

Abstract
Pseudomonas aeruginosa pneumonia usually results from a deficit of the innate immune system. To investigate whether inflammatory signalling by airway epithelial cells provides a pivotal line of defence against P. aeruginosa infection, we utilized two separate lines of inducible transgenic mice that express a constitutive activator of the nuclear factor kappa-B (NF-κB) pathway (IKTA) or a dominant inhibitor of NF-κB (DNTA) in airway epithelial cells. Compared with control mice, IKTA mice showed an enhanced host response to P. aeruginosa infection with greater neutrophil influx into the lungs, increased expression of Glu-Leu-Arg-positive (ELR+) CXC chemokines macrophage inflammatory protein-2 and keratinocyte chemoattractant (KC), superior bacterial clearance and improved survival at 24 h after infection. Neutrophil depletion abrogated the improvement in host defence identified in IKTA mice. In contrast, DNTA mice showed impaired responses to P. aeruginosa infection with higher bacterial colony counts in the lungs, decreased neutrophilic lung inflammation and lower levels of KC in lung lavage fluid. DNTA mice given recombinant KC at the time of P. aeruginosa infection demonstrated improved neutrophil recruitment to the lungs and enhanced bacterial clearance. Our data indicate that the NF-κB pathway in airway epithelial cells plays an essential role in defence against P. aeruginosa through generation of CXC chemokines and recruitment of neutrophils.
Keywords: chemokines, immune regulation, infections, lung immunology, transgenics
Introduction
Pseudomonas aeruginosa is a well-known opportunistic pathogen that causes significant morbidity and mortality in hospitalized and immunocompromised patients. This Gram-negative bacterium is frequently responsible for ventilator-associated pneumonia, which affects up to 28% of mechanically ventilated patients with an associated mortality ranging from 24 to 74% [1–5]. Gram-negative bacteria represent the majority of recovered organisms in ventilator-associated pneumonia, and of these, P. aeruginosa is the most commonly isolated pathogen [3]. Because of its flexible genome and variable phenotype, P. aeruginosa is adept at evading host defences and developing resistance to antibiotics, thus remaining a difficult infection to eradicate. Although the pathogenesis of P. aeruginosa infection has been studied extensively and many of its bacterial virulence factors identified, key cell types and signalling pathways that determine the host response to bacterial infection are incompletely understood.
In P. aeruginosa lung infections, airway epithelial cells contribute potentially to host defence in several ways, including initiation of nuclear factor kappa-B (NF-κB)-associated innate immune signalling in response to contact with intact bacteria or bacterial products. P. aeruginosa ligands [such as pili, flagella and lipopolysaccharide (LPS)] bind to cell surface Toll-like receptors (TLRs), which have been identified on a variety of cell types, including airway epithelium [6–14]. An important TLR-associated adaptor protein, MyD88, has been shown to be required for downstream activation of NF-κB by P. aeruginosa[8,12,14]. NF-κB is a heterodimer composed of two subunits, RelA (also known as p65) and p50, which usually exists in the cytoplasm in an inactive form bound to an inhibitor, IκB [15,16]. Activation of the IκB kinase (IKK) complex phosphorylates two specific serine residues on IκB, leading to selective ubiquination and degradation by the 26S proteosome. NF-κB then translocates into the nucleus and attaches to its target DNA binding sites, initiating the transcription of CXC chemokines and a variety of other inflammatory mediators. Direct interaction between P. aeruginosa and human airway epithelial cells in culture causes increased NF-κB reporter gene expression and interleukin (IL)-8 production, effects which are inhibited by co-transfecting the cells with a plasmid expressing defective MyD88 [11]. MyD88-deficient mice infected with P. aeruginosa have impaired bacterial clearance from the lung with reduced neutrophil recruitment and impaired NF-κB activation [12,14]. A study of bone marrow chimeras between MyD88 knock-out and wild-type (WT) mice treated with aerosolized P. aeruginosa found that non-bone marrow-derived cells, rather than bone marrow-derived inflammatory cells, were necessary for early control of P. aeruginosa infection through the MyD88 pathway [17]. Together, these studies suggest that activation of NF-κB pathway in a MyD88-dependent manner is involved in host defence against P. aeruginosa. However, the cell types responsible for the NF-κB-dependent innate immune response in vivo and the mechanisms by which NF-κB influences host defence against P. aeruginosa have not been determined.
Recently, we have generated two novel transgenic mouse lines which conditionally induce or block NF-κB activation exclusively in airway epithelium. Because our previous work suggests that NF-κB signalling is important for host defence against P. aeruginosa and intratracheal (i.t.) injection of this pathogen results in early activation of NF-κB in airway epithelium [8,18], we utilized these transgenic mouse lines to modulate NF-κB activation in airway epithelium in order to investigate further the function of this pathway in P. aeruginosa infection. Our results indicate that signalling through the NF-κB pathway in epithelium controls the protective host response to P. aeruginosa infection by production of CXC chemokines and recruitment of neutrophils.
Materials and methods
Mouse models
For these experiments, we used two separate inducible transgenic mouse models based on the tet-on system (FVB background) [19]. A detailed description of the generation and characterization of these transgenic lines, designated IKTA (for cIKK2 TransActivated) and DNTA (for IκBα-DN TransActivated), has been reported [20]. IKTA mice express a FLAG-tagged activated form of human IKK2 (NF-κB activator) under control of the tet-O7 enhancer/promoter, and DNTA mice express a Myc-His-tagged mutant avian IκBα (NF-κB inhibitor) with serine–adenine substitutions at two serine residues (S36,40) that cannot be phosphorylated or degraded [21,22]. To achieve selective expression in airway epithelium, mice containing tet-O7-FLAG-cIKK2 or tet-O7-IκBα-DN-Myc-His constructs were crossed to mice expressing reverse tetracycline transactivator under control of the rat CC10 promoter (gift from Dr Jeff Whitsett) [23]. Following construction of the IKTA and DNTA transgenic mouse lines, we identified selective, inducible transgene expression in airway epithelium [20].
For these experiments, transgene expression was induced by addition of 0·5 or 1 mg/ml doxycycline (Dox) (Sigma, St Louis, MO, USA) in 2% sucrose to drinking water for 2 days (IKTA) or 7 days (DNTA) prior to infection with P. aeruginosa. The water bottle was covered in aluminum foil to prevent light-induced Dox degradation; Dox water was replaced twice during the week. The mice were treated continuously with Dox for the duration of P. aeruginosa infection (24 h). All animal experiments were approved by the Vanderbilt University Institutional Animal Care and Utilization Committee (Nashville, TN, USA).
Pseudomonas aeruginosa (PA103 strain) infection
PA103 is a well-characterized, highly virulent strain of P. aeruginosa. Bacteria from frozen stocks (stored at −70°C) were streaked onto sheep blood agar and grown in a deferrated dialysate of tripticase soy broth supplemented with 10 mM nitrilotriacetic acid (Sigma), 1% glycerol and 100 mM monosodium glutamate. After growing at 33°C for 13 h in a shaking incubator, the cultures were centrifuged at 8500 g for 5 min. The bacterial pellet was washed twice in Ringers lactate and diluted into the appropriate number of colony-forming units (cfu) per ml in phosphate-buffered saline (PBS) determined by spectrophotometry. The bacterial concentration was confirmed by dilution and plating on sheep blood agar plates.
IKTA, DNTA and WT FVB control mice were treated with 1 × 104–5 × 106 cfu P. aeruginosa by i.t. injection. After sedation with inhaled isoflurane, mouse tracheas were exposed directly by surgical dissection, pierced with a 26-gauge needle and injected with 50 µl of P. aeruginosa in PBS. The neck wound was closed with sterile sutures under aseptic conditions. Twenty-four hours after infection, mice were euthanized and tracheas were cannulated for lung lavage in situ with sterile PBS instilled in two 0·8 ml aliquots and withdrawn gently with a 1 ml tuberculin syringe. The lungs and spleen were then removed using aseptic techniques, and the right middle lobe or whole right lung was separated and placed in 1 ml of sterile PBS. The remaining lung tissue was frozen at −80°C for future studies. The lung and spleen tissues were then homogenized in separate tissue homogenizers under sterile conditions. Serial dilutions of the homogenates were plated on sheep blood agar. The plates were incubated for 18–24 h at 37°C followed by colony counting. At the time of infection, the right lung of one WT mouse was harvested 30 min after i.t. injection of P. aeruginosa for determination of bacterial deposition. Baseline and post-infection peripheral blood total and differential cell counts were performed.
Bronchoalvolar lavage (BAL) fluid was centrifuged at 2500 g for 10 min at 4°C; the supernatant was removed and stored at −80°C for further studies. The cell pellet was resuspended in PBS, and the total cell count in the lavage fluid was acquired using a grid haemocytometer. Cell differentials were obtained by Wright–Giemsa-stained cytocentrifuged slides (Three Step Stain Set, Richard-Allen Scientific, Kalamazoo, MI, USA).
Cytokine measurements
Tumour necrosis factor (TNF)-α, regulated upon activation normal T cell expressed and secreted (RANTES), monocyte chemoattractant protein-1, macrophage inflammatory protein (MIP)-1α, keratinocyte chemoattractant (KC), granulocyte colony-stimulating factor (G-CSF), granulocyte–macrophage colony-stimulating factor (GM-CSF), IL-1α, IL-1β, IL-6 and IL-12 were measured in BAL fluid by Bio-Plex™ mouse cytokine 18-Plex and 23-Plex kits (Bio-Rad, Hercules, CA, USA) in conjunction with Luminex® technology, according to the manufacturer's protocol. STarStation software (Applied Cytometry Systems, Sheffield, UK) was used for data acquisition and analysis. MIP-2 expression in BAL fluid was measured by enzyme-linked immunosorbent assay (Quantikine® mouse MIP-2 immunoassay; R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions.
Neutrophil depletion
Neutrophil depletion was accomplished by intraperitoneal (i.p.) injections of rabbit anti-mouse neutrophil anti-serum (Accurate Chemicals, Westbury, NY, USA) [24]. Anti-neutrophil antibodies were diluted 1 : 15 in 0·3 ml of PBS and administered by i.p. injection on 4 consecutive days prior to infection.
KC replacement
Recombinant mouse KC (R&D Systems) was reconstituted in 1% bovine serum albumin in PBS to a concentration of 200 µg/ml. Five µg KC (in PBS) or vehicle (PBS) was injected i.t. into DNTA mice at the time of P. aeruginosa infection.
Statistical methods
For comparisons between experimental groups, Student's t-test or one-way analysis of variance with the Tukey–Kramer post-test was employed with P values < 0·05 considered statistically significant (In Stat; Graphpad Software, Inc., San Diego, CA, USA). Mortality data were analysed using the Pearson χ2 test. Results are presented as mean ± standard error of the mean.
Results
Increased NF-κB activation in airway epithelium is protective against P. aeruginosa pneumonia
To examine whether augmented induction of NF-κB activation in airway epithelial cells could influence the pathogenesis of P. aeruginosa infection we used IKTA transgenic mice, which express selectively an NF-κB activator (constitutively active human IKK2) in airway epithelium after addition of Dox to drinking water [20]. For these studies, a sublethal inoculum of P. aeruginosa (PA103 strain, 1 × 105 cfu) was chosen to study the effects of transgene-induced NF-κB activation on lung inflammation and bacterial clearance. Mice were treated with Dox for 48 h prior to infection to ensure adequate time for transgene expression, and this treatment was continued until harvest at 24 h after P. aeruginosa infection (a total of 3 days on Dox water). The direct addition of Dox to PA103 cultures (up to 200 µg/ml) did not affect bacterial viability (data not shown). Four groups of mice were used for these studies: IKTA transgenic mice treated with Dox (IKTA + Dox), IKTA mice without Dox treatment (IKTA), WT controls treated with Dox (WT + Dox) and WT controls without Dox treatment (WT).
IKTA mice treated with Dox had a higher number of neutrophils in BAL following P. aeruginosa infection (379·6 ± 28·8 × 104) compared with WT (92·3 ± 9·0 × 104), WT + Dox (85·1 ± 7·2 × 104) or IKTA mice (93·3 ± 4·9 × 104) infected with P. aeruginosa (P < 0·01) (Fig. 1a). In comparison, uninfected IKTA mice treated with Dox for 3 days had 69·2 ± 14·3 × 104 neutrophils in BAL while only rare neutrophils were present in BAL from WT mice (with or without Dox) or IKTA mice without Dox treatment. Following P. aeruginosa infection, IKTA + Dox mice also had a greater number of macrophages in BAL (73·1 ± 9·6 × 104) compared with WT (19·7 ± 2·1 × 104), WT + Dox (18·1 ± 3·2 × 104) or IKTA mice (16·2 ± 2·4 × 104) infected with P. aeruginosa (P < 0·01) (Fig. 1a). Uninfected IKTA mice treated with Dox for 3 days had 31·1 ± 5·3 × 104 macrophages in BAL compared with 7·9 ± 1·3 × 104 macrophages in BAL from IKTA mice without Dox treatment and 7·8 ± 1·3 × 104 macrophages in BAL of WT mice treated with Dox. In addition to increased numbers of inflammatory cells in BAL, IKTA + Dox mice showed a marked reduction in bacterial colony counts (250 ± 6) compared with WT (11·53 ± 0·80 × 103), WT + Dox (11·23 ± 0·75 × 103) and IKTA mice (11·18 ± 0·52 × 103) (P < 0·001) at 24 h after P. aeruginosa infection (Fig. 1b). These results indicate that enhanced NF-κB activation in IKTA mice increases inflammatory cell influx into the lungs following P. aeruginosa infection and improves bacterial clearance.
Fig. 1.
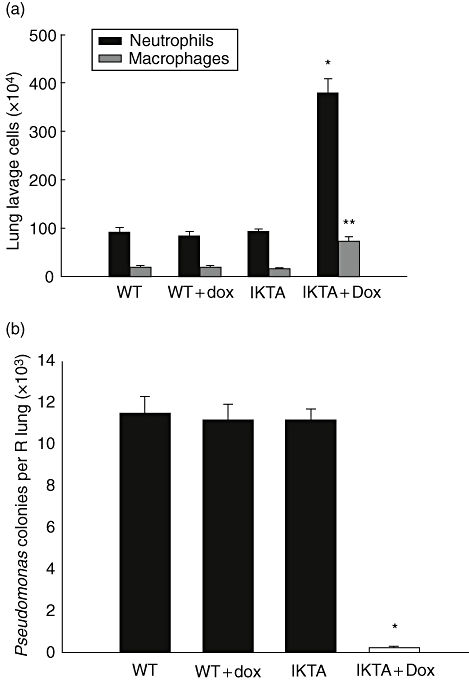
cIKK2 TransActivated (IKTA) mice have enhanced host defence against Pseudomonas aeruginosa. (a) Lung lavage neutrophil and macrophage counts from IKTA mice with or without doxycycline (Dox) treatment and wild-type (WT) controls with or without Dox treatment harvested at 24 h after intratracheal injection of a sublethal dose of PA103 [1 × 105 colony-forming units (cfu)] (n = 5 per group, *P < 0·01 and **P < 0·01 compared with neutrophils and macrophages in other groups respectively). (b) P. aeruginosa colony counts per lung from mice harvested 24 h after IT injection of 1 × 105 cfu of PA103. Dox was added to drinking water 2 days prior to infection (n = 5 per group, *P < 0·001 compared with other groups).
We evaluated the profile of cytokine and chemokine mediators present in BAL from: (i) WT mice and IKTA mice treated with Dox for 3 days; and (ii) WT and IKTA mice treated with Dox for 2 days prior to infection with P. aeruginosa and harvested 24 h later (Table 1). While a variety of mediators were increased in BAL from IKTA mice treated with Dox or WT mice infected with P. aeruginosa compared with WT mice treated with Dox alone, synergistic increases were found for several cytokines in BAL of Dox treated IKTA mice following P. aeruginosa infection. The combination of NF-κB activation induced by transgene expression and P. aeruginosa infection increased production markedly of IL-1α, IL-6, G-CSF, GM-CSF, MIP-1α, MIP-2, KC, RANTES and TNF-α in IKTA mice. Augmented production of the CXC neutrophil chemotactic chemokines KC and MIP-2 may be particularly important for host defence in this model by signalling increased neutrophil emigration into the airspaces. Over-production of other key host defence cytokines, including TNF-α, in Dox-treated IKTA mice infected with P. aeruginosa provides additional evidence for an improved innate immune response related to augmented NF-κB activation in airway epithelial cells, positively impacting bacterial clearance.
Table 1.
Mediator concentration in bronchoalveolar lavage (BAL) (pg/ml).
| WT + Dox | IKTA + Dox | WT + Dox + PA | IKTA + Dox + PA | |
|---|---|---|---|---|
| IL-1α | 0 | 0 | 33 (14) | 96 (5)* |
| IL-1β | 0 | 56 (6) | 100 (15) | 141 (16) |
| IL-6 | 0 | 342 (165) | 254 (111) | 1818 (341)* |
| IL-12 (p40) | 32 (4) | 37 (13) | 69 (2·3) | 128 (30) |
| G-CSF | 0 | 236 (81) | 713 (167) | 2961 (652)* |
| GM-CSF | 0 | 12 (10) | 30 (18) | 224 (34)* |
| MIP-1α | 0 | 0 | 286 (138) | 1143 (191)* |
| MIP-2 | 0 | 48 (15) | 26 (6) | 381 (90)* |
| KC | 12 (2) | 65 (11) | 196 (75) | 985 (117)* |
| RANTES | 0 | 356 (40) | 708 (231) | 4604 (2039)* |
| TNF-α | 0 | 35 (12) | 164 (53) | 510 (130)* |
| IFN-γ | 0 | 0 | 0 | 27 (17)* |
Values measured by luminex or enzyme-linked immunosorbent assay are presented as mean (± standard error of the mean); n = 3–6 per group.
P < 0·05 compared with all other groups by analysis of variance. Dox, doxycycline; G-CSG, granulocyte colony-stimulating factor; GM-CSF, granulocyte–macrophage colony-stimulating factor; IKTA, cIKK2 TransActivated; IL, interleukin; IFN, interferon; KC, keratinocyte chemoattractant; KC, keratinocyte chemoattractant; MIP, macrophage inflammatory protein; RANTES, regulated upon activation normal T cell expressed and secreted; TNF, tumour necrosis factor; WT, wild-type.
In addition to studies described above using a P. aeruginosa inoculum of 1 × 105 cfu, we performed studies using a higher bacterial inoculum (5 × 106 cfu). In these studies, WT controls and IKTA mice were treated with or without Dox for 48 h prior to IT injection with P. aeruginosa (Fig. 2). By 24 h after infection, all IKTA mice treated with Dox survived, but WT control mice (with or without Dox) and IKTA mice not treated with Dox experienced mortality rates of 80–100% (P < 0·05). These data indicate that augmented NF-κB activation in epithelium improves survival following lethal P. aeruginosa lung infection, probably through improved bacterial clearance.
Fig. 2.
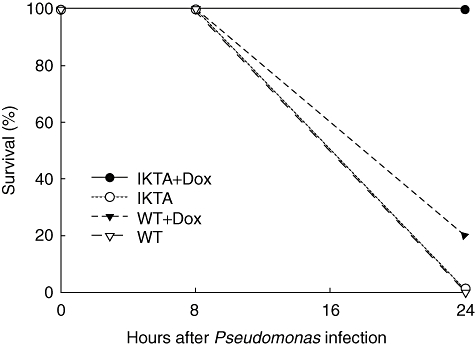
Survival of cIKK2 TransActivated (IKTA) mice with [IKTA + doxycycline (Dox)] or without (IKTA) Dox treatment and wild-type (WT) controls (WT and WT + Dox) at 24 h after after intratracheal administration of PA103 (5 × 106 colony-forming units). Dox was added to drinking water 2 days prior to infection (n = 5 per group, P < 0·05 for IKTA + Dox compared with other groups).
To assess directly the contribution of neutrophils to improved bacterial clearance by Dox-treated IKTA mice, we depleted neutrophils using i.p. injection of anti-neutrophil antibodies [25]. An 82 ± 3·1% reduction in peripheral blood neutrophil counts was found in anti-neutrophil antibody treated mice compared with baseline. After documenting neutrophil depletion, P. aeruginosa (1 × 105 cfu) was introduced by i.t. injection. Twenty-four hours after infection, BAL neutrophil counts were reduced similarly in all groups, while the macrophage population remained unaffected (Fig. 3a). In these studies, no differences were identified in bacterial colony counts among the four treatment groups (Fig. 3b). These studies show that the beneficial effects of NF-κB activation in airway epithelium on host defence against P. aeruginosa are transduced through increased recruitment of neutrophils in the lungs.
Fig. 3.
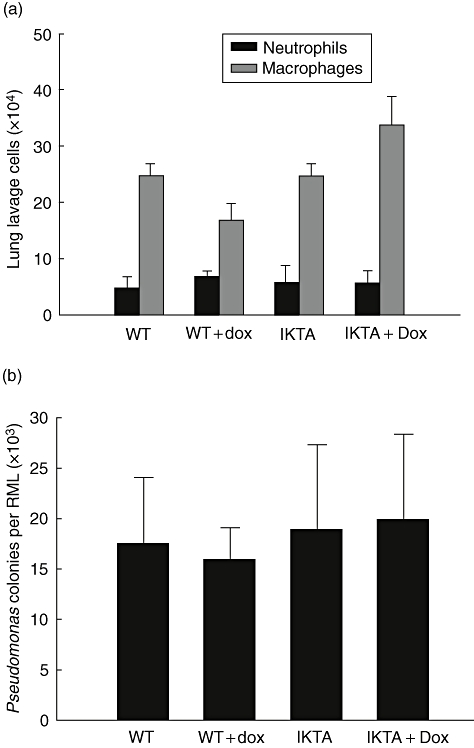
Neutrophil depletion eliminates the protective effects of increased epithelial cell nuclear factor kappa-B activation in Pseudomonas aeruginosa pneumonia. (a) Lung lavage neutrophil and macrophage counts from four groups of neutrophil depleted mice: wild-type (WT), WT + doxycycline (Dox), cIKK2 TransActivated (IKTA) and IKTA + Dox. Dox was added to drinking water 2 days prior to infection and mice were harvested at 24 h after after intratracheal injection of PA103 [1 × 105 colony-forming units (cfu)]. (b) P. aeruginosa colony counts per right middle lobe (RML) from mice harvested 24 h after i.t. injection of 1 × 105 cfu of PA103 (n = 5 per group).
Nuclear factor kappa-B inhibition in airway epithelium impairs clearance of P. aeruginosa pneumonia
After observing that augmented NF-κB activation in airway epithelium resulted in improved host defence against P. aeruginosa lung infection, we investigated whether epithelial NF-κB activation is necessary for a protective host response to P. aeruginosa infection. For these studies, we used DNTA transgenic mice, which express inducibly a dominant inhibitor of the NF-κB pathway in airway epithelial cells following Dox treatment [20]. As our previous studies indicated no significant differences in BAL neutrophil counts and bacterial colony counts in the three types of control animals (IKTA mice without Dox treatment and WT mice with and without Dox treatment), we used two groups of mice in these studies: DNTA mice treated with Dox (DNTA + Dox) and WT controls treated with Dox (WT + Dox). A lower dose of P. aeruginosa (1 × 104 cfu, PA103 strain) was chosen to prevent mortality in the DNTA + Dox group. DNTA mice were treated with Dox for 7 days prior to bacterial infection for maximal transgene expression and NF-κB inhibition.
As opposed to our findings in P. aeruginosa infected IKTA + Dox mice, DNTA mice treated with Dox had a lower number of neutrophils in BAL (14·1 ± 5·1 × 104) than WT + Dox mice (69·0 ± 11·5 × 104) at 24 h after infection (P < 0·01). DNTA + Dox mice also had fewer macrophages in BAL (13·3 ± 3·3 × 104) than WT + Dox mice (31·5 ± 5·7 × 104) (P < 0·05) (Fig. 4a). In addition, DNTA + Dox mice showed higher bacterial colony counts compared with WT + Dox mice (P < 0·05) (Fig. 4b). There were no significant differences in total peripheral white blood cell counts or neutrophils between WT + Dox and DNTA + Dox mice following P. aeruginosa infection (data not shown). These results show that blocking NF-κB activation in the airway epithelium of DNTA mice decreases neutrophil influx into the lungs following P. aeruginosa infection despite an increased bacterial burden, suggesting that impaired neutrophil recruitment is the primary abnormality in these mice.
Fig. 4.
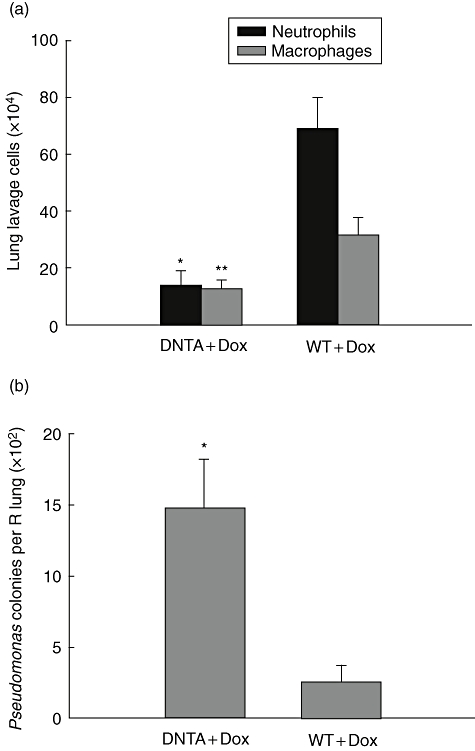
IκBα-DN TransActivated (DNTA) mice have impaired host defence against Pseudomonas aeruginosa. (a) Lung lavage neutrophils and macrophage counts from DNTA and wild-type (WT) mice with doxycycline (Dox) treatment at 24 h after after intratracheal injection of a sublethal dose of PA103 [1 × 104 colony-forming units (cfu)] (n = 6–7 per group, *P < 0·01 and **P < 0·05 compared with neutrophils and macrophages in WT + Dox group respectively). (b) P. aeruginosa colony counts per lung from mice harvested 24 h after i.t. injection of 1 × 104 cfu of PA103 (n = 7–9 per group, *P = 0·01 compared with WT + Dox). (c) Cytokine and chemokine expression in bronchoalveolar lavage fluid from DNTA and WT mice treated with Dox at 24 h after infection with PA103 (1 × 104 cfu) (n = 5–8 per group, *P < 0·01 compared with DNTA + Dox).
We measured cytokines and chemokines in BAL from DNTA and WT mice treated with Dox at 24 h after P. aeruginosa infection (Fig. 4c). In these studies, the production of KC differed significantly between the DNTA + Dox mice (103·0 ± 14·1 pg/ml) and WT + Dox mice (184·0 ± 34·3 pg/ml) (P < 0·05). The reduced expression of the chemokine KC in the DNTA mice treated with Dox was consistent with the finding of lower BAL neutrophil counts in this group.
To determine whether the selective reduction of KC in BAL of Dox-treated DNTA mice could be responsible for impaired neutrophil recruitment and reduced clearance of PA103 from the lungs, we administered recombinant KC by i.t. injection. WT mice treated with 5 µg of recombinant KC (without P. aeruginosa infection) showed an increased number of neutrophils in BAL (approximately 10 × 104 cells) compared with WT mice treated with vehicle (PBS), which had few neutrophils (<1 × 103). We then instilled KC or vehicle into the airway of Dox-treated DNTA mice at the time of P. aeruginosa infection and evaluated lung inflammation and bacterial clearance at 24 h. After P. aeruginosa infection, DNTA + Dox mice treated with KC had increased accumulation of neutrophils and macrophages in BAL (29·5 ± 8·1 × 104, 28·0 ± 4·1 × 104 respectively) compared with DNTA + Dox mice treated with vehicle (5·3 ± 1·1 × 104, 17·6 ± 3·5 × 104 respectively) (P < 0·001, neutrophils; P < 0·05, macrophages) (Fig. 5a). Bacterial colony counts were also significantly different between the groups. DNTA + Dox mice treated with KC had essentially cleared the P. aeruginosa lung infection, whereas vehicle-treated DNTA + Dox mice still harboured viable bacteria (Fig. 5b). With the addition of exogenous KC, DNTA mice developed enhanced pulmonary neutrophilia and cleared P. aeruginosa efficiently. These findings indicate that successful elimination of P. aeruginosa from the lungs requires NF-κB driven chemokine production by airway epithelial cells.
Fig. 5.
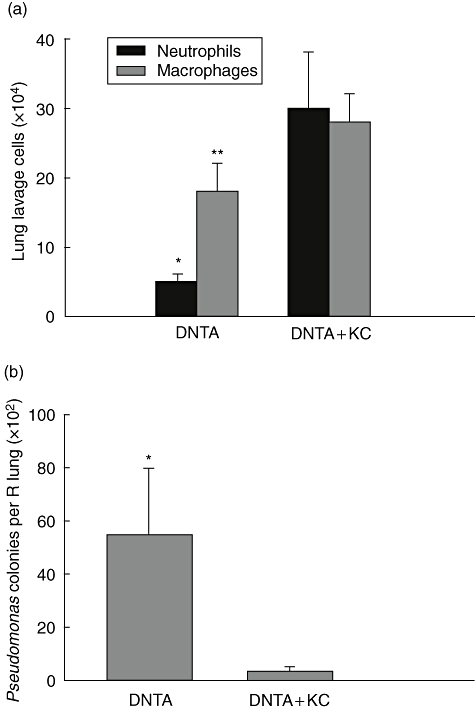
Keratinocyte chemoattractant (KC) replacement in IκBα-DN TransActivated (DNTA) mice improves host defence against Pseudomonas aeruginosa. (a) Lung lavage neutrophils and macrophage counts in doxycycline (Dox)-treated DNTA mice treated with KC (DNTA + KC) or vehicle (DNTA) at 24 h following after intratracheal injection of PA103 [1 × 104 colony-forming units (cfu)] (n = 8–12 per group, *P < 0·001 and **P < 0·05 compared with neutrophils and macrophages in DNTA + KC group respectively). (b) P. aeruginosa colony counts per lung from mice harvested 24h after i.t. injection of 1 × 104 cfu of PA103 (n = 8–12 per group, *P < 0·05 compared with DNTA + KC).
Discussion
Epithelial cells in the lungs are exposed constantly to the environment through the essential process of respiration. This unique feature imposes an important responsibility for these cells to protect the lungs from microorganisms that are introduced into the airway. In these studies, we demonstrate that the airway epithelium plays an active role in early host defence against P. aeruginosa pneumonia through activation of the NF-κB pathway. The level of NF-κB activity in airway epithelial cells corresponds directly with the resolution of infection at 24 h. Increased NF-κB activation improves bacterial clearance, whereas inhibition of NF-κB activity hinders host defence. The critical downstream event regulated by NF-κB activation in airway epithelial cells appears to be CXC chemokine-mediated neutrophil recruitment into the lungs.
Previous work by our group and others using transgenic mice with modified NF-κB activation in the airway has shown a strong link between epithelial NF-κB activation and lung inflammation. When Escherichia coli LPS is administered to transgenic mice with a CC10 promoter-driven mutated IκBα in the proximal airways, the inflammatory response is attenuated significantly in comparison with similarly treated WT mice [20,26]. This is manifested by reduced neutrophilic lung inflammation and decreased levels of chemokines and cytokines in BAL fluid. In addition, lung inflammation is reduced following exposure to E. coli LPS in another line of transgenic mice that express mutated IκBα in type 2 alveolar epithelial cells using a surfactant protein C promoter [27]. Collectively, these studies indicate that inhibition of NF-κB activity in both the proximal and distal lung epithelium limits the acute inflammatory response to LPS. The current study expands this work to include other features pneumonia, namely live bacterial infection and assessment of bacterial clearance. We provide evidence that modulating NF-κB activation in airway epithelium of transgenic mice alters P. aeruginosa-induced lung inflammation and impacts bacterial clearance and survival from infection. Together, these investigations indicate a non-redundant role for airway epithelium in protecting the lungs from bacterial infection.
Rapid recruitment of neutrophils to the lungs via production of Glu-Leu-Arg-positive (ELR+) CXC chemokines has been shown to be necessary for effective clearance of P. aeruginosa. Administration of a neutralizing antibody against the CXCR2 receptor (common receptor for murine ELR+ CXC chemokines MIP-2 and KC) prior to P. aeruginosa infection resulted in increased mortality compared with untreated mice [28]. Also, in vivo neutrophil depletion studies demonstrated that neutrophil-deficient mice have reduced survival after P. aeruginosa infection. Here, we show that that enhanced neutrophilic alveolitis in IKTA mice augments bacterial clearance and improves survival following lethal doses of P. aeruginosa at 24 h. In contrast, continued Dox treatment of IKTA mice for 7–14 days results in persistent inflammation, lung injury and substantial mortality [20]. Together with the current findings, these data suggest that a fine balance is required for rapid bacterial clearance without significant host injury and highlight the complexity in control of innate immunity by the NF-κB pathway.
While host defence against P. aeruginosa was improved in IKTA mice, inhibition of NF-κB signalling in DNTA mice resulted in impaired neutrophil recruitment and reduced bacterial clearance, which was reversed by treatment with recombinant KC. Interestingly, exogenous KC did not restore lung neutrophils in DNTA mice to the level seen in WT mice following P. aeruginosa infection, despite administration of a large chemokine dose. This may indicate that other NF-κB-dependent products are involved in neutrophil recruitment in these mice. The bacterial clearance phenotype, however, was completely restored by KC replacement, highlighting the importance of ELR+ CXC chemokines and neutrophils in pulmonary host defence against P. aeruginosa.
Alveolar macrophages, which normally comprise greater than 95% of the pulmonary leucocyte population, are important in innate immunity; however, the role of alveolar macrophages in the setting of P. aeruginosa pneumonia is not well understood [25]. Three separate in vivo studies of alveolar macrophage depletion in P. aeruginosa lung infection produced variable results in chemokine generation, neutrophil recruitment and bacterial clearance [29–31]. Although we observed differences in alveolar macrophage numbers in both IKTA and DNTA mice compared with controls after P. aeruginosa infection, it appears likely that activated neutrophils (rather than macrophages) are the principal inflammatory cells that mediate clearance of P. aeruginosa downstream of epithelial NF-κB signalling.
Epithelial cells from many organs (in addition to the lungs) have the capacity to produce cytokines and mediate neutrophil influx, suggesting that all cells at the interface of host and environment participate actively in innate immunity. P. aeruginosa-treated human corneal epithelial cells in culture showed an NF-κB-dependent early expression of TNF-α, IL-6 and IL-8 [32]. In vivo studies of P. aeruginosa corneal infections demonstrated increased neutrophil numbers associated with increased MIP-2 and KC expression in the corneal tissue [33]. In a murine model of P. aeruginosa urinary tract infection, increased MIP-2 expression was observed in urinary tract epithelial cells in conjunction with neutrophil recruitment [34,35]. These observations in non-respiratory epithelial cells provide support for the concept of a conserved mechanism by which epithelial cells interact directly with products of P. aeruginosa, activating NF-κB and leading to chemokine production and subsequent neutrophil recruitment.
Previous ideas regarding contributions of the airway epithelium to innate immunity have focused primarily on its function as a physical barrier between the host and environmental pathogens. The ciliated border of columnar epithelial cells provides mechanical clearance of inhaled organisms. Airway surface fluid bathes epithelial cell surfaces in a rich mixture of anti-microbial agents, including lysozyme, lactoferrin and anti-microbial peptides [36–38]. Until recently, emphasis has been placed on other members of innate immune cell community, such as macrophages and dendritic cells, as the major enablers of early host defence. Our studies indicate that airway epithelial cells are active and important participants in innate immune responses rather than passive bystanders in the presence of invading micro-organisms.
In summary, activation of the NF-κB pathway in airway epithelial cells in vivo is essential for an effective innate host response to acute P. aeruginosa pneumonia through chemokine production and subsequent neutrophil recruitment to the lungs. Alterations of this critical pathway in airway epithelial cells directly affect the pathogenesis and outcome of P. aeruginosa infection. Therefore, developing targeted immunomodulatory therapies which augment the NF-κB-mediated response in the airway may ultimately improve patient outcomes in the face of a growing prevalence of multi-drug-resistant P. aeruginosa strains.
Acknowledgments
This work was supported by the US Department of Veterans Affairs (J. W. C., R.T. S., T. S. B.) and NIH grants HL66196 (J. W. C., T. S. B.), HL61419 (T. S. B.) and HL07123 (S. M. C. training grant) from the National Heart Lung and Blood Institute.
References
- 1.Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med. 2005;171:388–416. doi: 10.1164/rccm.200405-644ST. [DOI] [PubMed] [Google Scholar]
- 2.Rello J, Ollendorf DA, Oster G, et al. Epidemiology and outcomes of ventilator-associated pneumonia in a large US database. Chest. 2002;122:2115–21. doi: 10.1378/chest.122.6.2115. [DOI] [PubMed] [Google Scholar]
- 3.Chastre J, Fagon JY. Ventilator-associated pneumonia. Am J Respir Crit Care Med. 2002;165:867–903. doi: 10.1164/ajrccm.165.7.2105078. [DOI] [PubMed] [Google Scholar]
- 4.Kollef MH. Prevention of hospital-associated pneumonia and ventilator-associated pneumonia. Crit Care Med. 2004;32:1396–405. doi: 10.1097/01.ccm.0000128569.09113.fb. [DOI] [PubMed] [Google Scholar]
- 5.Rello J, Diaz E. Pneumonia in the intensive care unit. Crit Care Med. 2003;31:2544–51. doi: 10.1097/01.CCM.0000089928.84326.D2. [DOI] [PubMed] [Google Scholar]
- 6.Martin TR, Frevert CW. Innate immunity in the lungs. Proc Am Thorac Soc. 2005;2:403–11. doi: 10.1513/pats.200508-090JS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abreu MT, Arditi M. Innate immunity and Toll-like receptors: clinical implications of basic science research. J Pediatr. 2004;144:421–9. doi: 10.1016/j.jpeds.2004.01.057. [DOI] [PubMed] [Google Scholar]
- 8.Sadikot RT, Blackwell TS, Christman JW, Prince AS. Pathogen–host interactions in Pseudomonas aeruginosa pneumonia. Am J Respir Crit Care Med. 2005;171:1209–23. doi: 10.1164/rccm.200408-1044SO. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tseng J, Do J, Widdicombe JH, Machen TE. Innate immune responses of human tracheal epithelium to Pseudomonas aeruginosa flagellin, TNF-alpha, and IL-1beta. Am J Physiol Cell Physiol. 2006;290:C678–90. doi: 10.1152/ajpcell.00166.2005. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Z, Louboutin JP, Weiner DJ, Goldberg JB, Wilson JM. Human airway epithelial cells sense Pseudomonas aeruginosa infection via recognition of flagellin by Toll-like receptor 5. Infect Immun. 2005;73:7151–60. doi: 10.1128/IAI.73.11.7151-7160.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greene CM, Carroll TP, Smith SG, et al. TLR-induced inflammation in cystic fibrosis and non-cystic fibrosis airway epithelial cells. J Immunol. 2005;174:1638–46. doi: 10.4049/jimmunol.174.3.1638. [DOI] [PubMed] [Google Scholar]
- 12.Power MR, Peng Y, Maydanski E, Marshall JS, Lin TJ. The development of early host response to Pseudomonas aeruginosa lung infection is critically dependent on myeloid differentiation factor 88 in mice. J Biol Chem. 2004;279:49315–22. doi: 10.1074/jbc.M402111200. [DOI] [PubMed] [Google Scholar]
- 13.Rastogi D, Ratner AJ, Prince A. Host–bacterial interactions in the initiation of inflammation. Paediatr Respir Rev. 2001;2:245–52. doi: 10.1053/prrv.2001.0147. [DOI] [PubMed] [Google Scholar]
- 14.Skerrett SJ, Liggitt HD, Hajjar AM, Wilson CB. Cutting edge: myeloid differentiation factor 88 is essential for pulmonary host defense against Pseudomonas aeruginosa but not Staphylococcus aureus. J Immunol. 2004;172:3377–81. doi: 10.4049/jimmunol.172.6.3377. [DOI] [PubMed] [Google Scholar]
- 15.Mercurio F, Manning AM. Multiple signals converging on NF-kappaB. Curr Opin Cell Biol. 1999;11:226–32. doi: 10.1016/s0955-0674(99)80030-1. [DOI] [PubMed] [Google Scholar]
- 16.Mercurio F, Manning AM. NF-kappaB as a primary regulator of the stress response. Oncogene. 1999;18:6163–71. doi: 10.1038/sj.onc.1203174. [DOI] [PubMed] [Google Scholar]
- 17.Hajjar AM, Harowicz H, Liggitt HD, Fink PJ, Wilson CB, Skerrett SJ. An essential role for non-bone marrow-derived cells in control of Pseudomonas aeruginosa pneumonia. Am J Respir Cell Mol Biol. 2005;33:470–5. doi: 10.1165/rcmb.2005-0199OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sadikot RT, Zeng H, Joo M, et al. Targeted immunomodulation of the NF-kappaB pathway in airway epithelium impacts host defense against Pseudomonas aeruginosa. J Immunol. 2006;176:4923–30. doi: 10.4049/jimmunol.176.8.4923. [DOI] [PubMed] [Google Scholar]
- 19.Perl AK, Tichelaar JW, Whitsett JA. Conditional gene expression in the respiratory epithelium of the mouse. Transgenic Res. 2002;11:21–9. doi: 10.1023/a:1013986627504. [DOI] [PubMed] [Google Scholar]
- 20.Cheng DS, Han W, Chen SM, et al. Airway epithelium controls lung inflammation and injury through the NF-kappa B pathway. J Immunol. 2007;178:6504–13. doi: 10.4049/jimmunol.178.10.6504. [DOI] [PubMed] [Google Scholar]
- 21.Mercurio F, Zhu H, Murray BW, et al. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science. 1997;278:860–6. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 22.Sadikot RT, Han W, Everhart MB, et al. Selective I kappa B kinase expression in airway epithelium generates neutrophilic lung inflammation. J Immunol. 2003;170:1091–8. doi: 10.4049/jimmunol.170.2.1091. [DOI] [PubMed] [Google Scholar]
- 23.Tichelaar JW, Lu W, Whitsett JA. Conditional expression of fibroblast growth factor-7 in the developing and mature lung. J Biol Chem. 2000;275:11858–64. doi: 10.1074/jbc.275.16.11858. [DOI] [PubMed] [Google Scholar]
- 24.Abraham E, Carmody A, Shenkar R, Arcaroli J. Neutrophils as early immunologic effectors in hemorrhage- or endotoxemia-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1137–45. doi: 10.1152/ajplung.2000.279.6.L1137. [DOI] [PubMed] [Google Scholar]
- 25.Happel KI, Bagby GJ, Nelson S. Host defense and bacterial pneumonia. Semin Respir Crit Care Med. 2004;25:43–52. doi: 10.1055/s-2004-822304. [DOI] [PubMed] [Google Scholar]
- 26.Poynter ME, Irvin CG, Janssen-Heininger YM. A prominent role for airway epithelial NF-kappa B activation in lipopolysaccharide-induced airway inflammation. J Immunol. 2003;170:6257–65. doi: 10.4049/jimmunol.170.12.6257. [DOI] [PubMed] [Google Scholar]
- 27.Skerrett SJ, Liggitt HD, Hajjar AM, Ernst RK, Miller SI, Wilson CB. Respiratory epithelial cells regulate lung inflammation in response to inhaled endotoxin. Am J Physiol Lung Cell Mol Physiol. 2004;287:L143–52. doi: 10.1152/ajplung.00030.2004. [DOI] [PubMed] [Google Scholar]
- 28.Tsai WC, Strieter RM, Mehrad B, Newstead MW, Zeng X, Standiford TJ. CXC chemokine receptor CXCR2 is essential for protective innate host response in murine Pseudomonas aeruginosa pneumonia. Infect Immun. 2000;68:4289–96. doi: 10.1128/iai.68.7.4289-4296.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheung DO, Halsey K, Speert DP. Role of pulmonary alveolar macrophages in defense of the lung against Pseudomonas aeruginosa. Infect Immun. 2000;68:4585–92. doi: 10.1128/iai.68.8.4585-4592.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kooguchi K, Hashimoto S, Kobayashi A, et al. Role of alveolar macrophages in initiation and regulation of inflammation in Pseudomonas aeruginosa pneumonia. Infect Immun. 1998;66:3164–9. doi: 10.1128/iai.66.7.3164-3169.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hashimoto S, Pittet JF, Hong K, et al. Depletion of alveolar macrophages decreases neutrophil chemotaxis to Pseudomonas airspace infections. Am J Physiol. 1996;270:L819–28. doi: 10.1152/ajplung.1996.270.5.L819. [DOI] [PubMed] [Google Scholar]
- 32.Zhang J, Wu XY, Yu FS. Inflammatory responses of corneal epithelial cells to Pseudomonas aeruginosa infection. Curr Eye Res. 2005;30:527–34. doi: 10.1080/02713680590968150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kernacki KA, Barrett RP, Hobden JA, Hazlett LD. Macrophage inflammatory protein-2 is a mediator of polymorphonuclear neutrophil influx in ocular bacterial infection. J Immunol. 2000;164:1037–45. doi: 10.4049/jimmunol.164.2.1037. [DOI] [PubMed] [Google Scholar]
- 34.Mittal R, Chhibber S, Sharma S, Harjai K. Macrophage inflammatory protein-2, neutrophil recruitment and bacterial persistence in an experimental mouse model of urinary tract infection. Microbes Infect. 2004;6:1326–32. doi: 10.1016/j.micinf.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 35.Chowdhury P, Sacks SH, Sheerin NS. Minireview: functions of the renal tract epithelium in coordinating the innate immune response to infection. Kidney Int. 2004;66:1334–44. doi: 10.1111/j.1523-1755.2004.00896.x. [DOI] [PubMed] [Google Scholar]
- 36.Beutler B. Innate immunity: an overview. Mol Immunol. 2004;40:845–59. doi: 10.1016/j.molimm.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 37.Diamond G, Legarda D, Ryan LK. The innate immune response of the respiratory epithelium. Immunol Rev. 2000;173:27–38. doi: 10.1034/j.1600-065x.2000.917304.x. [DOI] [PubMed] [Google Scholar]
- 38.Bals R. Epithelial antimicrobial peptides in host defense against infection. Respir Res. 2000;1:141–50. doi: 10.1186/rr25. [DOI] [PMC free article] [PubMed] [Google Scholar]