that RBCG simulations with ɛ = 1 furnished the best agreement with the all-atom simulations, in terms of the observed membrane curvature and the timescale over which it arises. Probably, the necessary value of ɛ is so low because the formation of curvature is driven by the electrostatic interactions between the protein and charged lipid heads at a short range, when no water or other atoms shield the charges. Longer-range electrostatics seems to be not so important for this problem, and, although in principle one should use higher values of ɛ for the long range, even with ɛ = 1 this long-range electrostatics is too weak to influence the system significantly. Thus, ɛ = 1 is used in the RBCG simulations.
RBCG simulations were run with one BAR domain (simulation 1BAR-RB) and with six BAR domains, for the nonstaggered arrangement (6BAR-1row-RB) as well as for the staggered one (6BAR-2row-RB), with the DOPC/DOPS membrane (30% DOPS, as in the all-atom simulation). RBCG systems with a single BAR domain involved 74,916 beads with an initial size of 470 × 98 × 160 Å3. To facilitate the global membrane curvature development in the six-BAR domain simulations, the membrane was ruptured in the longest dimension, namely, at both ends of the membrane; water paddings of 50 Å were added. The system in simulation 6BAR-1row-RB contained 223,017 CG beads, with a size of 1,110 × 124 × 202 Å3; the system in simulation 6BAR-2row-RB contained 181,873 CG beads, with a size of 736 × 180 × 167 Å3. All RBCG simulations were carried out in the NpT ensemble with the same parameters as those in the all-atom simulations. The cut-off distances for nonbonded interactions were also the same as in the all-atom simulations. Periodic boundary conditions were applied, using PME to compute the long-range electrostatic interaction. A timestep of 20 fs was used.
Shape-based CG simulations
The shape-based CG (SBCG) method has been developed to simulate proteins and their assemblies (52), and has been applied to study viral capsids (52) and the bacterial flagellum (53) on a timescale of tens of microseconds and with the ratio of 200 or 500 atoms per CG bead. The SBCG tools are available as a VMD (72) plug-in. In this study, we used 150 atoms per bead. The shape of a protein in an SBCG model is represented by several CG beads whose positions are assigned by a topology-conserving algorithm (76); the beads are connected by harmonic bonds to maintain the protein shape. The method was applied to coarse-grain BAR domain dimers, each dimer being represented by 50 CG beads. The SBCG method was also extended to describe lipid membranes.
Each SBCG bead describes a domain of atoms in the molecule. The mass of the domain and its charge are assigned to the bead, and time evolution of the whole system is described using classical MD. Interactions between beads are described by a CHARMM-like force field (70), i.e., bonded interactions are represented by harmonic bond and angle potentials (but no dihedral potentials), and the nonbonded potentials include 6-12 Lennard-Jones (LJ) and Coulomb terms. For the same reasons as in case of the RBCG model we set ɛ = 1.
The solvent is modeled through Langevin terms (fluctuating and frictional forces), representing water viscosity. Frictional and fluctuating forces are accounted for by a single parameter, the damping constant γ (see (52)). To select an appropriate value for γ, we ran simulations of a single free-floating BAR domain dimer with γ = 0.5, 2, and 5 ps−1. Ten 1-μs-long simulations were carried out for each value of γ and the diffusion constant D of the BAR domain was computed applying the Einstein relation to the protein's center of mass 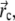
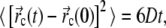 |
(1) |
where the average 〈…〉 was taken over all simulations for given γ. We found D = 21 ± 5, 5 ± 2, and (1.9 ± 0.5) × 10−7 cm2/s for γ = 0.5, 2, and 5 ps−1, respectively. For the protein approximately the size of BAR domain (55,000 amu), experimental values of the diffusion constant are ∼6 × 10−7 cm2/s (77, 78). Accordingly, we choose γ = 2 ps−1.
The SBCG model of the BAR domain is built as described in Arkhipov et al. (52). The BAR domain is a homodimer; each monomer was constructed identically. However, using the SBCG model it was impossible to preserve the stability and shape of the dimer by using nonbonded forces alone, since the dimerization depends on interactions between the monomers, such as specific hydrogen bonds and salt bridges, not accounted for in a straightforward SBCG model. The SBCG method in its present form is designed to reproduce gross effects from electrostatic and hydrophobic/hydrophilic interactions, but these are not sufficient to account for BAR domain dimerization properly. Therefore, bonds between the monomers are introduced and used in the SBCG simulations (Fig. 3 G). The same rule is applied for connecting beads by bonds within one monomer and between the two monomers: two beads are connected if the distance between them is <18 Å.
Interaction parameters were extracted from the all-atom structure and MD simulation of a single BAR domain system. The procedure described in Arkhipov et al. (52) was extended to introduce more specificity for each CG bead. The nonbonded interaction strength εij for the pair of beads i and j was computed as 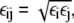 where εi and εj are the strengths for each bead. The value of εi was assigned for each bead i based on the hydrophobic solvent-accessible surface area (SASA) for the protein domain represented by the bead,
where εi and εj are the strengths for each bead. The value of εi was assigned for each bead i based on the hydrophobic solvent-accessible surface area (SASA) for the protein domain represented by the bead,
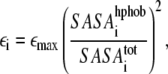 |
(2) |
where SASA and SASA
and SASA are the hydrophobic and total SASA of the domain i, and εmax = 10 kcal/mol (in (52), εi = const). SASA for a domain was computed in the context of the whole protein, i.e., atoms that are at the surface between two domains, but are buried inside the protein, do not contribute to the computed value. This approach, when used in SBCG simulations of single BAR domains on a membrane patch, provided results that are in the closest agreement with all-atom simulations; other approaches, such as using εmax greater or smaller than 10 kcal/mol and using other powers of SASA ratio instead of the square, or setting εi = const gave worse agreements between the all-atom and SBCG simulations. The idea behind using the SASA to determine εi is to let hydrophobic beads aggregate and hydrophilic beads dissolve in the solvent. For a pair of completely hydrophilic beads, Eij = 0 holds—in which case the two beads are free to dissociate unless they are bound to other particles; εij for two completely hydrophobic beads is 10 kcal/mol, which is significantly higher than the thermal energy (kBT ≈ 0.6 kcal/mol at 300 K), but still permits thermal fluctuations.
are the hydrophobic and total SASA of the domain i, and εmax = 10 kcal/mol (in (52), εi = const). SASA for a domain was computed in the context of the whole protein, i.e., atoms that are at the surface between two domains, but are buried inside the protein, do not contribute to the computed value. This approach, when used in SBCG simulations of single BAR domains on a membrane patch, provided results that are in the closest agreement with all-atom simulations; other approaches, such as using εmax greater or smaller than 10 kcal/mol and using other powers of SASA ratio instead of the square, or setting εi = const gave worse agreements between the all-atom and SBCG simulations. The idea behind using the SASA to determine εi is to let hydrophobic beads aggregate and hydrophilic beads dissolve in the solvent. For a pair of completely hydrophilic beads, Eij = 0 holds—in which case the two beads are free to dissociate unless they are bound to other particles; εij for two completely hydrophobic beads is 10 kcal/mol, which is significantly higher than the thermal energy (kBT ≈ 0.6 kcal/mol at 300 K), but still permits thermal fluctuations.
The terms for bonded interactions in the SBCG method (52) are described by potentials Vbond(r) = Kb(r – r0)2 and Va(θ) = Ka(θ – θ0)2 for bond-length r and angle θ, where Kb, r0, Ka, and θ0 are the force-field parameters. In many CG approaches, such parameters are derived from all-atom simulations using the Boltzmann inversion method: for each variable x (such as ith bond-length ri), one obtains the distribution ρ(x) from the all-atom simulation, and uses the Boltzmann relation 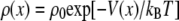 to obtain V(x). But, as the example with the BAR domain in Fig. 5 A shows, this approach can be grossly inadequate. The Boltzmann relation for a single variable holds only if x is an independent variable, not affected by other potentials. For a network of bonds, such as is often the case in SBCG protein models, the bond-lengths and angles are not independent, and when parameters for each of them are derived individually using Boltzmann inversion, the stiffness of the structure is overestimated (dotted in Fig. 5 A). Therefore, we used Boltzmann inversion only to obtain the initial values for force constants Kb and Ka, and then scaled Kb or Ka repeatedly (and uniformly) until the stiffness of the CG model became closer to that of the all-atom model (compare solid and shaded curves in Fig. 5 A). To judge how well the stiffness is represented for each set of force constants tried, we extracted the force constants from CG simulations using the Boltzmann relation above, and averaged them over all bonds (or angles). The scaling was repeated until this average 〈K〉CG reached the all-atom average 〈K〉AA, with an allowed deviation of ±25% for 〈K〉AA. Such scaled values were used in our actual simulations. This approach is similar to the iterative Boltzmann inversion method suggested in the literature (79–81). The latter method is used, for instance, in polymer coarse-graining (usually, for nonbonded potentials) by iteratively tuning the CG potentials to match the radial distribution functions observed in all-atom simulations. For r0 and θ0, Boltzmann inversion provides a very good guess, i.e., one which does not require any further modification.
to obtain V(x). But, as the example with the BAR domain in Fig. 5 A shows, this approach can be grossly inadequate. The Boltzmann relation for a single variable holds only if x is an independent variable, not affected by other potentials. For a network of bonds, such as is often the case in SBCG protein models, the bond-lengths and angles are not independent, and when parameters for each of them are derived individually using Boltzmann inversion, the stiffness of the structure is overestimated (dotted in Fig. 5 A). Therefore, we used Boltzmann inversion only to obtain the initial values for force constants Kb and Ka, and then scaled Kb or Ka repeatedly (and uniformly) until the stiffness of the CG model became closer to that of the all-atom model (compare solid and shaded curves in Fig. 5 A). To judge how well the stiffness is represented for each set of force constants tried, we extracted the force constants from CG simulations using the Boltzmann relation above, and averaged them over all bonds (or angles). The scaling was repeated until this average 〈K〉CG reached the all-atom average 〈K〉AA, with an allowed deviation of ±25% for 〈K〉AA. Such scaled values were used in our actual simulations. This approach is similar to the iterative Boltzmann inversion method suggested in the literature (79–81). The latter method is used, for instance, in polymer coarse-graining (usually, for nonbonded potentials) by iteratively tuning the CG potentials to match the radial distribution functions observed in all-atom simulations. For r0 and θ0, Boltzmann inversion provides a very good guess, i.e., one which does not require any further modification.
FIGURE 5.

Tuning bonded forces in SBCG models. (A) Bond constants Kb for all bonds in the SBCG model of the BAR domain. Using Boltzmann inversion, Kb values are extracted from an all-atom simulation (solid representation). These values of Kb are used in a SBCG simulation. The Boltzmann inversion is performed on the resulting SBCG trajectory, but the Kb values extracted are significantly higher (dotted) than those obtained from the all-atom simulation. Then, Kb constants used for the SBCG simulation are all multiplied by the same number (0.3 in this example), and a new SBCG simulation is carried out with these constants. The Boltzmann inversion performed on the new SBCG trajectory returns Kb values (shaded representation) that are much closer to those found in the all-atom simulation. Thus, the scaled constants are better suited for SBCG simulations. (B and C) DOPC leaflet thickness (defined as the distance between the centers of mass of the upper and lower parts of a lipid, averaged over the leaflet) and its RMSD recorded in all-atom (solid representation) and SBCG (shaded representation) simulations of a patch of DOPC bilayer. The all-atom and SBCG simulations produce matching results when the SBCG bond parameters are r0 = 12.0 Å and Kb = 0.2 kcal/(mol Å2), which are the values used in all further SBCG simulations. A variation of these parameters, such as r0 = 12.5 Å and Kb = 0.3 kcal/(mol Å2), produce a noticeable deviation.
Lipids have not been previously modeled with SBCG, although models in which a lipid is represented by just a few particles have been proposed before (see, e.g., (60)). However, we desire to employ hundreds of atoms per bead, while a single DOPC lipid consists of only 138 atoms. Since the lipid molecule is elongated, it is impossible to represent it by a single spherical bead. Using a few beads per lipid would reduce the maximal possible time step in the SBCG simulation, which would compromise the accessible timescales. Thus, we abandon representing single lipid molecules as separate entities, and, instead, represent each leaflet of a lipid bilayer by two layers of CG beads: one for the lipid heads and one for the lipid tails (Fig. 3, C and D, and Fig. 6), a head and a tail bead pair representing several lipid molecules. No head-tail pair is connected to other pairs (nonbonded interactions only), but the two beads within the pair are connected by a harmonic bond. Since the leaflet thickness (including furthest-reaching heads) is ∼25 Å, each bead accounts for the part of the leaflet that is 12.5 Å in height, or for the volume 12.5 × 12.5 × 12.5 Å3 (since the bead has to be symmetric in each dimension). Thus, a two-bead CG molecule (Fig. 6) stretching across the leaflet accounts for the area 12.5 × 12.5 Å2 and occupies the volume of 12.5 × 12.5 × 25 Å3. With the DOPC area per lipid being ∼70 Å2, each two-bead DOPC molecule represents 2.2 DOPC lipids on average, or ∼300 atoms. This dictates the choice of 150 atoms per CG bead that we are using in this work.
FIGURE 6.

SBCG model for lipids. A patch of DOPC membrane is shown from the top and from the side, in an all-atom as well as in SBCG representations. The head and tail halves of the all-atom lipids are shown in cyan and white, respectively. Each SBCG molecule represents ∼2.2 lipids; the head SBCG beads are in green, and the tail beads are in pink.
The lipids are parameterized based on simulations of a DOPC membrane patch. The length of the bond between the head and tail beads, r0, and the bond strength, Kb, control the average membrane leaflet (or bilayer) thickness, as well as root mean-square deviations (RMSDs) of the thickness (r0 mainly accounts for the thickness and Kb for the RMSD). As shown in Fig. 5, B and C, these values are quite sensitive to r0 and Kb used in SBCG simulations, even within the variation of 0.5 Å for r0 and 0.1 kcal/(mol Å2) for Kb. The values for r0 and Kb used in the actual simulations are 12 Å and 0.2 kcal/(Å2mol). Note that the leaflet thickness is defined here as a distance between the centers of mass of the upper and lower parts of a lipid, averaged over the leaflet, which is described by the distance between the head and tail beads in the SBCG model (Fig. 5 B).
The LJ parameters for lipid beads are chosen to reproduce the area per lipid (∼70 Å2), but they also influence the bilayer thickness, due to the interactions between the two leaflets. The LJ energy εi for a tail bead is set to 10 kcal/mol, and that for head beads to 0.1 kcal/mol; LJ radii are set to 6.8 Å for both. However, it appears that for the head beads, values of εi in the range 3.0–20.0 kcal/mol (with LJ radii being 6.8 Å) result in approximately the same area per lipid and bilayer thickness. The LJ radius has a much stronger effect on these properties, e.g., an LJ radius of 6.3 Å results in an area per lipid of ∼60 Å2. The area per lipid cited here was computed from the area per two-bead SBCG molecule, assuming that such a molecule represents 2.2 lipids on average.
Each bead in the DOPC model has zero charge. For DOPS lipids, a charge of −2.2|e| is assigned to the head bead. The masses are 864.75 amu for the DOPC head and tail beads, and 866.76 amu for the DOPS head bead. To match the charge of the DOPS beads, we introduce ions, each with the charge of ±2.2|e| and mass of 1000 amu, roughly corresponding to eight ions of mixed nature (such as both Na+ and Cl−) with their hydration shells. For the initial conditions in our simulations, ions (either positive or negative) are distributed uniformly in the simulated volume (excluding the areas occupied by lipids and proteins).
SBCG simulations were carried out with a time step of 100 fs. A 30 Å cutoff was used for the nonbonded interactions. Periodic conditions were used in all SBCG simulations; the membrane was ruptured in the longer dimension at the periodic cell boundaries, similarly to the RBCG simulations 6BAR-1row-RB and 6BAR-2row-RB. The membrane was a mixture of DOPC and DOPS, with 30% of DOPS as in the all-atom and RBCG simulations.
Bending rigidity of the model SBCG membrane
To estimate the bending rigidity of the SBCG model membrane, we employ an approach recently suggested by Harmandaris et al. (45). In this approach, simulations mimic a typical experimental setup, which obtains membrane bending rigidity by measuring the force required to pull a membrane tube from a vesicle. In the approximation of an elastic membrane described by the Helfrich Hamiltonian (64), the curvature energy E of a cylinder of radius R and length L is given by
 |
(3) |
where kc is the bending modulus. The axial force F required to hold the cylinder at given length and radius is assumed to be equal to the derivative of E with respect to L, with the total area 2πLR being fixed, namely, F = 2πkc/R. This force can be directly measured in an experiment or in a simulation (see (45) for details), and used to estimate the bending modulus as
 |
(4) |
We simulated various membrane tubes, prepared as bilayer cylinders with hollow ends, with radii varying between 50 and 200 Å (each tube contained ∼5000 beads, no periodic conditions were used; see Lip-Bend simulations in Table 1). The tubes were first equilibrated for 1 μs. Then, the simulations were continued, while harmonic constraints were applied in the axial direction to the beads at the tips of the tubes (see Fig. 7), with the spring constant 0.01 kcal/(mol Å2). The axial forces from the constraints acting at both ends of a tube were computed. The direction of force that corresponds to extending the tube (the force acting against the shrinking of the tube's length) was chosen as the positive one. The forces from both ends, projected on the appropriate direction for each end, were summed together, providing the full force acting to extend the tube. The first 1 μs of the simulations was disregarded, to allow for equilibration. Our approach differs slightly from that of Harmandaris et al. (45); these authors extended simulated tubes across a periodic box, and only the axial strain in the tubes was measured to estimate F.
TABLE 1.
Simulated systems
| Name | Method | Nrun | NBAR | N | Time | Result |
|---|---|---|---|---|---|---|
| Lip-Bend | SBCG | 10 | 0 | 5000 | 4 μs | Measurements of bending rigidity |
| Lip-SB | SBCG | 20 | 0 | 600 | 10 μs | Self-assembly |
| 1BAR-AA | All-atom | 1 | 1 | 671,331 | 25 ns | Local curvature |
| 1BAR-RB | RBCG | 3 | 1 | 74,916 | 50 ns | Local curvature |
| 1BAR-SB | SBCG | 5 | 1 | 1404 | 50 ns | Local curvature |
| 6BAR-1row-RB | RBCG | 1 | 6 | 223,017 | 20 ns | Local, no global curvature |
| 6BAR-2row-RB | RBCG | 1 | 6 | 181,873 | 50 ns | Global curvature |
| 6BAR-1row-SB | SBCG | 5 | 6 | 4436 | 5 μs | Local curvature at first, global at ∼1 μs |
| 6BAR-2row-SB | SBCG | 5 | 6 | 3265 | 5 μs | Global curvature |
| 6BAR-EM | EM | ∼100 | 6 | — | ∼10 μs | Global curvature |
Name corresponds to a series of similar simulations, and Nrun stands for the number of simulations. Time is the simulated time for a single simulation in the series. The only differences between the simulations listed under the same name were the initial velocities (which were randomly generated according to the Maxwell distribution); for series Lip-Bend and Lip-SB, initial structures for some of the runs were different as well. NBAR is the number of BAR domains in the simulated system, and N is the total number of particles (does not apply for EM). Lip-Bend are the simulations for measuring bending rigidity of the SBCG DOPC membrane, Lip-SB are the simulations of DOPC lipids self-assembly, starting from a random mixture of SBCG lipids. 6BAR-EMs are the EM computations reproducing the results of simulations 6BAR-2row-RB and 6BAR-2row-SB.
FIGURE 7.

Determination of the bending rigidity of the SBCG membrane. Shown at the top is an example bilayer tube that emulates a tether pulled from the membrane in experiments measuring the bending rigidity. Tubes of various radii were simulated with harmonic restraints applied to the ends of the tube (darker CG beads at the tips of the tube). The force experienced by the restrained beads was computed and used to estimate the bending rigidity, according to Eq. 4. Results of these measurements are shown at the bottom, where the estimated bending rigidity (in units of temperature, T = 300 K) is plotted versus the average tube radius.
We used Eq. 4 to determine kc, employing average force 〈F〉 and average radius 〈R〉 from each simulation (average over time for F, and over both time and axial dimension for R). The obtained kc values are presented in Fig. 7 in units of kBT (T = 300 K). The experimental value of kc for pure membranes (see, e.g., (82)) is ∼0.5–1.0 × 10−19 J, or 10–20 kBT; the overall average for kc obtained from the simulations is 19.6 kBT, the average values from each simulation being clustered in the range of 10–30 kBT with significant fluctuations. The SBCG membrane model overestimates slightly kc, but, taking into account the coarseness of the model, the agreement is satisfactory. The applicability of the SBCG membrane model for simulations of membrane reshaping phenomena is also indicated by the good agreement between the all-atom and SBCG simulations of membrane bending by BAR domains, described in Results.
Continuum elastic membrane model
Simulation of BAR domains and membranes can be speeded up even beyond the SBCG models if one uses a continuum elastic membrane (EM) model. We developed a simple string model, where the profile of the membrane along its center line is represented by a continuous string in two dimensions (instead of full three dimensions; the membrane is supposed to be fully periodic in the third dimension). The string dynamics is described by classical mechanics and is solved numerically. For that purpose, the string of length L is represented as a chain of pointlike elements i = 1, …, N, with the elements' positions s being distributed along the string length (s ∈ [0, L]). Positions of each element at each time moment t are described by vectors 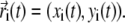 The forces acting on each element arise due to three causes: membrane stretching (
The forces acting on each element arise due to three causes: membrane stretching (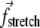 ), membrane bending (
), membrane bending (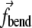 ), and damping from the environment. Thus, the dynamics of each element is described as
), and damping from the environment. Thus, the dynamics of each element is described as
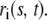 |
(5) |
Here  and
and 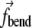 are not forces, but are proportional to forces (with the proportionality being defined by the linear density of the string, which does not enter the final equations).
are not forces, but are proportional to forces (with the proportionality being defined by the linear density of the string, which does not enter the final equations).  and
and 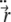 are the 1st and 2nd derivatives with respect to time t.
are the 1st and 2nd derivatives with respect to time t.
The stretching term is described by a harmonic spring acting between any two adjacent elements in the chain,
 |
(6) |
where  is the vector connecting the two elements, Δs = L/(N – 1) is the distance between the elements along the straight string at rest, and Astretch is a constant.
is the vector connecting the two elements, Δs = L/(N – 1) is the distance between the elements along the straight string at rest, and Astretch is a constant.
The bending term is designed to maintain a straight membrane if no BAR domains are present, or sculpt the membrane to acquire a certain curvature, if BAR domains are present. The bending term is defined for each triple of consecutive elements, with the absolute value of 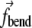 being the same for the edge elements in the triple,
being the same for the edge elements in the triple,
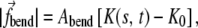 |
(7) |
where K0 is the assigned intrinsic curvature (K0 = 0 if no BAR domain is present), Abend is a constant, and
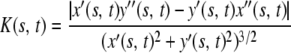 |
(8) |
is the local curvature (x′ and x″ being the first and second derivatives with respect to s). The values for K(s, t) are obtained numerically using Eq. 8 and three-point centered finite difference approximations for x′, x″, y′, and y″. The surface on which BAR domains are located is defined in the beginning of a calculation, thereby determining the direction of a positive or negative curvature. Directions of 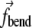 applied to the edge elements of the triple are defined identically to the case of a bonded angle potential in an MD force field, i.e.,
applied to the edge elements of the triple are defined identically to the case of a bonded angle potential in an MD force field, i.e., 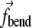 acts to close the angle formed by the triple if K(s, t) < K0, and to open the angle if K(s, t) > K0. The term
acts to close the angle formed by the triple if K(s, t) < K0, and to open the angle if K(s, t) > K0. The term 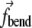 for the central element is a negative of the sum of
for the central element is a negative of the sum of 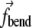 from the two edges. Sums in Eq. 5 indicate that each element is acted upon by more than a single
from the two edges. Sums in Eq. 5 indicate that each element is acted upon by more than a single 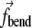 and
and  : the terms acting on each element are two
: the terms acting on each element are two 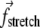 (one from the link with the previous element and another from the link with the next one) and three
(one from the link with the previous element and another from the link with the next one) and three 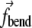 (each from the three triples that the element is a part of). The edge elements experience fewer forces since they have fewer neighbors.
(each from the three triples that the element is a part of). The edge elements experience fewer forces since they have fewer neighbors.
The EM model uses four parameters: K0, Astretch, Abend, and γEM. In general, K0 can depend on s and t; in our computations, we set it to uniform values chosen to reproduce the final curvatures of the membrane obtained in RBCG and SBCG simulations of six BAR domains. The stretching parameter Astretch can be connected with the Young modulus for the membrane extensibility; the experimental value (82) of the Young modulus of a membrane at room temperature is approximately the same (within ∼3%) for such lipids as DMPC, SOPC, etc., and we find that it corresponds to Astretch ≈ 104 ns−2. The other two parameters, Abend and γEM, have to be chosen to reproduce our CG simulations. In principle, Abend could be obtained using experimental data for the membrane bending elasticity, but it may differ significantly for the free membrane and the one covered with BAR domains. One simulation with six BAR domains (6BAR-2row-RB) is chosen for the parameterization, using the curvature values and the time dependence of the membrane end-to-end distance for reference. The best-matching values, used for further EM computations, are Abend ≈ 7 nm2/ns2 and γEM ≈ 0.06 ns−1.
Assessing the simulation timescales
Since the BAR domain systems are described at four levels of resolution, one may wonder how times reported in simulations at different levels compare with each other, and whether some type of time scaling links the dynamics at different levels. The times reported are given by the number of integration steps simulated, multiplied by the integration time step (in terms of the chosen units of time). In fact, the convention of fixed units (t0 = 1 fs, l0 = 1 Å, m0 = 1 amu) is used in NAMD (71), the program we employed for all-atom, RBCG, and SBCG simulations. For EM computations, we used units t0 = 1 ns, l0 = 1 nm, and m0 = 1 amu. Thus, the times reported for each simulation (and across the resolution levels) can be compared directly.
RESULTS
We performed a number of all-atom, RBCG, SBCG, and EM simulations, as listed in Table 1. In an all-atom simulation of a single BAR domain with a patch of planar membrane, 1BAR-AA, the protein was found to curve the membrane, in agreement with previous observations (30). Multiple RBCG and SBCG simulations of the same process (1BAR-RB and 1BAR-SB) were found in good agreement with all-atom simulations. RBCG simulations of multiple-BAR domain systems showed that, over the timescale of ∼50 ns, the nonstaggered arrangement of BAR domains does not produce a global curvature, while the staggered arrangement does. The SBCG simulations confirmed these conclusions, but also allowed us to reach times of ∼5 μs, at which the global curvature developed for both arrangements, although to a different extent (radius of curvature 1000 vs. ∼300 Å).
Self-assembly properties of the model SBCG membrane
For a validation of the new SBCG lipid model, we carried out self-assembly simulations with DOPC, starting from a random lipid mixture and using a number of different sizes of the periodic cell (Lip-SB simulations). Depending on the size of the periodic cell, the SBCG DOPC molecules assemble into well-defined structures, as expected for lipid phases. For example, 300 SBCG lipid molecules in a 100 × 100 × 100 Å3 periodic cell form multilamellar structures (Fig. 8 A). For smaller box sizes (higher lipid concentration), the self-assembly results in inverted hexagonal phase-like structures. For larger cells, a single bilayer forms across the periodic cell, often with a metastable lipid bridge (stalk) connecting the periodic images of the bilayer. Such bridges have been observed in lipid self-assembly simulations using, e.g., Marrink's CG model (37). It may often take 100 nanoseconds to several microseconds to dissolve these bridges. For even larger cell size (such as 183 × 122 × 218 Å3 shown in Fig. 8 B) the lipids aggregate into a number of micelles. In the simulation demonstrated in Fig. 8 B, a relatively rare event happened when several micelles fused quickly into a single complex that formed a bilayer spanning the whole cell. The initial formation of the structures described here happens usually on timescales of 10–100 ns, which is then followed by a slower stabilization of the structure and elimination of defects, taking hundreds of nanoseconds to tens of microseconds.
FIGURE 8.

SBCG simulations of DOPC self-assembly. Formation of a multilamellar structure is shown in panel A, and of a single bilayer in panel B. In both cases, the system consists of 300 SBCG two-bead molecules, corresponding to ∼660 DOPC lipids, and the simulations start from a randomized mixture of SBCG molecules. The periodic cell is 100 × 100 × 100 Å3 in panel A (shown as an open square), and 183 × 122 × 218 Å3 in panel B (one cell is shown). In panel A, lamellar-like structures form quickly and then stabilize with time, producing eventually stacks of bilayers. In panel B, a number of large and small micelles forms first, but then almost all lipids aggregate into a single bilayer extending over the entire size of the periodic cell. A large micelle that has not fused with the bilayer can be seen at the snapshots at the bottom.
Thus, the SBCG model qualitatively captures the self-assembly properties of lipids, with correct phases forming depending on the lipid concentration. Notably, the lipid SBCG model has been parameterized to reproduce the bilayer properties, namely, thickness, and area per lipid. The chosen parameters allow one to capture other properties of lipid systems as well, such as the self-assembly dynamics and bending elasticity of the membrane.
Single BAR domain simulations
In simulation 1BAR-AA we observed that a single BAR domain induces local curvature on a patch of membrane (Fig. 9 A), in agreement with previous all-atom simulations (30). All three RBCG simulations of a single BAR domain (1BAR-RB) exhibited the same behavior (Fig. 9 B); the induced curvature varied in each simulation, again in agreement with previous studies (30,31). The same behavior was observed in the SBCG simulations (1BAR-SB, Fig. 9 C). In all cases, a bending mode developed underneath the BAR domain-occupied region of the membrane and stabilized after ∼25 ns. The bending mode in the RBCG and SBCG simulations was more pronounced than that in our single all-atom simulation 1BAR-AA; however, some of the earlier all-atom simulations (30) generated a higher membrane curvature, similar or even higher than those observed in our CG simulations (Fig. 9 D).
FIGURE 9.

Single BAR domain simulations. (A–C) Snapshots from all-atom simulation 1BAR-AA, one of RBCG simulations 1BAR-RB, and one of the SBCG simulations 1BAR-SB. Negatively charged PS lipid headgroups are shown in red, and neutral PC headgroups are shown in cyan. The protein consists of two monomers, shown in green and purple. (D) Time evolution of membrane curvature. The two black dots are the curvatures from two all-atom simulations reported in Blood and Voth (30), averaged from time 20–27 ns. The black curve is from simulation 1BAR-AA; red, orange, and magenta are from simulations 1BAR-RB; the rest (turquoise, blue, green, deep green, cyan) are from simulations 1BAR-SB.
The time evolution of the membrane curvatures is shown in Fig. 9 D. The curvature displayed is the overall curvature of the membrane underneath the BAR domain, calculated by fitting the profile of the corresponding membrane section to a circle. This definition of curvature is different from that used in Blood and Voth (30), which employed the point curvature for the membrane directly beneath the center of the BAR domain. The two black circles in Fig. 9 D illustrate the curvature reached in the simulations reported in Blood and Voth (30), when calculated by our method; the data were taken from the membrane profiles in Fig. 3b of Blood and Voth (30), averaged from 20 to 27 ns. One can see that earlier all-atom simulations produced higher curvatures than 1BAR-AA (orange curve). However, in other simulations by Blood et al. (31) the authors observed a curvature significantly lower than that found in Blood and Voth (30), suggesting that single BAR domains induce a wide range of membrane curvatures, varying from case to case, as well as over time for a single case. The curvature observed in our simulations 1BAR-AA, 1BAR-RB, and 1BAR-SB falls in the same range and varies among the simulations. Thus, the RBCG and SBCG models reproduce the phenomenon that a single-BAR domain induces a membrane curvature, with the magnitude and variation of the curvatures, as well as the timescale of curvature generation, agreeing well with characteristics seen in all-atom simulations.
Simulations of six-BAR domain systems
Systems with six BAR domains considered here would amount to a few million atoms in the all-atom representation, making long-enough all-atom MD unfeasible. Thus, we employed RBCG and SBCG simulations. Six BAR domains were placed on an initially planar membrane (Fig. 10), in a nonstaggered arrangement (simulations 6BAR-1row-RB and 6BAR-1row-SB; see Fig. 2 A), and in a staggered arrangement (6BAR-2row-RB and 6BAR-2row-SB; Fig. 2 B).
FIGURE 10.

Membrane curvature induced by multiple BAR domains. (A) Six BAR domains in the nonstaggered arrangement in RBCG simulation 6BAR-1row-RB. (B) Six BAR domains in the staggered arrangement in RBCG simulation 6BAR-2row-RB. Upper and middle panels in panels A and B show side- and top-views of the initial setup. Lower panels are snapshots after 20 or 50 ns. (C) BAR domains in the SBCG simulations 6BAR-1row-SB. (D) BAR domains in the SBCG simulations 6BAR-2row-SB.
Results of these simulations are shown in Fig. 10. The RBCG systems were composed of ∼200,000 particles. The simulations were stopped at 20 ns for 6BAR-1row-RB and at 50 ns for 6BAR-2row-RB, since the membrane edges from the neighboring periodic images came within the cutoff distance due to large changes in the dimensions of the periodic cell, kept flexible to maintain constant pressure.
In the simulations with the nonstaggered arrangement (6BAR-1row-RB, 6BAR-1row-SB), BAR domains induced a local, but no global curvature. In the simulations with the staggered formation (6BAR-2row-RB, 6BAR-2row-SB), a global bending mode developed, with the radius of curvature reaching ∼400 Å. This radius is comparable to experimentally observed values (21). Fig. 11 shows the membrane curvature versus time as seen in the RBCG and SBCG simulations. The plotted curvatures are those developed under individual BAR domains in RBCG simulations and overall curvatures. Curvatures under individual BAR domains in SBCG simulations show the same trend as those in RBCG simulations, and are not depicted. The local curvatures observed are close to those in simulations of single BAR domains (Fig. 9 D), and exhibit a similar scatter from one simulation to another. In simulation 6BAR-1row-RB (Fig. 11 A), the highest local membrane curvature (radius of 150 Å) occurs at the edge of the membrane patch, i.e., beneath BAR domains 1 and 6 shown in Fig. 10 A. This is probably due to the fact that the edge of the membrane is more flexible than the middle since the edge is unrestrained and does not interact with other BAR domains. In simulation 6BAR-2row-RB (Fig. 11 B), high local curvatures also occur at the edge of the membrane beneath BAR domains 1 and 5.
FIGURE 11.

Time evolution of membrane curvatures in the simulated six-BAR domain systems. The shaded number n (1, 2, 3, 4, 5, or 6) denotes that the curve is for the membrane beneath BAR domain n shown in Fig. 10, A and B. (A) Local curvatures in simulation 6BAR-1row-RB. (B) Local and global curvatures in the staggered arrangement of BAR domain systems at t < 50 ns. Numbered curves are for local curvatures in the RBCG simulation 6BAR-2row-RB. The thick curves (deep green, cyan, blue, sage, and turquoise) are global curvatures observed in each of the five SBCG simulations 6BAR-2row-SB, and the black curve is the global curvature from 6BAR-2row-RB.
In case of the nonstaggered arrangement of BAR domains, the membrane directly beneath each BAR domain is pulled in by the concave charged surface of a protein, while the membrane between two adjacent BAR domains remains in its original, planar conformation. Thus, a ripple-shaped membrane (Fig. 10, A and C) develops in ∼15 ns and remains stable. For BAR domains in the staggered arrangement, the membrane beneath each BAR domain is also bent locally, but because the membrane between two adjacent BAR domains in one row is covered by another BAR domain from the next row, the bending results in a uniform curvature (Fig. 10, B and D).
Due to the reduction in the number of particles and the increase of the integration time step in going from the RBCG to the SBCG model, we were able to simulate the dynamics of six BAR domains on a longer timescale, namely, up to 5 μs, as shown in Fig. 12 (see also Movie S1 and Movie S2 in Supplementary Material). The SBCG simulations reproduce the results of the RBCG simulations over 50 ns ideally, but reaching further in time reveal global curvature arising also in the case of the nonstaggered BAR domains arrangement. Up to 100–400 ns, the membrane remains globally flat, but then starts to curve (Fig. 12 C). The bending reaches its maximum at 1.5 μs, with a radius of global curvature of ∼700–1200 Å. The radius converges to a value of ∼1000 Å at 2.7 μs when the membrane in all five simulations 6BAR-1row-SB stops bending. In contrast, the membrane in the staggered system reaches a global curvature radius of 300–400 Å during the first 300 ns of simulation, and does not exhibit significant further changes thereafter.
FIGURE 12.

Six-BAR domain systems at t < 5 μs. (A and B) Snapshots at 5 μs for 6BAR-1row-SB (A) and 6BAR-2row-SB (B). (C) Time evolution of global curvatures. The five curves at the bottom are for simulation 6BAR-1row-SB, the five at the top for 6BAR-2row-SB.
The differences between the curvatures reached and the dynamics of bending between simulations with a single BAR domain, with nonstaggered six BAR domains, and staggered six BAR domains indicate that the distribution of BAR domains on the membrane determines the membrane curvature. The observed values of the radius of curvature range from 100 to 150 Å for local bending produced by individual BAR domains to 1000 Å in the case of global curvature in simulations 6BAR-1row-SB. This agrees well with experimental observations that BAR domains may produce tubes with a wide spectrum of curvatures (21). In simulations 6BAR-2row-SB, the global curvature radius reaches ∼250 Å, equal to the value observed in one of the micrographs in Peter et al. (21). Interestingly, simulations of the systems with exactly the same initial structure produce different membrane shapes, which is true for all our simulations as well as previous single-BAR domain all-atom simulations. The final structure is always slightly, but noticeably different from one simulation to another, and the radii of bent membranes, although close to each other (within 30–50%), are never exactly equal. However, the qualitative behavior is found to be the same in simulations starting from the same structure, indicating that certain arrangements of BAR domains can robustly bend membranes into curved structures with a relatively well-defined radius.
EM computations
The continuum EM model (see Methods) was parameterized to reproduce the dynamics of the six-BAR domain system with the staggered arrangements of the proteins. Typical results of the EM computations are shown in Fig. 13. Snapshots of the membrane shape are shown in Fig. 13 A and correspond well to the membrane evolution observed in simulations 6BAR-2row-SB. The membrane starts to bend gradually and reaches a radius of ∼250–400 Å at ∼300 ns, at which point the curvature stabilizes. Fig. 13 B compares the end-to-end distances recorded in the five simulations 6BAR-2row-SB with the end-to-end distances obtained from the EM computations (only the first 1 μs is shown since after 300–500 ns the distance remains rather constant). Overall, the EM model captures very well the membrane curvature dynamics observed in the simulations.
FIGURE 13.

Membrane dynamics in EM computations. (A) Snapshots of the membrane shape in a typical EM computation. (B) End-to-end distance of the membrane over time. The fluctuating curves are taken from simulations 6BAR-2row-SB, and smooth curves are from EM computations.
In the EM model, four parameters are used: intrinsic curvature K0, force constants Astretch and Abend, and a damping coefficient γEM. We worked with the uniform values of K0 set for each element of the EM model; these values were tuned according to the curvatures observed in each of the SBCG simulation to reproduce their individual end-to-end distances, as shown in Fig. 13 B. Parameters Astretch, Abend, and γEM were also set uniformly, but were not changed from one computation to another. The final end-to-end distances obtained in each simulation are apparently determined by the final curvature, and are matched well by the EM model when K0 is tuned, but the dynamics of the bending (the speed of decay of the end-to-end distance) is reproduced well by the EM model even without changing the other three parameters. This means that the membrane stretching elasticity (described by Astretch), as well as bending elasticity and bending forces exerted by BAR domains (described together by Abend), are the same in each simulation, and the variety of curvatures observed in the simulations is due to small differences accumulating throughout the trajectory (and arising because of the thermal fluctuations and randomness of initial velocities), rather than large-scale variations. The radii of curvature observed in simulations 6BAR-2row-SB are the same within ∼40% of the value, and the end-to-end distances vary within ∼100 Å around the average of 500 Å. These difference can be tolerated in an EM model and, thus, one can set the value of K0 to an average constant. With such a choice, namely, K0 = 1/300 Å−1, and the other three parameters being the same, we obtain a specific model that results in a robust description of the staggered arrangement used in the SBCG simulations.
Extensive computations were performed with the EM model, sampling many possible values of parameters Astretch, Abend, and γEM, with K0 being fixed. We found that multiple sets of these three parameters, differing by orders of magnitude, could result in the same dynamics (same end-to-end distance versus time curve). However, Astretch could be fixed at ≈104 ns−2 by matching the experimentally known membrane stretching elasticity, and then the values of Abend and γEM that result in the correct dynamics are unique. In this case, variation of Abend results in changing the speed with which the end-to-end distance (or curvature) reaches a constant value, or, in other words, the decay time of the curves in Fig. 13 B. Deviation of γEM from the correct value results in under- or overdamping. In the case of overdamping, the correct values of the end-to-end distance and of the curvature cannot be reached, even though K0 matches the desired curvature, because the system loses too much energy too fast. In the case of underdamping, correct curvature and end-to-end distance are reached, but do not remain constant; instead, these values oscillate with time, reflecting the oscillations of the membrane shape around its equilibrium position. The values that we found to match the results of SBCG simulations well are Abend ≈ 7 ± 1 nm2/ns2 and γEM ≈ 0.06 ± 0.01 ns−1. In the vicinity of K = K0, Abend can be related to the membrane bending rigidity, or bending modulus, kc, which determines the membrane free energy per unit area as EF = kc(K – K0)2/2. The value of kc can be estimated as kc ∼3ρaAbend/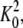 where ρa is the area density of the membrane. For Abend = 7 nm2/ns2, we get kc ≈ 0.9 × 10−19 J. The experimental values of kc for free membranes (82), such as DMPC, are kc ≈ 0.5–1.0 × 10−19 J. Thus, our estimation suggests that the rigidity of the membrane with staggered arrangement of BAR domains is approximately the same, or at the most twice higher than that of a free membrane. The damping parameter γEM accounts for the solvent viscosity and temperature T. Thus, the value γEM = 0.06 ns−1 corresponds to the membrane bending in water at T = 300 K, in the case when the intrinsic membrane curvature (due to the action of BAR domains) is K0 = 1/300 Å−1.
where ρa is the area density of the membrane. For Abend = 7 nm2/ns2, we get kc ≈ 0.9 × 10−19 J. The experimental values of kc for free membranes (82), such as DMPC, are kc ≈ 0.5–1.0 × 10−19 J. Thus, our estimation suggests that the rigidity of the membrane with staggered arrangement of BAR domains is approximately the same, or at the most twice higher than that of a free membrane. The damping parameter γEM accounts for the solvent viscosity and temperature T. Thus, the value γEM = 0.06 ns−1 corresponds to the membrane bending in water at T = 300 K, in the case when the intrinsic membrane curvature (due to the action of BAR domains) is K0 = 1/300 Å−1.
DISCUSSION
We have developed and tested models describing interactions of a membrane-sculpting protein, BAR domain (using amphiphysin BAR domain from Drosophila), with a lipid bilayer membrane, at four scales: atomic level, RBCG level with ∼10 atoms represented by a CG bead and single-residue resolution, SBCG level with ∼150 atoms per CG bead and a group of beads per protein, and a mesoscopic continuum level with the membrane and proteins represented through an elastic membrane model. Each description level is parameterized based on the more detailed description of simple systems. The all-atom MD used the CHARMM force field (70,75), derived from quantum chemistry calculations. The RBCG model is parameterized using experimental data (this is done for lipids in the original Marrink model (37)), structural considerations (57), and all-atom simulations (in this study). The SBCG model is based on results of all-atom simulations and on available experimental data, such as area per lipid for membrane bilayers. The EM model is parameterized using SBCG simulations and experimental data (Young modulus for membrane extensibility).
Our results reproduce, whenever available, experimentally known features of membrane bending by BAR domains and observations of alternative simulations, i.e., our all-atom, RBCG, and SBCG simulations of a single BAR domain on a membrane patch agree well with each other and with previous all-atom simulations (30,31), in terms of the value and range of the induced membrane curvature and the timescale of the bending. Our SBCG simulations of six BAR domains reproduce the behavior observed in the analogous RBCG simulations. The SBCG model has been used before for studies of protein complexes (52,53), and here we have described a new SBCG model for lipids and showed that the model captures many features of lipid systems, such as the characteristics of a bilayer (its thickness, area per lipid, and fluctuations in these values), and self-assembly properties, including the formation of different phases depending on the lipid concentration. Finally, the continuous EM model captures the membrane sculpting events observed in the simulations with more detailed descriptions.
The simplified models used provide significant speed-ups for computations. The RBCG model reduces the number of particles in the system 10-fold, and allows one to use an integration time step of 20–25 fs, vs. 1 or 2 fs common for all-atom MD. The SBCG model uses ∼150 atoms per CG bead and describes the solvent implicitly, thus reducing the system size further, approximately fivefold (in our all-atom simulations, ∼80% of the atoms belong to water); thus, the overall reduction in the number of particles is 750-fold. The EM model employs a continuum description, typically with ∼100 elements being enough to do computations with the membranes of the size explored here (600–1000 Å long). The speed-ups provided by the CG and EM models allowed us to perform several simulations to sample the observed dynamics. The simulated systems behaved somewhat differently in each simulation, although overall trends remained the same; this sampling resulted in a range of observed curvatures. The models used in this study are not specific to the BAR domain-membrane system and can be adapted to many other protein-lipid systems, which can be accomplished through the coarse-graining tools available in VMD (72).
Results of this study show that individual BAR domains produce local membrane curvatures with the radii being in the range from 100 to 500 Å, while multiple BAR domains acting together may induce global curvatures with radii 250–1000 Å. We sampled only two arrangements of BAR domains, and other arrangements may result in a wider range of values for the radius of global curvature. The two arrangements studied (see Fig. 2) result in two distinct curvatures of the sculpted membrane, ∼1000 Å in the case of the nonstaggered formation, and ∼250–400 Å in the case of the staggered one. For the former arrangement, BAR domains first induce local, but no global curvature, and the system remains in this conformation for 100–400 ns. The SBCG model allowed us to observe the dynamics on longer timescales, and to find that further on, the global curvature slowly emerges, and stabilizes at t∼3 μs. For the latter arrangement, the global curvature starts to develop from the beginning. In both cases, the systems do not change much after t∼3 μs.
The two arrangements studied were chosen to reproduce qualitatively formations of BAR domains observed experimentally (see Fig. 1). Such observations (29) show that BAR domains form spiraling rows on the surface of membrane tubules, and, together with our study, suggest that the lateral alignment of these rows within the spiral determines the size of the tubule. Then, the BAR domain arrangement that is more favorable for forming narrow tubules (such as the staggered arrangement in our study) may be exploited by the cell for sculpting membranes, and one expects that the amino-acid composition of BAR domains evolves to account for this function. Indeed, at least F-BAR domains form the spiraling rows by polymerizing through interactions between very highly conserved residues on their surface (26,29). These interactions seem to favor the staggered formation, where centers of BAR domains in one lateral row are aligned (longitude-wise) with ends of BAR domains in the neighboring rows (see Figs. 1 and 2). In addition, the end-to-center polymerization of BAR domains (as that in Fig. 1) enforces a single row with no gaps left between the ends of BAR domains, which may also favor stronger tubulation. Thus, the experimental evidence and our study suggest that the collective action of BAR domains to bend membranes is enhanced by arrangement in specific lattices. Such lattices (see Fig. 1) are employed by the cell in many instances of membrane morphogenesis.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
This work is supported by National Institutes of Health grant No. P41-RR05969 and R01-GM067887. The authors gladly acknowledge supercomputer time provided by Pittsburgh Supercomputing Center and the National Center for Supercomputing Applications via Large Resources Allocation Committee grant No. MCA93S028 and by the Turing Xserve Cluster. A.A. was supported by the Edelheit fellowship.
Anton Arkhipov and Ying Yin contributed equally to this work.
Editor: Helmut Grubmüller.
References
- 1.Zimmerberg, J., and M. M. Kozlov. 2006. How proteins produce cellular membrane curvature. Nat. Rev. Mol. Cell Biol. 7:9–19. [DOI] [PubMed] [Google Scholar]
- 2.Rothman, J. E., and L. Orci. 1996. Budding vesicles in living cells. Sci. Am. 274:70–75. [DOI] [PubMed] [Google Scholar]
- 3.Lecuit, T., and F. Pilot. 2003. Developmental control of cell morphogenesis: a focus on membrane growth. Nat. Cell Biol. 5:103–108. [DOI] [PubMed] [Google Scholar]
- 4.Marsh, M., and H. T. McMahon. 1999. The structural era of endocytosis. Science. 285:215–220. [DOI] [PubMed] [Google Scholar]
- 5.Kirchhausen, T. 2000. Clathrin. Annu. Rev. Biochem. 69:699–727. [DOI] [PubMed] [Google Scholar]
- 6.McMahon, H. T., and I. G. Hills. 2004. COP and clathrin-coated vesicle budding: different pathways, common approaches. Curr. Opin. Cell Biol. 16:379–391. [DOI] [PubMed] [Google Scholar]
- 7.McMahon, H. T., and J. L. Gallop. 2005. Membrane curvature and mechanisms of dynamic cell membrane remodeling. Nature. 438:590–596. [DOI] [PubMed] [Google Scholar]
- 8.Itoh, T., K. S. Erdmann, A. Roux, B. Habermann, H. Werner, and P. D. Camilli. 2005. Dynamin and the actin cytoskeleton cooperatively regulate plasma membrane invagination by BAR and F-BAR proteins. Dev. Cell. 9:791–804. [DOI] [PubMed] [Google Scholar]
- 9.Cho, W., and R. V. Stahelin. 2005. Membrane-protein interactions in cell signaling and membrane trafficking. Annu. Rev. Biophys. Biomol. Struct. 296:153–161. [DOI] [PubMed] [Google Scholar]
- 10.Gurkan, C., S. M. Stagg, P. LaPointe, and W. E. Balch. 2006. The COPII cage: unifying principles of vesicle coat assembly. Nat. Rev. Mol. Cell Biol. 7:727–738. [DOI] [PubMed] [Google Scholar]
- 11.Römer, W., L. Berland, V. C. K. Gaus, B. Windschieg, D. Tenza, M. R. E. Aly, V. Fraisier, J.-C. Florent, D. Perrais, C. Lamaze, G. Raposo, C. Steinem, P. Sens, P. Bassereau, and L. Johannes. 2007. Shiga toxin induces tubular membrane invaginations for its uptake into cells. Nature. 450:670–675. [DOI] [PubMed] [Google Scholar]
- 12.Lemmon, M. A. 2008. Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 9:99–111. [DOI] [PubMed] [Google Scholar]
- 13.Chandler, D., J. Hsin, C. B. Harrison, J. Gumbart, and K. Schulten. 2008. Intrinsic curvature properties of photosynthetic proteins in chromatophores. Biophys. J. 95:2822–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Botelho, A. V., N. J. Gibson, R. L. Thurmond, Y. Wang, and M. F. Brown. 2002. Conformational energetics of rhodopsin modulated by nonlamellar-forming lipids. Biochemistry. 41:6354–6368. [DOI] [PubMed] [Google Scholar]
- 15.Huber, T., A. Botelho, K. Beyer, and M. Brown. 2004. Membrane model for the G-protein-coupled receptor rhodopsin: hydrophobic interface and dynamical structure. Biophys. J. 86:2078–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Botelho, A. V., T. Huber, T. P. Sakmar, and M. F. Brown. 2006. Curvature and hydrophobic forces drive constitutive association and modulate activity of rhodopsin in membranes. Biophys. J. 91:4464–4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Periole, X., T. Huber, S.-J. Marrink, and T. P. Sakmar. 2007. G protein-coupled receptors self-assemble in dynamics simulations of model bilayers. J. Am. Chem. Soc. 129:10126–10132. [DOI] [PubMed] [Google Scholar]
- 18.Zimmerberg, J., and S. McLaughlin. 2004. Membrane curvature: how BAR domains bend bilayers. Curr. Biol. 14:R250–R252. [DOI] [PubMed] [Google Scholar]
- 19.Ren, G., P. Vajjhala, J. S. Lee, B. Winsor, and A. L. Munn. 2006. The BAR domain proteins: molding membranes in fission, fusion, and phagy. Microbiol. Mol. Biol. Rev. 70:37–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takei, K., V. I. Slepnev, V. Haucke, and P. De Camilli. 1999. Functional partnership between amphiphysin and dynamin in clathrin-mediated endocytosis. Nat. Cell Biol. 1:33–39. [DOI] [PubMed] [Google Scholar]
- 21.Peter, B. J., H. M. Kent, I. G. Mills, Y. Vallis, P. J. G. Butler, P. R. Evans, and H. T. McMahon. 2004. BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science. 303:495–499. [DOI] [PubMed] [Google Scholar]
- 22.Weissenhorn, W. 2005. Crystal structure of the endophilin-A1 BAR domain. J. Mol. Biol. 351:653–661. [DOI] [PubMed] [Google Scholar]
- 23.Millard, T. H., G. Bompard, M. Y. Heung, T. R. Dafforn, D. J. Scott, L. M. Machesky, and K. T. Futterer. 2005. Structural basis of filopodia formation induced by the IRSp53/MIM homology domain of human IRSp53. EMBO J. 24:240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Masuda, M., S. Takeda, M. Sone, T. Ohki, H. Mori, Y. Kamioka, and N. Mochizuki. 2006. Endophilin BAR domain drives membrane curvature by two newly identified structure-based mechanisms. EMBO J. 25:2889–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gallop, J. L., C. C. Jao, H. M. Kent, P. J. Butler, P. R. Evans, R. Langen, and H. T. McMahon. 2006. Mechanism of endophilin N-BAR domain-mediated membrane curvature. EMBO J. 25:2898–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shimada, A., H. Niwa, K. Tsujita, S. Suetsugu, K. Nitta, K. Hanawa-Suetsugu, R. Akasaka, Y. Nishino, M. Toyama, L. Chen, Z. Liu, B. Wang, M. Yamamoto, T. Terada, A. Miyazawa, A. Tanaka, S. Sugano, M. Shirouzu, K. Nagayama, T. Takenawa, and S. Yokoyama. 2007. Curved EFC/F-BAR-domain dimers are joined end to end into a filament for membrane invagination in endocytosis. Cell. 129:761–772. [DOI] [PubMed] [Google Scholar]
- 27.Henne, W. M., H. M. Kent, M. G. J. Ford, B. G. Hegde, O. Daumke, P. J. G. Butler, R. Mittal, R. Langen, P. R. Evans, and H. T. McMahon. 2007. Structure and analysis of FCHo2 F-BAR domain: a dimerizing and membrane recruitment module that effects membrane curvature. Structure. 15:1–14. [DOI] [PubMed] [Google Scholar]
- 28.Mattila, P. K., A. Pykäläinen, J. Saarikangas, V. O. Paavilainen, H. Vihinen, E. Jokitalo, and P. Lappalainen. 2007. Missing-in-metastasis and IRSp53 deform PI(4,5)P2-rich membranes by an inverse BAR domain-like mechanism. J. Cell Biol. 176:953–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frost, A., R. Perera, A. Roux, K. Spasov, O. Destaing, E. H. Egelman, P. D. Camilli, and V. M. Unger. 2008. Structural basis of membrane invagination by F-BAR domains. Cell. 132:807–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blood, P. D., and G. A. Voth. 2006. Direct observation of Bin/amphiphysin/Rvs (BAR) domain-induced membrane curvature by means of molecular dynamics simulations. Proc. Natl. Acad. Sci. USA. 103:15068–15072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blood, P. D. 2007. Computer simulation studies of membrane remodeling by shear flow and N-BAR domain protein modules. PhD thesis. University of Utah, Department of Bioengineering, Salt Lake City, UT.
- 32.Leach, A. R. 2001. Molecular Modeling: Principles and Applications, 2nd Ed. Prentice-Hall, Upper Saddle River, NJ.
- 33.Karplus, M., and J. McCammon. 2002. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 265:652–654. [DOI] [PubMed] [Google Scholar]
- 34.Sanbonmatsu, K. Y., and C.-S. Tung. 2007. High performance computing in biology: multimillion atom simulations of nanoscale systems. J. Struct. Biol. 157:470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smit, B., P. A. J. Hilbers, K. Esselink, L. A. M. Rupert, N. M. van Os, and A. G. Schlijper. 1990. Computer simulations of a water/oil interface in the presence of micelles. Nature. 348:624. [Google Scholar]
- 36.Shelley, J. C., M. Y. Shelley, R. C. Reeder, S. Bandyopadhyay, and M. L. Klein. 2001. A coarse grain model for phospholipid simulations. J. Phys. Chem. B. 105:4464–4470. [Google Scholar]
- 37.Marrink, S. J., A. H. de Vries, and A. E. Mark. 2004. Coarse grained model for semiquantitative lipid simulations. J. Phys. Chem. B. 108:750–760. [Google Scholar]
- 38.Stevens, M. J. 2004. Coarse-grained simulations of lipid bilayers. J. Chem. Phys. 121:11942–11948. [DOI] [PubMed] [Google Scholar]
- 39.Ayton, G. S., and G. A. Voth. 2004. Mesoscopic lateral diffusion in lipid bilayers. Biophys. J. 87:3299–3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marrink, S. J., J. Risselada, and A. E. Mark. 2005. Simulation of gel phase formation and melting in lipid bilayers using a coarse grained model. Chem. Phys. Lipids. 135:223–244. [DOI] [PubMed] [Google Scholar]
- 41.Izvekov, S., and G. A. Voth. 2005. Multiscale coarse graining of liquid-state systems. J. Chem. Phys. 123:134105. [DOI] [PubMed] [Google Scholar]
- 42.Shillcock, J. C., and R. Lipowsky. 2005. Tension-induced fusion of bilayer membranes and vesicles. Nat. Mater. 4:225–228. [DOI] [PubMed] [Google Scholar]
- 43.Shi, Q., S. Izvekov, and G. A. Voth. 2006. Mixed atomistic and coarse-grained molecular dynamics: simulation of a membrane-bound ion channel. J. Phys. Chem. B. 110:15045–15048. [DOI] [PubMed] [Google Scholar]
- 44.Kasson, P. M., N. W. Kelley, N. Singha, M. Vrljic, A. T. Brunger, and V. S. Pande. 2006. Ensemble molecular dynamics yields submillisecond kinetics and intermediates of membrane fusion. Proc. Natl. Acad. Sci. USA. 103:11916–11921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harmandaris, V. A., and M. Deserno. 2006. A novel method for measuring the bending rigidity of model lipid membranes by simulating tethers. J. Chem. Phys. 125:204905. [DOI] [PubMed] [Google Scholar]
- 46.Marrink, S. J., H. J. Risselada, S. Yefimov, D. P. Tieleman, and A. H. de Vries. 2007. The Martini forcefield: coarse grained model for biomolecular simulations. J. Phys. Chem. B. 111:7812–7824. [DOI] [PubMed] [Google Scholar]
- 47.Marrink, S. J., A. H. de Vries, T. A. Harroun, J. Katsaras, and S. R. Wassall. 2008. Cholesterol shows preference for the interior of polyunsaturated lipid membranes. J. Am. Chem. Soc. 130:10–11. [DOI] [PubMed] [Google Scholar]
- 48.Tama, F., O. Miyashita, and C. L. Brooks III. 2004. Flexible multi-scale fitting of atomic structures into low-resolution electron density maps with elastic network normal mode analysis. J. Mol. Biol. 337:985–999. [DOI] [PubMed] [Google Scholar]
- 49.Tozzini, V. 2005. Coarse-grained models for proteins. Curr. Opin. Struct. Biol. 15:144–150. [DOI] [PubMed] [Google Scholar]
- 50.Chu, J.-W., and G. A. Voth. 2005. Allostery of actin filaments: molecular dynamics simulations and coarse-grained analysis. Proc. Natl. Acad. Sci. USA. 102:13111–13116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shih, A. Y., A. Arkhipov, P. L. Freddolino, and K. Schulten. 2006. Coarse grained protein-lipid model with application to lipoprotein particles. J. Phys. Chem. B. 110:3674–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arkhipov, A., P. L. Freddolino, and K. Schulten. 2006. Stability and dynamics of virus capsids described by coarse-grained modeling. Structure. 14:1767–1777. [DOI] [PubMed] [Google Scholar]
- 53.Arkhipov, A., P. L. Freddolino, K. Imada, K. Namba, and K. Schulten. 2006. Coarse-grained molecular dynamics simulations of a rotating bacterial flagellum. Biophys. J. 91:4589–4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bond, P. J., and M. S. P. Sansom. 2006. Insertion and assembly of membrane proteins via simulation. J. Am. Chem. Soc. 128:2697–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bond, P. J., and M. Sansom. 2007. Bilayer deformation by the Kv channel voltage sensor domain revealed by self-assembly simulations. Proc. Natl. Acad. Sci. USA. 104:2631–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bond, P. J., J. Holyoake, A. Ivetac, S. Khalid, and M. Sansom. 2007. Coarse-grained molecular dynamics simulations of membrane proteins and peptides. J. Struct. Biol. 157:593–605. [DOI] [PubMed] [Google Scholar]
- 57.Shih, A. Y., P. L. Freddolino, A. Arkhipov, and K. Schulten. 2007. Assembly of lipoprotein particles revealed by coarse-grained molecular dynamics simulations. J. Struct. Biol. 157:579–592. [DOI] [PubMed] [Google Scholar]
- 58.Shih, A. Y., A. Arkhipov, P. L. Freddolino, S. G. Sligar, and K. Schulten. 2007. Assembly of lipids and proteins into lipoprotein particles. J. Phys. Chem. B. 111:11095–11104. [DOI] [PubMed] [Google Scholar]
- 59.Shih, A. Y., P. L. Freddolino, S. G. Sligar, and K. Schulten. 2007. Disassembly of nanodiscs with cholate. Nano Lett. 7:1692–1696. [DOI] [PubMed] [Google Scholar]
- 60.Reynwar, B. J., G. Illya, V. A. Harmandaris, M. M. Müller, K. Kremer, and M. Deserno. 2007. Aggregation and vesiculation of membrane proteins by curvature-mediated interactions. Nature. 447:461–464. [DOI] [PubMed] [Google Scholar]
- 61.Mirijanian, D. T., J. W. Chu, G. S. Ayton, and G. A. Voth. 2007. Atomistic and coarse-grained analysis of double spectrin repeat units: the molecular origins of flexibility. J. Mol. Biol. 365:523–534. [DOI] [PubMed] [Google Scholar]
- 62.Zhou, J., I. F. Thorpe, S. Izvekov, and G. A. Voth. 2007. Coarse-grained peptide modeling using a systematic multiscale approach. Biophys. J. 92:4289–4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rueda, M., P. Chacon, and M. Orozco. 2007. Thorough validation of protein normal modes analysis: a comparative study with essential dynamics. Structure. 15:565–575. [DOI] [PubMed] [Google Scholar]
- 64.Helfrich, W. 1973. Elastic properties of lipid bilayers: theory and possible experiments. Z. Naturforsch. 28:693–703. [DOI] [PubMed] [Google Scholar]
- 65.Kozlovsky, Y., L. V. Chernomordik, and M. M. Kozlov. 2002. Lipid intermediates in membrane fusion: formation, structure, and decay of hemifusion diaphragm. Biophys. J. 83:2634–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chernomordik, L. V., and M. M. Kozlov. 2003. Protein-lipid interplay in fusion and fission of biological membranes. Annu. Rev. Biochem. 72:175–207. [DOI] [PubMed] [Google Scholar]
- 67.Katsov, K., M. Müller, and M. Schick. 2004. Field theoretic study of bilayer membrane fusion. I. Hemifusion mechanism. Biophys. J. 87:3277–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Katsov, K., M. Müller, and M. Schick. 2006. Field theoretic study of bilayer membrane fusion. II. Mechanism of a stalk-hole complex. Biophys. J. 90:915–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ayton, G. S., P. D. Blood, and G. A. Voth. 2007. Membrane remodeling from N-BAR domain interactions: insights from multi-scale simulation. Biophys. J. 92:3595–3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.MacKerell, A., Jr., D. Bashford, M. Bellott, R. L. Dunbrack, Jr., J. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, I. W. E. Reiher, B. Roux, M. Schlenkrich, J. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, and M. Karplus. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- 71.Phillips, J. C., R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale, and K. Schulten. 2005. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD—visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- 73.Feller, S. E., D. Yin, R. W. Pastor, and A. D. MacKerell, Jr. 1997. Molecular dynamics simulation of unsaturated lipids at low hydration: parameterization and comparison with diffraction studies. Biophys. J. 73:2269–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 75.Feller, S. E. 2000. Molecular dynamics simulations of lipid bilayers. Curr. Opin. Colloid Interface Sci. 5:217–223. [Google Scholar]
- 76.Ritter, H., T. Martinetz, and K. Schulten. 1992. Neural Computation and Self-Organizing Maps: An Introduction, revised English edition. Addison-Wesley, New York.
- 77.Fasman, G. D., and H. A. Sober. 1976. Handbook of Biochemistry and Molecular Biology. CRC Press, New York.
- 78.Liu, M., P. Li, and J. C. Giddings. 1993. Rapid protein separation and diffusion coefficient measurement by frit inlet flow field-flow fractionation. Protein Sci. 2:1520–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lyubartsev, A. P., and A. Laaksonen. 1995. Calculation of effective interaction potentials from radial distribution functions: a reverse Monte Carlo approach. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Topics. 52:3730–3737. [DOI] [PubMed] [Google Scholar]
- 80.Faller, R., and D. Reith. 2003. Properties of poly(isoprene): model building in the melt and in solution. Macromolecules. 36:5406–5414. [Google Scholar]
- 81.Reith, D., M. Pütz, and F. Müller-Plathe. 2003. Deriving effective mesoscale potentials from atomistic simulations. J. Comput. Chem. 24:1624–1636. [DOI] [PubMed] [Google Scholar]
- 82.Evans, E., and W. Rawicz. 1990. Entropy-driven tension and bending elasticity in condensed-fluid membranes. Phys. Rev. Lett. 64:2094–2097. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.