

Abstract
Most of the decisive molecular events in biology take place at the protein-water interface. The dynamical properties of the hydration layer are therefore of fundamental importance. To characterize the dynamical heterogeneity and rotational activation energy in the hydration layer, we measured the 17O spin relaxation rate in dilute solutions of three proteins in a wide temperature range extending down to 238 K. We find that the rotational correlation time can be described by a power-law distribution with exponent 2.1–2.3. Except for a small fraction of secluded hydration sites, the dynamic perturbation in the hydration layer is the same for all proteins and does not differ in any essential way from the hydration shell of small organic solutes. In both cases, the dynamic perturbation factor is <2 at room temperature and exhibits a maximum near 262 K. This maximum implies that, at low temperatures, the rate of water molecule rotation has a weaker temperature dependence in the hydration layer than in bulk water. We attribute this difference to the temperature-independent constraints that the protein surface imposes on the water H-bond network. The free hydration layer studied here differs qualitatively from confined water in solid protein powder samples.
INTRODUCTION
The biological functions of most proteins depend critically on the dynamical properties of the protein-water interface. It is therefore of fundamental importance to obtain a quantitative description of water dynamics in the hydration layer enveloping the protein. Many experimental techniques have been applied to this problem, but our understanding is still incomplete (1). Studies of protein hydration dynamics are challenging for several reasons. First, few techniques can probe water molecules selectively. Second, at physiologically relevant hydration levels, the measured property tends to be dominated by the large excess of bulk water. Third, the mobility of water molecules varies by orders of magnitude among different hydration sites on the structurally heterogeneous protein surface.
Oxygen-17 spin relaxation has been used extensively to study single-molecule rotation in bulk water (2,3) and in the hydration shells of small organic solutes (4,5) and proteins (1,6,7) in dilute aqueous solution. In the case of protein solutions, frequency-dependent 17O magnetic relaxation dispersion (MRD) measurements allow detailed characterization of internal water molecules (6,8), but provide only limited information about the rapidly exchanging water molecules in the external hydration layer (1,7). Essentially, the 17O spin relaxation rate R1 at high resonance frequency (∼100 MHz) yields the average rotational correlation time 〈τ〉 for all water molecules in the hydration layer, often reported as the dynamic perturbation factor (DPF) ξH = 〈τ〉/τ0, where τ0 is the bulk-water correlation time. (The DPF is sometimes called the rotational retardation factor (1).) Because of the topographical and chemical heterogeneity of the protein surface, the τ-distribution ranges from picoseconds to nanoseconds (1,9,10). In principle, information about this distribution could be obtained from measurements of the intermolecular cross-relaxation between water protons and spectrally resolved protons on the protein surface (11). However, because the cross-relaxation rate is usually dominated by long-range dipole couplings to remote bulk-water protons (7,12), this approach has not provided useful information about dynamics in the protein hydration layer.
Our principal aim here is to use 17O spin relaxation to elucidate the generic dynamical behavior of the protein hydration layer in the presence of excess bulk water. Current magnet technology sets an upper limit of ∼100 MHz for the 17O resonance frequency, so the picosecond-nanosecond hydration layer dynamics cannot be resolved directly in the frequency domain, as for internal water molecules (8). Our strategy is instead to characterize the dynamically heterogeneous hydration layer via the temperature dependence of the 17O spin relaxation rate at a fixed high frequency. For this strategy to be effective, measurements must be performed over a wide temperature range. High temperatures are precluded by thermal denaturation and dominant internal water contributions to the 17O relaxation rate even at high frequencies. On the low temperature side, water freezing presents an obstacle. However, by dispersing the protein solution in the form of emulsion droplets, the (metastable) liquid state can be maintained down to the homogeneous nucleation temperature (13,14). We have thus studied dilute solutions of three proteins in a 50 K temperature range extending down to 238 K. Accurate relaxation measurements in the deeply supercooled regime are technically challenging and until recently such data were not available even for small organic solutes (5). For small solutes, the data can be analyzed in a straightforward and essentially model-independent way. For proteins, data analysis is complicated by the strong dynamical heterogeneity. Before presenting the experimental results, we therefore outline the theoretical framework needed to analyze this kind of data.
The three proteins studied here, bovine pancreatic trypsin inhibitor (BPTI), ubiquitin, and β-lactoglobulin (BLG), were selected because they do not undergo cold denaturation in the investigated temperature range and because results from previous MRD studies (8,15) of the long-lived internal water molecules in these proteins allow us to reliably correct for the (small) contribution from internal water molecules to R1 at high frequencies.
MATERIALS AND METHODS
Sample preparation
Bovine pancreatic trypsin inhibitor (BPTI, batch 9104, 97% purity by HPLC) from Bayer HealthCare AG (Wuppertal, Germany) was exhaustively dialyzed to remove residual salt. Ubiquitin was expressed in Escherichia coli and was purified to >99% as described (8). Bovine β-lactoglobulin (BLG) isoform A (cat. No. L-7880; Sigma, St. Louis, MO) was purified by anion exchange and size-exclusion chromatography as described in Qvist et al. (15). Protein solutions were prepared by dissolving the purified lyophilized protein in 17O-enriched H2O (19 atom % 17O; Isotec, St. Louis, MO) and adjusting pH to the desired value (Table 1). Protein concentrations, expressed as the water/protein mole ratio NW (Table 1), were determined with ∼1% accuracy by complete amino-acid analysis. Nuclear magnetic resonance (NMR) measurements were made down to 238 K on emulsions of protein solution in heptane, with sorbitan tristearate as emulsifier (5). In a typical emulsion droplet of 10 μm diameter, only 0.3% of the protein molecules in the solution are within 5 nm of the interface. Furthermore, polyols (like the sorbitan headgroup of the emulsifier) are preferentially excluded from protein surfaces, so the protein should not interact strongly with the interface. Indeed, no effect of the interface could be detected in control experiments where 2H and 17O MRD profiles (1–100 MHz) were recorded at temperatures above 277 K from identical BPTI (16) or β-lactoglobulin (M. Davidovic, C. Mattea, J. Qvist, and B. Halle, unpublished) solutions with and without confinement to emulsion droplets.
TABLE 1.
Sample characteristics and results derived from 17O relaxation data
| Property | BPTI | Ubiquitin | BLG |
|---|---|---|---|
| MP (kDa) | 6.5 | 8.6 | 18.4 |
| τP(T*) (ns) | 3.9 | 4.8 | 11.0 |
| CP (mM) | 9.46 | 5.10 | 0.98 |
| pH | 5.2 | 5.0 | 2.7 |
| NW | 5590 | 10500 | 55730 |
| NH | 380 | 443 | 735 |
| ν | 2.32 ± 0.02 | 2.32 ± 0.01 | 2.15 ± 0.01 |
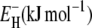 |
26.5 ± 0.5 | 27.3 ± 0.3 | 27.0 ± 0.3 |
| TX(p = 1) | 263.1 | 261.6 | 261.0 |
| TX(p = 0.5 or 0.9) | 264.1 | 262.4 | 263.0 |
Spin relaxation measurements
The relaxation rate R1 of the water 17O longitudinal magnetization was measured at 81.3 MHz on a Varian Unity Plus 600 spectrometer (Varian Cary, Palo Alto, CA). R1 was determined with 0.5–1.0% accuracy from a three-parameter fit to the single-exponential magnetization curve obtained with the inversion recovery pulse sequence with 30 delay times in nonmonotonic order. The small scatter indicated that no freezing occurred during relaxation measurements down to 238 K. Reported R1 values are averages of several measurements. At each temperature, measurements of 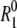 on a pure water reference sample were alternated with solution R1 measurements. The samples were carefully equilibrated at each temperature, which was regulated to within 0.1 K with a precooled stream of dry air and determined before and after R1 measurements with a copper-constantan thermocouple in an NMR tube containing a water-ethanol mixture.
on a pure water reference sample were alternated with solution R1 measurements. The samples were carefully equilibrated at each temperature, which was regulated to within 0.1 K with a precooled stream of dry air and determined before and after R1 measurements with a copper-constantan thermocouple in an NMR tube containing a water-ethanol mixture.
Hydration numbers
The number of water molecules in the first hydration layer around the protein was computed as NH = AS/aW, where AS is the solvent-accessible surface area (SASA) of the protein and aW is the amount of SASA occupied by one water molecule on average. The SASA was computed with GetArea 1.1 (17), using 1.7 Å probe radius and standard van der Waals radii (18). We used the value aW = 10.75 Å2, established for peptides and other organic solutes (5) by requiring that the SASA-derived hydration number NH matches the hydration number computed from a molecular dynamics (MD) simulation as the mean number of water molecules satisfying at least one of the following criteria: R(OW–O) < 3.3 Å; R(OW–N) < 3.5 Å; and R(OW–C) < 5.0 Å. These cutoff distances are close to the first minimum in the radial distribution function (19). Averaging over 2000 configurations from a 4-ns MD trajectory of BLG at NW = 6249 (15), we obtain with the same cutoff distances NH = 745 ± 13, close to AS/aW = 7906/10.75 = 735. This agreement indicates that the adopted aW value is valid for proteins as well as for small organic solutes. Previous MRD studies of protein hydration have used aW = 15 Å2 (1,20), leading to smaller NH values that agree with MD simulations when a uniform cutoff R(OW–X) < 3.5 Å is used to define the hydration layer (21). However, this more conservative definition of the hydration layer excludes most of the water in the primary hydration shells of apolar groups. The value aW = 10.75 Å2 adopted here yields NH values that correspond more closely to the first layer of water molecules covering the protein. Note that the previously used aW value of 15 Å2 yields a somewhat larger DPF since the observed effect is attributed to a smaller number of water molecules. The DPF can be converted between the different aW conventions according to 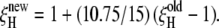
THEORY
Water 17O spin relaxation in protein solutions
The water 17O spin relaxation rate R1 in a protein solution exceeds the bulk-water value 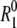 because the water molecules that interact with the protein rotate more slowly. The exceptionally high cohesive energy density of liquid water, stemming from the small size and fourfold hydrogen-bonding capacity of the water molecule, essentially limits the range of the protein-induced perturbation to the first hydration layer (and to the few water molecules that penetrate the protein). Support for this view comes from NMR relaxation studies of model systems (22,23) and from MD simulations (21,24). (Note that, in MD simulations, the first hydration shell of apolar groups is sometimes included in the second hydration layer of the protein (21,25).)
because the water molecules that interact with the protein rotate more slowly. The exceptionally high cohesive energy density of liquid water, stemming from the small size and fourfold hydrogen-bonding capacity of the water molecule, essentially limits the range of the protein-induced perturbation to the first hydration layer (and to the few water molecules that penetrate the protein). Support for this view comes from NMR relaxation studies of model systems (22,23) and from MD simulations (21,24). (Note that, in MD simulations, the first hydration shell of apolar groups is sometimes included in the second hydration layer of the protein (21,25).)
Accordingly, we attribute the observed relaxation enhancement to the NH water molecules in the first hydration layer (Table 1) and to the NI internal water molecules. The 17O spin relaxation rate R1(ω0,T) for a protein solution with water/protein mole ratio NW can then be expressed as (6)
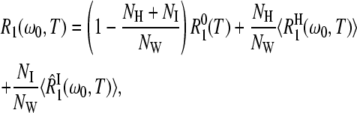 |
(1) |
where 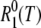 is the 17O spin relaxation rate for a pure-water reference sample.
is the 17O spin relaxation rate for a pure-water reference sample.
In the second term, the angular brackets represent an average over the NH sites in the hydration layer. The form of this term assumes that all hydration sites are in the fast-exchange limit, which is the case at the high resonance frequency used here, ω0/(2π) = 81.3 MHz. The intrinsic relaxation rate at a given hydration site is
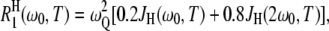 |
(2) |
where ωQ is the 17O nuclear quadrupole frequency. The spectral density function,
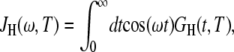 |
(3) |
is the cosine transform of the time correlation function (TCF),
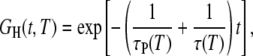 |
(4) |
where τP is the rotational correlation time of the protein (Table 1) and τ is the rotational correlation time of a water molecule in the hydration site. This form of the TCF follows from the statistical independence of protein tumbling and water motions in the hydration layer.
The third term in Eq. 1 describe the contribution to R1 from NI internal water molecules. Here, we do not assume fast exchange but only dilute conditions (NI ≪ NW). The effective average intrinsic relaxation rate for the internal-water class is (6)
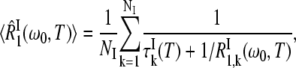 |
(5) |
where 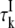 is the mean water residence time in internal site k. The intrinsic relaxation rate
is the mean water residence time in internal site k. The intrinsic relaxation rate 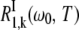 is given by expressions analogous to Eqs. 2–4, except that the TCF is now
is given by expressions analogous to Eqs. 2–4, except that the TCF is now
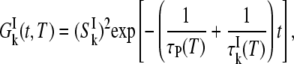 |
(6) |
where 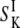 is the orientational order parameter of a water molecule in site k (6).
is the orientational order parameter of a water molecule in site k (6).
By measuring R1 for a protein solution and 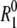 for a pure-water reference sample at the same temperature, we can obtain the apparent dynamic perturbation factor (ADPF), defined as
for a pure-water reference sample at the same temperature, we can obtain the apparent dynamic perturbation factor (ADPF), defined as
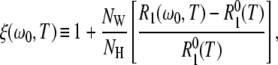 |
(7) |
where NW is the water/protein mole ratio in the sample and NH is the number of water molecules in the hydration layer (Table 1). By substituting R1(ω0,T) from Eq. 1, we can express the ADPF as a sum of two terms associated with the hydration layer and with internal water molecules:
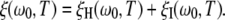 |
(8) |
These terms are given by
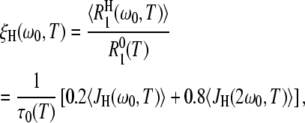 |
(9) |
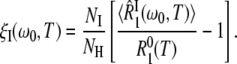 |
(10) |
Our aim here is to study the hydration-layer ADPF ξH(ω0,T), so we minimize the internal-water contribution ξI(ω0,T) by measuring R1 at a high resonance frequency, ω0/(2π) = 81.3 MHz. The remaining small ξI(ω0,T) contribution was computed from Eq. 10 with known parameter values (8,15). After this minor correction, we thus obtain ξH(ω0,T) from the experimental data. The central problem, which we now address, is to interpret the temperature dependence of ξH(ω0,T).
Dynamic heterogeneity in the hydration layer
The angular brackets in Eq. 9 signify an average over the temperature-dependent distribution f(τ,T) of correlation times τ in the hydration layer. To calculate this average, we need to know the mathematical form of the distribution function f(τ,T). From previous MRD studies (1,7), we know that the longest water correlation times in the hydration layer are in the nanosecond range at room temperature. The shortest correlation times, at exposed hydration sites, should be similar to the bulk-water correlation time of ∼2 ps at room temperature. The correlation time distribution is therefore very wide, spanning three orders of magnitude. The precise functional form of the distribution has not been determined experimentally, but the MRD data (1,7) require it to be highly skewed toward shorter τ-values, with only a small number of hydration sites in the nanosecond range. This limited experimental information is consistent with the more detailed results of molecular dynamics (MD) simulations (9,10,26,27). The simulations indicate that the distribution of correlation times in the hydration layer of proteins can be described by a power law. We therefore adopt the power-law form
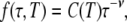 |
(11) |
where C(T) is a normalization constant and the exponent ν is assumed to be independent of temperature, as indicated by MD simulations (9).
At a given temperature, the correlation times of the NH = 380–735 water molecules (Table 1) in the hydration layer fall in a broad but finite range: τ–(T) ≤ τ ≤ τ+(T). Within this temperature-dependent range, the correlation times are taken to be distributed according to Eq. 11. Outside this range, f(τ,T) = 0. We must now specify the limits of the distribution. From previous MRD studies (1,6,7,28) and MD simulations (9,10,21,24,26,27,29–31) of protein solutions and from studies of the hydration dynamics of small organic solutes (5,32–35), we know that, at room temperature, the lower limit τ– is close to the bulk-water correlation time τ0 and that the upper limit τ+ is a few nanoseconds. As a natural choice for the upper limit, we choose the protein tumbling time τP, which is in the nanosecond range for the investigated proteins (Table 1). This choice is not critical, because the averages in Eq. 9 are quite insensitive to the upper limit τ+ (Appendix A). This is so because 1), the power-law distribution is highly skewed toward shorter correlation times; and 2), protein tumbling acts to filter-out longer correlation times (see Eq. 4). We introduce a reference temperature T*, where these limits apply. This temperature should be close to room temperature and we choose the temperature T* = 293.2 K, where many of the previous studies have been performed. At this temperature, τ0 = 1.8 ps (2,3). At the reference temperature, we thus have
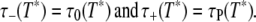 |
(12) |
To obtain the distribution f(τ,T) at an arbitrary temperature T, we must know how the limits τ–(T) and τ+(T) vary with temperature. From 17O spin relaxation measurements on supercooled bulk water, we know the temperature dependence of τ0 (2,3). Furthermore, the protein tumbling time τP is known to scale as η0(T)/T (36) and the temperature dependence of the bulk-water viscosity η0 is known (37). At low temperatures, and especially in the supercooled regime, both τ0 and η0 exhibit an anomalously strong (super-Arrhenius) temperature dependence, caused by interference with the cooperative rotation mechanism (38) as the bulk liquid structure becomes more open and tetrahedrally ordered, that is, more icelike (39). However, in the hydration layer, the fixed (temperature-independent) geometric and H-bonding constraints imposed by the protein surface prevent the structural changes responsible for the anomalous temperature dependence of bulk-water dynamics. We therefore expect water dynamics in the hydration layer to exhibit a weaker, more normal (Arrhenius-like) temperature dependence than in bulk water. This has recently been shown to be the case for peptides and other small organic solutes (5). We thus assume that the temperature dependence of the shortest and longest water correlation time in the protein hydration layer follow the Arrhenius law, but with different activation energies and preexponential factors. (Note that it is not necessary to assume that all water molecules in the hydration layer follow the Arrhenius law.) The limits of the power-law distribution, given by Eq. 12 at T*, then vary with temperature as
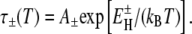 |
(13) |
Since the upper limit τ+(T) is unimportant (see above), the effect of the assumed Arrhenius law essentially enters via the lower limit τ–(T). We have tested Eq. 13 by analyzing the relaxation data with an extended model, where the activation energy 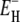 is allowed to depend linearly on temperature. The results indicate that Eq. 13 is an excellent approximation in the investigated temperature range (Appendix A).
is allowed to depend linearly on temperature. The results indicate that Eq. 13 is an excellent approximation in the investigated temperature range (Appendix A).
The width of the correlation time range is essentially determined by the activation energies 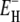 and
and 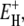 rather than by the preexponential factors A– and A+. In other words: |ln(A+/A−)|≪
rather than by the preexponential factors A– and A+. In other words: |ln(A+/A−)|≪ 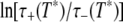 so that Eqs. 12 and 13 yield
so that Eqs. 12 and 13 yield
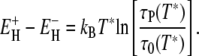 |
(14) |
The model thus contains only two free parameters, which we choose as 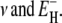
Averaging the TCF in Eq. 4 over the correlation time distribution in Eq. 11, we obtain
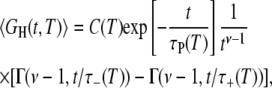 |
(15) |
with the incomplete γ-function defined as
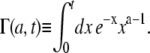 |
(16) |
The hydration-layer-averaged spectral density function in Eq. 9 can now be obtained from Eqs. 3 and 13–15. The protein tumbling time τP(T) in Eq. 15 is obtained from experimentally determined values at T* (Table 1) and the hydrodynamic η0(T)/T scaling (36). In general, the cosine transform in Eq. 3 must be evaluated numerically, but for the special case ν = 2 we obtain the simple analytical result
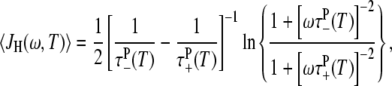 |
(17) |
where
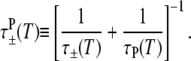 |
(18) |
At high frequencies and low temperatures, where 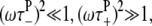 and τ− ≪ τ+,τp, Eq. 17 reduces to
and τ− ≪ τ+,τp, Eq. 17 reduces to
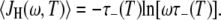 |
(19) |
Substitution of this result into Eq. 9 yields for the ADPF
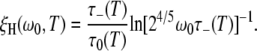 |
(20) |
Once the model parameters ν and 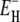 have been determined from a fit to the experimental ξH(ω0,T), we can calculate the temperature variation of the mean correlation time 〈τ〉 and the true DPF
have been determined from a fit to the experimental ξH(ω0,T), we can calculate the temperature variation of the mean correlation time 〈τ〉 and the true DPF 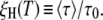 We can also calculate these quantities for subpopulations, such as the 50% most mobile water molecules in the hydration layer. To calculate the partial DPF ξH(T,p), we need the average correlation time 〈τ(T,p)〉 for the fraction p of hydration sites with shortest correlation times. For the power-law distribution in Eq. 11, we obtain
We can also calculate these quantities for subpopulations, such as the 50% most mobile water molecules in the hydration layer. To calculate the partial DPF ξH(T,p), we need the average correlation time 〈τ(T,p)〉 for the fraction p of hydration sites with shortest correlation times. For the power-law distribution in Eq. 11, we obtain
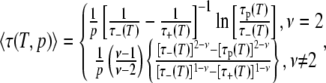 |
(21) |
with
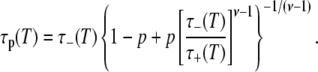 |
(22) |
For p = 1 (the entire hydration layer), Eq. 22 reduces to τp(T) = τ+(T). If τ+ ≫ τ–, Eq. 21 yields for ν = 2 and p = 1,
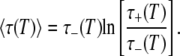 |
(23) |
For this case, the effective activation energy of 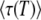 becomes
becomes
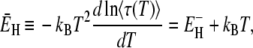 |
(24) |
where we have used Eq. 13.
A related, but more general, result can be obtained if we assume that the correlation times at all hydration sites obey the Arrhenius law with the same preexponential factor but with different activation energies. The power-law τ-distribution in Eq. 11 then corresponds to an exponential distribution of activation energies in the range 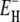 ≤
≤ 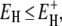
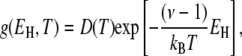 |
(25) |
with the normalization constant D(T). From this distribution, we can calculate the mean activation energy for the hydration layer. For τ+ ≫ τ–, we obtain the simple result
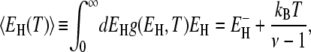 |
(26) |
which coincides with Eq. 24 for ν = 2.
EXPERIMENTAL RESULTS
For dilute aqueous solutions of the three proteins BPTI, ubiquitin, and β-lactoglobulin (BLG), we have measured the water 17O spin relaxation rate R1(ω0,T) at a resonance frequency of 81.3 MHz and in a 50 K temperature range extending down to 238 K. At each temperature, we also measured the relaxation rate  or a pure-water reference sample. To prevent the samples from freezing, the aqueous phase was dispersed as ∼10 μm-sized droplets in an emulsion (13). Because of the extremely strong temperature dependence of water dynamics at these low temperatures (2,3), high-precision relaxation measurements and careful temperature control are required (see Materials and Methods) to extract the hydration-layer dynamics from the small difference between
or a pure-water reference sample. To prevent the samples from freezing, the aqueous phase was dispersed as ∼10 μm-sized droplets in an emulsion (13). Because of the extremely strong temperature dependence of water dynamics at these low temperatures (2,3), high-precision relaxation measurements and careful temperature control are required (see Materials and Methods) to extract the hydration-layer dynamics from the small difference between 
The measured R1 is an average over all rapidly exchanging water molecules in the sample, comprising bulk water, hydration water (in direct contact with the protein surface), and internal water (buried in cavities inside the protein). The contributions from these water classes, described by the three terms in Eq. 1, are shown in Fig. 1 for the ubiquitin sample. The internal-water contribution, which is very small, was calculated (at the experimental temperatures) from Eqs. 1–6 with parameter values from Table 1 and Persson and Halle (8). The bulk-water contribution was obtained from the relaxation rate 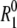 measured on a pure-water reference sample, using NW and NH values from Table 1. Finally, the hydration-layer contribution was obtained by subtracting the internal-water and bulk-water contributions from the relaxation rate R1 measured on the protein solution. The data in Fig. 1 show 1), that the hydration-layer contribution is an order-of-magnitude smaller than the bulk-water contribution at a protein concentration of 5 mM; and 2), that, at low temperatures, the hydration-layer contribution has a weaker temperature dependence than the bulk-water contribution.
measured on a pure-water reference sample, using NW and NH values from Table 1. Finally, the hydration-layer contribution was obtained by subtracting the internal-water and bulk-water contributions from the relaxation rate R1 measured on the protein solution. The data in Fig. 1 show 1), that the hydration-layer contribution is an order-of-magnitude smaller than the bulk-water contribution at a protein concentration of 5 mM; and 2), that, at low temperatures, the hydration-layer contribution has a weaker temperature dependence than the bulk-water contribution.
FIGURE 1.

Temperature dependence of different contributions to the water 17O relaxation rate R1 at 81.3 MHz for a 5.1 mM ubiquitin solution at pH 5.0, plotted on linear (left) and logarithmic (right) scales. The depicted R1 contributions correspond to the three terms in Eq. 1, representing 10,000 bulk water molecules (○), 443 water molecules in the hydration layer (•), and a single internal water molecule (▪).
For a quantitative comparison of the temperature dependencies of the hydration-layer and bulk-water contributions, we focus on their ratio, that is, on the apparent dynamic perturbation factor (ADPF) for the hydration layer, ξH(ω0,T), defined in Eq. 9. This quantity is obtained by subtracting the small (negligible at low temperatures) internal-water contribution ξI(ω0,T) from the total ADPF ξ(ω0,T), obtained from the measured R1(ω0,T) and 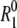 values by means of Eq. 7. The internal-water contribution is calculated with the aid of Eqs. 2, 3, 5, 6, and 10. For this calculation, we need to know, for each internal site, the order parameter
values by means of Eq. 7. The internal-water contribution is calculated with the aid of Eqs. 2, 3, 5, 6, and 10. For this calculation, we need to know, for each internal site, the order parameter 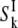 and the residence time
and the residence time 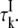 For BPTI, these quantities have been determined for all four internal water molecules at the reference temperature T* (8). The order parameters are governed by the protein structure and should therefore be essentially independent of temperature. The temperature dependence of the residence times is described by activation energies that have been determined experimentally (8). For ubiquitin (NI = 1) and BLG (NI = 3), the internal-water contribution is negligibly small at all investigated temperatures. Nevertheless, this small contribution was computed from the known or estimated internal-water parameters (8,15) and subtracted from the total ADPF.
For BPTI, these quantities have been determined for all four internal water molecules at the reference temperature T* (8). The order parameters are governed by the protein structure and should therefore be essentially independent of temperature. The temperature dependence of the residence times is described by activation energies that have been determined experimentally (8). For ubiquitin (NI = 1) and BLG (NI = 3), the internal-water contribution is negligibly small at all investigated temperatures. Nevertheless, this small contribution was computed from the known or estimated internal-water parameters (8,15) and subtracted from the total ADPF.
The measured total ADPF and its hydration-layer and internal-water contributions, separated as described above, are shown in Fig. 2 for the three proteins. Evidently, the internal-water contribution (dark shaded area) is significant only for BPTI, and then only at the higher temperatures. The dominant hydration-layer ADPF ξH(ω0,T) (light shaded area) exhibits a broad maximum at 267–271 K for the three proteins. Such a maximum has not been observed previously for proteins. The ξH(ω0,T) curves in Fig. 2 were obtained from fits where the two free parameters in the model were adjusted: the exponent ν in the power-law correlation time distribution (Eq. 11) and the activation energy 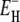 at the short-τ end of the distribution (Eq. 13). The resulting parameter values are given in Table 1. The exponent ν has the same value for BPTI and ubiquitin. The slightly smaller value for BLG implies a broader τ-distribution, consistent with an unusually large (but still ≪1) fraction of hydration sites with long correlation times (comparable to, or longer than, 1/ω0). This conclusion is consistent with the observation of an unusually strong frequency dependence in the 10–100 MHz range of the room-temperature MRD profile of BLG (15). The minimum activation energy
at the short-τ end of the distribution (Eq. 13). The resulting parameter values are given in Table 1. The exponent ν has the same value for BPTI and ubiquitin. The slightly smaller value for BLG implies a broader τ-distribution, consistent with an unusually large (but still ≪1) fraction of hydration sites with long correlation times (comparable to, or longer than, 1/ω0). This conclusion is consistent with the observation of an unusually strong frequency dependence in the 10–100 MHz range of the room-temperature MRD profile of BLG (15). The minimum activation energy 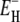 is 27 kJ mol−1 for all three proteins. From Eq. 14, we obtain
is 27 kJ mol−1 for all three proteins. From Eq. 14, we obtain 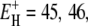 and 48 kJ mol−1 for BPTI, ubiquitin, and BLG, respectively. From Eq. 26, we obtain for all three proteins 〈EH(T*)〉 = 28 kJ mol−1 at the reference temperature T* = 293.2 K. The fitted values of ν and
and 48 kJ mol−1 for BPTI, ubiquitin, and BLG, respectively. From Eq. 26, we obtain for all three proteins 〈EH(T*)〉 = 28 kJ mol−1 at the reference temperature T* = 293.2 K. The fitted values of ν and 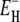 are insensitive to reasonable variations in τ–(T*) and τ+(T*) (Appendix A).
are insensitive to reasonable variations in τ–(T*) and τ+(T*) (Appendix A).
FIGURE 2.

Temperature dependence of the ADPF ξ(ω0,T) at 81.3 MHz for BPTI, ubiquitin, and BLG. The contributions from hydration water (light shading) and internal water (dark shading) are indicated. The curves were obtained by fitting the two model parameters ν and 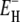 (Table 1) to the data (solid circles).
(Table 1) to the data (solid circles).
From the model parameters ν and 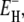 we can calculate the temperature variation of the true (rather than apparent) DPF ξH(T), which relates hydration-layer dynamics to bulk-water dynamics in an essentially method-independent and model-independent way. The temperature dependence of the DPF ξH(T) exhibits a maximum at 262 ± 1 K for all three proteins (Table 1, Fig. 3, left panel). Formally,
we can calculate the temperature variation of the true (rather than apparent) DPF ξH(T), which relates hydration-layer dynamics to bulk-water dynamics in an essentially method-independent and model-independent way. The temperature dependence of the DPF ξH(T) exhibits a maximum at 262 ± 1 K for all three proteins (Table 1, Fig. 3, left panel). Formally,
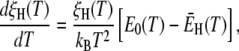 |
(27) |
where E0 and 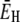 are the apparent Arrhenius activation energies of τ0 and 〈τ〉, respectively, defined as in Eq. 24. The DPF maximum thus occurs at the crossover temperature TX where the weakly temperature-dependent (Eq. 24) activation energy
are the apparent Arrhenius activation energies of τ0 and 〈τ〉, respectively, defined as in Eq. 24. The DPF maximum thus occurs at the crossover temperature TX where the weakly temperature-dependent (Eq. 24) activation energy 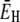 of the mean hydration-layer correlation time 〈τ〉 matches the strongly temperature-dependent activation energy E0 of the bulk-water correlation time τ0:
of the mean hydration-layer correlation time 〈τ〉 matches the strongly temperature-dependent activation energy E0 of the bulk-water correlation time τ0:
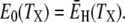 |
(28) |
The reduction of ξH(T) at lower temperatures is thus primarily caused by the drastic slowing down of bulk-water motions. The ADPF curves (Fig. 2) do not differ much from the DPF curves (Fig. 3, left panel), suggesting that the reason for the reduction of ξH(ω0,T) at low temperatures is the same as for ξH(T).
FIGURE 3.

Temperature dependence of the hydration-layer DPF ξH(T,p) for BPTI (solid curves), ubiquitin (dash), and BLG (dash-dot). The three panels show the DPF for all NH hydration waters (left), the 90% most mobile waters (middle), and the 50% most mobile waters (right).
At first sight, one might be tempted to interpret the ADPF maxima in Fig. 2 in terms of a maximum in the spectral density function 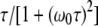 with increasing correlation time τ. However, this cannot be the case because nearly all water molecules in the hydration layer have subnanosecond correlation times even at the lowest investigated temperature. Furthermore, if this explanation were correct, then
with increasing correlation time τ. However, this cannot be the case because nearly all water molecules in the hydration layer have subnanosecond correlation times even at the lowest investigated temperature. Furthermore, if this explanation were correct, then 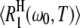 would exhibit a maximum, which is not the case (Fig. 1). The frequency dependence in
would exhibit a maximum, which is not the case (Fig. 1). The frequency dependence in 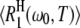 reduces the magnitude of ξH(ω0,T) and shifts the temperature of its maximum slightly, but it does not give rise to the ξH(ω0,T) maximum. This frequency dependence explains why the difference between the ADPF curves in Fig. 2 and the DPF curves in Fig. 3 (left panel) is largest for BLG, which contains the largest fraction hydration sites with correlation times of order 1/ω0 or longer (15). This fraction of relatively slow hydration sites also accounts for the substantially larger maximum DPF for BLG (7.4) as compared to BPTI (4.7) and ubiquitin (4.9) in Fig. 3 (left panel). (The experimentally determined power-law exponent ν is close to 2, where equal intervals of log(τ) make equal contributions to 〈τ〉.)
reduces the magnitude of ξH(ω0,T) and shifts the temperature of its maximum slightly, but it does not give rise to the ξH(ω0,T) maximum. This frequency dependence explains why the difference between the ADPF curves in Fig. 2 and the DPF curves in Fig. 3 (left panel) is largest for BLG, which contains the largest fraction hydration sites with correlation times of order 1/ω0 or longer (15). This fraction of relatively slow hydration sites also accounts for the substantially larger maximum DPF for BLG (7.4) as compared to BPTI (4.7) and ubiquitin (4.9) in Fig. 3 (left panel). (The experimentally determined power-law exponent ν is close to 2, where equal intervals of log(τ) make equal contributions to 〈τ〉.)
From the model parameters ν and 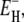 we can also calculate the partial DPF ξH(T,p) for the most mobile fraction p of the NH water molecules in the hydration layer (Eq. 21). Whereas the DPF can be strongly affected by a minor fraction of protein-specific hydration sites, the partial DPF for the 90 or 50% most mobile water molecules in the hydration layer should reflect the generic dynamics for most of the hydration layer. Although based on only three proteins, the convergence of the 50% DPF curves (Fig. 3, right panel) supports this view.
we can also calculate the partial DPF ξH(T,p) for the most mobile fraction p of the NH water molecules in the hydration layer (Eq. 21). Whereas the DPF can be strongly affected by a minor fraction of protein-specific hydration sites, the partial DPF for the 90 or 50% most mobile water molecules in the hydration layer should reflect the generic dynamics for most of the hydration layer. Although based on only three proteins, the convergence of the 50% DPF curves (Fig. 3, right panel) supports this view.
The different temperature dependence of bulk and hydration water dynamics is displayed in the form of an Arrhenius plot in Fig. 4. Whereas the τ0 curve is strongly curved (super-Arrhenius temperature dependence), the 〈τ〉 curves are nearly linear. The high degree of linearity is to some extent a result of assuming that τ–(T) obeys the Arrhenius law (Eq. 13), since for a broad distribution (with τ+≫ τ−), 〈τ〉 is approximately proportional to τ–(T) (Eq. 23). However, the data in Fig. 2 demonstrate in a model-independent way that the activation energy has a weaker temperature dependence for 〈τ〉 than for τ0. Moreover, three-parameter fits to the data in Fig. 2 allowing for a linear temperature dependence in  show that this dependence is insignificant (Appendix A). The data do not uniquely determine the functional form of the distribution f(τ,T), but they are consistent with a power-law distribution. Given this functional form, the data indicate that the temperature dependence of 〈τ〉 does not deviate much from the Arrhenius law in the range 238–288 K, where τ0 deviates strongly (Fig. 4).
show that this dependence is insignificant (Appendix A). The data do not uniquely determine the functional form of the distribution f(τ,T), but they are consistent with a power-law distribution. Given this functional form, the data indicate that the temperature dependence of 〈τ〉 does not deviate much from the Arrhenius law in the range 238–288 K, where τ0 deviates strongly (Fig. 4).
FIGURE 4.

Arrhenius plot showing the mean correlation time 〈τ〉 for the 50% (narrow shaded band) and 90% (wide shaded band) most mobile hydration waters and the correlation time τ0 in bulk water (solid curve). The shaded bands each contain three nearly linear curves for BPTI, ubiquitin, and BLG.
DISCUSSION
Proteins versus peptides
The DPF at 300 K has been determined previously from the MRD profiles for the three proteins studied here and several others (1,20). Extrapolating the curves in Fig. 3 (left panel) to 300 K, we find ξH values of 3.6 for BPTI and ubiquitin, and 5.3 for BLG, consistent with previous results (15,40). The mean and standard deviation for 11 monomeric proteins was reported as ξH = 1 + (0.30 ± 0.04) aW (1). For the mean solvent-accessible area occupied per water molecule, aW = 10.75 Å2, adopted here (see Materials and Methods), this yields ξH = 4.2 ± 0.4.
To what extent, and why, does the hydration layer differ from one protein to another? The majority of the several hundred water molecules that constitute the hydration layer of a protein are associated with convex protrusions on the surface and only a minority of the hydration waters occupy concave depressions or pockets on the protein surface. It is in the latter, more secluded, locations that we find the water molecules with the longest correlation times (1,10,26,27). The substantial variation of the full DPF ξH(T, p = 1) among different proteins (Fig. 3, left panel) reflects protein-specific variations in the relative abundance of such secluded hydrations sites. The convergence of the partial DPFs ξH(T, p) as the secluded hydrations sites are removed from the average (p = 1 → 0.9 → 0.5) suggests that the remaining major part of the hydration layer has the same average dynamics for most proteins (Fig. 3, middle and right panels).
Disregarding the protein-specific small subset of secluded hydration sites, does the dominant generic part of the protein hydration layer differ dynamically from the hydration shell of small organic solutes? The hydration structure of a small apolar solute (like methane) differs from that at a flat apolar surface (41), but solvent-exposed protein surfaces are not flat on the scale of a water molecule. Water NMR relaxation data over a wide temperature range have recently been reported for dilute aqueous (D2O) solutions of two small peptides: N-acetyl-glycine-N′-methylamide (NAGMA) and N-acetyl-leucine-N′-methylamide (NALMA) (5). Such small solutes do not contain the type of secluded hydration sites that are responsible for the long-τ tail of the τ-distribution for the protein hydration layer. Therefore, we have ω0τ ≪ 1 for all hydration sites at all investigated temperatures and ξH(ω0,T) = ξH(T) (Eq. 9). We thus obtain the DPF ξH(T) without having to specify the form of the correlation time distribution f(τ,T). (As explained in the Theory section, this is not possible for proteins.) Just as for the proteins studied here, NAGMA and NALMA yield a maximum in ξH(T). The observation of a ξH(T) maximum for small solutes supports our conclusion that the ξH(ω0,T) maximum observed here for proteins is caused by a crossover of activation energies rather than by a R1 maximum associated with correlation times of order 1/ω0. For all three proteins, we obtain TX = 263 ± 1 K for p = 0.9 and for p = 0.5 (Table 1, Fig. 3, middle and right panels), only 7 K above the TX value for the peptides (5). The maximum ξH (at TX) is 1.55 for NAGMA and 2.40 for the more hydrophobic NALMA (5). Very similar maximum ξH values are obtained for the proteins: 2.38 (1.55), 2.47 (1.61), and 2.76 (1.65) for BPTI, ubiquitin, and BLG, respectively, at p = 0.9 (0.5). From the similar ξH and TX values obtained for proteins and peptides, we conclude that the dynamics, and presumably also the structure, at the vast majority of protein hydration sites are essentially the same as in the hydration shell of small peptides (and other organic solutes (5)). The only essential difference between protein and small-solute hydration is the presence of a minority of secluded hydration sites on protein surfaces, responsible for the long-τ tail of the correlation time distribution.
The similar hydration dynamics of proteins and small solutes was noted in previous work from this laboratory (1,7), but the limited amount of temperature-dependent NMR relaxation data available at that time did not allow a quantitative comparison. While previously published low-temperature MRD profiles for BPTI (7) are consistent with the present findings, the nonmonotonic temperature dependence of the DPF was not anticipated. Instead, the observed ADPF reduction at low temperatures (for BPTI) was attributed to the gradual removal (when τ becomes comparable to 1/ω0) of the contribution to the ADPF from a few strongly perturbed water molecules in secluded hydration sites. That this is not the main reason for the ADPF reduction at low temperatures is demonstrated by the present finding of a similar reduction of the DPF for three proteins (Fig. 3, left panel) and by similar results for four small organic solutes (5). Therefore, the previous conclusion (1,7) that 〈τ〉, for most of the protein hydration layer, has a significantly weaker temperature dependence than 〈τ〉 for the hydration shell of small organic solutes must be revised. A quantitative comparison with low-temperature data for small organic solutes, not available until recently (5), reveals that the hydration dynamics of proteins and small solutes depend on temperature in essentially the same way (see below).
Dynamical heterogeneity in the protein hydration layer
The correlation time τ probed by spin relaxation is closely related to the mean residence time (MRT) of a water molecule in a hydration site. In exposed sites with short τ-values, the water molecule is more strongly coupled to water than to protein. Rotation (τ) and translation (MRT) should then occur on the same timescale, since they are both governed by rearrangement of water-water H-bonds (1,30,42). In secluded sites with long τ-values, the water molecule is more strongly coupled to protein than to water. Rotation is then hindered (that is, only librations are allowed) until the water molecule is released by the protein so, again, τ should be close to the MRT.
Two MD simulation studies have reported the MRT distribution for the protein hydration layer; in both cases, the power-law distribution in Eq. 11 was found to describe the MRT data over a wide range (9,10). For cytochrome c (9), the exponent ν ≈ 2.3 (for the range 1 ps−1 ns) at 300 K agrees closely with our results (Table 1). (Since the data shown in Fig. 6 of García and Hummer (9) were not corrected for logarithmic binning, the slope yields ν − 1; G. Hummer, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, personal communication, 2007.) The smaller exponent, ν = 0.84 (for the range ∼20 ps−20 ns), obtained for acetylcholinesterase (10) may reflect a broader τ-distribution for this 60 kDa protein, or it may simply be a consequence of the particular definition of time-averaged hydration sites used in that study (10).
Rotational correlation times are ideal for probing the dynamics in the hydration layer since rotation is a localized motion. In contrast, translational motion is inherently nonlocal, making it difficult to uniquely define a translational diffusion coefficient for the hydration layer. It is sometimes asserted that water diffusion in the hydration layer is anomalous in the sense that 〈(Δr)2〉 ∼ tα with α < 1 (29,31). For a continuous-time random walk with a power-law waiting time distribution, as in Eq. 11, one can show that α = ν – 1 (43). However, the subdiffusive (for α < 1) motion in this model results from the scale invariance of an unrestricted (valid for all τ) power-law distribution, leading to a divergent 〈τ〉. In contrast, in the protein hydration layer, the τ values are limited to a finite range [τ–, τ+]. Furthermore, a water molecule that starts out in the hydration layer will rarely visit more than a few hydration sites before reaching the bulk water region. The α-exponents <1 deduced from MD simulations (29,31) may thus be artifacts of treating an inhomogeneous system as homogeneous, rather than being an indication of a fundamental change in the diffusion mechanism.
The spin relaxation rate R1 reports on an orientational TCF 〈GH(t)〉 that is averaged over all hydration sites on the heterogeneous protein surface. After the ubiquitous subpicosecond damped librational oscillation, the TCF GH(t) for a given hydration site should decay in an approximately exponential fashion, as in Eq. 4. However, because of the broad correlation time distribution, the average TCF decays nonexponentially, as seen from Eq. 15. In simulation studies, the average TCF is sometimes fitted (in a limited time interval) to a stretched exponential function, 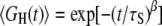 (24,29). The correlation time distribution f(τ) obtained by inverting this empirical function (44) does not exhibit the algebraic long-τ tail indicated by our data (and by MD simulations). The β-exponent is reported to be ∼0.5 or larger (24,29). For β = 0.5, the distribution implied by the stretched exponential TCF has the form f(τ) ∝
(24,29). The correlation time distribution f(τ) obtained by inverting this empirical function (44) does not exhibit the algebraic long-τ tail indicated by our data (and by MD simulations). The β-exponent is reported to be ∼0.5 or larger (24,29). For β = 0.5, the distribution implied by the stretched exponential TCF has the form f(τ) ∝ 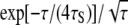 (44), which is very different from a power-law. The stretched exponential function may provide a decent fit in a limited time interval, but it has no theoretical basis and it misses the protein-specific algebraic tail of the correlation time distribution (which is rarely sampled with sufficient statistical accuracy in MD simulations).
(44), which is very different from a power-law. The stretched exponential function may provide a decent fit in a limited time interval, but it has no theoretical basis and it misses the protein-specific algebraic tail of the correlation time distribution (which is rarely sampled with sufficient statistical accuracy in MD simulations).
Temperature dependence of hydration dynamics
In our analysis, the observed temperature dependence of R1 is attributed entirely to water dynamics. We thus assume that the protein structure is independent of temperature so that the number NH of water molecules in the hydration layer is constant. At the low temperatures studied here, protein cold denaturation (45) is a potential complication. For proteins that are known to cold-denature, we have found that cold denaturation is accompanied by a characteristic increase in the ADPF ξ(ω0,T) at low temperatures (M. Davidovic, C. Mattea, J. Qvist, and B. Halle, unpublished). Because this feature is not seen for the three proteins investigated here, we conclude that these proteins do not cold-denature down to 238 K. This conclusion is consistent with the finding that the solution structure of BPTI is virtually identical at 258 and 309 K (46).
BLG is predominantly dimeric at neutral pH, but the large net charge (+20) at pH 2.7 reduces the dimer fraction to <10% under our salt-free conditions (47). Since the dimer interface buries only 6% of the protein's SASA (48), dimerization should not produce a significant temperature dependence in NH. Furthermore, we neglect the small contraction of the protein at low temperatures. The thermal expansivity of most proteins is ∼1 × 10−4 K−1 (49). Over a 50 K temperature interval, the protein volume thus changes by ∼0.5%, so NH changes by merely (1.005)2/3 − 1 = 0.3%.
The Arrhenius-like temperature dependence of the average correlation time 〈τ〉 for the protein hydration layer (Fig. 4) may seem inconsistent with the wide range of activation energies, from 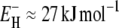 to
to 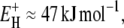 of the individual hydration sites on the protein surface. But the exponential EH distribution decays with a characteristic energy of kBT /(ν −1) so that the activation energy of 〈τ〉 is essentially
of the individual hydration sites on the protein surface. But the exponential EH distribution decays with a characteristic energy of kBT /(ν −1) so that the activation energy of 〈τ〉 is essentially 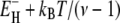 (Eqs. 24–26), which differs little from
(Eqs. 24–26), which differs little from 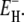 In other words: whereas 〈τ〉 has substantial contributions from the long-τ tail of the correlation time distribution, the temperature dependence of 〈τ〉 is almost completely determined by the more numerous hydration sites with τ close to the lower limit τ–. To understand the Arrhenius-like behavior of 〈τ〉, we must thus explain why
In other words: whereas 〈τ〉 has substantial contributions from the long-τ tail of the correlation time distribution, the temperature dependence of 〈τ〉 is almost completely determined by the more numerous hydration sites with τ close to the lower limit τ–. To understand the Arrhenius-like behavior of 〈τ〉, we must thus explain why 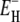 is constant whereas the bulk-water activation energy E0 varies strongly with temperature (2,3).
is constant whereas the bulk-water activation energy E0 varies strongly with temperature (2,3).
In bulk water, molecular rotation is a highly cooperative process where large energy barriers are circumvented by a concerted interchange of hydrogen-bonding partners (38,50,51). Decreasing temperature favors low-density configurations with high (icelike) tetrahedral order, which interfere with the cooperative rotation mechanism. This progressive structural change accounts for the dramatic increase of the apparent activation energy E0 in supercooled bulk water. In the protein hydration layer, the slowing down of water rotation can also be attributed to interference with the cooperative rotation mechanism, partly because of the reduced number of nearby water molecules with which to swap H-bonds and partly because H-bond partners in the protein are either absent (at apolar sites) or else are geometrically constrained (at polar sites). Because these constraints are essentially temperature-independent, the activation energy 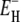 should not change much with temperature. In a sense, the hydration layer can be regarded as a defect in the surrounding, increasingly tetrahedrally ordered, supercooled solvent. At sufficiently low temperatures, we therefore expect most of the hydration layer to have higher mobility than bulk water. Indeed, our analysis predicts that 〈τ〉 for the 50 and 90% most mobile hydration waters falls below τ0 at 239 ± 1 K and 234 ± 1 K, respectively (Fig. 3). For the peptides NAGMA and NALMA, the corresponding temperature is 237 K (5). Thus, again we see that most of the water molecules in the protein hydration layer behave in the same way as water molecules in the hydration shell of small organic solutes.
should not change much with temperature. In a sense, the hydration layer can be regarded as a defect in the surrounding, increasingly tetrahedrally ordered, supercooled solvent. At sufficiently low temperatures, we therefore expect most of the hydration layer to have higher mobility than bulk water. Indeed, our analysis predicts that 〈τ〉 for the 50 and 90% most mobile hydration waters falls below τ0 at 239 ± 1 K and 234 ± 1 K, respectively (Fig. 3). For the peptides NAGMA and NALMA, the corresponding temperature is 237 K (5). Thus, again we see that most of the water molecules in the protein hydration layer behave in the same way as water molecules in the hydration shell of small organic solutes.
Coupling of protein and water dynamics
The solvent plays a dual role by fundamentally altering the protein's conformational free energy landscape and by providing much of the thermal energy and frictional damping that together govern diffusive conformational motions in the protein. In addressing the latter dynamical aspect, the solvent may be modeled, to a first approximation, as a homogeneous viscous continuum. This description is implicit in conventional hydrodynamic treatments of global dynamics, domain movements, and protein folding.
At the next level of approximation, the dynamic perturbation of the solvent by the protein may be incorporated by assigning a local viscosity ηH to the hydration layer, different from the bulk-water viscosity η0 (36). In bulk water, the Stokes-Einstein-Debye radius is constant over a wide temperature range, demonstrating that molecular rotation samples dynamical heterogeneities in the same way as the viscosity (J. Qvist, C. Mattea, and B. Halle, unpublished). The rotational correlation time of hydration water is therefore a reasonable proxy for the local viscosity in the hydration layer (36). In other words: the ratio ηH/η0 is essentially given by the DPF ξH. The relative importance of hydration layer and bulk water depends on the fraction of the viscous energy dissipation that occurs in each region. We therefore expect that the hydration layer is more important for localized motions than for large-scale motions and that it is more important for rotational motions than for translational motions (36). Our results show that most of the hydration layer is highly mobile, being retarded by a factor 2 or less (Fig. 3), as for the hydration shell of small organic solutes (5). While large-scale protein motions are only marginally affected by this perturbation (36), localized protein motions and ligand binding events that involve water displacement should be retarded to nearly the same extent as the hydration water. In view of the results in Fig. 4, we also expect that the frictional damping of localized protein motions has a weaker and more Arrhenius-like temperature dependence than if they were governed by bulk-water viscosity.
Free hydration layer versus confined water
In the protein solutions studied here, only 1–7% of the water molecules are in contact with the protein surface (Table 1). The water molecules in this free hydration layer interact with bulklike water as well as with the protein surface. Only by studying such dilute systems can we characterize the perturbation of bulk-water dynamics at the interface between liquid water and protein. Our results show that this perturbation has generic as well as protein-specific aspects (Fig. 3). This type of study represents a logical starting point for understanding protein-water interactions in vivo. It has been estimated that ∼15% of the water in an E. coli cell belongs to the first hydration layer of proteins or other macromolecular structures (52). It is therefore likely that most solvent-exposed protein surfaces in vivo are surrounded by multiple water layers. As a model system, a dilute protein solution is thus not only well defined but also of direct biological relevance.
A substantial literature has accumulated on the study of protein-water interactions in solid samples at low water content, such as rehydrated protein powders (53,54). Such samples have been studied for their biotechnological relevance or simply because of the difficulty of separating the hydration-layer response from the dominant bulk-water background in a protein solution. Here, we want to caution against uncritical extrapolation of results obtained on solid water-poor samples to in vivo or dilute-solution conditions.
In a protein powder at a typical hydration level of 0.3 g H2O (g protein)−1, the protein molecules are densely packed at 70% volume fraction. For hen egg white lysozyme (HEWL), this water content corresponds to NW = 240 or 40% of the 600 water molecules required to cover the protein surface (calculated as described in Materials and Methods). Some of the water molecules in this sample occupy subnanometer-sized interstitial spaces, others are buried in small cavities between contacting protein molecules. Few, if any, water molecules experience an environment that resembles a free hydration layer. Water dynamics in a protein powder may therefore be dominated by confinement effects and its temperature dependence may reflect structural and dynamic changes in the protein component more than intrinsic water dynamics.
These expectations are confirmed by a recent study of water diffusion (on a μm-length scale) in a HEWL powder sample with 0.3 g H2O (g protein)−1 (55). From the water self-diffusion coefficients measured in the powder sample (Dpowder) and in bulk water (D0), the DPF ξpowder = D0/Dpowder is found to vary from ∼20 at 288 K to ∼60 at 238 K (55). These values are 1–2 orders-of-magnitude larger than the DPF ξH for the free hydration layer (Fig. 3). (Note that the measured average diffusion coefficient Dpowder is dominated by the most mobile water molecules, whereas the average correlation time 〈τ〉 that we measure is biased toward the least mobile water molecules. This methodological difference should make ξpowder smaller than ξH, not larger.) Furthermore, the diffusion coefficient Dpowder was found to have an even more anomalous (super-Arrhenius) temperature dependence than the bulk-water diffusion coefficient D0, so ξpowder(T) increases monotonically at least down to 238 K (55). In contrast, for the free hydration layer, we find that 〈τ〉 has a much weaker temperature dependence than τ0 (Fig. 4), resulting in a crossover of activation energies and a decrease of ξH(T) at low temperatures (Fig. 3).
A series of recent studies indicate that water dynamics in a HEWL powder with 0.3 g H2O (g protein)−1 exhibit a weaker, Arrhenius-like temperature dependence to <∼220 K (55–58). It was suggested (55–58) that this change in the temperature dependence of water dynamics triggers the glasslike dynamical transition that is observed in incompletely hydrated protein samples at 170–230 K and below which conformational fluctuations in the protein are suppressed (54,59,60). Moreover, it was proposed (56–58) that this transition has the same underlying cause as the apparent power-law singularity in bulk water properties at a similar temperature (61), namely a metastable liquid-liquid critical point in bulk water (62).
We are skeptical to these proposals. If water in protein powders differs qualitatively from the free hydration layer, why should it resemble bulk water? Although neither the apparent singularity nor the metastable low-temperature phase behavior of bulk water are understood, it is widely believed that these and other anomalies are linked to the tetrahedral H-bond network in liquid water. In a HEWL powder sample with 0.3 g H2O (g protein)−1, there is not enough water to cover even half of the protein surface and few, if any, water molecules are likely to be fully coordinated by other water molecules. It is for this reason that water in protein powders does not form ice at any temperature; it remains in a thermodynamically stable state down to the glasslike dynamical transition. In contrast, bulk water, and the protein solutions studied here, are metastable with respect to ice Ih at subzero temperatures. Whereas the free hydration layer can be regarded as the perturbed interfacial region of a bulk liquid water phase, the water molecules in a protein powder do not participate in a bulklike H-bond network. In a protein powder, water and protein are so strongly coupled that it may be meaningless to say that either component triggers the dynamical transition, where water and protein motions are simultaneously suppressed.
CONCLUSIONS
The 17O NMR relaxation data presented here provide a quantitative view of water dynamics in the hydration layer that constitutes the protein-water interface. Our main conclusions are as follows:
The strong dynamical heterogeneity of the protein hydration layer can be described by a power-law distribution of rotational correlation times, f(τ) ∝ 1/τν, with exponent ν = 2.1–2.3 for the three proteins examined here. The hydration-layer-averaged correlation time 〈τ〉 obtained from the data thus has roughly equal contributions from equal logarithmic intervals within the broad τ-distribution.
The long-τ tail of the power-law distribution is associated with a protein-specific small population of slowly rotating water molecules, consistent with the finding from MD simulations that the most strongly perturbed water molecules reside in secluded sites (9,10,26,27).
The majority of the water molecules in the hydration layer exhibit a weak and generic (same for all proteins) dynamic perturbation. At room temperature, the average dynamic perturbation factor is ∼2 for 90% of the hydration layer and only ∼1.3 for the most mobile half of the layer.
At room temperature, the hydration-layer-averaged correlation time 〈τ〉 has a stronger temperature dependence than the bulk-water correlation time τ0, but at low temperatures the reverse is true. As a result, the dynamic perturbation factor ξH = 〈τ〉/τ0 has a nonmonotonic temperature dependence, with a maximum at the crossover temperature TX = 262 ± 1 K where the apparent activation energies of hydration layer and bulk water are equal. Below 239 K, most water molecules in the hydration layer are more mobile than in bulk water.
The hydration layer can be regarded as a defect in the predominantly tetrahedrally coordinated H-bond network of bulk water, induced by a protein surface that provides fewer and less flexible H-bonding opportunities for the adjacent water molecules. These constraints slow down water rotation because they interfere with the cooperative mechanism that facilitates rotation in bulk water. Because the constraints are essentially temperature-independent, hydration water does not follow the strongly super-Arrhenius temperature dependence of bulk water. In this sense, hydration water is less anomalous than bulk water.
With the exception of a small fraction of secluded hydration sites, the protein hydration layer differs little from the hydration shell of peptides and other small organic solutes (5). In both cases, the dynamic perturbation factor is <2 at room temperature and exhibits a maximum near 260 K.
The free hydration layer at the protein-water interface differs fundamentally from confined water in partially hydrated solid protein samples, where the dynamic perturbation factor is 1–2 orders-of-magnitude larger and has a qualitatively different temperature dependence.
Acknowledgments
We thank Hanna Nilsson for protein purification, Hans Lilja for NMR spectrometer maintenance, Erik Persson for NMR advice and discussions, and Bayer Healthcare AG for a generous supply of BPTI.
This work was supported by the Swedish Research Council, the Crafoord Foundation, and the Knut & Alice Wallenberg Foundation.
APPENDIX A
Robustness of model fit
The two free parameters ν and 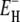 in the model described in the Theory section were fitted to the relaxation data in Fig. 2. The resulting parameter values are given in Table 1. We also performed fits with different choices for the limits τ−(T*) and τ+(T*) of the correlation time distribution at the reference temperature T*. The fit is insensitive to the upper limit. In particular, τ+(T*) can be increased without limit above τP(T*) without affecting the fit, because R1 depends on the effective correlation time (1/τ + 1/τP)−1 rather than on τ itself (Eq. 4). The upper limit τ+(T*) can also be reduced from τP(T*) by as much as a factor 4 without changing the parameters ν and
in the model described in the Theory section were fitted to the relaxation data in Fig. 2. The resulting parameter values are given in Table 1. We also performed fits with different choices for the limits τ−(T*) and τ+(T*) of the correlation time distribution at the reference temperature T*. The fit is insensitive to the upper limit. In particular, τ+(T*) can be increased without limit above τP(T*) without affecting the fit, because R1 depends on the effective correlation time (1/τ + 1/τP)−1 rather than on τ itself (Eq. 4). The upper limit τ+(T*) can also be reduced from τP(T*) by as much as a factor 4 without changing the parameters ν and 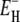 outside the error limits quoted in Table 1. The fit is more sensitive to the lower limitτ−(T*). Increasing or decreasing τ−(T*) by a factor 2 from τ0(T*) changes ν by 10–20% and
outside the error limits quoted in Table 1. The fit is more sensitive to the lower limitτ−(T*). Increasing or decreasing τ−(T*) by a factor 2 from τ0(T*) changes ν by 10–20% and 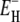 by 10–15%. Three-parameter fits, where τ−(T*) was also freely adjustable, gave τ−(T*) values well within this range.
by 10–15%. Three-parameter fits, where τ−(T*) was also freely adjustable, gave τ−(T*) values well within this range.
To examine the temperature dependence of 〈τ〉 in a less biased way, we removed the assumption of a strict Arrhenius temperature dependence for τ−(T*). (The temperature dependence of τ+(T*) is unimportant.) Specifically, we allowed the activation energy in Eq. 13 to depend on temperature according to 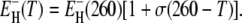 We then performed three-parameter fits where
We then performed three-parameter fits where 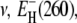 and σ were adjusted. The values for ν and
and σ were adjusted. The values for ν and 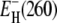 differed by <0.02 and <0.4 kJ mol−1, respectively, from the values given in Table 1. The temperature coefficient σ was not significantly different from zero for any of the three proteins. Taking the error limits into account, the fit indicates that |σ| < 0.001, corresponding to, at most, 5% variation of
differed by <0.02 and <0.4 kJ mol−1, respectively, from the values given in Table 1. The temperature coefficient σ was not significantly different from zero for any of the three proteins. Taking the error limits into account, the fit indicates that |σ| < 0.001, corresponding to, at most, 5% variation of  over the examined 50 K interval.
over the examined 50 K interval.
Carlos Mattea's present address is Institute of Physics, Technical University of Ilmenau, Ilmenau D-98684, Germany.
Editor: Arthur G. Palmer 3rd.
References
- 1.Halle, B. 2004. Protein hydration dynamics in solution: a critical survey. Philos. Trans. R. Soc. Lond. B Biol. Sci. 359:1207–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hindman, J. C. 1974. Relaxation processes in water: viscosity, self-diffusion, and spin-lattice relaxation. A kinetic model. J. Chem. Phys. 60:4488–4496. [Google Scholar]
- 3.Lang, E. W., and H.-D. Lüdemann. 1982. Anomalies of liquid water. Angew. Chem. Int. Ed. Engl. 21:315–329. [Google Scholar]
- 4.Bagno, A., F. Rastrelli, and G. Saielli. 2005. NMR techniques for the investigation of solvation phenomena and non-covalent interactions. Prog. Nucl. Magn. Reson. Spectrosc. 47:41–93. [Google Scholar]
- 5.Qvist, J., and B. Halle. 2008. Thermal signature of hydrophobic hydration dynamics. J. Am. Chem. Soc. In press. DOI: 10.1021/ja802668w. [DOI] [PubMed]
- 6.Halle, B., V. P. Denisov, and K. Venu. 1999. Multinuclear relaxation dispersion studies of protein hydration. In Biological Magnetic Resonance. N. R. Krishna, and L. J. Berliner, editors. Kluwer Academic/Plenum, New York.
- 7.Modig, K., E. Liepinsh, G. Otting, and B. Halle. 2004. Dynamics of protein and peptide hydration. J. Am. Chem. Soc. 126:102–114. [DOI] [PubMed] [Google Scholar]
- 8.Persson, E., and B. Halle. 2008. Nanosecond to microsecond protein dynamics probed by magnetic relaxation dispersion of buried water molecules. J. Am. Chem. Soc. 130:1774–1787. [DOI] [PubMed] [Google Scholar]
- 9.García, A. E., and G. Hummer. 2000. Water penetration and escape in proteins. Proteins. 38:261–272. [PubMed] [Google Scholar]
- 10.Henchman, R. H., and J. A. McCammon. 2002. Structural and dynamic properties of water around acetylcholinesterase. Protein Sci. 11:2080–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Otting, G. 1997. NMR studies of water bound to biological molecules. Prog. Nucl. Magn. Reson. Spectrosc. 31:259–285. [Google Scholar]
- 12.Halle, B. 2003. Cross-relaxation between macromolecular and solvent spins: the role of long-range dipole couplings. J. Chem. Phys. 119:12372–12385. [Google Scholar]
- 13.Rasmussen, D. H., and A. P. MacKenzie. 1973. Clustering in supercooled water. J. Chem. Phys. 59:5003–5013. [Google Scholar]
- 14.Angell, C. A. 1983. Supercooled water. Annu. Rev. Phys. Chem. 34:593–630. [Google Scholar]
- 15.Qvist, J., M. Davidovic, D. Hamelberg, and B. Halle. 2008. A dry ligand-binding cavity in a solvated protein. Proc. Natl. Acad. Sci. USA. 105:6296–6301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jóhannesson, H., and B. Halle. 1998. Minor groove hydration of DNA in solution: dependence on base composition and sequence. J. Am. Chem. Soc. 120:6859–6870. [Google Scholar]
- 17.Fraczkiewicz, R., and W. Braun. 1998. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Comput. Chem. 19:319–333. [Google Scholar]
- 18.Shrake, A., and J. A. Rupley. 1973. Environment and exposure to solvent of protein atoms. Lysozyme and insulin. J. Mol. Biol. 79:351–371. [DOI] [PubMed] [Google Scholar]
- 19.Kim, B., T. Young, E. Harder, R. A. Friesner, and B. J. Berne. 2005. Structure and dynamics of the solvation of bovine pancreatic trypsin inhibitor in explicit water: a comparative study of the effects of solvent and protein polarizability. J. Phys. Chem. B. 109:16529–16538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Denisov, V. P., and B. Halle. 1996. Protein hydration dynamics in aqueous solution. Faraday Discuss. 103:227–244. [DOI] [PubMed] [Google Scholar]
- 21.Schröder, C., T. Rudas, S. Boresch, and O. Steinhauser. 2006. Simulation studies of the protein-water interface. I. Properties at the molecular resolution. J. Chem. Phys. 124:234907. [DOI] [PubMed] [Google Scholar]
- 22.Woessner, D. E. 1980. An NMR investigation into the range of the surface effect on the rotation of water molecules. J. Magn. Reson. 39:297–308. [Google Scholar]
- 23.Carlström, G., and B. Halle. 1988. Water dynamics in microemulsion droplets. A nuclear spin relaxation study. Langmuir. 4:1346–1352. [Google Scholar]
- 24.Abseher, R., H. Schreiber, and O. Steinhauser. 1996. The influence of a protein on water dynamics in its vicinity investigated by molecular dynamics simulation. Proteins. 25:366–378. [DOI] [PubMed] [Google Scholar]
- 25.Smolin, N., and R. Winter. 2004. Molecular dynamics simulations of Staphylococcal nuclease: properties of water at the protein surface. J. Phys. Chem. B. 108:15928–15937. [Google Scholar]
- 26.Makarov, V. A., B. K. Andrews, P. E. Smith, and B. M. Pettitt. 2000. Residence times of water molecules in the hydration sites of myoglobin. Biophys. J. 79:2966–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luise, A., M. Falconi, and A. Desideri. 2000. Molecular dynamics simulation of solvated azurin: correlation between surface solvent accessibility and water residence times. Proteins. 39:56–67. [DOI] [PubMed] [Google Scholar]
- 28.Halle, B. 1998. Water in biological systems: the NMR picture. In Hydration Processes in Biology. M.-C. Bellisent-Funel, editor. IOS Press, Dordrecht, The Netherlands.
- 29.Bizzarri, A. R., and S. Cannistraro. 2002. Molecular dynamics of water at the protein-solvent interface. J. Phys. Chem. B. 106:6617–6633. [Google Scholar]
- 30.Marchi, M., F. Sterpone, and M. Ceccarelli. 2002. Water rotational relaxation and diffusion in hydrated lysozyme. J. Am. Chem. Soc. 124:6787–6791. [DOI] [PubMed] [Google Scholar]
- 31.Pizzitutti, F., M. Marchi, F. Sterpone, and P. J. Rossky. 2007. How protein surfaces induce anomalous dynamics of hydration water. J. Phys. Chem. B. 111:7584–7590. [DOI] [PubMed] [Google Scholar]
- 32.Ishimura, M., and H. Uedaira. 1990. Natural abundance O-17 magnetic relaxation in aqueous solutions of apolar amino acids and glycine peptides. Bull. Chem. Soc. Jpn. 63:1–5. [Google Scholar]
- 33.Bagno, A., G. Lovato, G. Scorrano, and J. W. Wijnen. 1993. Solvation of nonelectrolytes in water probed by 17O NMR relaxation of the solvent. J. Phys. Chem. 97:4601–4607. [Google Scholar]
- 34.Ishihara, Y., S. Okouchi, and H. Uedaira. 1997. Dynamics of hydration of alcohols and diols in aqueous solution. J. Chem. Soc., Faraday Trans. 93:3337–3342. [Google Scholar]
- 35.Shimizu, A., K. Fumino, K. Yukiyasu, and Y. Taniguchi. 2000. NMR studies on the dynamic behavior of water molecules in aqueous denaturant solutions at 25°C: effects of guanidine hydrochloride, urea and alkylated ureas. J. Mol. Liq. 85:269–278. [Google Scholar]
- 36.Halle, B., and M. Davidovic. 2003. Biomolecular hydration: from water dynamics to hydrodynamics. Proc. Natl. Acad. Sci. USA. 100:12135–12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hallett, J. 1963. The temperature dependence of the viscosity of supercooled water. Proc. Phys. Soc. 82:1046–1050. [Google Scholar]
- 38.Laage, D., and J. T. Hynes. 2006. A molecular jump mechanism of water reorientation. Science. 311:832–835. [DOI] [PubMed] [Google Scholar]
- 39.Debenedetti, P. G. 2003. Supercooled and glassy water. J. Phys. Cond. Mat. 15:R1669–R1726. [Google Scholar]
- 40.Denisov, V. P., and B. Halle. 1995. Protein hydration dynamics in aqueous solution: a comparison of bovine pancreatic trypsin inhibitor and ubiquitin by oxygen-17 spin relaxation dispersion. J. Mol. Biol. 245:682–697. [DOI] [PubMed] [Google Scholar]
- 41.Chandler, D. 2005. Interfaces and the driving force of hydrophobic assembly. Nature. 437:640–647. [DOI] [PubMed] [Google Scholar]
- 42.Geiger, A., M. Kleene, D. Paschek, and A. Rehtanz. 2003. Mechanisms of the molecular mobility of water. J. Mol. Liq. 106:131–146. [Google Scholar]
- 43.Klafter, J., G. Zumofen, and A. Blumen. 1993. Non-Brownian transport in complex systems. Chem. Phys. 177:821–829. [Google Scholar]
- 44.Lindsey, C. P., and G. D. Patterson. 1980. Detailed comparison of the Williams-Watts and Cole-Davidson functions. J. Chem. Phys. 73:3348–3357. [Google Scholar]
- 45.Privalov, P. L. 1990. Cold denaturation in proteins. Crit. Rev. Biochem. Biophys. 25:281–305. [DOI] [PubMed] [Google Scholar]
- 46.Shen, Y., and T. Szyperski. 2008. Structure of the protein BPTI derived with NOESY in supercooled water: validation and refinement of solution structures. Angew. Chem. Int. Ed. 47:324–326. [DOI] [PubMed] [Google Scholar]
- 47.Sakurai, K., M. Oobatake, and Y. Goto. 2001. Salt-dependent monomer-dimer equilibrium of bovine β-lactoglobulin at pH 3. Protein Sci. 10:2325–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brownlow, S., J. H. M. Cabral, R. Cooper, D. R. Flower, S. J. Yewdall, I. Polikarpov, A. C. T. North, and D. W. Sawyer. 1997. Bovine β-lactoglobulin at 1.8 Å resolution—still an enigmatic lipocalin. Structure. 5:481–495. [DOI] [PubMed] [Google Scholar]
- 49.Chalikian, T. V. 2003. Volumetric properties of proteins. Annu. Rev. Biophys. Biomol. Struct. 32:207–235. [DOI] [PubMed] [Google Scholar]
- 50.Sciortino, F., A. Geiger, and H. E. Stanley. 1991. Effect of defects on molecular mobility in liquid water. Nature. 354:218–221. [Google Scholar]
- 51.Rodríguez Fris, J. A. R., G. A. Appignanesi, E. La Nave, and F. Sciortino. 2007. Metabasin dynamics and local structure in supercooled water. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 75:041501. [DOI] [PubMed] [Google Scholar]
- 52.Persson, E., and B. Halle. 2008. Cell water dynamics on multiple time scales. Proc. Natl. Acad. Sci. USA. 105:6266–6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rupley, J. A., and G. Careri. 1991. Protein hydration and function. Adv. Protein Chem. 41:37–172. [DOI] [PubMed] [Google Scholar]
- 54.Gregory, R. B. 1995. Protein hydration and glass transition behavior. In Protein-Solvent Interactions. R. B. Gregory, editor. Marcel Dekker, New York.
- 55.Mallamace, F., S.-H. Chen, M. Broccio, C. Corsaro, V. Crupi, D. Majolino, V. Venuti, P. Baglioni, E. Fratini, C. Vannucci, and H. E. Stanley. 2007. Role of the solvent in the dynamical transitions of proteins: the case of the lysozyme-water system. J. Chem. Phys. 127:045104. [DOI] [PubMed] [Google Scholar]
- 56.Chen, S.-H., L. Liu, E. Fratini, P. Baglioni, A. Faraone, and E. Mamontov. 2006. Observation of fragile-to-strong dynamic crossover in protein hydration water. Proc. Natl. Acad. Sci. USA. 103:9012–9016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kumar, P., Z. Yan, L. Xu, M. G. Mazza, S. V. Buldyrev, S.-H. Chen, S. Sastry, and H. E. Stanley. 2006. Glass transition in biomolecules and the liquid-liquid critical point of water. Phys. Rev. Lett. 97:177802. [DOI] [PubMed] [Google Scholar]
- 58.Lagi, M., X. Chu, C. Kim, F. Mallamace, P. Baglioni, and S.-H. Chen. 2008. The low-temperature dynamic crossover phenomenon in protein hydration water: simulations vs experiments. J. Phys. Chem. B. 112:1571–1575. [DOI] [PubMed] [Google Scholar]
- 59.Daniel, R. M., R. V. Dunn, J. L. Finney, and J. C. Smith. 2003. The role of dynamics in enzyme activity. Annu. Rev. Biophys. Biomol. Struct. 32:69–92. [DOI] [PubMed] [Google Scholar]
- 60.Doster, W. 2008. The dynamical transition of proteins, concepts and misconceptions. Eur. Biophys. J. 37:591–602. [DOI] [PubMed] [Google Scholar]
- 61.Speedy, R. J., and C. A. Angell. 1976. Isothermal compressibility of supercooled water and evidence for a thermodynamic singularity at −45°C. J. Chem. Phys. 65:851–858. [Google Scholar]
- 62.Poole, P. H., F. Sciortino, U. Essmann, and H. E. Stanley. 1992. Phase behavior of metastable water. Nature. 360:324–328. [Google Scholar]