

Abstract
In purple bacteria, photosynthesis is carried out on large indentations of the bacterial plasma membrane termed chromatophores. Acting as primitive organelles, chromatophores are densely packed with the membrane proteins necessary for photosynthesis, including light harvesting complexes LH1 and LH2, reaction center (RC), and cytochrome bc1. The shape of chromatophores is primarily dependent on species, and is typically spherical or flat. How these shapes arise from the protein-protein and protein-membrane interactions is still unknown. Now, using molecular dynamics simulations, we have observed the dynamic curvature of membranes caused by proteins in the chromatophore. A membrane-embedded array of LH2s was found to relax to a curved state, both for LH2 from Rps. acidophila and a homology-modeled LH2 from Rb. sphaeroides. A modeled LH1-RC-PufX dimer was found to develop a bend at the dimerizing interface resulting in a curved shape as well. In contrast, the bc1 complex, which has not been imaged yet in native chromatophores, did not induce a preferred membrane curvature in simulation. Based on these results, a model for how the different photosynthetic proteins influence chromatophore shape is presented.
INTRODUCTION
Protein-induced shaping of cellular membranes has enjoyed a great deal of interest in recent years (1,2). Common mechanisms of membrane shaping by proteins involve either protein scaffolds, e.g., BAR domains (3,4), or the shape and oligomerization of integral membrane proteins, such as reticulons and DP1/Yop1p in the endoplasmic reticulum (5). An elegant example of membrane reshaping by membrane proteins is seen in the chromatophores of purple photosynthetic bacteria (6,7). Chromatophores are the bacterial photosynthetic pseudo-organelles which form as extensions of the cytoplasmic membrane upon transition to phototrophic growth and house the photosynthetic proteins (see Fig. 1). The shape of these chromatophores varies among species, the two most common forms being stacks of flat lamellar folds seen in Rhodopseudomonas (Rps.) acidophila and Rhodospirillum (Rs.) molischianum and small spherical vesicles in Rhodobacter (Rb.) sphaeroides and Rb. capsulatus (8–11). It is thought that the shape of the chromatophore is determined by the proteins it contains, light harvesting complexes I and II (LH1 and LH2), cytochrome bc1 (bc1), and possibly ATP synthase, and their interactions with the membrane and with each other (6,7,12,13).
FIGURE 1.

Schematic of the vesicular photosynthetic chromatophore. Shown here is a drawing of a chromatophore from Rb. sphaeroides. The LH1-RC dimers and LH2 complexes are closely packed in the bulb of the chromatophore, as seen in AFM images, and the bc1 complex and ATP synthase, which are absent from AFM images, are tentatively placed near the neck of the chromatophore.
While the evolution of photosynthesis is quite complex (14,15), purple bacteria are likely the earliest photosynthetic organisms and as such have one of the simplest photosynthetic organelles (16,17). The so-called photosynthetic unit of purple bacteria consists of only five types of membrane proteins: reaction center (RC), LH1, LH2, bc1, and ATP synthase. Photons are absorbed by LH2, large arrays of which serve as broad light-harvesting antennae. After light absorption, LH2 passes its excitation energy on to LH1, which then passes the excitation energy to the RC, where electron transfer across the membrane is initiated. Within the RC, electrons and protons from the cytoplasm are given to molecules of quinone, forming quinols. The quinols diffuse through the membrane to bc1, where the electrons and protons are released into the periplasm, resulting in a proton gradient. This proton gradient, in turn, drives the synthesis of ATP in ATP synthase.
Structurally, LH2 is a ring-shaped oligomer which scaffolds an array of bacteriochlorophylls and carotenoids. LH1 is an oligomeric protein which directly surrounds the RC, either forming a ring around the RC in its monomeric form, or forming an S-shaped structure in its dimeric form, a choice which appears to be determined by the absence or presence of the protein PufX in the Rhodobacter (Rb.) species (7,12). There is a wealth of structural information on these photosynthetic proteins. Crystal structures of RC have been published for a variety of species, including Rb. sphaeroides (18,19), and crystal structures of LH2 exist for Rps. acidophila and Rs. molischianum (20,21). The structure of bc1 from Rb. sphaeroides has also become available recently (22). There are presently no fully resolved structures available for a complete LH1 complex, although the Rb. sphaeroides LH1 β-subunit and the dimerizing protein PufX have been individually resolved (23,24).
On a larger scale, AFM imaging has provided some insight into the organization of the photosynthetic proteins in the chromatophore. Since the lamellar chromatophores of some species, such as Rs. molischianum and Rps. palustris, are naturally flat, they are amenable to AFM imaging in their native form (8,10,25). The small vesicular chromatophores of species such as Rb. sphaeroides and Rb. blasticus cannot be imaged in their native form, but fractured or freeze-thawed membranes have been successfully examined (13,26). Images of native chromatophores from Rs. molischianum and Rps. palustris show large regions of closely-packed LH2 and LH1 monomers (27,28). Fractured Rb. sphaeroides membranes reveal distinctive rows of LH1-RC-PufX interspersed with LH2s. Farther away from the LH1-RC-PufX dimers, large regions of closely-packed LH2 complexes are seen (13). Images of freeze-thawed Rb. blasticus membranes also show LH1-RC-PufX dimers and LH2, but they lack the domain organization seen in Rb. sphaeroides (26). Whether this is caused by membrane preparation or is representative of the natural state of Rb. blasticus membranes is not known. ATP synthase and bc1 are enigmatically absent in all AFM images of chromatophore membranes (13,27). Their absence has prompted hypotheses placing ATP synthase and bc1 at either the neck of the chromatophore, where it connects to the cytoplasmic membrane (see Fig. 1), or at the pole of the chromatophore vesicle, as these portions of the membrane are possibly lost in sample preparation (29,30); however, these hypotheses do not explain the absence of the proteins in the untreated Rs. molischianum membranes (27).
Several articles have discussed how single integral membrane proteins may individually affect membrane curvature (31,32). Simulations have previously demonstrated collective lipid responses to the transmembrane α-helices of the nicotinic acetylcholine receptor. Differently reacting leaves of the bilayer and a correlation between degree of lipid response and distance from the α-helices were found, indicating that specific protein-lipid interactions may be crucial to understanding both the structure and function of membrane-bound protein complexes (33). In addition, theoretical studies have previously been used successfully to study the structure of photosynthetic proteins as related to their function (34,35). On a larger scale, the cooperative action of many integral membrane proteins can induce protein aggregation (36–38). A recent coarse-grained simulation study observed the aggregation of protein caps in a membrane, and the subsequent production of a vesicle from their combined action on the membrane (39). A similar Monte Carlo study explored the effects of LH1 and LH2 aggregation, modeling LH1 and LH2 as beads with intrinsic curvature properties (7) and found that vesiculation is induced by the packing of the proteins, forming a vesicle not unlike those postulated by Sener et al. (29) and Geyer and Helms (30). This idea is supported by radiolabeling experiments on Rb. sphaeroides chromatophores, which indicate that aggregation of LH1-RC-PufX dimers and LH2s in the membrane directly precedes the formation of the chromatophores (6), as well as the observed close-packing of the light-harvesting proteins in AFM images (13,26–28).
Presented in this article are the first set of simulations on the three chromatophore proteins, LH2, LH1-RC-PufX dimer, and bc1, designed to study their curvature properties. Systems of seven LH2s pack and induce curvature in a way that depends on their packing density. A single LH1-RC-PufX dimer spontaneously bends at its dimerizing interface in a way that also induces curvature. bc1 does not induce curvature in simulation, neither singly nor in pairs. These simulations are the beginning of a study of much broader scope, which seeks to explore the interactions between many light harvesting proteins that lead to the creation of a chromatophore.
METHODS
The absence of complete structures for LH2 and LH1-RC-PufX from Rb. sphaeroides necessitated the use of homology modeling. Once the protein models were completed, the proteins were placed in noncontinuous rectangular patches of lipid membrane and equilibrated. The details of the protein modeling and system construction are described in this section.
Homology model construction
LH2 models
The Rps. acidophila LH2 model was taken from the 2.0 Å crystal structure (Protein Data Bank entry 1NKZ (21)). The Rb. sphaeroides LH2 was modeled based on the Rps. acidophila crystal structure by mapping the homologous Rb. sphaeroides amino-acid sequence onto the Rps. acidophila structure. Rps. acidophila LH2 was chosen over Rs. molischianum LH2, which also has a high-resolution crystal structure available (20), because both Rps. acidophila and Rb. sphaeroides are known to have nine subunits whereas Rs. molischianum has eight (20,21,40).
Since the sequence similarity between the protein subunits of Rps. acidophila and Rb. sphaeroides LH2s is very high (see Fig. 2), the mapping is straightforward. The only part of the Rb. sphaeroides sequence that could not be modeled in this way was an 11-residue insertion on the N-terminus of the β-subunit. Secondary structure predictions using PSIPRED (41) for this sequence (MTDDLNKVW) showed some affinity for a helical arrangement, and modeling of the sequence as an α-helix with the VMD (42) plug-in Molefacture revealed an amphipathic helix. Several other species of purple bacteria (Rb. capsulatus, Rd. sulfidophilum, and Rbv. gelatinosus) contain a similar N-terminal extension of the β-subunit. Experiments have established that at least part of the homologous N-terminal extension in Rb. capsulatus is inaccessible to solvent, suggesting that it may be submerged in the membrane (43). An EM map of Rd. sulfidophilum LH2 shows a density between adjacent β-subunits which is not present in the EM map of Rps. acidophila; the density may be attributable to the N-terminal extension lying between them (44). Based on these observations, the Rb. sphaeroides N-terminal region was modeled as an α-helix, and was placed parallel to the membrane with its hydrophobic face submerged, in a typical snorkeling amphipathic helix position. These helices were packed against the LH2 mass so that they bridged the space between adjacent β-subunits. During simulation, the helices tended to stay near their starting position, suggesting that this is a favorable placement.
FIGURE 2.

Comparison of LH2 from Rps. acidophila and Rb. sphaeroides. (Upper) Sequence alignment of the α- and β-subunits from Rps. acidophila and Rb. sphaeroides LH2. The α- and β- (mainly helical) subunits show 44% and 52% sequence identities, respectively. Conserved residues are highlighted in blue. The most notable difference between the two species is an N-terminal insertion in the β-helix of Rb. sphaeroides, highlighted in green. (Lower) Structural comparison of the Rps. acidophila LH2 crystal structure and the Rb. sphaeroides LH2 model. The proteins are shown in a surface representation, colored according to residue type (blue is basic, red is acidic, green is polar, and white is nonpolar). The modeled N-terminal portion is indicated.
The placement of the chromophore molecules in the Rb. sphaeroides model was based on that in the Rps. acidophila structure. The bacteriochlorophylls are ligated by the same residues in both Rps. acidophila and Rb. sphaeroides (45), so the bacteriochlorophylls from the Rps. acidophila structure were retained in the Rb. sphaeroides model. The carotenoid present in Rb. sphaeroides is primarily spheroidene (46). Placement was done by mutating the rhodopsin glucosides in the Rps. acidophila structure into spheroidenes, to preserve the contacts that the carotenoids make with the protein and bacteriochlorophylls.
LH1-RC-PufX model
Currently, no complete high resolution crystal structure is available for the LH1 of any photosynthetic purple bacteria, so it was necessary to construct a model for the Rb. sphaeroides LH1-RC-PufX dimer based on available solution structures and EM data. For the LH1 β-apoprotein we used the NMR solution structure (PDB entry 1DX7) (23). Lacking an LH1 α-apoprotein structure for Rb. sphaeroides, we used the solution structure from Rsp. rubrum reported in Wang et al. (47) (PDB entry 1XRD), noting that the two polypeptides have a high sequence identity (43.10%), making homology modeling feasible. A sequence alignment was performed between the α-apoproteins from Rb. sphaeroides (58 residues) and Rsp. rubrum (52 residues) (see Supplementary Material in Data S1). The Rsp. rubrum LH1 α-apoprotein residues 1–48 were mutated to form residues 4–51 of Rb. sphaeroides LH1 α-apoprotein. The three-residue sequence (MSK) at the N-terminus of Rb. sphaeroides α-apoproteins was added as a coil. The seven-residue sequence (NRVAVAE) at the C-terminus was modeled to be a short α-helix with the VMD (42) plug-in Molefacture. Two bacteriochlorophylls and one carotenoid (spheroidene) were then placed in the space between the α/β-apoprotein pair to form an LH1 subunit, their positions chosen to be similar to the chromophore placement in Rs. molischianum LH2 (20). Energy minimization was performed on the resulting LH1 protein-pigment subunit to ensure proper ligation between the Mg atoms of the bacteriochlorophylls and the highly-conserved histidine residues in the α- and β-apoproteins. The resulting LH1 subunit model was replicated into 28 subunits; these subunits were then rotated and arranged into the S-shaped dimer with subunit-subunit distances as observed in the cryo-EM data (48).
The structure of Rb. sphaeroides RC was provided in several x-ray diffraction studies (18,19). Two RCs (PDB entry 1PCR) were placed in the LH1 dimer model in the same location and orientation as observed in Qian et al. (48). We then used the recently available solution structure of PufX (24) and placed two PufX proteins inside the LH1-RC dimer at the locations suggested in Qian et al. (48). A PDB file of the LH-RC-PufX dimer used in this study is available upon request.
Simulated systems
Noncontinuous membranes
All systems were simulated in a noncontinuous lipid bilayer. Typically in membrane simulations, the membrane extends to the lateral edges of the unit cell, and is thus continuous through the periodicity of the system. However, the continuity of the bilayer at the unit cell edge makes it difficult to observe curvature of the membrane. One way to circumvent this problem is to make a significantly longer bilayer such that local curvature effects do not need to involve the entire membrane (49). Since our system sizes are already quite large, we instead chose to truncate the bilayer short of the unit cell boundary, as shown in Fig. 3. When simulated for ∼3 ns, we see lipids move to cap the edge of the bilayer exposed to water in a micellar fashion. This rearrangement of the simulated lipids occurs on a timescale similar to that of lateral lipid diffusion (∼3 ± 0.6 Å2/ns) previously observed experimentally and reproduced in simulations of phospholipid bilayers (50).
FIGURE 3.

Pure membrane in a water box with extra water padding around the membrane edge. A short equilibration was performed for a 50% POPE/50% POPG lipid patch with patch size 100 Å × 200 Å. A 10 Å gap is left between the edge of the lipid and the edge of the water box in all three directions. The phosphorus atoms are shown as spheres, and the water box is transparent. (a) Starting arrangement (t = 0 ns) in top (top), and side (bottom) view. (b) Lipid system at the end of equilibration at t = 2.5 ns, in top (top), and side (bottom) view.
LH2 systems
Three separate LH2 systems were constructed; all three contain seven LH2s placed in a 300 Å × 300 Å noncontinuous membrane patch composed of 50% POPE and 50% POPG lipids. The first simulation contained seven Rb. sphaeroides LH2s arranged in a hexagonal array, allowing enough space between neighboring LH2s for a single layer of lipids. The protein/lipid system was placed in a water box of size 350 Å × 350 Å × 82 Å and the system charge was neutralized by the addition of sodium ions, giving an ion concentration of 257 mM. The Rb. sphaeroides system contained 909,830 atoms total. The second simulation contained seven Rps. acidophila LH2s that were arranged in the same manner as in the first simulation, and placed in the same membrane patch; simulated were a total of 988,391 atoms, at an ion concentration of 208 mM. The third simulation also contained seven Rps. acidophila LH2s arranged in a hexagonal array, but in this simulation the LH2s were packed so closely that there was no continuous layer of lipid between neighboring LH2s. This more closely-packed Rps. acidophila system contained 996,535 atoms and had an ion concentration of 223 mM.
In addition to the three LH2 simulations, a control simulation was performed containing 19 copies of the protein rhodopsin. The 19 rhodopsins were arranged in a hexagonal array such that the area covered by the rhodopsins was comparable to that covered by the seven LH2s in the previous simulations, and they were placed in the same 300 Å × 300 Å membrane patch used in the LH2 simulations. This system was placed in a water box of size 350 Å × 350 Å × 77 Å, contained 880,243 atoms, and had an ion concentration of 315 mM.
All systems were first minimized and equilibrated in an NVT ensemble for 0.5 ns with all atoms except the lipid tails restrained. The system was then equilibrated in an NPT ensemble with only the protein and pigments restrained for ∼150 ps to allow the water molecules to equilibrate and the water box size to stabilize. Finally, the system was equilibrated in an NPT ensemble without restraints.
LH1-RC-PufX system
The completed LH1-RC-PufX model was placed in a 172 Å × 320 Å membrane patch composed of 50% POPE and 50% POPG lipids. The protein/membrane system was placed in a water box of size 196 Å × 344 Å × 119 Å. The system charge was neutralized by the addition of Na+ and Cl− ions at a concentration of 320 mM. The final system contained 688,373 atoms. Fig. 4 a provides a schematic view of the LH1-RC-PufX system.
FIGURE 4.

Simulation of the LH1-RC-PufX dimer. The S-shaped LH1 is colored in gray, surrounding the two RCs in white, and PufX is shown in black. (a) Snapshots at the beginning of the simulation, viewed from the cytoplasm (top), and in the plane of the membrane (bottom). (b) Snapshot of the simulated system at the end of the 20-ns equilibration. The LH1 protein exhibits a slight bending toward the periplasmic side. The membrane adapts to the LH1's change in shape. (c) Snapshot of the simulated system at the end of the 20-ns equilibration viewed in the direction perpendicular to that of panel b. No visible curvature is observed for the protein or the membrane in this direction.
The membrane-protein system was first minimized and equilibrated for 0.5 ns in an NVT ensemble, with only the lipid tails released to ensure proper lipid packing. The system was then minimized and equilibrated for 1 ns in an NPT ensemble with the protein harmonically constrained to allow equilibration of the lipid, water, and ion molecules. The production run then began in an NPT ensemble.
Cytochrome bc1-complex systems
The model of a membrane-embedded dimer of the bc1-complex was constructed using the crystal structure of the bc1-complex from Rb. sphaeroides (PDB entry 2FYN (22)). In simulations containing only a single dimer of the bc1-complex, the protein structure was positioned in the center of a 162 Å × 160 Å patch of 50% POPE and 50% POPG lipids. The membrane-protein system was subsequently solvated in a 162 Å × 160 Å × 120 Å box of water. The overall system charge was neutralized by the addition of Na+ and Cl− ions to a concentration of 200 mM. The 276,904 atom system was subsequently minimized and equilibrated in an NVT ensemble for 1 ns and an NPT ensemble for 4 ns to ensure proper packing of lipids against the protein. A 12 Å water buffer was then added around the system to break the continuous periodicity of the membrane and produce a periodic system containing the bc1-dimer embedded in a noncontinuous membrane patch surrounded by a water buffer on all sides. Additional Na+ and Cl− ions were added to maintain an ion concentration of 200 mM. The final system was 186 Å × 184 Å × 120 Å in size and contained 376,989 atoms. The system was then reminimized and again equilibrated under NVT conditions for 1 ns and NPT conditions for 2 ns before production sampling of the system was begun. The process of constructing this model of a single membrane-embedded bc1-dimer was repeated for the creation of a second similar system in which the membrane consisted purely of POPC lipids.
Two similar models containing two bc1-dimers embedded in a membrane were constructed using the protein structure from the equilibrated system of the single bc1-dimer in an uninterrupted, periodic membrane layer. The two bc1-dimers were positioned ∼30 Å distant from one another in a 222 Å × 379 Å rectangular bilayer patch of 50% POPG and 50% POPE lipids. In the first model containing two bc1-dimers, the dimers were oriented such that the four cytochrome-c1 subunits (two per bc1 dimer) were in-line with the long 379 Å axis of the membrane patch. The system was solvated in a 222 Å × 379 Å × 123 Å waterbox. The system was then neutralized with a 200 mM concentration of Na+ and Cl− ions, minimized, and equilibrated for 1 ns assuming an NVT ensemble and for 3 ns assuming an NPT ensemble. A water buffer of 12 Å containing sufficient ions to maintain the 200 mM ion concentration was then added to the system resulting in a final system size of 246 Å × 403 Å × 123 Å with 1,122,340 atoms. The system was reminimized and equilibrated under NVT conditions for 1 ns and NPT conditions for 2 ns. Production sampling of the system was conducted for 10 ns of simulation time under NPT conditions. The second model of two bc1-dimers embedded in a membrane was prepared with identical methods as the first, but in this second model, the bc1-dimers were oriented such that the Rieske subunits, instead of the cytochrome-c1 subunits, were in-line with the long axis of the periodic box. This resulted in a final system size of 356 Å × 176 Å × 123 Å containing a 332 Å × 152 Å membrane patch. This system of 709,877 atoms was also sampled under NPT conditions for 10 ns in an effort to observe spontaneous curvature of the lipid bilayer in response to the bc1 proteins.
Molecular dynamics
All simulations were performed using NAMD 2.6 (51) and the CHARMM27 force field with the CMAP correction (52,53). The TIP3P water model was used (54). Long-range electrostatic forces were evaluated using the particle-mesh Ewald summation method with a grid spacing of <1 Å. An integration time step of 1 fs was used with a multiple time-stepping algorithm (55,56). Bonded terms were evaluated every time step, with short-range nonbonded terms evaluated every second time step, and long range (>12 Å) nonbonded terms evaluated every fourth time step. Constant temperature (T = 310 K) was maintained using Langevin dynamics (damping coefficient = 1.0 ps−1). A constant pressure of 1 atm was enforced independently in each dimension using a Nosé-Hoover Langevin piston (decay period = 100 fs, time constant = 50 fs). The total simulation effort in units of million atoms × nanoseconds (Mans) was measured to be 86.3 Mans; i.e., was one of the largest such efforts to-date.
RESULTS
The protein-membrane systems described in Methods were equilibrated to investigate the possibility of spontaneous curvature formation. Multiple LH2s were found to induce curvature via packing, while single LH1-RC-PufX dimers were seen to induce curvature via bending at the dimer interface. The cytochrome bc1-dimer was not observed to induce curvature either alone or in pairs, prompting further questions about its possible role in curvature and position in the chromatophore. A summary of the simulations performed is given in Table 1.
TABLE 1.
Summary of simulations performed
| System | Number of atoms | Simulation time |
|---|---|---|
| 7 Rps. acidophila LH2s | 909,830 | 13.5 ns |
| 7 Rb. sphaeroides LH2s | 988,391 | 13.5 ns |
| 7 Rps. acidophila LH2s (closely packed) | 996,535 | 18 ns |
| LH1-RC-PufX dimer | 688,373 | 20 ns |
| single bc1 | 376,989 | 15 ns |
| 2 bc1s—alignment 1 | 1,122,340 | 10 ns |
| 2 bc1s—alignment 2 | 709,877 | 10 ns |
| pure membrane patch | 235,676 | 3 ns |
| 19 rhodopsins in membrane | 880,243 | 10 ns |
LH2
Mutant studies have implicated LH2 in producing the spherical shape of Rb. sphaeroides chromatophores: Rb. sphaeroides mutants that express LH2, but not LH1, produce normal, spherical chromatophores, while mutants that express LH1, but not LH2, produce abnormal, tubular structures (57). Further studies found that chromatophore size was inversely proportional to LH2 concentration (58), suggesting that more LH2 leads to stronger curvature. This suggestion is supported by radiolabeling experiments revealing that aggregation and close-packing of the light-harvesting complexes in the membrane is linked to chromatophore formation (6). AFM images of native Rb. sphaeroides chromatophore sections show regions of highly concentrated, closely-packed LH2s (13). AFM images of native membranes of other species, such as Rs. molischianum (27) and Rps. palustris (28), which have lamellar chromatophores, also show a close packing of light harvesting complexes, and a tendency of LH2 to pack hexagonally with its neighbors. We sought to investigate via MD simulation whether closely-packed LH2s would induce curvature, and whether LH2s from a species with vesicular chromatophores (Rb. sphaeroides) would have curvature properties different from LH2s from a species with lamellar chromatophores (Rps. acidophila).
Seven LH2s were placed in the membrane in a hexagonally packed arrangement, allowing just enough space between LH2s for a single line of lipids. The edges of the membrane patch were padded with water to disconnect the patch from its neighboring images (see Methods). This was done out of concern that a continuous, connected membrane as produced by the periodic boundary conditions would resist curvature. The simulation was performed once with Rps. acidophila LH2s and once with Rb. sphaeroides LH2s constructed via homology modeling to determine whether LH2 curvature properties would differ between the two species, possibly due to the small structural differences between the two LH2 models. In both simulations, the LH2s packed together and tilted with respect to adjacent LH2s, producing a curved patch within a few nanoseconds, and stabilizing to a final curvature after ∼10 ns. The curvature then remained relatively stable until the end of the simulation, as shown in Fig. 5. Additionally, the lipids in contact with the LH2s adopted the same curvature as the LH2s, and the membrane as a whole adopted an overall curvature after ∼10 ns and retained that shape until the end of the simulation. The net curvature of the membrane was shallower than that of the proteins, and was directional, in contrast to the roughly isotropic protein curvature, possibly because the membrane requires a longer relaxation time. No substantial differences were observed in the curvature of Rps. acidophila versus Rb. sphaeroides LH2s.
FIGURE 5.

LH2-induced curvature. (a–c) Rps. acidophila system at t = 0 ns from the top (a) and side (b) and at t = 13.5 ns (c). (d) Plot of average tilt angle versus time for the three simulations. The more sparsely packed Rps. acidophila and Rb. sphaeroides systems converged to final tilt angles of ∼6°. The closely packed Rps. acidophila system converged to a final tilt angle of ∼8.5°. (e) Schematic figure showing LH2 tilt angles. The tilt angle between each of the outer six LH2s and the central LH2 was calculated, and the six angles were averaged to compute an average tilt angle at each time step.
The curvature of the LH2 protein patch was analyzed by calculating the average tilt of the six peripheral LH2s with respect to the central one. Both the Rps. acidophila and Rb. sphaeroides systems equilibrated to a final average tilt angle of ∼6° (6.1 ± 0.4° for Rps. acidophila and 5.5 ± 0.3° for Rb. sphaeroides) within 10 ns.
Using the relation
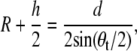 |
(1) |
where h is the height of LH2 (h = 50 Å), d is the distance between the centers of two adjacent LH2s (d = 85 Å for this packing), θt is the tilt angle, and R is the radius of curvature, a tilt angle of 6° corresponds to a radius of curvature of ∼790 Å. A typical radius for a spherical chromatophore, by comparison, is 150–400 Å (8), smaller than the curvature radius observed in the simulations. However, it is expected that the inclusion of more LH2s would lead to greater curvature (58). That both species adopted approximately the same curvature was unexpected, as the chromatophores of Rps. acidophila are lamellar and not highly curved. The results suggest that the curvature properties of LH2 arise not from the structural differences, but rather from some other property that is conserved in both species.
It is not certain how many lipids should be present around the LH2s; AFM images of native Rs. molischianum show LH2s packed so closely that there is no continuous layer of lipids between them (27), and images of reconstituted Rb. blasticus membranes also show LH2s in direct contact (59). AFM images of native Rb. sphaeroides membranes also show LH2s in direct contact, but they may also accommodate a single layer of lipids between some LH2s (13). For this reason, a third simulation was performed with Rps. acidophila LH2s in which adjacent LH2s were in direct contact. This simulation with tighter packing allowed us to investigate whether the curvature is sensitive to closer protein-protein packing and to explore any specific protein-protein contacts between adjacent LH2s. The system exhibited curvature more quickly than the previous ones with looser packing, and equilibrated to a final average tilt angle of 8.5° (8.4 ± 0.3°), corresponding to a radius of curvature of ∼495 Å (d = 77 Å for this packing). The smaller radius suggests that the curvature properties of LH2 are strengthened by close crowding of the proteins.
To ensure that the curvature of the protein-membrane system was not an artifact of the simulation parameters or of the edge effects caused by the water surrounding the membrane patch, a fourth simulation was run as a control. This simulation contained 19 copies of the membrane protein rhodopsin (PDB entry 1H68) (60), which is not thought to be involved in membrane curvature as it inhabits large, flat regions of retinal cells (61). The rhodopsins were placed in a membrane patch identical to that used in the LH2 simulations and were arranged in a hexagonal array whose area was comparable to that covered by the seven LH2s. This system was equilibrated for 10 ns (the time required for the formation of curvature in the LH2 systems), and no curvature, either of the protein region, or of the membrane as a whole, was observed (see Data S1). The absence of curvature in the rhodopsin system suggests that no portion of the curvature seen in the LH2 simulations was caused by a simulation artifact, but was instead caused by protein-protein and/or protein-lipid contacts specific to LH2.
The results of these simulations imply that LH2 complexes can induce curvature via packing, and that this behavior does not depend strongly on the small sequence or structural differences between LH2s from different species. However, LH2-induced curvature is amplified by closer packing of the proteins. It is interesting to note that the Rps. acidophila LH2 complexes are not wedge-shaped such that their packing would produce the curvature seen in the simulation, suggesting that the curvature is not caused by the sterical shape of LH2. Although there may be several factors contributing to LH2 curvature, simulations of modified LH2s suggest that one of those factors is the presence of charged residues on the cytoplasmic side of the protein whose electrostatic repulsion appears to contribute to curvature formation. Future work will explore the details of protein-protein interactions in LH2 curvature.
LH1-RC-PufX
Mutant studies have shown that LH2-deficient mutants of Rb. sphaeroides form tubular instead of spherical chromatophores (57,62–64), suggesting that the LH1-RC-PufX dimers generate unidirectional curvature in tubular chromatophores. To test this hypothesized membrane-bending property of the Rb. sphaeroides LH1-RC-PufX dimer, a model dimer was constructed, positioned in a noncontinuous lipid patch, and solvated in a waterbox. The resulting system was equilibrated for 20 ns, as described in Methods. Initially the dimer was flat when viewed along the membrane plane, but during the simulation, a bend at the dimerization junction was seen to develop (Fig. 4 b and Fig. 6 a). The outer edges of the two LH1 monomers tilted toward the periplasmic side of the membrane, and as a result, the angle at the periplasmic surface of the dimer was smaller than 180° at the end of the equilibration. Similar bending of the Rb. sphaeroides LH1-RC-PufX dimer has also been observed in single particle electron microscopy analysis (C. N. Hunter, University of Sheffield, personal communication, 2007). When viewed from the direction perpendicular to the side-view in Fig. 4 b, the dimer and membrane show no apparent curvature (Fig. 4 c), confirming the expected unidirectionality. Additionally, an equivalent simulation without PufX also generated similar dimer bending (see Data S1).
FIGURE 6.

Bending of the LH1-RC-PufX dimer. (a) The initially flat LH1-RC-PufX dimer exhibits bending at the dimerizing junction after equilibration. LH1β is not shown for clarity; the α-apoprotein rings are colored purple and orange for the left and right monomers, respectively; the RCs are colored in green and PufX in red. (b) Measurement of dimer bending angles θperi and θcyto. (c) RMSD plot for the protein components of the LH1-RC-PufX dimer. (d) Tilting of the RC periplasmic surfaces and hydrophobic belt. PufX and LH1 β-apoproteins are not shown, and the LH1 α-apoproteins are drawn transparent, with one monomer in pink and one in purple. RCs are colored according to residue type (white, hydrophobic; green, polar; red, negatively charged; blue, positively charged). (e) Response of LH1 α. The initially flat LH1 α-apoproteins hydrophobic region became tilted by the end of the simulation. The α-apoprotein is colored by residue type, and only the central helical region is shown. The dotted lines mark example curves formed by the His32 residues.
The observed LH1-RC-PufX bending angle was characterized by measuring the dimer bending angle θb at the dimerizing junction as defined in Fig. 6 a. Center-of-mass (COM) positions were measured for chosen residues at the outer edge of each LH1 monomer, and compared to the COM positions of the same residues at the dimerizing junction (Fig. 6 b, top). The angle was measured on the cytoplasmic as well as periplasmic side of LH1, and is labeled θcyto and θperi, respectively. For θcyto, the COM positions of α-apoprotein residues 21 and 22 were used, and for θperi the positions of residues 45 and 46 were used. These residues are chosen because residues 21–46 form a structurally stable segment within the α-apoproteins' transmembrane helix. As shown in Fig. 6 b, the two angles, θcyto and θperi, are equivalent throughout the 20-ns equilibration, with the dimer continuously bending during the first 10 ns, after which the two angles fluctuate at ∼172° for the rest of the equilibration. One can approximate the radius of curvature from the measured bending angle via Eq. 1, noting that the relation between tilting angle θt and bending angle θb is θt = 180° – θb. Also, given that each LH1 monomer has a diameter of ∼100 Å, the term d in Eq. 1 can be substituted by 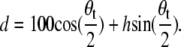 Together these substitutions give the simple relation
Together these substitutions give the simple relation
 |
(2) |
Accordingly, θb = 172° corresponds to a radius of curvature of ∼720 Å. It is worth noting that the radius of the tubular chromatophores has been estimated to be 250–550 Å (57,62–64), of the same order as the radius of native Rb. sphaeroides chromatophores, which corresponds to θb = 160°–170°.
An RMSD plot (Fig. 6 c) of the separate protein components (RCs, PufX, LH1α, and LH1β) over time shows that the RCs have much lower RMSD values throughout the simulation in comparison to PufX, LH1α, and LH1β, which all have similar RMSD values, suggesting that the RCs were relatively stationary during the simulation while other protein components underwent a similar degree of reorientation. When viewed along the membrane plane, the periplasmic surfaces of both RCs are tilted with respect to the membrane plane, making an angle of ∼160° at the dimerizing junction (Fig. 6 d). The hydrophobic region of the RCs is also tilted, forming a curved hydrophobic belt that is preserved throughout the simulation (Fig. 6 d). Together with the RMSD measurements, the tilted hydrophobic region of the RCs suggests that the shape and the relative orientations of the RCs drive the dimer bending via hydrophobic interactions within the LH1-RC-PufX dimer: LH1α first matches its hydrophobic region with that of the RCs, and the rearrangement of LH1β then follows similarly, driven by LH1α, causing the dimer to bend overall. The surrounding lipid then relaxes around the bent dimer to form a curved state. This effect is seen in the structural change of LH1α before and after the equilibration, as shown in Fig. 6 e. The hydrophobic region in LH1α is originally flat with respect to the membrane plane, but forms over time a tilted hydrophobic belt much like that of the RCs (Fig. 6 e).
Fig. 6 e also shows that after equilibration, each LH1 monomer bends slightly, such that the residues in each monomer do not line up, forming curves instead. Such internal flexibility within each LH1 monomer reduces the overall bending achieved by the entire LH1-RC-PufX-dimer and may explain why the simulated dimer bending is not sufficient to reproduce the observed smaller radius of Rb. sphaeroides tubular chromatophores.
Cytochrome bc1-complex
AFM and electron microscopy images reveal location and interprotein organization of the LH2 (27,65) and LH1-RC complexes (13,64) in Rb. sphaeroides chromatophores, but do not identify the locations of the bc1-complex. As suggested above, the determination of the lipid bilayer curvature surrounding a functional dimer of bc1-complexes offers potential insight into the location of bc1 within the photosynthetic chromatophores. If the bc1-complex is located in a curved region of the lipid bilayer, there are two potential scenarios to explain any curvature found in the surrounding membrane. In the first scenario, the bc1-dimer actively shapes the membrane by inducing local bilayer curvature through protein-lipid interactions. In the second scenario, the bc1-dimer passively accommodates a curved bilayer. Simulations of single and multiple bc1-dimers were conducted to examine the first of these two scenarios, and establish whether the bc1-dimer actively induces spontaneous bilayer curvature.
A single bc1-dimer embedded in a periodic continuous membrane was equilibrated to establish a properly packed lipid-protein complex with a lipid density of 66.9 Å2/lipid. A water buffer was subsequently added to separate the bilayer from its image in the reciprocal periodic cell and permit bulk membrane fluctuations that may lead to the formation of curvature. After equilibration, the resulting system with a noncontinuous bilayer exhibited a lipid density of 75.3 Å2/lipid. After 10 ns of simulation, as seen in Fig. 7, the lipid bilayer surrounding the single bc1-dimer remained flat. This simulation was also conducted in a bilayer of pure POPC lipids as typically used in the in vitro experiments involving membrane proteins. As in the heterogeneous POPG/POPE bilayer, the POPC lipid bilayer was seen to remain flat over the course of 10 ns of simulation.
FIGURE 7.

Results from simulation of the bc1 dimers from Rb. sphaeroides in a mixed POPE/POPG membrane patch. (a) Schematic view of simulated system. The dotted lines show the two principal axes of curvature: ISP axis and Cyt-B1 axis. (b) Side-view at t = 0 ns along the Cyt-c1 axis. The bc1-complex/membrane system exhibits zero membrane curvature at t = 0 ns along both the ISP and Cyt-B1 axes. (c) Side-view along ISP axis after 10 ns of equilibrium molecular dynamics. (d) Side-view along Cyt-c1 axis after 10 ns of equilibrium molecular dynamics. No significant membrane curvature is observed.
A second simulation was conducted to investigate the possibility that multiple bc1-dimers cooperatively promote curvature formation in the membrane region separating the proteins. Two bc1-dimers were simulated in a finite rectangular membrane patch that was prepared from a continuous membrane system as described in Methods. The bc1-dimers were oriented such that all Cyt-C1 subunits lay in-line along the long axis of the surrounding bilayer with an interdimer distance of 35 Å. This significant distance between the two bc1-dimers does not offer them the opportunity to pack during the simulation. It is anticipated that tight protein-protein packing of the bc1-dimers interferes with bc1 activity by restricting movement of the Rieske subunits or obstructing entrance to the dimer's two quinone and two quinol binding sites. Equilibration of the membrane/protein system yielded a lipid density of 76.9 Å2/lipid. As in the prior simulation of a single bc1-dimer and as seen in Fig. 8, spontaneous formation of bilayer curvature was not observed in 10 ns of simulation.
FIGURE 8.

Curvature results from two bc1 dimers in separated arrangements. (a) Top view of system containing two bc1 dimers oriented with Cyt-c1 in-line along the long axis of the membrane patch. (b) Short side-view of system containing two bc1 dimers with Cyt-c1 subunits aligned, after 10 ns of equilibration. (c) Long side-view of system containing two bc1 dimers with Cyt-c1 subunits aligned, after 10 ns equilibration. (d) Plot of tilt angles versus time for two bc1 dimers with Cyt-c1 subunits aligned (shown above in c). (e) Plot of tilt angles versus time for two bc1 dimers with ISP subunits aligned (shown below in f). (f) Long side-view of system containing two bc1 dimers with ISP subunits aligned, after 10 ns equilibration. No curvature was observed in simulations with either orientation of the bc1 dimers.
A third simulation was performed to investigate whether an alternative orientation of the two bc1-dimers would generate bilayer curvature. This system included two bc1-dimers again arranged within a finite lipid bilayer. As in the previous simulation, the dimers were positioned 35 Å apart with the key difference being that the two dimers were each rotated 90° about their respective z axes such that all four ISP subunits were oriented in-line with the bilayer's long axis. After equilibration of the system and verification of a 75.7 Å2/lipid membrane density, 10 ns of simulation were completed. No observable curvature of the lipid bilayer occurred during the course of this simulation as seen in Fig. 8.
Finally, a fourth simulation was performed to investigate more tightly packed bc1-dimers. The two bc1-dimers were arranged in a membrane according to the interdimer orientation suggested by Esser et al.'s crystal structure (PDB entry 2QJP) (66). This orientation does not occlude any of the four binding sites in each dimer and has minimal interdimer contacts at the ISP subunit to minimize steric hindrance of the ISP's catalytically relevant conformational transition. The interdimer contacts are shown in Fig. 9 and involve direct contact between one ISP subunit of each dimer which may significantly hinder catalysis in a single bc1-dimer. After equilibration, 10 ns of simulation were completed. As in the above described simulations of multiple bc1-dimers, no curvature of the lipid bilayer was observed, suggesting again that although bc1-dimers may accommodate curved membranes, they do not themselves directly induce membrane curvature through bc1-to-bc1 interactions.
FIGURE 9.

Curvature results from two bc1 dimers in packed arrangement. (a) Top view of system containing two packed bc1 dimers oriented with ISP subunits in-line along the long axis of the membrane patch. (b) Short side-view of system containing two bc1 dimers with ISP subunits aligned, after 10 ns of equilibration. (c) Long side-view of system containing two packed bc1 dimers with ISP subunits aligned, after 10-ns equilibration. (d) Plot of tilt angles versus time for two packed bc1 dimers. As in simulations of separated bc1 dimers, no curvature was observed in simulations of packed bc1 dimers.
DISCUSSION
By using large-scale molecular dynamics simulations of the primary components of the photosynthetic chromatophore of purple bacteria, we have taken the first steps toward modeling the interactions and behavior of a key multiprotein system at atomic resolution. The simulations illustrate that integral membrane proteins are capable of inducing membrane curvature and suggest key factors that may drive the development of curvature in these specific systems. It is worth noting that the curvature observed in our simulations is solely a result of the atomic-scale interactions. No assumption of curvature was put into our model, distinguishing it from coarse-grained and Monte Carlo simulations where each curvature-inducing element was assigned, a priori, either a curved shape or a specific local curvature (7,39). In multiple simulations, an array of seven LH2s developed curvature on the nanosecond timescale. This curvature was sensitive to the degree of protein packing, but not to the species of LH2. Additionally, the LH1-RC-PufX dimer, a unique S-shaped protein complex, exhibited a bend at its dimerizing interface, and can thereby induce membrane curvature similar to that seen for LH2. In contrast, cytochrome bc1 did not induce curvature of its surrounding bilayer. However, bc1 may be found to sense and respond to the curvature of the chromatophore to find its own natural placement.
It was initially surprising to find nearly identical curvature in small patches of LH2s from two different species, one producing vesicular and one producing flat chromatophores. Given the appearance of vesicular chromatophores in LH1-deficient mutants as well as the dependence of chromatophore radius on LH2 concentration (57,58), it is tempting to single out LH2 as the primary curvature-inducing element that determines whether chromatophores are vesicular or flat. Yet we see in our simulations that LH2s from different species produce the same curvature, and that LH1-RC-PufX dimers also generate a net curvature of their membranes. An explanation might be that while aggregates of LH2s always produce a net curvature, such curvature can be disrupted by breaking up the LH2 domains. The connection between domain formation and curvature is well supported by AFM studies of native photosynthetic membranes, which show separate domains for vesicular chromatophores as opposed to relatively more homogeneous distributions for flat chromatophores (13,27,67). LH1 monomers interspersed randomly in LH2-rich regions may produce flat chromatophores. However, LH1 dimers and LH2s separated into mostly distinct domains may form a nonzero global curvature and, thus, produce vesicular chromatophores. Recent Monte Carlo simulations have also demonstrated that LH2 and LH1 dimers spontaneously self-organize during membrane budding to form distinct domains (7). In those simulations, it was assumed that LH1 develops a local curvature only through interaction with LH2. While this mechanism still results in both proteins contributing to the overall curvature, it does not explain the experimentally observed formation of tubular or vesicular chromatophores in LH2-deficient mutants and species (57,62–64). Our results instead suggest that the LH1 dimer alone is capable of bending at the dimer interface perpendicular to the membrane, adding to the already known flexibility of the LH1 rings (68,69). Bending at the dimer interface is required for an LH1 dimer to properly fit within a vesicular chromatophore, and is also supported by recent single-particle EM reconstruction (C. N. Hunter, University of Sheffield, personal communication, 2007). It remains to be seen how interactions between LH1-RC monomers differ from those of dimers as well as how multiple dimers may collectively curve the membrane.
While our results indicate how LH2 and LH1-RC-PufX individually shape the chromatophore, extensive simulations of single and multiple bc1 complexes did not display any curvature, leaving the question of the bc1's location in the chromatophore open. Neither bc1 nor ATP synthase have been imaged by AFM of native photosynthetic membranes (27,70). Since as few as five bc1 complexes and one ATP synthase are required per chromatophore, they may be clustered in a region not retained during fractionation before imaging (71,72). Suggestions put forth for their location include localization of both complexes to the pole of the vesicular chromatophore (30) or the neck of the chromatophore and the surrounding membrane (29). Both locations have structural and functional consequences, particularly for the rate of migration of quinones from the RC to the bc1. It has recently been observed that proteins can sense membrane curvature and even that curvature sensing can be caused by enzymatic activity, giving the intriguing possibility that bc1 relies on the shape of the membrane to find its optimal location (2,73). Although it does not actively curve the surrounding bilayer, bc1 may still exhibit a preference for a particular curvature, such as the positive curvature of the vesicle pole or the saddle-shaped curvature of the neck.
Our simulations also clearly demonstrate a relationship between protein packing and curvature. In simulations in which LH2s were in close contact, curvature formation occurred earlier and the degree of curvature was greater than when LH2s were separated by a layer of lipids. The packing dependence of curvature is supported by both theoretical and experimental studies. Coarse-grained simulations of curved particles in a bilayer demonstrated that protein aggregation and curvature reinforce each other (39); similar results have recently been observed in Monte Carlo simulations of LH1 and LH2 (7). AFM images of native chromatophore membranes have also shown them to be well packed (13,26,27,67). The tight packing serves a functional purpose as it is known to be a necessary condition for efficient energy transfer (29,74).
Investigating the structure and organization of large, multiprotein complexes has only recently become accessible to all-atom MD simulations. With the accessibility of such large systems also comes new, large-scale questions, such as the shaping of membranes by proteins. While we have focused on the curvature-inducing properties of individual photosynthetic proteins, the current results point to many other avenues of exploration such as the interplay between heterogeneous mixtures of LH1, LH2, and bc1.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
We thank C. Neil Hunter, Pu Qian, and Melih Şener for many helpful discussions. Molecular images in this article were generated with the molecular graphics program VMD (42).
This work was supported by National Science Foundation grant No. MCB02034938. and National Institutes of Health grant No. P41-RRO5969. Computer time was provided at the National Center for Supercomputing Applications and the Texas Advanced Computing Center via Large Resources Allocation Committee grant No. MCA93S028.
Danielle Chandler, Jen Hsin, Christopher B. Harrison, and James Gumbart contributed equally to this work.
Editor: Helmut Grubmüller.
References
- 1.McMahon, H. T., and J. L. Gallop. 2005. Membrane curvature and mechanisms of dynamic cell membrane remodeling. Nature. 438:590–596. [DOI] [PubMed] [Google Scholar]
- 2.Zimmerberg, J., and M. M. Kozlov. 2006. How proteins produce cellular membrane curvature. Nat. Rev. Mol. Cell Biol. 7:9–19. [DOI] [PubMed] [Google Scholar]
- 3.Gallop, J. L., and H. T. McMahon. 2005. Bar domains and membrane curvature: bringing your curves to the BAR. Biochem. Soc. Symp. 72:223–231. [DOI] [PubMed] [Google Scholar]
- 4.Arkhipov, A., Y. Yin, and K. Schulten. 2008. Four-scale description of membrane sculpting by BAR domains. Biophys. J. 95:2806–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu, J., Y. Shibata, C. Voss, T. Shemesh, Z. Li, M. Coughlin, M. M. Kozlov, T. A. Rapoport, and W. A. Prinz. 2008. Membrane proteins of the endoplasmic reticulum induce high-curvature tubules. Science. 319:1247–1250. [DOI] [PubMed] [Google Scholar]
- 6.Hunter, C. N., J. D. Tucker, and R. A. Niederman. 2005. The assembly and organization of photosynthetic membranes in Rhodobacter sphaeroides. Photochem. Photobiol. Sci. 4:1023–1027. [DOI] [PubMed] [Google Scholar]
- 7.Frese, R. N., J. C. Pàmies, J. D. Olsen, S. Bahatyrova, C. D. van der Weij-de Wit, T. J. Aartsma, C. Otto, C. N. Hunter, D. Frenkel, and R. van Grondelle. 2008. Protein shape and crowding drive domain formation and curvature in biological membranes. Biophys. J. 94:640–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oelze, J., and G. Drews. 1972. Membranes of photosynthetic bacteria. Biochim. Biophys. Acta. 265:209–239. [DOI] [PubMed] [Google Scholar]
- 9.Pfennig, N. 1969. Rhodopseudomonas acidophila, sp. n., a new species of the budding purple nonsulfur bacteria. J. Bacteriol. 99:597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hickman, D. D., and A. W. Frenkel. 1965. Observations on the structure of Rhodospirillum molischianum. J. Cell Biol. 25:261–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaplan, S., B. D. Cain, T. J. Donohue, W. D. Shepherd, and G. S. L. Yen. 1983. Biosynthesis of the photosynthetic membranes of Rhodopseudomonas sphaeroides. J. Cell Biol. 22:15–29. [DOI] [PubMed] [Google Scholar]
- 12.Frese, R. N., C. A. Siebert, R. A. Niederman, C. N. Hunter, C. Otto, and R. van Grondelle. 2004. The long-range organization of a native photosynthetic membrane. Proc. Natl. Acad. Sci. USA. 101:17994–17999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bahatyrova, S., R. N. Frese, C. A. Siebert, J. D. Olsen, K. O. van der Werf, R. van Grondelle, R. A. Niederman, P. A. Bullough, C. Otto, and C. N. Hunter. 2004. The native architecture of a photosynthetic membrane. Nature. 430:1058–1062. [DOI] [PubMed] [Google Scholar]
- 14.Blankenship, R. E. 1992. Origin and early evolution of photosynthesis. Photosynth. Res. 33:91–111. [PubMed] [Google Scholar]
- 15.Olson, J. M., and R. E. Blankenship. 2004. Thinking about the evolution of photosynthesis. Photosynth. Res. 80:373–386. [DOI] [PubMed] [Google Scholar]
- 16.Xiong, J., W. M. Fischer, K. Inoue, M. Nakahara, and C. E. Bauer. 2000. Molecular evidence for the early evolution of photosynthesis. Science. 289:1724–1730. [DOI] [PubMed] [Google Scholar]
- 17.Hu, X., T. Ritz, A. Damjanović, F. Autenrieth, and K. Schulten. 2002. Photosynthetic apparatus of purple bacteria. Q. Rev. Biophys. 35:1–62. [DOI] [PubMed] [Google Scholar]
- 18.Ermler, U., G. Fritzsch, S. K. Buchanan, and H. Michel. 1994. Structure of the photosynthetic reaction center from Rhodobacter sphaeroides at 2.65 Å resolution: cofactors and protein-cofactor interactions. Structure. 2:925–936. [DOI] [PubMed] [Google Scholar]
- 19.Camara-Artigas, A., D. Brune, and J. P. Allen. 2002. Interactions between lipids and bacterial reaction centers determined by protein crystallography. Proc. Natl. Acad. Sci. USA. 99:11055–11060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koepke, J., X. Hu, C. Muenke, K. Schulten, and H. Michel. 1996. The crystal structure of the light harvesting complex II (B800–850) from Rhodospirillum molischianum. Structure. 4:581–597. [DOI] [PubMed] [Google Scholar]
- 21.Papiz, M. Z., S. M. Prince, T. Howard, R. J. Cogdell, and N. W. Isaacs. 2003. The structure and thermal motion of the B800–850 LH2 complex from Rps. acidophila at 2.0 Å resolution and 100 K: new structural features and functionally relevant motions. J. Mol. Biol. 326:1523–1538. [DOI] [PubMed] [Google Scholar]
- 22.Esser, L., X. Gong, S. Yang, L. Yu, C. A. Yu, and D. Xia. 2006. Surface-modulated motion switch: capture and release of iron-sulfur protein in the cytochrome bc1 complex. Proc. Natl. Acad. Sci. USA. 103:13045–13050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Conroy, M. J., W. Westerhuis, P. S. Parkes-Loach, P. A. Loach, C. N. Hunter, and M. P. Williamson. 2000. The solution structure of the Rhodobacter sphaeroides LH1 β reveals two helical domains separated by a more flexible region: structural consequences for the LH1 complex. J. Mol. Biol. 298:83–94. [DOI] [PubMed] [Google Scholar]
- 24.Tunnicliffe, R. B., E. C. Ratcliffe, C. N. Hunter, and M. P. Williamson. 2006. The solution structure of the PufX peptide from Rhodobacter sphaeroides. FEBS Lett. 580:6967–6971. [DOI] [PubMed] [Google Scholar]
- 25.Firsow, N. N., and G. Drews. 1977. Differentiation of the intracytoplasmic membrane of Rhodopseudomonas palustris induced by variations of oxygen partial pressure or light intensity. Arch. Microbiol. 115:299–306. [DOI] [PubMed] [Google Scholar]
- 26.Scheuring, S., J. Busselez, and D. Lévy. 2005. Structure of the dimeric PufX-containing core complex of Rhodobacter blasticus by in situ atomic force microscopy. J. Biol. Chem. 280:1426–1431. [DOI] [PubMed] [Google Scholar]
- 27.Gonçalves, R. P., A. Bernadac, J. N. Sturgis, and S. Scheuring. 2005. Architecture of the native photosynthetic apparatus of Phaeopirillum molischianum. J. Struct. Biol. 152:221–228. [DOI] [PubMed] [Google Scholar]
- 28.Scheuring, S., R. P. Gonçalves, V. Prima, and J. N. Sturgis. 2006. The photosynthetic apparatus of Rhodopseudomonas palustris: structures and organization. J. Mol. Biol. 358:83–96. [DOI] [PubMed] [Google Scholar]
- 29.Sener, M. K., J. D. Olsen, C. N. Hunter, and K. Schulten. 2007. Atomic level structural and functional model of a bacterial photosynthetic membrane vesicle. Proc. Natl. Acad. Sci. USA. 104:15723–15728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geyer, T., and V. Helms. 2006. A spatial model of the chromatophore vesicles of Rhodobacter sphaeroides and the position of the cytochrome bc1 complex. Biophys. J. 91:921–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weikl, T. R., M. M. Kozlov, and W. Helfrich. 1998. Interactions of conical membrane inclusions: effect of lateral tension. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Topics. 57:6988–6995. [DOI] [PubMed] [Google Scholar]
- 32.Fošnarič, M., A. Iglič, and S. May. 2006. Influence of rigid inclusions on the bending elasticity of a lipid membrane. Phys. Rev. E. 74:051503-1–051503-12. [DOI] [PubMed] [Google Scholar]
- 33.Saiz, L., S. Bandyopadhyay, and M. L. Klein. 2004. Effect of the pore region of a transmembrane ion channel on the physical properties of a simple membrane. J. Phys. Chem. B. 108:2608–2613. [Google Scholar]
- 34.Jolley, C., S. A. Wells, B. M. Hespenheide, M. F. Thorpe, and P. Fromme. 2006. Docking of photosystem I subunit C using a constrained geometric simulation. J. Am. Chem. Soc. 128:8803–8812. [DOI] [PubMed] [Google Scholar]
- 35.Jolley, C., A. Ben-Shem, N. Nelson, and P. Fromme. 2005. Structure of plant photosystem I revealed by theoretical modeling. J. Biol. Chem. 280:33627–33636. [DOI] [PubMed] [Google Scholar]
- 36.Kim, K. S., J. Neu, and G. Oster. 1998. Curvature-mediated interactions between membrane proteins. Biophys. J. 75:2274–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marčelja, S. 1976. Lipid-mediated protein interaction in membranes. Biochim. Biophys. Acta. 455:1–7. [DOI] [PubMed] [Google Scholar]
- 38.Botelho, A. V., T. Huber, T. P. Sakmar, and M. F. Brown. 2006. Curvature and hydrophobic forces drive oligomerization and modulate activity of rhodopsin in membranes. Biophys. J. 91:4464–4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reynwar, B. J., G. Illya, V. A. Harmandaris, M. M. Müller, K. Kremer, and M. Deserno. 2007. Aggregation and vesiculation of membrane proteins by curvature-mediated interactions. Nature. 447:461–464. [DOI] [PubMed] [Google Scholar]
- 40.Walz, T., S. J. Jamieson, C. M. Bowers, P. A. Bullough, and C. N. Hunter. 1998. Projection structures of three photosynthetic complexes from Rhodobacter sphaeroides: LH2 at 6 Å, LH1 and RC-LH1 at 25 Å. J. Mol. Biol. 282:833–845. [DOI] [PubMed] [Google Scholar]
- 41.Jones, D. T. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292:195–202. [DOI] [PubMed] [Google Scholar]
- 42.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD—visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- 43.Tadros, M. H., R. Frank, and G. Drews. 1986. Localization of the exposed N-terminal region of the B800–850 α and β light-harvesting polypeptides on the cytoplasmic surface of Rhodopseudomonas capsulata chromatophores. J. Bacteriol. 167:96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Savage, H., M. Cyrklaff, G. Montoya, W. Kühlbrandt, and I. Sinning. 1996. Two-dimensional structure of light harvesting complex II (LHII) from the purple bacterium Rhodovulum sulfidophilum and comparison with LHII from Rhodopseudomonas acidophila. Structure. 4:243–252. [DOI] [PubMed] [Google Scholar]
- 45.Olsen, J. D., J. N. Sturgis, W. H. J. Westerhuis, G. J. S. Fowler, C. N. Hunter, and B. Robert. 1997. Site-directed modification of the ligands to the bacteriochlorophylls of the light-harvesting LH1 and LH2 complexes of Rhodobacter sphaeroides. Biochemistry. 36:12625–12632. [DOI] [PubMed] [Google Scholar]
- 46.Cogdell, R. J., A. Gall, and J. Köhler. 2006. The architecture and function of the light-harvesting apparatus of purple bacteria: from single molecules to in vivo membranes. Q. Rev. Biophys. 39:227–324. [DOI] [PubMed] [Google Scholar]
- 47.Wang, Z. Y., K. Gokan, M. Kobayashi, and T. Nozawa. 2005. Solution structures of the core light-harvesting α and β polypeptides from Rhodospirillum rubrum: implications for the pigment-protein and protein-protein interactions. J. Mol. Biol. 347:465–477. [DOI] [PubMed] [Google Scholar]
- 48.Qian, P., C. N. Hunter, and P. A. Bullough. 2005. The 8.5 Å projection structure of the core RC-LH1-PufX dimer of Rhodobacter sphaeroides. J. Biol. Chem. 349:948–960. [DOI] [PubMed] [Google Scholar]
- 49.Blood, P. D., and G. A. Voth. 2006. Direct observation of Bin/amphiphysin/Rvs (BAR) domain-induced membrane curvature by means of molecular dynamics simulations. Proc. Natl. Acad. Sci. USA. 103:15068–15072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Essman, U., and M. L. Berkowitz. 1999. Dynamical properties of phospholipid bilayers from computer simulation. Biophys. J. 76:2081–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Phillips, J. C., R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale, and K. Schulten. 2005. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.MacKerell, A. D., Jr., D. Bashford, M. Bellott, R. L. Dunbrack, Jr., J. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, I. W. E. Reiher, B. Roux, M. Schlenkrich, J. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, and M. Karplus. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- 53.MacKerrell, A. D., Jr., M. Feig, and C. L. Brooks III. 2004. Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 25:1400–1415. [DOI] [PubMed] [Google Scholar]
- 54.Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 55.Schlick, T., R. Skeel, A. Brünger, L. Kalé, J. A. Board Jr., J. Hermans, and K. Schulten. 1999. Algorithmic challenges in computational molecular biophysics. J. Comput. Phys. 151:9–48. [Google Scholar]
- 56.Grubmüller, H., H. Heller, A. Windemuth, and K. Schulten. 1991. Generalized Verlet algorithm for efficient molecular dynamics simulations with long-range interactions. Mol. Simul. 6:121–142. [Google Scholar]
- 57.Kiley, P. J., A. Varga, and S. Kaplan. 1988. Physiological and structural analysis of light-harvesting mutants of Rhodobacter sphaeroides. J. Bacteriol. 170:1103–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sturgis, J. N., and R. A. Niederman. 1996. The effect of different levels of the B800–B850 light-harvesting complex on intracytoplasmic membrane development in Rhodobacter sphaeroides. Arch. Microbiol. 165:235–242. [DOI] [PubMed] [Google Scholar]
- 59.Scheuring, S., D. Lévy, and J.-L. Rigaud. 2005. Watching the components of photosynthetic bacterial membranes and their in situ organization by atomic force microscopy. Biochim. Biophys. Acta. 1712:109–127. [DOI] [PubMed] [Google Scholar]
- 60.Royant, A., P. Nollert, K. Edman, R. Neutze, E. M. Landau, E. Pebay-Peyroula, and J. Navarro. 2001. X-ray structure of sensory rhodopsin II at 2.1 Å resolution. Proc. Natl. Acad. Sci. USA. 98:10131–10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Spudich, J. L., C.-S. Yang, K.-H. Jung, and E. N. Spudich. 2000. Retinylidene proteins: structures and functions from archaea to humans. Annu. Rev. Cell Dev. Biol. 16:365–392. [DOI] [PubMed] [Google Scholar]
- 62.Hunter, C. N., J. D. Pennoyer, J. N. Sturgis, D. Farrelly, and R. A. Niederman. 1988. Oligomerization states and associations of light-harvesting pigment protein complexes of Rhodobacter sphaeroides as analyzed by lithium dodecyl sulfate-polyacrylamide gel electrophoresis. Biochemistry. 27:3459–3467. [Google Scholar]
- 63.Jungas, C., J. Ranck, J. Rigaud, P. Joliot, and A. Verméglio. 1999. Supramolecular organization of the photosynthetic apparatus of Rhodobacter sphaeroides. EMBO J. 18:534–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Siebert, C. A., P. Qian, D. Fotiadis, A. Engel, C. N. Hunter, and P. A. Bullough. 2004. Molecular architecture of photosynthetic membranes in Rhodobacter sphaeroides: the role of PufX. EMBO J. 23:690–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Scheuring, S., F. Reiss-Husson, A. Engel, J.-L. Rigaud, and J.-L. Ranck. 2001. High resolution AFM topographs of Rubrivivax gelatinosus light-harvesting complex LH2. EMBO J. 20:3029–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Esser, L., M. Elberry, F. Zhou, C.-A. Yu, L. Yu, and D. Xia. 2008. Inhibitor-complexed structures of the cytochrome bc1 from the photosynthetic bacterium Rhodobacter sphaeroides. J. Biol. Chem. 283:2846–2847. [DOI] [PubMed] [Google Scholar]
- 67.Scheuring, S., J. Rigaud, and J. Sturgis. 2004. Variable LH2 stoichiometry and core clustering in native membranes of Rhodospirillum photometricum. EMBO J. 23:4127–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fotiadis, D., P. Qian, A. Philippsen, P. A. Bullough, A. Engel, and C. N. Hunter. 2004. Structural analysis of the reaction center light-harvesting complex I photosynthetic core complex of Rhodospirillum rubrum using atomic force microscopy. J. Biol. Chem. 279:2063–2068. [DOI] [PubMed] [Google Scholar]
- 69.Bahatyrova, S., R. N. Frese, K. O. van der Werf, C. Otto, C. N. Hunter, and J. D. Olsen. 2004. Flexibility and size heterogeneity of the LH1 light harvesting complex revealed by atomic force microscopy. J. Biol. Chem. 279:21327–21333. [DOI] [PubMed] [Google Scholar]
- 70.Sturgis, J. N., and R. A. Niederman. 2007. Atomic force microscopy reveals multiple patterns of antenna organization in purple bacteria: implications for energy transduction mechanisms and membrane modeling. Photosynth. Res. 95:269–278. [DOI] [PubMed] [Google Scholar]
- 71.Feniouk, B. A., D. A. Cherepanov, W. Junge, and A. Y. Mulkidjanian. 1999. ATP-synthase of Rhodobacter capsulatus: coupling of proton flow through F0 to reactions in F1 under the ATP synthesis and slip conditions. FEBS Lett. 445:409–414. [DOI] [PubMed] [Google Scholar]
- 72.Geyer, T., and V. Helms. 2006. Reconstruction of a kinetic model of the chromatophore vesicles from Rhodobacter sphaeroides. Biophys. J. 91:927–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bigay, J., P. Gounon, S. Robineau, and B. Antonny. 2003. Lipid packing sensed by ArfGAP1 couples COPI coat disassembly to membrane bilayer curvature. Nature. 426:563–566. [DOI] [PubMed] [Google Scholar]
- 74.Westerhuis, W. H. J., M. Vos, R. van Grondelle, J. Amesz, and R. A. Niederman. 1998. Altered organization of light-harvesting complexes in phospholipid-enriched Rhodobacter sphaeroides chromatophores as determined by fluorescence yield and singlet-singlet annihilation measurements. Biochim. Biophys. Acta Bioenerg. 1366:317–329. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.