

Abstract
Genome organization within the cell nucleus is a result of chromatin condensation achieved by histone tail-tail interactions and other nuclear proteins that counter the outward entropic pressure of the polymeric DNA. We probed the entropic swelling of chromatin driven by enzymatic disruption of these interactions in isolated mammalian cell nuclei. The large-scale decondensation of chromatin and the eventual rupture of the nuclear membrane and lamin network due to this entropic pressure were observed by fluorescence imaging. This swelling was accompanied by nuclear softening, an effect that we quantified by measuring the fluctuations of an optically trapped bead adhered onto the nucleus. We also measured the pressure at which the nuclear scaffold ruptured using an atomic force microscope cantilever. A simple theory based on a balance of forces in a swelling porous gel quantitatively explains the diffusive dynamics of swelling. Our experiments on decondensation of chromatin in nuclei suggest that its compaction is a critical parameter in controlling nuclear stability.
INTRODUCTION
The nuclear architecture in eukaryotic cells is maintained by the nuclear membrane, lamin scaffold, and chromatin organization (1). This organization and its dynamic compaction states are central to cellular functions (2). The nucleosomal structure comprising 146 basepairs of DNA wound around the histone octamer (3) is further compacted to a 30-nm chromatin fiber as a result of histone tail-tail interactions (4–6). The radius of gyration, Rg = (lpl0)0.5 of genomic DNA of contour length l0 = 1 m, and with persistence length lp = 50 nm is ∼220 μm; hence, further compaction is required to organize the chromatin in the cell nucleus with a diameter of a few μm. Here, the outward entropic forces are balanced by the inward forces resulting from histone tail-tail interactions and other nonhistone proteins to further condense the chromatin fiber, resulting in a maximum packing ratio of ∼104 in mitotic cells. Further, we recently showed that the nuclear size and shape within living cells are the result of opposing tension between histone tail-tail interactions complexed with nuclear lamina and cytoplasmic filaments (7). Functional alterations of specific histone tail residues affect the compaction state of chromatin (5,8–10). The highly compacted heterochromatin regions are organized along the nuclear periphery and are known to be anchored to the lamin network at distinct foci and to the inner nuclear membrane (11). This anchorage is mediated by the interactions of HP1 (heterochromatin protein 1) with lamin A and LBR (lamin B receptor) in the nuclear membrane (1). The lamin proteins in nuclear matrix preparations from human cell lines, such as CaSki, form 10-nm filaments (12) and provide a highly extensible structural scaffolding within the nucleus with an elastic stiffness as high as 25 mN/m in the Xenopus oocyte lamin network (13). Therefore, the chromatin interaction with the lamin network and nuclear membrane makes up the scaffold that defines nuclear architecture. Although there have been attempts to link chromatin assembly anchored to the nuclear scaffold as a primary load-bearing element of the nucleus, direct evidence for such mechanical anchorage remain elusive. To probe this mechanical coupling of chromatin with the nuclear scaffold, we studied the effect of chromatin decompaction on the stability of nuclear architecture using fluorescence imaging and micromechanical methods. In our approach, the decompaction was achieved by enzymatic cleavage of histone tails in isolated nuclei using trypsin (14–17) and clostripain (18). Trypsin is a serine protease that cuts next to the lysine and arginine residues of proteins. Clostripain is yet another protease that cleaves at arginine residues, and is more specific for the terminal regions of the histone proteins (18). In earlier work in our lab, we were able to directly map viscoelastic changes from trypsin digestion of isolated chromatin fiber in real time using an optical tweezer (19).
MATERIALS AND METHODS
Isolation of intact nuclei from HeLa cells
HeLa cells were stably transfected with histone H2B-EGFP fusion protein using Lipofectamine-2000 (Invitrogen, Carlsbad, CA) selection by blasticidin (Calbiochem, San Diego, CA). The cells were grown in DMEM-F12 (Gibco BRL, Invitrogen, Carlsbad, CA) with 5% FBS (Gibco BRL, Invitrogen), in a 5% CO2 incubator maintained at 37°C. Freshly harvested cells were washed in PBS (pH 7.4) buffer and resuspended in TM2 buffer (10 mM Tris-Hcl, pH 7.4, 2 mM MgCl2, 0.5 mM (PMSF added fresh before use)). The cells were incubated for 5 min each in room temperature and on ice. Triton-X-100 (0.5% volume/volume) was added and mixed well before the cells were incubated on ice for an additional 5 min. The cells were sheared by passing them through a syringe needle (22 gauge) 10 times and centrifuged at 12,000 rpm for 5 min to obtain nuclei. The nuclei were observed under the microscope and Triton-X-100 treatment was repeated if the nuclei were found to have cellular debris sticking to them. Clean, isolated nuclei completely free of cellular debris were used for experiments, and all experiments were performed in PBS buffer at pH 7.4.
Nuclear membrane staining
The isolated nuclei were stained with FM4-64 dye (Molecular Probes, Invitrogen, Carlsbad, CA) as follows: The nuclei were allowed to stick to poly-D-lysine (PDL)-coated coverslips and washed with PBS 7.4 buffer. FM4-64 was added to a final concentration of 0.3 μg/mL to a 0.1% solution of BSA (HiMedia Laboratories, Mumbai, India) in PBS 7.4. The above solution was added to the nuclei and incubated at room temperature for 30 min.
Fluorescence confocal imaging
Fluorescence confocal images were taken on a Zeiss ConfoCor laser scanning microscope, using the 488-nm line of an argon-ion laser (Lasos, Jena, Germany) and 543 nm of an He-Ne laser (Spectra Physics). A 40× 1.3 NA, or a 63× 1.4 NA oil immersion objective was used to acquire all images. Dual-color imaging was performed to visualize chromatin using H2B-EGFP fluorescence, and the membrane using FM4-64 fluorescence. Excitation of the Hoechst stain to image DNA was carried out using a pulsed Ti-Sapphire multiphoton laser (Spectra Physics, Mountain View, CA) mode-locked at 750 nm.
Optical trap measurements
The optical trap was built on an inverted microscope (model IX70; Olympus, Tokyo, Japan) using a current controlled diode laser (wavelength 830 nm, GaAlAs diode; SDL Inc., San Jose, CA). A 5-mW diode laser, 635 nm (5-mW power; Coherent Inc., Santa Clara, CA) was used to track the bead position in the trap by imaging the back-scattered light onto a photodetector. The photodetector current was amplified by two low-noise current preamplifiers (model SR570; Stanford Research Systems, Sunnyvale, CA), with the difference in the voltages giving the position of the bead from the trap center. Data acquisition and analysis were done using DAQ (PCI-MIO-16XE-10; National Instruments, Austin, TX) and LabView (National Instruments, TX).
RESULTS
Nuclear swelling and rupture upon enzymatic decondensation of chromatin
The proteases used in our studies initially act on unstructured terminal domains of proteins and later degrade the structured regions. Decondensation of the chromatin structure in nuclei isolated from live HeLa cells stably expressing histone H2B tagged with enhanced green fluorescence protein (EGFP) fusion protein was achieved by protease digestion of histone tails. The nuclei were adhered onto PDL-coated coverslips mounted on the microscope, and clostripain was added to final concentrations ranging between 0.5 and 4 mU/μL. The enzyme was activated using DTT (2.5 mM) and calcium acetate (1 mM) for ∼30 min in room temperature before addition into the sample. The size of the nucleus was followed by confocal fluorescence imaging of the H2B-EGFP fusion protein. The cross-sectional area of the nucleus was measured as a function of time from the time-lapse fluorescence images. The panel in Fig. 1 A shows the slow expansion kinetics of isolated H2B-EGFP expressing HeLa nuclei when digested with 0.5 mU/μL clostripain. The dynamics of this swelling was dependent on the concentration of the enzyme used, as shown in the representative curves in Fig. 1 B for three different enzyme concentrations (1, 2, 4 mU/μL). Both the initial lag phase and the final fold change in nuclear size at a particular time point varied with enzyme concentration (Fig. 1 C). At the same enzyme concentration, the variability in nuclear expansion is captured by the error bars in Fig. 1 C. To further establish that such swelling of the chromatin occurs due to the disruption of the higher-order structure maintained by histone tail interactions and other nuclear scaffold proteins, we also digested similar nuclei with enzymes targeted for nucleic acids to verify whether disruption of the chromatin alone is enough to cause such entropic swelling. Even at high concentrations, neither DNase 1 (50, 500 mU/μL) nor RNase (2 mg/mL) caused a swelling of the nuclei (Fig. 1 D). At high DNase 1 concentrations (500 mU/μL), fragmented bits of chromatin gradually surrounded the nucleus with time, showing that the enzyme was indeed acting, though there was no entropic swelling in this case (Supplementary Material Fig. 1, Data S1). Thus, disruption of the chromatin is not enough. Only when the protein interactions that maintain the higher-order chromatin structure are violated is there an entropic swelling of the chromatin. Further, neither the detergent extraction of the nuclei nor the PDL coating of the coverslip altered the dynamics of nuclear swelling (Supplementary Material Figs. 2 and 3, Data S1).
FIGURE 1.

(A) Panel of images shows the entropic swelling of an isolated H2B-EGFP nucleus digested with 0.5 mU/μL clostripain. The corresponding time points (minutes) are indicated. The scale bar is equal to 10 μm. (B) Expansion kinetics at different clostripain concentrations—1 mU/μL (squares), 2 mU/μL (circles), and 4 mU/μL (triangles)—normalized to the initial area in each case. (C) The fold change in cross-sectional area with respect to the initial ∼750 s after addition of the enzyme (or until the nucleus bursts), shown for two different clostripain concentrations: 0.5 mU/μL and 2 mU/μL. The mean and standard deviations for six different experiments each are plotted. (D) Nucleic acid digestions of isolated nuclei. Normalized area plots as in B are shown for 50 mU/μL (triangles), 500 mU/μL (circles) DNase 1, or 2 mg/mL RNase (squares). No entropic expansion is observed in any of the cases.
We went on to do similar experiments with a general protease, trypsin, which also caused a similar entropic swelling of isolated nuclei. Clostripain is a more specific enzyme than trypsin; however, it is true in either case that the enzymes are likely to first act on unstructured histone tails rich in lysine and arginine residues, eventually leading to the disruption of the proteinaceous nuclear lamin scaffold. In Fig. 2, we plot a typical increase in the area of the nuclei obtained by quantifying images such as that shown in Fig. 1 A. Fig. 2 A shows an ∼4-fold increase in the area of the isolated nuclei in ∼15 min (trypsin concentration of 4 mU in 100 μL), suggesting a volume increase of at least eightfold. As expected, the kinetics of the nuclear swelling is altered with different trypsin concentrations (data not shown). As demonstrated in Fig. 1, even for any one enzyme, the dynamics of swelling is very much dependent on the concentration of the enzyme used; thus, whereas both trypsin and clostripain cause an entropic swelling of the chromatin, the dynamics of swelling may not exactly be always comparable. The fact that protease digestion of isolated nuclei causes entropic swelling of the chromatin is a general feature of digestion by both of the enzymes. Trypsin digestion of nuclei fixed with glutaraldehyde (1% glutaraldehyde for 15′) completely abolished the nuclear swelling. Also, the observed swelling was not due to adsorption on the PDL-coated coverslip, since such a nucleus left to itself did not show any entropic expansion (Fig. 2 A). For further experiments in this work, we consistently used trypsin digestion of isolated H2B-EGFP HeLa nuclei unless otherwise stated.
FIGURE 2.

(A) Area of the nucleus obtained from confocal images, taken as described before, as a function of time. The plots show the increase in area obtained from confocal images with trypsin digestion (squares, ∼4 mU of trypsin) and clostripain digestion (triangles, ∼4 mU of clostripain). The area remains constant with trypsin digestion (4 mU) when the nuclei are fixed with glutaraldehyde (solid circles, 1%, for ∼15′) or just left on a PDL-coated coverslip without any added enzyme (open circles). Confocal fluorescence images of nuclear swelling during trypsin-induced chromatin decondensation are shown in B–E. The figure shows images of (B) chromatin using H2B-EGFP fusion protein, (C) nuclear membrane using the lipophilic dye FM4-64, (D) DNA stained with Hoescht dye, and (E) lamin scaffold using the EGFP-laminB1 fusion protein. The Z-slice with the largest area of the nucleus was selected from each stack. Scale bar for all images is 5 μm, and time “t” is in minutes.
In the panel of images in Fig. 2 B, the chromatin was imaged using H2B-EGFP fluorescence, and the DNA was imaged with Hoescht dye staining. To observe the nuclear membrane swelling and rupture, a lipophilic dye was used as a membrane marker. The H2B-EGFP fluorescence images in Fig. 2 B show the chromatin swelling due to trypsin digestion. There was an initial increase in the nuclear membrane area (stained with a lipophilic dye FM4-64) before its rupture, as shown in Fig. 2 C. Images of Hoechst dye staining of DNA (Fig. 2 D) show that the DNA is intact after trypsinization and is decondensed. Next, we studied the lamin reorganization and rupture independently during chromatin decondensation. The HeLa nuclei with the LaminB protein fused with EGFP were used for fluorescence imaging of the lamin scaffold. The nuclei were isolated from the HeLa cells that were transfected with the EGFP-laminB1 plasmid. The EGFP-laminB1 images in Fig. 2 E show the increase in the circumference of the lamin scaffold and its eventual rupture due to digestion of the nuclei with trypsin. The large-scale decondensation of chromatin due to trypsin digestion was further confirmed by fluorescence anisotropy imaging method as described previously (20). The mean anisotropy value decreased with time of digestion, as shown in Supplementary Material Fig. 4 (Data S1), suggesting decompaction of the chromatin assembly. The control fixed nuclei showed a marginal decrease in anisotropy values. The nuclear swelling induced by trypsin digestion is a combined effect of chromatin decompaction, breakages in chromatin anchoring to the lamin scaffold and disruption of the lamin scaffold, and entropic swelling once the structure of the chromatin has been disrupted. In addition, since DNA is a charged polymer, the entropic swelling of the chromatin assembly also includes the differential contribution that arises due to the electrostatic repulsion.
In Fig. 3, the kinetics of nuclear swelling under varying conditions of cross-linking was measured. We observe a critical cross-linking concentration regime of ∼0.006% glutaraldehyde, below which the kinetics was similar to the unfixed condition, and above which the nuclei did not show a significant expansion upon trypsin digestion. At this critical concentration, the kinetics of expansion was significantly slowed down compared to control unfixed nuclei, as seen in Fig. 3, which is an average of three experiments for each condition. We further contrasted the protease-induced nuclear swelling with findings from previous studies that investigated swelling under conditions of low divalent salt concentration (11). This swelling, which happens without any apparent loss of histones from the chromatin, and can be attributed to the loss of screening effects, is perfectly reversible. A similar result was reproduced in our hands (Supplementary Material Fig. 5, Data S1). However, histone-DNA interactions are disrupted at high salt concentrations (21). In keeping with this, there was a total loss of H2B-EGFP fluorescence with 1 M MgCl2, and a corresponding partial swelling of isolated nuclei. As expected for loss of histones, this swelling was not reversed when the nuclei were returned to normal buffer conditions, and further swelling occurred in low salt concentrations, which was reversible. Clostripain digestion of such nuclei caused a far larger swelling and eventual rupture of the nuclei. (See Supplementary Material Fig. 6 A, Data S1). Despite the loss of H2B-EGFP fluorescence, the H3-H4 tetramer core probably is not lost in these nuclei because they still stain with an antibody to acetylated H3 (a common activation mark on histone H3) (Supplementary Material Fig. 6 B, Data S1), and they also have an intact lamin scaffold, as shown by staining for Lamin B1 (Supplementary Material Fig. 6 C, Data S1). Thus, under conditions of partial loss of histones, there is irreversible swelling that cannot be explained by screening effects, and can be attributed to partial swelling of the chromatin resisted by the lamin scaffold. With protease digestion, as condensing forces (histone and other interactions) are lost, there is a swelling and rupture of the nucleus.
FIGURE 3.

Expansion kinetics for control unfixed nuclei (solid circles) and nuclei fixed with 0.006% glutaraldehyde (open circles). The error bars are standard deviations for three different nuclei for each case.
Pressure generated on the nuclear membrane due to chromatin decompaction
The decondensation of the higher-order chromatin structure by trypsin digestion leads to initial expansion of the nuclear scaffold, as seen in the fluorescence images (Fig. 2 E). To estimate the magnitude of the pressure due to this expansion, we directly measured the force during the swelling using an atomic force microscope (AFM) cantilever mounted on an Alpha-SNOM microscope (WiTEC, Germany). A soft rectangular cantilever (stiffness = kcant = 0.02 N/m; Veeco Instruments Inc., NY) was positioned on the nucleus stuck on a PDL-coated coverslip, as shown in the schematic in Fig. 4. The deflection due to nuclear expansion was measured as a function of time after trypsin addition. A high trypsin concentration was used to reduce the time of measurement and hence avoid long-term mechanical drifts in the AFM experiment. The force on the cantilever was obtained by using 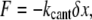 where kcant is the cantilever stiffness, and δx is the cantilever deflection. The pressure as a function of time is plotted in Fig. 4, which shows an increase of ∼3 kPa before the nucleus disintegrates, beyond which the cantilever deflects back to its initial position. The pressure on the membrane before rupture was calculated using a 300-nN change in force as measured by the cantilever deflection and an area of contact of 100 μm2 between the nucleus and the cantilever. Since the nucleus is covered by the cantilever, we calculated the pressure using the area of cross-section of the nucleus. The chromatin swelling results in a surprisingly high force on the membrane before rupture. The subsequent fall in cantilever deflection corresponds to nuclear scaffold breakdown, as also seen in the fluorescence images of EGFP-Lamin B1 (Fig. 2 E). The chromatin continues to swell beyond this point, but cannot exert pushing forces on the cantilever. The rupture of the nuclear scaffold may be due to both the entropic expansion of the chromatin, and direct protease activity on it. From this, the work done on the cantilever by the entropic forces resulting in chromatin swelling was estimated to be ∼108 kBT. This number is not unreasonable since DNA histone binding energies of a single nucleosome measured using single-molecule experiments are on the order of a few kBT (22).
where kcant is the cantilever stiffness, and δx is the cantilever deflection. The pressure as a function of time is plotted in Fig. 4, which shows an increase of ∼3 kPa before the nucleus disintegrates, beyond which the cantilever deflects back to its initial position. The pressure on the membrane before rupture was calculated using a 300-nN change in force as measured by the cantilever deflection and an area of contact of 100 μm2 between the nucleus and the cantilever. Since the nucleus is covered by the cantilever, we calculated the pressure using the area of cross-section of the nucleus. The chromatin swelling results in a surprisingly high force on the membrane before rupture. The subsequent fall in cantilever deflection corresponds to nuclear scaffold breakdown, as also seen in the fluorescence images of EGFP-Lamin B1 (Fig. 2 E). The chromatin continues to swell beyond this point, but cannot exert pushing forces on the cantilever. The rupture of the nuclear scaffold may be due to both the entropic expansion of the chromatin, and direct protease activity on it. From this, the work done on the cantilever by the entropic forces resulting in chromatin swelling was estimated to be ∼108 kBT. This number is not unreasonable since DNA histone binding energies of a single nucleosome measured using single-molecule experiments are on the order of a few kBT (22).
FIGURE 4.

Estimation of pressure on the membrane due to chromatin decompaction. A soft cantilever (k = 0.02 N/m) was positioned on the nucleus stuck onto a PDL-coated coverslip, and the cantilever deflection due to nuclear swelling was measured by using a laser reflected off the cantilever surface and a quadrant photodiode, as shown in the inset. The maximum force of ∼300 nN was measured for nuclear swelling due to trypsin digestion (∼100 mU), after which the force fell to the initial value due to complete rupture of the nucleus.
Measurement of nuclear stiffness during chromatin decondensation
To investigate whether the observed chromatin decondensation leads to softening of the nucleus, we probed the change in stiffness of the nucleus using an optical tweezer as described previously (23). A 2-μm-diameter bead was nonspecifically adhered onto the nuclear membrane and confined to the center of the optical trap. The position fluctuations of the bead were measured by imaging the red laser back-scattered from the trapped bead onto a photodiode (see Materials and Methods). The change in the standard deviation of position fluctuation distributions of the bead is then a direct measure of the change in the stiffness of the isolated nucleus. Fig. 5 shows the increase in the bead fluctuations with time due to trypsin digestion. The standard deviation increased from ∼10 nm to ∼30 nm. For a trapped bead in solution (and not adhered to the nucleus), it increased to ∼50 nm. The increase in the bead fluctuations reflects the nuclear softening due to the chromatin decondensation in the nucleus. As a control, nuclei fixed with glutaraldehyde were treated with trypsin, and in this case the standard deviations of the position histograms remained unaltered with time (Fig. 5).
FIGURE 5.

Softening of the nucleus measured using an optically trapped bead adhered onto the nucleus stuck to a PDL-coated coverslip. In an optical trap measurement, the width of position histograms of the bead fluctuations about the trap center is plotted as a function of time for trypsin digestion (concentration ∼4 mU in a sample volume of 100 μL) of untreated (circles) and glutaraldehyde-fixed (squares, 1% for 15′) nuclei. The trap stiffness in all cases was ∼10−5 N/m, and the position time series was acquired at a sampling rate of 5000 Hz.
Diffusive dynamics of swelling
To understand the dynamics of swelling, we first estimate the time required for trypsin, clostripain, and glutaraldehyde to diffuse into the nucleus. Using typical values for the diffusion constants on the order of 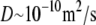 and a nuclear size on the order of
and a nuclear size on the order of 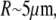 we find that the time required is four orders of magnitude less than the observed time. Since the dynamics of trypsin binding is also relatively fast, we must look elsewhere to explain the very slow dynamics of swelling. Once the trypsin has digested the histone tails and eventually other nuclear proteins, the chromatin swells into the ambient fluid. The dynamics of this process is limited by the rate of water movement through the nuclear membrane and the interstices of the porous higher-order chromatin assembly, as well as the rate at which the elastically prestrained chromatin and nuclear lamina expand into the water. The latter is the slower of the two processes when the nuclear membrane and the interstices do not limit the diffusion of small protein molecules as here. Since the excess pressure driving the expansion is roughly the prestress in the chromatin network after the breakdown of the lamin scaffold, where the pressure gradient scales as
we find that the time required is four orders of magnitude less than the observed time. Since the dynamics of trypsin binding is also relatively fast, we must look elsewhere to explain the very slow dynamics of swelling. Once the trypsin has digested the histone tails and eventually other nuclear proteins, the chromatin swells into the ambient fluid. The dynamics of this process is limited by the rate of water movement through the nuclear membrane and the interstices of the porous higher-order chromatin assembly, as well as the rate at which the elastically prestrained chromatin and nuclear lamina expand into the water. The latter is the slower of the two processes when the nuclear membrane and the interstices do not limit the diffusion of small protein molecules as here. Since the excess pressure driving the expansion is roughly the prestress in the chromatin network after the breakdown of the lamin scaffold, where the pressure gradient scales as 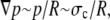 where
where 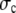 is the stress in the chromatin network of bulk size R, the nuclear radius. The dominant resistance to flow is due to viscous forces at the scale of interstices (pores) in the network, so that
is the stress in the chromatin network of bulk size R, the nuclear radius. The dominant resistance to flow is due to viscous forces at the scale of interstices (pores) in the network, so that 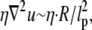 where
where 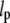 is the typical pore size,
is the typical pore size, 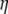 is the viscosity of the nucleoplasm, and the fluid velocity is approximately the rate of increase of the nucleus,
is the viscosity of the nucleoplasm, and the fluid velocity is approximately the rate of increase of the nucleus, 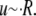 Substituting these relations into the Stokes equations
Substituting these relations into the Stokes equations 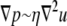 that embody the balance of forces yields a simple relation for the time required for the swelling of the chromatin network, given by
that embody the balance of forces yields a simple relation for the time required for the swelling of the chromatin network, given by
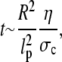 |
(1) |
which may be effectively rewritten as 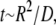 i.e., the relaxation is diffusive with a stress (pressure) diffusion constant D given by
i.e., the relaxation is diffusive with a stress (pressure) diffusion constant D given by
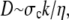 |
(2) |
where 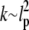 is the hydraulic permeability. This simple relation is of course relevant only in the limit when the network stress is approximately constant and the pore size is constant, but it suffices to explain our experimental observations fairly well. Substituting in typical parameter values, with
is the hydraulic permeability. This simple relation is of course relevant only in the limit when the network stress is approximately constant and the pore size is constant, but it suffices to explain our experimental observations fairly well. Substituting in typical parameter values, with 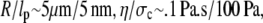 we get t ∼ 1000s, which is qualitatively consistent with our experimental observations of the swelling induced by both trypsin and clostripain. Here we use estimates for viscosity from Bhattacharya et al. (29) and stiffness of the nucleoplasm from Dahl et al. (13).
we get t ∼ 1000s, which is qualitatively consistent with our experimental observations of the swelling induced by both trypsin and clostripain. Here we use estimates for viscosity from Bhattacharya et al. (29) and stiffness of the nucleoplasm from Dahl et al. (13).
DISCUSSION
The organization of a genome in a eukaryotic nucleus and its anchoring to the nuclear scaffold are critical to its function (24,25). This organization is a balance between outward forces arising due to entropic nature of the genome, as well as inward forces arising due to histone tail-tail interactions stabilized by other nuclear proteins. A schematic of the force balance is shown in Fig. 6. Here we have shown that the compaction of chromatin within the nucleus is a key factor in maintaining the structural integrity and stability of the nucleus. The structural destabilization of isolated nuclei during chromatin decondensation was probed using both imaging and micromechanical methods. Decondensation led to entropic expansion of chromatin, and the enormous pressure (∼3 kPa) resulted in the expansion and eventual rupture of the nuclear membrane and lamin scaffold. The area enclosed by the lamin network increased by <2-fold just before it ruptured. The chromatin, however, continued to expand even after the lamin scaffold ruptured, to a >10-fold increase in area. Our results clearly show that enzymatic disruption of histone tail-tail interactions and other nuclear proteins results in entropic expansion of chromatin assembly. This is in line with previous observations that the chromatin organization and nuclear lamina in an interphase nucleus is a load-bearing factor of the nucleus, as measured in both intact nuclei and nuclei swollen in buffer with chelation of divalent salt concentration (11,13,26). Given that DNA is a good example of a semiflexible polymer, the outward entropic forces result in a coil size with a radius of gyration Rg ∼ 220 μm. However, the inward forces arising from histone tail-tail interactions and other nuclear proteins drive this organization to a condensed, closely packed chromatin structure, as in the metaphase chromosome. The isolated cell nucleus is held in equilibrium under a balance of these two opposing forces. Therefore, enzymatic disruption of the inward forces in isolated nuclei results in the chromatin assembly tilting toward the entropic limit, resulting in nuclear swelling and eventual rupture of the higher-order chromatin structure.
FIGURE 6.

Schematic of the force balance that stabilizes chromatin assembly in an intact isolated nucleus. Protease digestion results in chromatin decondensation, nuclear swelling, and eventual rupture.
The nuclear swelling led to a sharp deflection (corresponding to ∼300 nN force) in the cantilever position, and this was followed by a similar sharp relaxation. The relaxation suggests a sudden rupture of the nuclear scaffold that might have supported the cantilever deflection due to swelling. The results indicate that the nuclear scaffold could sustain a pressure of a few kilopascals, and a further increase in pressure would disrupt it. Rheology experiments on lamin networks reconstituted in vitro showed strain hardening, with the network stiffening under stress (26). Our experiments also show that the lamin network can undergo large deformations before it breaks down. Further, we have measured the softening of the nucleus during decondensation using an optical trap. The measured nuclear softening suggests that chromatin compaction and the integrity of the proteinaceous lamin scaffold has a bearing on nuclear stiffness. Anchorage of chromatin to the lamin scaffold at numerous distinct foci has been reported in previous works (12,24,25). Experiments on isolated TC7 epithelium cells showed that the aspiration of the nuclei resulted in buckling of lamin external to the aspirated regions (11). This indicates that the tension is coupled to lamin through distinct internal foci provided by the chromatin. Our results suggest that the reduction in the nuclear stiffness may be due to cleavage of these chromatin anchorages, as well as rupture of the nuclear scaffold during decondensation.
CONCLUSIONS
A simple phenomenological model has been presented to account for the timescales involved in nuclear expansion. The swelling timescale determined by a balance between the viscous forces associated with water flow through the nucleus and the elastic forces due to the expansion of the prestressed chromatin assembly after enzymatic disruption, ∼1000 s, is qualitatively consistent with the experimentally measured dynamics of nuclear swelling. Clearly, a quantitative description of the dynamics will depend on better measurements of the hydraulic permeability of the nucleus, its elasticity, and the viscosity of the nucleoplasm during swelling.
There have been numerous reports that the lamin networks are required for chromatin organization, DNA replication, nuclear assembly, and nuclear positioning (27); however, our work shows that chromatin organization is a critical parameter for the structural integrity of the nucleus. In addition, recent work on stem cells lacking lamin-A/C, as well as epithelial cells knocked down for lamin-A/C, also indicates that chromatin is the major contributor to nuclear rheology (28). Thus, the picture of the nucleus that seems to be emerging is not one in which the functions of the chromatin and structural elements, such as the lamin networks and nuclear membranes, are physically distinct, but rather is one in which they are mechanically coupled in a way that is essential to cellular function. Compaction of chromatin, driven by the histone tail-tail interactions and other nuclear proteins, is essential for the condensation of the genome and determines the structural stability of the nucleus. Local entropic swelling induced by destabilizing histone interactions may be exploited by cells to effect the onset of DNA-related genomic processes. For example, region-specific acetylation of histone tails, which decondense chromatin fiber, make regulatory sites on the genome accessible to the transcription machinery.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
We thank the National Centre for Biological Sciences imaging facility and the National Nanoscience Initiative, Department of Science and Technology, India, for financial assistance, and Harvard University for cosponsoring the Bangalore-Harvard symposium in August 2006, which stimulated some of this research.
Aprotim Maumder and T. Roopa contributed equally to this work.
Editor: Marileen Dogterom.
References
- 1.Gruenbaum, Y., A. Margalit, R. D. Goldman, D. K. Shumaker, and K. L. Wilson. 2005. The nuclear lamina comes of age. Nat. Rev. Mol. Cell Biol. 6:21–31. [DOI] [PubMed] [Google Scholar]
- 2.Widom, J. 1998. Structure, dynamics, and function of chromatin in vitro. Annu. Rev. Biophys. Biomol. Struct. 27:285–327. [DOI] [PubMed] [Google Scholar]
- 3.Luger, K., A. W. Mader, R. K. Richmond, D. F. Sargent, and T. J. Richmond. 1997. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 389:251–260. [DOI] [PubMed] [Google Scholar]
- 4.Fletcher, T. M., and J. C. Hansen. 1995. Core histone tail domains mediate oligonucleosome folding and nucleosomal DNA organization through distinct molecular mechanisms. J. Biol. Chem. 270:25359–25362. [DOI] [PubMed] [Google Scholar]
- 5.Brower-Toland, B., D. A. Wacker, R. M. Fulbright, J. T. Lis, W. L. Kraus, and M. D. Wang. 2005. Specific contributions of histone tails and their acetylation to the mechanical stability of nucleosomes. J. Mol. Biol. 346:135–146. [DOI] [PubMed] [Google Scholar]
- 6.Placek, B. J., and L. M. Gloss. 2002. The N-terminal tails of the H2A–H2B histones affect dimer structure and stability. Biochemistry. 41:14960–14968. [DOI] [PubMed] [Google Scholar]
- 7.Mazumder, A., and G. V. Shivashankar. 2007. Gold-nanoparticle-assisted laser perturbation of chromatin assembly reveals unusual aspects of nuclear architecture within living cells. Biophys. J. 93:2209–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khorasanizadeh, S. 2004. The nucleosome: from genomic organization to genomic regulation. Cell. 116:259–272. [DOI] [PubMed] [Google Scholar]
- 9.Iizuka, M., and M. M. Smith. 2003. Functional consequences of histone modifications. Curr. Opin. Genet. Dev. 13:154–160. [DOI] [PubMed] [Google Scholar]
- 10.Dorigo, B., T. Schalch, K. Bystricky, and T. J. Richmond. 2003. Chromatin fiber folding: requirement for the histone H4 N-terminal tail. J. Mol. Biol. 327:85–96. [DOI] [PubMed] [Google Scholar]
- 11.Dahl, K. N., A. J. Engler, J. D. Pajerowski, and D. E. Discher. 2005. Power-law rheology of isolated nuclei with deformation mapping of nuclear substructures. Biophys. J. 89:2855–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nickerson, J. A., G. Krockmalnic, K. M. Wan, and S. Penman. 1997. The nuclear matrix revealed by eluting chromatin from a cross-linked nucleus. Proc. Natl. Acad. Sci. USA. 94:4446–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dahl, K. N., S. M. Kahn, K. L. Wilson, and D. E. Discher. 2004. The nuclear envelope lamina network has elasticity and a compressibility limit suggestive of a molecular shock absorber. J. Cell Sci. 117:4779–4786. [DOI] [PubMed] [Google Scholar]
- 14.Leuba, S. H., C. Bustamante, J. Zlatanova, and K. van Holde. 1998. Contributions of linker histones and histone H3 to chromatin structure: scanning force microscopy studies on trypsinized fibers. Biophys. J. 74:2823–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leuba, S. H., C. Bustamante, K. van Holde, and J. Zlatanova. 1998. Linker histone tails and N-tails of histone H3 are redundant: scanning force microscopy studies of reconstituted fibers. Biophys. J. 74:2830–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krajewski, W. A., and J. Ausio. 1996. Modulation of the higher-order folding of chromatin by deletion of histone H3 and H4 terminal domains. Biochem. J. 316:395–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bertin, A., A. Leforestier, D. Durand, and F. Livolant. 2004. Role of histone tails in the conformation and interactions of nucleosome core particles. Biochemistry. 43:4773–4780. [DOI] [PubMed] [Google Scholar]
- 18.Protacio, R. U., G. Li, P. T. Lowary, and J. Widom. 2000. Effects of histone tail domains on the rate of transcriptional elongation through a nucleosome. Mol. Cell. Biol. 20:8866–8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roopa, T., and G. V. Shivashankar. 2006. Direct measurement of local chromatin fluidity using optical trap modulation force spectroscopy. Biophys. J. 91:4632–4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Banerjee, B., D. Bhattacharya, and G. V. Shivashankar. 2006. Chromatin structure exhibits spatio-temporal heterogeneity within the cell nucleus. Biophys. J. 91:2297–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eickbush, T. H., and E. N. Moudrianakis. 1978. The histone core complex: an octamer assembled by two sets of protein-protein interactions. Biochemistry. 17:4955–4964. [DOI] [PubMed] [Google Scholar]
- 22.Soni, G. V., L. Brar, F. M. Hameed, A. K. Raychaudhuri, and G. V. Shivashankar. 2007. Distinct levels in the nanoscale organization of DNA-histone complex revealed by its mechanical unfolding. Appl. Phys. Lett. 90:163904. [Google Scholar]
- 23.Roopa, T., and G. V. Shivashankar. 2003. Nanomechanics of membrane tubulation and DNA assembly. Appl. Phys. Lett. 82:1631–1633. [Google Scholar]
- 24.Marshall, W. F. 2002. Order and disorder in the nucleus. Curr. Biol. 12:R185–R192. [DOI] [PubMed] [Google Scholar]
- 25.Makatsori, D., N. Kourmouli, H. Polioudaki, L. D. Shultz, K. McLean, P. A. Theodoropoulos, P. B. Singh, and S. D. Georgatos. 2004. The inner nuclear membrane protein lamin B receptor forms distinct microdomains and links epigenetically marked chromatin to the nuclear envelope. J. Biol. Chem. 279:25567–25573. [DOI] [PubMed] [Google Scholar]
- 26.Panorchan, P., B. W. Schafer, D. Wirtz, and Y. Tseng. 2004. Nuclear envelope breakdown requires overcoming the mechanical integrity of the nuclear lamina. J. Biol. Chem. 279:43462–43467. [DOI] [PubMed] [Google Scholar]
- 27.Mattout, A., T. Dechat, S. A. Adam, R. D. Goldman, and Y. Gruenbaum. 2006. Nuclear lamins, diseases and aging. Curr. Opin. Cell Biol. 18:335–341. [DOI] [PubMed] [Google Scholar]
- 28.Pajerowski, J. D., K. N. Dahl, F. L. Zhong, P. J. Sammak, and D. E. Discher. 2007. Physical plasticity of the nucleus in stem cell differentiation. Proc. Natl. Acad. Sci. USA. 104:15619–15624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhattacharya, D., A. Mazumder, S. A. Miriam, and G. V. Shivashankar. 2006. EGFP-tagged core and linker histones diffuse via distinct mechanisms within living cells. Biophys. J. 91:2326–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.