

Abstract
Solubility plays a major role in protein purification, and has serious implications in many diseases. We studied the effects of pH and mutations on protein solubility by calculating the transfer free energy from the condensed phase to the solution phase. The condensed phase was modeled as an implicit solvent, with a dielectric constant lower than that of water. To account for the effects of pH, the protonation states of titratable side chains were sampled by running constant-pH molecular dynamics simulations. Conformations were then selected for calculations of the electrostatic solvation energy: once for the condensed phase, and once for the solution phase. The average transfer free energy from the condensed phase to the solution phase was found to predict reasonably well the variations in solubility of ribonuclease Sa and insulin with pH. This treatment of electrostatic contributions combined with a similar approach for nonelectrostatic contributions led to a quantitative rationalization of the effects of point mutations on the solubility of ribonuclease Sa. This study provides valuable insights into the physical basis of protein solubility.
INTRODUCTION
The ability of a protein to dissolve in aqueous solutions is an important property. This ability is measured by the solubility, i.e., the equilibrium concentration of the protein in the solution phase when a saturation amount of the protein is present. Most biochemical experiments rely on this ability, such as protein expression and purification, high-resolution structural determination, and quantitative binding assays. Low solubility is found to enhance amyloid fibril formation (1), and is expected to play a major role in diseases associated with protein aggregation. A number of techniques were proposed to increase protein solubility. These include time-consuming screening strategies for optimal solvent conditions (2,3), coexpression with molecular chaperones (4), attachment of a small protein (fusion tag) (5,6) or peptide (poly-Lys or poly-Arg) (7) as a solubility enhancement tag, and rational mutations of surface-exposed hydrophobic residues to polar residues (8–10), as reviewed by Trevino et al. (11). In addition, charge mutations have been introduced to manipulate the isoelectric point (pI), which is an important determinant of protein solubility (12–15).
Although experimental techniques have led to improvements in protein solubility, it is highly desirable to develop theoretical methods for predicting protein relative and absolute solubility. Such methods may be used directly to obtain desired levels of solubility for particular proteins, and may provide the necessary guidance for refining experimental techniques to achieve optimal solvent conditions for protein solubility. Theoretical models were developed to predict aqueous solubility for drug and druglike compounds (16–19), but developments for proteins are still very limited. Of great interest are methods that would predict how protein solubility varies with solvent conditions. A first step in that direction was taken recently (20), and accounted for the effect of salt concentrations on protein solubility by considering electrostatic interactions of salt ions with the charges and the low-dielectric region of a protein in the solution phase, while neglecting the impact of salts in the condensed phase.
Here we continue our efforts at quantitative predictions of protein solubility. An important advance beyond the previous study (20) involves a specific account of the condensed phase. The chemical potential of a protein in the solution phase can be written in the form
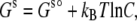 |
(1) |
where C is the protein concentration, kBT is the thermal energy, and Gs° is the chemical potential at a “standard” concentration (e.g., at C = 1 M). In the condensed phase, the individual protein molecules do not mix, so there is no concentration-dependent term as in Eq. 1, and the chemical potential will be denoted as Gc. The solubility, S, is the concentration at which Gs and Gc are equal. Hence
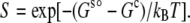 |
(2) |
The difference Gs° − Gc ≡ ΔGc→s can be viewed as the free energy of transfer from the condensed phase to the solution phase. The more favorable it is for a protein to transfer from the condensed phase to the solution phase, the lower the transfer free energy, and correspondingly the higher the solubility. We modeled the condensed phase as an implicit solvent, and decomposed the transfer free energy ΔGc→s into electrostatic and nonelectrostatic components:
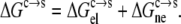 |
(3) |
The electrostatic component was obtained by solving the Poisson-Boltzmann (PB) equation (21–31), and the nonelectrostatic component was obtained based on the solvent-accessible surface area (22,28,32–35). Using this approach, we specifically investigated the effects of pH and point mutations on the solubility of ribonuclease Sa, motivated by the availability of experimental data by Pace et al. (10) and Shaw et al. (14). As an independent test of our approach, the pH dependence of the solubility of insulin (12,36,37) was also calculated. To the best of our knowledge, this work marks the first time that the effects of pH and mutations on protein solubility were calculated. Our treatment of the condensed phase may prove inspiring for developing models of other complex environments, such as the crowded milieu inside cells.
To gain insights into the implicit model of the condensed phase, we also calculated the transfer free energies, from octanol (an organic solvent) to water, of pentapeptides AcWL-X-LL, where X is any of the 20 amino acids. Experimental results for these transfer free energies are available from Wimley et al. (38) Our parallel studies of protein solubility and peptide transfer free energy clearly demonstrate that these two properties can be calculated from very similar approaches, and confirm the expectation that the implicit model for the condensed phase has a dielectric constant and nonelectrostatic solvation parameters intermediate between water and an organic solvent.
METHODS
Constant-pH molecular dynamics simulations of RNase Sa and insulin
Starting from the x-ray structure of RNase Sa given by Protein Data Bank entry 1rgg (chain A), missing hydrogen atoms were added by the LEAP program in the AMBER package. Constant-pH molecular dynamics simulations followed the protocol established by Mongan et al. (39), as implemented in the AMBER package (40). At each pH, 400 cycles of energy minimization preceded 1 ns of constant-pH equilibration. From the next 2 ns of simulation, 100 snapshots (separated by 20 ps) were taken as representative conformations of the protein at the given pH, to be used for calculating electrostatic solvation energies (described below). In the simulations, the generalized Born model for the implicit solvent (with the “igb” variable set to 2) was used, along with the modified parm99 force field (41). The simulation time step was 2 fs. The protonation states of titratable groups were assigned by a Monte Carlo procedure, with one titratable group at every 5 time steps. Nonbonded interactions were evaluated with a 30-Å cutoff. The monovalent salt concentration in the generalized Born model was 0.1 M, and the temperature was set to 300 K.
Corresponding to experimental conditions (14), independent molecular dynamics (MD) simulations at nine pH values were carried out to study the effects of pH on the solubility of RNase Sa. The pH values were 2.3, 2.9, 3.6, 4.0, 4.5, 4.8, 5.0, 5.2, and 5.4. In total, 13 residues were titrated (Fig. 1 A). These included 11 carboxyls (E14, D17, D25, D33, E41, E54, E74, E78, D79, D84, and D93) and two histidines (H53 and H85). The N-terminals and C-terminals and the side chain of the N-terminal residue (D1) were not titrated because of the lack of this option in the AMBER package.
FIGURE 1.

(A) Structure of RNase Sa. Eleven carboxyl side chains, two histidines, and residue T76 are labeled and displayed as red, cyan, and yellow sticks, respectively. The N-terminal residue happens to be an aspartate. The side chain of this residue was not titrated in the constant-pH simulations, and is not shown here. (B) Structure of zinc-insulin hexamer. The two trimers are shown in the foreground and background in green and gray, respectively. The two zinc ions and the coordinating histidines and water molecules are shown. In addition, the five titrated side chains of one monomer are labeled and shown as sticks.
In addition, a constant-pH simulation was performed at pH 4.25, to study the effects of point mutations at position 76 on the solubility of RNase Sa. On each of the 100 snapshots taken, residue T76 was replaced with the LEAP program by the 19 other types of amino acids. The new side chains were energy-minimized in a vacuum, whereas the rest of the protein was fixed. Similarly, a constant-pH simulation was performed at pH 7, and mutations of T76 to D, R, S, K, and A were created to compare against the experimental data (10) on these mutations.
Constant-pH simulations of zinc-insulin were performed in a similar fashion. A hexamer, which is the known oligomeric state of zinc-insulin in solution, was constructed by symmetric operations listed in Protein Data Bank entry 4ins. The hexamer consists of two trimers. With each trimer, a zinc ion is coordinated to H10 of chain B from the three monomers and one water molecule (Fig. 1 B). In each monomer, five residues were titrated: E4 and E17 of chain A, and H5, E13, and E21 of chain B. Simulations were performed at pH 4.0, 4.5, 5.0, 5.5, 5.75, 6.25, 6.75, and 7.0 up to 1.5 ns, with 100 snapshots from the last 0.5 ns saved for calculating electrostatic solvation energies.
MD simulations of pentapeptides
The 20 pentapeptides AcWL-X-LL were built with the LEAP program and energy-minimized before MD simulations in a vacuum. During the simulations, the attractive part of van der Waals interactions was turned off, to generate expanded conformations. Protonation states were fixed at those appropriate for pH 9 (with R and K protonated, and D, E, H, and the C-terminal unprotonated). No cutoff for nonbonded interactions was imposed. The temperature was 300 K. The simulation time step was 1 fs, and the total simulation time was 45 ns. One hundred conformations from the last 1 ns were uniformly sampled for later use.
From these 100 conformations of each pentapeptide appropriate for pH 9, corresponding conformations appropriate for pH 1 were generated by neutralizing the C-terminal, and when X = D, E, or H, adding a proton to the side chain of the guest residue. Neutralization of the C-terminal was modeled by reassigning partial charges of −0.4 to the two carboxyl O atoms, and modifying the charge of the carboxyl C atom to 0.5928 and those on the two Cδ atoms of the side chain of the C-terminal residue to −0.2163. Conformations of the groups undergoing charge reassignment upon pH change were optimized by 5000 cycles of energy minimization.
Calculation of electrostatic solvation energies
The electrostatic free energy of a solute molecule inside an implicit solvent can be separated into a Coulomb term and the solvation term:
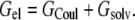 |
(4) |
The Coulomb term is given by
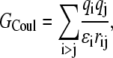 |
(5) |
where qi represents solute charges, rij represents intercharge distances, and ɛi represents the solute dielectric constant. We calculated electrostatic solvation energies by solving the PB equation; our default PB solver was the UHBD program (23). Unless otherwise noted, the values of electrostatic solvation energies are averages over 100 conformations, generated as described above. Following earlier work (25,29), the dielectric boundary between the solute low dielectric and the solvent high dielectric was set to the solute van der Waals surface. When a protein was in the solution phase or when a peptide was dissolved in water, the solvent dielectric constant was set to 78.5. For the condensed phase (or using octanol as solvent), a range of solvent dielectric constants centering around 55 (or 15) was tested. Each solvent dielectric constant was used in combination with a solute dielectric constant of 4. Note that for a fixed solute conformation, the Coulomb term is the same when the solute molecule is in either the solution or the condensed phase. Therefore, only the solvation term contributes to the transfer free energy ΔGc→s.
For modeling the effects of pH on the solubility of RNase Sa and insulin, the solution of the PB equation started with a coarse grid with dimensions of 100 × 100 × 100 and a spacing of 1.5 Å, followed by a fine grid with dimensions of 140 × 140 × 140 and a spacing of 0.5 Å, both centered at the center of the protein. For both proteins, the ionic strength was 0.1 M. For modeling the effects of point mutations at position 76 of RNase Sa, the dimensions of both the coarse and fine grids were increased to 200 × 200 × 200. We added a second fine grid centered on Cβ of residue 76, with dimensions of 140 × 140 × 140 and a spacing of 0.25 Å. The ionic strength was increased to 1.1 M. All calculations on the pentapeptides used a coarse grid with dimensions of 40 × 40 × 40 and a spacing of 1.5 Å, followed by a fine grid with dimensions of 80 × 80 × 80 and a spacing of 0.4 Å (both centered at the peptide center). The ionic strength was 0.06 M.
To gain further insights into the condensed phase, we modeled the condensed phase of RNase Sa as a crystalline array of monomers. In the crystal structure of RNase Sa, each unit cell contained eight monomers. We replicated the unit cell in three dimensions (Fig. 2 A). The electrostatic solvation free energy of a single monomer was calculated in the background of the crystalline replicas. The single monomer and all its replicas were assigned a dielectric constant of 4, and the rest of space was assigned the dielectric constant of water (i.e., 78.5). Because we were interested in the electrostatic solvation energy of a single monomer, only that monomer was charged (i.e., the charges on all the replicas were zeroed out; these replicas only served to modify the dielectric function in the environment outside the single monomer). Let this electrostatic solvation energy be 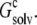 We then asked: if the inhomogeneous dielectric environment, consisting of the low-dielectric replicas embedded in the high-dielectric solvent, was mimicked by a uniform dielectric medium, what would the optimal dielectric constant be? We answered this question by reproducing the value of
We then asked: if the inhomogeneous dielectric environment, consisting of the low-dielectric replicas embedded in the high-dielectric solvent, was mimicked by a uniform dielectric medium, what would the optimal dielectric constant be? We answered this question by reproducing the value of 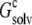 when the single monomer was placed in a uniform dielectric medium with dielectric constant ɛc (Fig. 2 B), and the value of ɛc was adjusted. In the calculation inside either the inhomogeneous dielectric environment or the uniform dielectric medium, the dielectric constant of the single monomer was maintained at 4.
when the single monomer was placed in a uniform dielectric medium with dielectric constant ɛc (Fig. 2 B), and the value of ɛc was adjusted. In the calculation inside either the inhomogeneous dielectric environment or the uniform dielectric medium, the dielectric constant of the single monomer was maintained at 4.
FIGURE 2.

(A) Crystalline phase of RNase Sa, and (B) its modeling as an effective dielectric medium. In the crystalline phase, a single RNase Sa monomer, shown in red, is surrounded by its crystalline replicas. The space between the replicas is filled by water. In the implicit model for the crystalline phase, the inhomogeneous environment of the red monomer is approximated by a uniform dielectric medium (depicted by the blue “cloud”), with a dielectric constant intermediate between a high value for water and a low value expected for protein molecules. The red monomer is the solute, which is always assigned a low dielectric constant (specifically, 4).
The value of 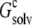 in principle involves an infinite number of replicas. We determined
in principle involves an infinite number of replicas. We determined 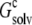 by including more and more replicas around the single monomer. The plateau value, after a sufficient number of replicas was included, was taken to be
by including more and more replicas around the single monomer. The plateau value, after a sufficient number of replicas was included, was taken to be 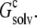 Because of the large system sizes involved, for these calculations we used a PB solver, the APBS program (26), which was designed to deal with large systems. The single monomer and all its replicas had a single conformation, taken to be the structure of RNase Sa at the start of constant-pH MD simulations.
Because of the large system sizes involved, for these calculations we used a PB solver, the APBS program (26), which was designed to deal with large systems. The single monomer and all its replicas had a single conformation, taken to be the structure of RNase Sa at the start of constant-pH MD simulations.
In all PB calculations, the ion exclusion radius was 2 Å, and the temperature was 300 K. Solute atoms were assigned Bondi radii (42) and AMBER94 partial charges (43), except for the charge modification on the C-terminal of pentapeptides at pH 1, as described above.
Nonpolar and polar surface areas of side chains
For modeling both the effects of point mutations on protein solubility and the variation of octanol-to-water transfer free energies among AcWL-X-LL peptides, the nonelectrostatic component was based on the areas of the nonpolar and polar portions of the solvent-accessible surface calculated on the side chain of a particular residue (the residue under mutation or the guest residue). Values for Anp and Ap in this study were taken directly from Wimley et al. (38) (as listed under the heading “ASA of X in AcWL-X-LL (Å2)” in their Table 1).
RESULTS
pH dependence of RNase Sa solubility
The pH affects the protonation/deprotonation of titratable groups in a protein. We sampled the protonation states by running constant-pH MD simulations. At each pH, conformations with different protonation states were selected, and the electrostatic component, 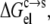 of the transfer free energy from the condensed phase to the solution phase was calculated as the average over the selected conformations. The pH dependence of protein solubility was then predicted from
of the transfer free energy from the condensed phase to the solution phase was calculated as the average over the selected conformations. The pH dependence of protein solubility was then predicted from
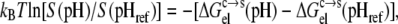 |
(6) |
where S(pH) and 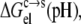 respectively, are the solubility and electrostatic component of ΔGc→s at a given pH, and pHref is a reference pH. Equation 6 is derived from Eq. 2 by assuming that the nonelectrostatic component of
respectively, are the solubility and electrostatic component of ΔGc→s at a given pH, and pHref is a reference pH. Equation 6 is derived from Eq. 2 by assuming that the nonelectrostatic component of 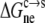 is not affected by pH. The electrostatic free energy of the protein in either phase consists of the Coulomb term and the solvation term (see Eq. 4). In our calculations, each conformation with a particular set of protonation states was used for both the solution phase and the condensed phase. Therefore, the Coulomb contributions of each conformation in the two phases were identical, and only the difference in solvation energy contributed to
is not affected by pH. The electrostatic free energy of the protein in either phase consists of the Coulomb term and the solvation term (see Eq. 4). In our calculations, each conformation with a particular set of protonation states was used for both the solution phase and the condensed phase. Therefore, the Coulomb contributions of each conformation in the two phases were identical, and only the difference in solvation energy contributed to 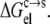
Generally, the electrostatic solvation energy of a protein in the solution phase increases in magnitude with the increasing magnitude of the net charge on the protein. A similar trend is expected in the condensed phase, except that the magnitude of the electrostatic solvation energy is reduced relative to its counterpart in the solution phase (under the assumption that the solvent dielectric constant in the condensed phase is lower than in the solution phase). Taken together, 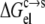 is expected to increase in magnitude with an increasing magnitude of the net charge on the protein. The net charge can be changed by varying the pH. At the pI, the net charge on the protein is zero. As pH moves away from the pI in either direction, the net charge increases in magnitude. Therefore, the pH dependence of
is expected to increase in magnitude with an increasing magnitude of the net charge on the protein. The net charge can be changed by varying the pH. At the pI, the net charge on the protein is zero. As pH moves away from the pI in either direction, the net charge increases in magnitude. Therefore, the pH dependence of 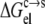 is expected to have a bell shape, with the maximum occurring around the pI. Correspondingly (see Eq. 6), the pH dependence of lnS should follow an inverted bell shape, as is usually observed (1,12,14,36,37).
is expected to have a bell shape, with the maximum occurring around the pI. Correspondingly (see Eq. 6), the pH dependence of lnS should follow an inverted bell shape, as is usually observed (1,12,14,36,37).
The RNase Sa has a total of 12 carboxyl side chains and two histidines (Fig. 1 A). Its isoelectric point is 3.5 (14). Our results for the pH dependence of 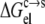 obtained in RNase Sa conform to the expected bell shape. By allowing the carboxyl side chains and histidines to titrate in the constant-pH MD simulations, the solubility data of Shaw et al. (14) in the pH range of 2.3–5.4 can be quantitatively rationalized by our calculation, when the solvent dielectric constant of the condensed phase is assigned to 55. That value is to be compared with the value of 78.5 for the solution phase. (Note that, in calculating the electrostatic solvation energy in either the condensed phase or the solution phase, the dielectric constant of the protein solute was kept at 4.) As Fig. 3 shows, the overall experimental pH dependence of lnS is well-reproduced by our calculation. Our calculation underestimated lnS at the lowest pH value (2.3), probably because we did not allow for the side chain of the N-terminal residue (which happens to be an aspartate) and the C-terminal carboxyl to titrate because of a limitation in the AMBER package that we used for constant-pH MD simulations.
obtained in RNase Sa conform to the expected bell shape. By allowing the carboxyl side chains and histidines to titrate in the constant-pH MD simulations, the solubility data of Shaw et al. (14) in the pH range of 2.3–5.4 can be quantitatively rationalized by our calculation, when the solvent dielectric constant of the condensed phase is assigned to 55. That value is to be compared with the value of 78.5 for the solution phase. (Note that, in calculating the electrostatic solvation energy in either the condensed phase or the solution phase, the dielectric constant of the protein solute was kept at 4.) As Fig. 3 shows, the overall experimental pH dependence of lnS is well-reproduced by our calculation. Our calculation underestimated lnS at the lowest pH value (2.3), probably because we did not allow for the side chain of the N-terminal residue (which happens to be an aspartate) and the C-terminal carboxyl to titrate because of a limitation in the AMBER package that we used for constant-pH MD simulations.
FIGURE 3.

Comparison of calculated and experimental results for the pH dependence of the solubility of RNase Sa.
We also performed a similar calculation with a solvent dielectric constant of 60 assigned to the condensed phase. The results did not differ significantly from those shown in Fig. 3 (with the root mean-square deviation between the calculation and the experiment changing from 0.18 to 0.22 kcal/mol). Our calculations thus suggest that the dielectric property of the condensed phase can be modeled with a solvent dielectric constant in the range of 55–60.
pH dependence of insulin solubility
In addition to the five titratable groups shown in Fig. 1 B, each monomer contains one arginine and one lysine (R22 and K29, both on chain B), and shares one third of a bound zinc ion. The pI of insulin would be expected to occur at a pH where H5 is still protonated, but the four glutamates have nearly completed deprotonation. The pI of insulin in our modeled structure is thus expected to be ∼6, which is just the result obtained from the constant-pH MD simulations. Experimentally, the pI of zinc-insulin is determined between 5–5.5 (12,36,37). We attribute the discrepancy of ∼0.75 in pI to anion binding, which is known to occur (36). We did not model anion binding explicitly, but to account crudely for its effect, we moved the nominal pH downward by 0.75 units when comparing our calculated pH dependence of insulin solubility against experimental data.
Fig. 4 shows this comparison. Below pH 5, our calculations are in good agreement with the results from three experimental studies (12,36,37). Above pH 5, the experimental results themselves differ significantly. Our calculations exhibit an increase in solubility with increasing pH, as seen in the experimental studies. In the calculations, the solvent dielectric constant of the condensed phase was again set to 55.
FIGURE 4.

Comparison of calculated and experimental results for the pH dependence of the solubility of insulin. The pH values of the calculated results were shifted downward by 0.75 units to account for anion binding. The experimental results of Desbuquois and Aurbach (37) and Fischel-Ghodsian et al. (12) were for porcine insulin, whereas those of Fredericq and Neurath (36) were for bovine insulin. The two species differ at only two positions, involving uncharged residues, in sequence. Our calculations used the structure of porcine insulin.
Effective dielectric constant of the condensed phase
In solubility measurements, the condensed phase is either crystalline or amorphous (11). In a strict thermodynamic sense, solubility is defined with respect to a crystalline condensed phase. (The solubility measurements of Trevino et al. (10) for RNase Sa were performed in the presence of an amorphous condensed phase that presumably features irregularly arranged RNase Sa monomers instead of a regular array, as found in a crystalline phase. See Trevino et al. (11) for a discussion of the subtle differences between amorphous and crystalline condensed phases.) We generated the crystalline phase of RNase Sa by replicating a single monomer to a crystalline array (Fig. 2 A). We then calculated the electrostatic solvation energy, 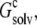 of the single monomer in the background of the crystalline replicas. In this calculation, the single monomer and all its replicas were assigned the low dielectric constant of 4, and the space between replicas, which should be filled with water, was assigned the high dielectric constant of water (i.e., 78.5). Fig. 5 A shows that
of the single monomer in the background of the crystalline replicas. In this calculation, the single monomer and all its replicas were assigned the low dielectric constant of 4, and the space between replicas, which should be filled with water, was assigned the high dielectric constant of water (i.e., 78.5). Fig. 5 A shows that  reaches its plateau value when ∼100 replicas around the single monomer are included. The plateau value can be taken as the value of
reaches its plateau value when ∼100 replicas around the single monomer are included. The plateau value can be taken as the value of 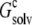 for a single monomer in an infinite crystalline array.
for a single monomer in an infinite crystalline array.
FIGURE 5.

(A) Electrostatic solvation energy, 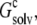 of a single RNase Sa monomer in the presence of a crystalline array of replicas. The replicas serve to change the dielectric environment of the single monomer. The infinite array is approached by including more and more distant replicas (n = total number of replicas included). The dashed curve through the data points provides guidance for the eye. (B) Reproduction of
of a single RNase Sa monomer in the presence of a crystalline array of replicas. The replicas serve to change the dielectric environment of the single monomer. The infinite array is approached by including more and more distant replicas (n = total number of replicas included). The dashed curve through the data points provides guidance for the eye. (B) Reproduction of 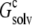 when the inhomogeneous dielectric environment is replaced by a uniform dielectric medium. The effective dielectric constant, ɛc, of the uniform dielectric medium is varied to match
when the inhomogeneous dielectric environment is replaced by a uniform dielectric medium. The effective dielectric constant, ɛc, of the uniform dielectric medium is varied to match 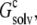 as indicated by the horizontal line.
as indicated by the horizontal line.
The infinite array of crystalline replicas embedded in water presents an inhomogeneous dielectric environment for the single monomer. In our model of the condensed phase, this inhomogeneous environment was replaced by a uniform dielectric medium, with an effective dielectric constant ɛc. The value of ɛc can be directly determined by matching the electrostatic solvation energy for a single monomer inside this uniform dielectric medium with the corresponding value, 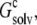 as determined above in the crystalline array. Fig. 5 B shows that
as determined above in the crystalline array. Fig. 5 B shows that 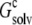 is reproduced with ɛc ∼ 53.5. This result demonstrates that the solvent dielectric constant of 55, as used above for calculating the pH dependence of protein solubility, is not merely a result of fitting experimental data, but has a solid physical basis.
is reproduced with ɛc ∼ 53.5. This result demonstrates that the solvent dielectric constant of 55, as used above for calculating the pH dependence of protein solubility, is not merely a result of fitting experimental data, but has a solid physical basis.
Effects of mutating residue T76
Trevino et al. (10) measured the changes in solubility of RNase Sa at pH 4.25 when residue T76 was mutated into the 19 other types of amino acids. We modeled the point mutations by accounting for their effects on both the electrostatic and nonelectrostatic components of the transfer free energy ΔGc→s. The mutational effect, 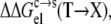 on the electrostatic component was obtained by calculating twice the change in electrostatic solvation energy from the condensed phase to the solution phase (once for the wild-type protein, and once for the mutant), and then taking the difference. The mutational effect
on the electrostatic component was obtained by calculating twice the change in electrostatic solvation energy from the condensed phase to the solution phase (once for the wild-type protein, and once for the mutant), and then taking the difference. The mutational effect 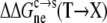 on the nonelectrostatic component was assumed to have the form
on the nonelectrostatic component was assumed to have the form
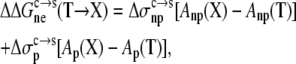 |
(7) |
where Anp(X) and Ap(X) are the areas of the nonpolar and polar portions, respectively, of the solvent-accessible surface calculated over the side chain of residue X, and 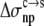 and
and 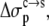 respectively, are nonpolar and polar solvation parameters for the transfer of a solute from the condensed phase to the solution phase. From
respectively, are nonpolar and polar solvation parameters for the transfer of a solute from the condensed phase to the solution phase. From 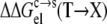 and
and 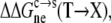 we predicted the relative solubility, S(X)/S(T), of the mutant in reference to the wild-type protein from
we predicted the relative solubility, S(X)/S(T), of the mutant in reference to the wild-type protein from
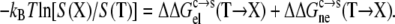 |
(8) |
Strong support for our approach to modeling mutational effects is given by simply examining the ordering of the solubility levels of the 20 RNase Sa variants, each with a different amino acid occupying position 76. As Fig. 6 shows, the solubility levels are high when position 76 is occupied by charged amino acids (D, E, R, and K), moderate when occupied by polar amino acids (H, N, Q, and T), and low when occupied by nonpolar amino acids (M, V, L, I, Y, F, and W). Initially, we performed a multilinear regression of the experimental data for −kBTln[S(X)/S(T)] against 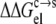 (T→X), Anp(X) – Anp(T), and Ap(X) – Ap(T). The correlation R2 of this analysis is 0.83. The coefficient of the
(T→X), Anp(X) – Anp(T), and Ap(X) – Ap(T). The correlation R2 of this analysis is 0.83. The coefficient of the 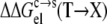 term was found to be very close to 1, without further adjusting the value (at 55) of the dielectric constant of the implicit solvent modeling the condensed phase. This finding offers solid validation for our treatment of the condensed phase as an effective dielectric medium in calculating the electrostatic component of transfer free energy.
term was found to be very close to 1, without further adjusting the value (at 55) of the dielectric constant of the implicit solvent modeling the condensed phase. This finding offers solid validation for our treatment of the condensed phase as an effective dielectric medium in calculating the electrostatic component of transfer free energy.
FIGURE 6.

Comparison of calculated and experimental results for the variation of solubility among 20 RNase Sa variants with point mutations at position 76. The calculated results for T76 are shown in lighter shades to indicate the wild-type protein.
By adjusting 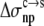 and
and 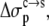 Eqs. 7 and 8 were found to reproduce well the experimental data for the variation of the solubility of RNase Sa when T76 is replaced by the 19 other types of amino acids. As Fig. 6 shows, the results of the calculation correctly rank the D and E variants as having the highest solubility levels, and the F and W variants as having the lowest solubility levels. In addition, variants with measured solubility levels either higher or lower than the T variant are all correctly ranked as such by the calculation. Trevino et al. (10) observed that there is a good correlation between solubility and hydrophobicity for some of the variants with nonpolar residues occupying position 76. By combining the electrostatic component with the nonelectrostatic component, as modeled based on polar and nonpolar surface areas, we show here that the variation in solubility among all 20 RNase Sa variants can be qualitatively rationalized. The optimized values of the adjustable parameters
Eqs. 7 and 8 were found to reproduce well the experimental data for the variation of the solubility of RNase Sa when T76 is replaced by the 19 other types of amino acids. As Fig. 6 shows, the results of the calculation correctly rank the D and E variants as having the highest solubility levels, and the F and W variants as having the lowest solubility levels. In addition, variants with measured solubility levels either higher or lower than the T variant are all correctly ranked as such by the calculation. Trevino et al. (10) observed that there is a good correlation between solubility and hydrophobicity for some of the variants with nonpolar residues occupying position 76. By combining the electrostatic component with the nonelectrostatic component, as modeled based on polar and nonpolar surface areas, we show here that the variation in solubility among all 20 RNase Sa variants can be qualitatively rationalized. The optimized values of the adjustable parameters 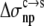 and
and 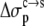 were 8.6 and 1.1 cal/mol/Å2, respectively.
were 8.6 and 1.1 cal/mol/Å2, respectively.
Trevino et al. (10) also measured the changes in solubility of RNase Sa at pH 7 when residue T76 was mutated into the five other types of amino acids: D, S, A, K, and R. We used this set of data as an important check for our approach. By repeating the calculations for mutational effects at pH 7 without further adjustment of any parameters, the solubility levels of the D, S, A, K, and R mutants were calculated to be 196, 61, 45, 23, and 28 g/L (on the basis of a solubility level of 41 g/L for the wild-type protein). These values can be compared with the experimental results of 160, 130, 92, 60, and 50 g/L. Our calculations correctly predict that the D and S mutants are more soluble than the A mutant, whereas the K and R mutants are less soluble.
Octanol-to-water transfer free energies of pentapeptides
We calculated the octanol-to-water transfer free energies, ΔGo→w, of the pentapeptides AcWL-X-LL in nearly the same way that we modeled the effects of point mutations on the solubility of RNase Sa. Specifically, ΔGo→w was assumed to consist of an electrostatic component 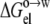 and a nonelectrostatic component
and a nonelectrostatic component 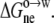 We obtained
We obtained 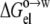 as the difference in electrostatic solvation energy between two implicit solvents: one modeling water with a dielectric constant of 78.5, and one modeling octanol with an adjustable dielectric constant (see below). The variation of
as the difference in electrostatic solvation energy between two implicit solvents: one modeling water with a dielectric constant of 78.5, and one modeling octanol with an adjustable dielectric constant (see below). The variation of 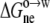 among the 20 pentapeptides was based on the nonpolar and polar portions of the solvent-accessible surface areas, calculated over the side chain of guest residue X:
among the 20 pentapeptides was based on the nonpolar and polar portions of the solvent-accessible surface areas, calculated over the side chain of guest residue X:
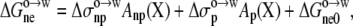 |
(9) |
where 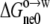 represents the contribution of the common moiety of the 20 pentapeptides to the nonelectrostatic component of the transfer free energy.
represents the contribution of the common moiety of the 20 pentapeptides to the nonelectrostatic component of the transfer free energy.
The dielectric constant of the octanol solvent was adjusted so that a multilinear regression of the experimental data of Wimley et al. (38) for ΔGo→w (at pH 9) against 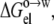 Anp(X), and Ap(X) resulted in a unity coefficient for the
Anp(X), and Ap(X) resulted in a unity coefficient for the 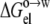 term. The unit coefficient was achieved when a dielectric constant of 15 was assigned to the octanol solvent, with the regression analysis reaching a correlation R2 of 0.91. Wimley et al. (38) performed regression analysis against Anp(X) and Ap(X) by restricting X to amino acids with nonpolar side chains. Our analysis, incorporating the electrostatic component, accounts for the variation of ΔGo→w among all 20 types of amino acids (Fig. 7). The resulting values of the nonpolar and polar solvation parameters
term. The unit coefficient was achieved when a dielectric constant of 15 was assigned to the octanol solvent, with the regression analysis reaching a correlation R2 of 0.91. Wimley et al. (38) performed regression analysis against Anp(X) and Ap(X) by restricting X to amino acids with nonpolar side chains. Our analysis, incorporating the electrostatic component, accounts for the variation of ΔGo→w among all 20 types of amino acids (Fig. 7). The resulting values of the nonpolar and polar solvation parameters 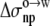 and
and 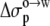 for the octanol-to-water transfer are 18.1 and 13.5 cal/mol/Å2, respectively.
for the octanol-to-water transfer are 18.1 and 13.5 cal/mol/Å2, respectively.
FIGURE 7.

Comparison of calculated and experimental results for the octanol-to-water transfer free energies of pentapeptides AcWL-X-LL. The identity of the guest residue X is shown on the horizontal axis.
Wimley et al. (38) also obtained octanol-to-water transfer free energies for nine of the 20 pentapeptides (with guest residues D, E, A, G, S, T, H, K, and R) at pH 1. We modeled the decrease from pH 9 to pH 1 by recalculating the electrostatic component 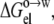 this time with all carboxyl groups and the histidine side chain protonated. The experimental data for the change in octanol-to-water transfer free energy from pH 9 to pH 1 were reasonably well-reproduced by
this time with all carboxyl groups and the histidine side chain protonated. The experimental data for the change in octanol-to-water transfer free energy from pH 9 to pH 1 were reasonably well-reproduced by 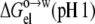 −
− 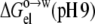 + 3 kcal/mol. It is not clear why a constant offset of 3 kcal/mol was required to fit the experimental data. Possible reasons include potential differences in experimental conditions at the two pH values, and neglect of pH-dependent conformational changes in our calculations.
+ 3 kcal/mol. It is not clear why a constant offset of 3 kcal/mol was required to fit the experimental data. Possible reasons include potential differences in experimental conditions at the two pH values, and neglect of pH-dependent conformational changes in our calculations.
DISCUSSION
We present a computational approach for predicting relative solubility. The power of this approach was demonstrated by quantitative rationalization of experimental results for the effects of pH and point mutations on protein solubility. The basic tenet of the approach involves the modeling of the condensed phase as an implicit solvent. As a result, the calculation of solubility becomes equivalent to the calculation of transfer free energy. The treatment of pH effects presented here, along with a previous computational study of salt effects (20), shows that solvent conditions can be quantitatively modeled. Physics-based computations thus open a new avenue for determining the dependence of protein solubility on solvent conditions, and for investigating optimal protein crystallization conditions. Likewise, our modeling of mutational effects on protein solubility demonstrates the ability to predict the contributions of individual residues to protein solubility. This ability has a potentially significant impact on the selection of protein targets in structural genomics projects, and on the design of therapies for diseases associated with protein aggregation.
Our calculation of protein solubility was performed in parallel with a calculation of octanol-to-water transfer free energies of peptides. This parallel study underscores the basic tenet of our approach, which involves the modeling of the condensed phase as an implicit solvent. Our results for protein solubility confirm that the condensed phase is amenable to such modeling. Transfer free energies of small solutes are widely used for parameterizing solvation models. Such data are difficult to obtain for proteins but are highly desirable, because there is evidence that the parameterization of solvation models may be sensitive to solute size (44). Our study suggests that protein solubility data can serve the role of transfer free energies for parameterizing solvation models for proteins.
Our expectation for the condensed phase as an implicit solvent is that its properties are intermediate between those of water and organic solvents. This expectation is matched by the values of the dielectric constant and nonelectrostatic solvation parameters required to reproduce experimental data on protein solubility. A calculation of the electrostatic solvation energy of RNase Sa in the crystalline condensed phase specifically confirms the value ∼55 for the dielectric constant when the condensed phase is modeled as a uniform dielectric medium. Although that calculation was for RNase Sa, a solvent dielectric constant of 55 for the condensed phase is 70% of the value for water. This percentage is close to the water content in many protein crystals. The nonelectrostatic solvation parameters, 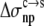 and
and 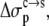 also have values (8.6 and 1.1 cal/mol/Å2, respectively) lower than those for the transfer from octanol to water (18.1 and 13.5 cal/mol/Å2, respectively).
also have values (8.6 and 1.1 cal/mol/Å2, respectively) lower than those for the transfer from octanol to water (18.1 and 13.5 cal/mol/Å2, respectively).
A number of details in our treatment of electrostatic and nonelectrostatic contributions can be improved. For example, in our electrostatic calculations, the solute charges were fixed upon transferring from one phase to another, or from one solvent to another. In reality, electronic polarization of the solute can be different in different solvent environments. A neglect of solute electronic polarization may partly explain why a dielectric constant of 15 was required to model the octanol solvent, which is known to have a dielectric constant of 10.3. (In this context, the interpretation of the value of 55 for the dielectric constant of the condensed phase is also subject to the error caused by the neglect of solute electronic polarization.) Likewise, our treatment of the nonelectrostatic component relied on the use of nonpolar and polar accessible surface areas. More sophisticated formulations were proposed (32,34,35). Protein solubility data, coupled with improved treatments of electrostatic and nonelectrostatic contributions, hold the promise of leading to better solvation models.
Finally, our modeling of such a complex environment as the condensed phase in terms of a simple implicit solvent may prove inspiring for the modeling of other complex environments, such as the crowded milieu inside cells. Such modeling will move computational studies of biomolecules closer to physiological conditions.
Acknowledgments
This work was supported in part by grant GM058187 from the National Institutes of Health.
Editor: Bertrand Garcia-Moreno.
References
- 1.Schmittschmitt, J. P., and J. M. Scholtz. 2003. The role of protein stability, solubility, and net charge in amyloid fibril formation. Protein Sci. 12:2374–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Howe, P. W. A. 2004. A straight-forward method of optimising protein solubility for NMR. J. Biomol. NMR. 30:283–286. [DOI] [PubMed] [Google Scholar]
- 3.Jancarik, J., R. Pufan, C. Hong, S.-H. Kim, and R. Kim. 2004. Optimum solubility (OS) screening: an efficient method to optimize buffer conditions for homogeneity and crystallization of proteins. Acta Crystallogr. D Biol. Crystallogr. 60:1670–1673. [DOI] [PubMed] [Google Scholar]
- 4.Machida, S., Y. Yu, S. P. Singh, J.-D. Kim, K. Hayashi, and Y. Kawata. 1998. Overproduction of β-glucosidase in active form by an Escherichia coli system coexpressing the chaperonin GroEL/ES. FEMS Microbiol. Lett. 159:41–46. [DOI] [PubMed] [Google Scholar]
- 5.Zhou, P., A. A. Lugovskoy, and G. Wagner. 2001. A solubility-enhancement tag (SET) for NMR studies of poorly behaving proteins. J. Biomol. NMR. 20:11–14. [DOI] [PubMed] [Google Scholar]
- 6.Waugh, D. S. 2005. Making the most of affinity tags. Trends Biotechnol. 23:316–320. [DOI] [PubMed] [Google Scholar]
- 7.Kato, A., K. Maki, T. Ebina, K. Kuwajima, K. Soda, and Y. Kuroda. 2007. Mutational analysis of protein solubility enhancement using short peptide tags. Biopolymers. 85:12–18. [DOI] [PubMed] [Google Scholar]
- 8.Bianchi, E., S. Venturini, A. Pessi, A. Tramontano, and M. Sollazzo. 1994. High level expression and rational mutagenesis of a designed protein, the minibody: from an insoluble to a soluble molecule. J. Mol. Biol. 236:649–659. [DOI] [PubMed] [Google Scholar]
- 9.Mosavi, L. K., and Z.-Y. Peng. 2003. Structure-based substitutions for increased solubility of a designed protein. Protein Eng. 16:739–745. [DOI] [PubMed] [Google Scholar]
- 10.Trevino, S. R., J. M. Scholtz, and C. N. Pace. 2007. Amino acid contribution to protein solubility: Asp, Glu, and Ser contribute more favorably than the other hydrophilic amino acids in RNase Sa. J. Mol. Biol. 366:449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trevino, S. R., J. M. Scholtz, and C. N. Pace. 2008. Measuring and increasing protein solubility. J. Pharm. Sci. In press. [DOI] [PubMed]
- 12.Fischel-Ghodsian, F., L. Brown, E. Mathiowitz, D. Brandenburg, and R. Langer. 1988. Enzymatically controlled drug delivery. Proc. Natl. Acad. Sci. USA. 85:2403–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan, P. H., V. Chu, J. E. Stray, D. K. Hamlin, D. Pettit, D. S. Wilbur, R. L. Vessella, and P. S. Stayton. 1998. Engineering the isoelectric point of a renal cell carcinoma targeting antibody greatly enhances scFv solubility. Immunotechnology. 4:107–114. [DOI] [PubMed] [Google Scholar]
- 14.Shaw, K. L., G. R. Grimsley, G. I. Yakovlev, A. A. Makarov, and C. N. Pace. 2001. The effect of net charge on the solubility, activity, and stability of ribonuclease Sa. Protein Sci. 10:1206–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kohn, W. D., R. Micanovic, S. L. Myers, A. M. Vick, S. D. Kahl, L. Zhang, B. A. Strifler, S. Li, J. Shang, J. M. Beals, J. P. Mayer, and R. D. DiMarchi. 2007. pI-shifted insulin analogs with extended in vivo time action and favorable receptor selectivity. Peptides. 28:935–948. [DOI] [PubMed] [Google Scholar]
- 16.Jorgensen, W. L., and E. M. Duffy. 2000. Prediction of drug solubility from Monte Carlo simulations. Bioorg. Med. Chem. Lett. 10:1155–1158. [DOI] [PubMed] [Google Scholar]
- 17.Livingstone, D. J., M. G. Ford, J. J. Huuskonen, and D. W. Salt. 2001. Simultaneous prediction of aqueous solubility and octanol/water partition coefficient based on descriptors derived from molecular structure. J. Comput. Aided Mol. Des. 15:741–752. [DOI] [PubMed] [Google Scholar]
- 18.Yang, G., Y. Ran, and S. H. Yalkowsky. 2002. Prediction of the aqueous solubility: comparison of the general solubility equation and the method using an amended solvation energy relationship. J. Pharm. Sci. 91:517–533. [DOI] [PubMed] [Google Scholar]
- 19.Delaney, J. S. 2005. Predicting aqueous solubility from structure. Drug Discov. Today. 10:289–295. [DOI] [PubMed] [Google Scholar]
- 20.Zhou, H.-X. 2005. Interactions of macromolecules with salt ions: an electrostatic theory for the Hofmeister effect. Proteins. 61:69–78. [DOI] [PubMed] [Google Scholar]
- 21.Gilson, M. K., and B. Honig. 1988. Calculation of the total electrostatic energy of a macromolecular system: solvation energies, binding energies, and conformational analysis. Proteins. 4:7–18. [DOI] [PubMed] [Google Scholar]
- 22.Sitkoff, D., K. A. Sharp, and B. Honig. 1994. Accurate calculation of hydration free energies using macroscopic solvent models. J. Phys. Chem. 98:1978–1988. [Google Scholar]
- 23.Madura, J. D., J. M. Briggs, R. Wade, M. E. Davis, B. A. Luty, A. Ilin, J. Antosiewicz, M. K. Gilson, B. Bagheri, L. R. Scott, and J. A. McCammon. 1995. Electrostatic and diffusion of molecules in solution: simulations with the University of Houston Brownian Dynamics program. Comput. Phys. Commun. 91:57–95. [Google Scholar]
- 24.Roux, B., and T. Simonson. 1999. Implicit solvent models. Biophys. Chem. 78:1–20. [DOI] [PubMed] [Google Scholar]
- 25.Vijayakumar, M., and H.-X. Zhou. 2001. Salt bridges stabilize the folded structure of barnase. J. Phys. Chem. B. 105:7334–7340. [Google Scholar]
- 26.Baker, N. A., D. Sept, S. Joseph, M. J. Holst, and J. A. McCammon. 2001. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA. 98:10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo, R., L. David, and M. K. Gilson. 2002. Accelerated Poisson-Boltzmann calculations for static and dynamic systems. J. Comput. Chem. 23:1244–1253. [DOI] [PubMed] [Google Scholar]
- 28.Bordner, A. J., C. N. Cavasotto, and R. A. Abagyan. 2002. Accurate transferable model for water, n-octanol, and n-hexadecane solvation free energies. J. Phys. Chem. B. 106:11009–11015. [Google Scholar]
- 29.Dong, F., M. Vijayakumar, and H.-X. Zhou. 2003. Comparison of calculation and experiment implicates significant electrostatic contributions to the binding stability of barnase and barstar. Biophys. J. 85:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feig, M., and C. L. Brooks III. 2004. Recent advances in the development and application of implicit solvent models in biomolecule simulations. Curr. Opin. Struct. Biol. 14:217–224. [DOI] [PubMed] [Google Scholar]
- 31.Baker, N. A. 2005. Improving implicit solvent simulations: a Poisson-centric view. Curr. Opin. Struct. Biol. 15:137–143. [DOI] [PubMed] [Google Scholar]
- 32.Levy, R. M., L. Y. Zhang, E. Gallicchio, and A. K. Felts. 2003. On the nonpolar hydration free energy of proteins: surface area and continuum solvent models for the solute-solvent interaction energy. J. Am. Chem. Soc. 125:9523–9530. [DOI] [PubMed] [Google Scholar]
- 33.Dzubiella, J., J. M. Swanson, and J. A. McCammon. 2006. Coupling nonpolar and polar solvation free energies in implicit solvent models. J. Chem. Phys. 124:084905. [DOI] [PubMed] [Google Scholar]
- 34.Wagoner, J. A., and N. A. Baker. 2006. Assessing implicit models for nonpolar mean solvation forces: the importance of dispersion and volume terms. Proc. Natl. Acad. Sci. USA. 103:8331–8336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan, C., Y. H. Tan, and R. Luo. 2007. Implicit nonpolar solvent models. J. Phys. Chem. B. 111:12263–12274. [DOI] [PubMed] [Google Scholar]
- 36.Fredericq, E., and H. Neurath. 1950. The interaction of insulin with thiocyanate and other anions. The minimum molecular weight of insulin. J. Am. Chem. Soc. 72:2684–2691. [Google Scholar]
- 37.Desbuquois, B., and G. D. Aurbach. 1974. Effects of iodination on the distribution of peptide hormones in aqueous two-phase polymer systems. Biochem. J. 143:83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wimley, W. C., T. P. Creamer, and S. H. White. 1996. Solvation energies of amino acid side chains and backbone in a family of host-guest pentapeptides. Biochemistry. 35:5109–5124. [DOI] [PubMed] [Google Scholar]
- 39.Mongan, J., D. A. Case, and J. A. McCammon. 2004. Constant pH molecular dynamics in generalized Born implicit solvent. J. Comput. Chem. 25:2038–2048. [DOI] [PubMed] [Google Scholar]
- 40.Case, D. A., T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, K. M. Merz, B. Wang, D. A. Pearlman, M. Crowley, S. Brozell, V. Tsui, H. Gohlke, J. Mogan, V. Hornak, G. Cui, P. Beroza, C. Schafmeister, J. W. Caldwell, W. S. Ross, and P. A. Kollman. 2004. AMBER 8. University of California, San Francisco.
- 41.Simmerling, C., B. Strockbine, and A. E. Roitberg. 2002. All-atom structure prediction and folding simulations of a stable protein. J. Am. Chem. Soc. 124:11258–11259. [DOI] [PubMed] [Google Scholar]
- 42.Bondi, A. 1964. Van der Waals volumes and radii. J. Phys. Chem. 68:441–451. [Google Scholar]
- 43.Cornell, W. D., P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell, and P. A. Kollman. 1995. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117:5179–5197. [Google Scholar]
- 44.Tjong, H., and H.-X. Zhou. 2008. On the dielectric boundary in Poisson-Boltzmann calculations. J. Chem. Theory Comput. 4:507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]