

Abstract
XAGE-1b is regarded as one of the most immunogenic antigens and the most promising targets for lung adenocarcinoma immunotherapy. In this study, we sought to determine whether monocyte-derived dendritic cells (DCs) pulsed with purified full-length XAGE-1b could induce specific cytotoxic T lymphocytes (CTLs) against tumour cells from patients with non-small cell lung cancer (NSCLC) in vitro. XAGE-1b mRNA expression was examined in primary cultures of lung cancer cells and normal lung epithelial cells established from fresh tissues surgically resected from 30 patients with NSCLC using reverse transcription–polymerase chain reaction (RT–PCR). XAGE-1b mRNA expression was observed in 11 of 18 (61·1%) adenocarcinomas and one of 12 (8·3%) lung cancers of other histological types (P = 0·015). The 246-base pairs XAGE-1b gene was inserted into a recombinant expression vector. Full-length XAGE-1b was then expressed in BL21 (DE3) Escherichia coli and purified by AKTA-fast performance liquid chromatography (FPLC). DCs generated from peripheral blood mononuclear cells were pulsed with XAGE-1b by incubation with the protein at an immature stage. The XAGE-1b-pulsed DCs induced CTLs following 14 days of co-culture. Finally, an adherent target detachment (ATD) assay was performed to test the cytotoxicity of the XAGE-1b-specific CTLs against cancer cells and normal lung epithelial cells. The XAGE-1b-specific CTLs had a stronger lytic effect on autologous XAGE-1b mRNA-positive cancer cells than on autologous XAGE-1b mRNA-negative cancer cells or allogenous XAGE-1b mRNA-positive cancer cells. The CTLs had no lytic activity against normal lung epithelial cells. These results can be used to develop simple and effective cancer/testis antigen-based immunotherapies for NSCLC.
Keywords: cancer/testis antigen, dendritic cells, non-small cell lung cancer, tumour immunity, XAGE-1b
Introduction
Dendritic cell (DC)-based tumour immunotherapy is one of the most promising therapeutic modalities because DCs are highly effective antigen-presenting cells that play a critical role in the induction of primary immune responses against tumour-associated antigens [1]. The tumour-associated antigens taken up and presented by DCs stimulate T cells to kill tumour cells expressing the same antigens. Thus, finding tumour-associated antigens that are expressed highly and specifically on tumour cells is essential for developing therapeutic anti-cancer vaccines. Most current DC-based tumour vaccines focus upon specific tumours, such as melanoma, myeloma, renal carcinoma and prostatic carcinoma, because many antigens associated with these tumours have already been identified. With the development of immunological and genetic techniques, increasing numbers of cytotoxic T cell-recognized tumour antigens [2–4] and antibody-defined antigens [5,6] have been identified. Cancer/testis antigens (CTAs) comprise the largest family of tumour antigens. Their expression is restricted to select human cancers and to gonadal germ cells and trophoblasts in healthy individuals [7]. The expression pattern and immunogenicity of CTAs make them ideal targets for tumour immunotherapy. Some CTAs, including the MAGE, BAGE and GAGE series, in addition to NY-ESO-1, have been detected in several tumours [8,9], and vaccines based on these CTAs have produced beneficial effects in vitro and in vivo[10,11]. In recent years several CTAs, including MAGE-A1, MAGE-A3, NY-ESO-1, SSX2 and XAGE-1b, were found in lung tumours, making it possible to apply CTA-based immunotherapy to the treatment of lung cancer [12,13].
XAGE-1 was identified originally as a PAGE/GAGE-related gene on the X chromosome by expressed sequence tag (EST) analysis [14]. Studies of XAGE-1 expression revealed that the gene is a CTA [15,16], and it has been shown since to be expressed in metastatic melanoma, Ewing's sarcoma and some epithelial tumours, including those of the breast and lung [16,17]. Among the four splice variants of XAGE-1 (XAGE-1a, b, c and d), XAGE-1b is regarded as the most immunogenic, capable of eliciting cellular and humoral immune responses. It is also highly expressed in lung adenocarcinoma; thus, XAGE-1b is one of the most promising targets for lung adenocarcinoma immunotherapy [12,18–20]. To determine whether monocyte-derived DCs pulsed with purified full-length XAGE-1b could induce specific cytotocic T lymphocytes (CTLs) against lung cancer cells in vitro, we amplified full-length XAGE-1b from lung tissue samples collected from patients with non-small cell lung cancer (NSCLC), and constructed a recombinant plasmid vector for the expression of XAGE-1b. By pulsing monocyte-derived DCs with XAGE-1b, we induced XAGE-1b-specific CTLs, whose cytotoxicity was tested in vitro using tumour cells from patients with NSCLC.
Materials and methods
Primary culture of lung cancer cells and normal lung epithelial cells
Surgically resected tumour tissues and corresponding adjacent normal lung tissues were obtained from 30 patients with NSCLC in Guangdong Provincial People's Hospital. Written informed consent was obtained from each patient and the study was approved by the hospital's ethics committee. Portions of the specimens were cut into small pieces (0·2–0·3 g) and frozen immediately in liquid nitrogen; the samples were stored at −80°C until RNA extraction. The remaining pieces of the specimens were placed in RPMI-1640 (Gibco–BRL, Grand Island, NY, USA) containing 50 µg/ml streptomycin and 50 U/ml penicillin for primary cell culture. The tumour tissues were then digested with 0·05% IV collagenase (Hyclone, Logan, UT, USA) and 0·001% DNA enzyme (Hyclone) for 4–6 h. The lung cancer cells were isolated by sedimentation using 75% Ficoll–Hypaque and cultured subsequently in RPMI-1640 supplemented with 200 mM l-glutamine, 50 µg/ml streptomycin, 50 U/ml penicillin and 10% fetal calf serum (FCS). Passage was performed every 2–3 days. The normal lung tissues were processed with 0·25% trypsase (pH 7·4; Hyclone) for 10–20 min to isolate the epithelial cells, which were then cultured as described above. After steady passage, the cells were stored in liquid nitrogen for the cytotoxicity assay.
Total RNA extraction and reverse transcription–polymerase chain reaction (RT–PCR)
Total cellular RNA was extracted from the frozen lung cancer and normal lung specimens using TRIZOL (Gibco–BRL), according to the manufacturer's instructions. The RNA was then reverse-transcribed using a SuperScript III First-Strand Synthesis System for RT–PCR (Invitrogen, Carlsbad, CA, USA). The integrity of the cDNAs was tested by amplifying GAPDH[308 base pairs (bp)]. Full-length XAGE-1b (246 bp) was amplified using the following primers: forward primer, 5′-GGAAGGAGTTCGAACCATGGAGAGCCCCAAAAAGAAGAA-3′; reverse primer, 5′-TGCGGCCGCACTCGAGCTAAACTTGTGGTTGCTCTTCACCTG-3′. The primers were designed to create an XmnI site at the 5′-end and a XhoI site at the 3′-end of the product. The products were confirmed by electrophoresis and sequencing.
Plasmid vector construction
The amplified XAGE-1b fragments were subcloned into pDRIVE T (Qiagen, Hilden, Germany), then excised with XmnI and XhoI and cloned into the pReceiver-B01a expression vector (GeneCopoeia & FulenGen, Inc., Germantown, MD, USA) (5050 bp) using T4 DNA ligase. The recombinant vector, pReceiver-B01a/XAGE-1b, is referred to hereafter as B01/XAGE. Escherichia coli BL21 (DE3) competent cells were then transformed with B01/XAGE. Three methods were used to identify the B01/XAGE plasmid: agarose gel electrophoresis together with pReceiver-B01a, amplification of XAGE-1b by PCR using transformed BL21 (DE3) or B01/XAGE as templates, and restriction digest analysis of B01/XAGE and pReceiver-B01a using XmnI and XhoI. Correct insertion of the cDNA was confirmed by sequencing.
Expression and purification of the recombinant XAGE-1b protein
BL21 (DE3) E. coli transformed with B01/XAGE were cultured in Luria–Bertani (LB) medium containing 100 µg/ml ampicillin. Once the optical density (OD)600 of the starter cultures reached 0·5–0·6, 1 mM isopropylthio-β-D-galactoside (IPTG; Sigma, St Louis, MO, USA) was added and the cells were further cultured at 37°C and 230 rpm for 4 h (the expression conditions were optimized in a preliminary experiment). The bacterial cells were then pelleted and lysed by sonication (JY92-II Sonic Dismembrator, Ningbo, China). The lysates were then centrifuged, and the supernatants and pellets were analysed by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE). Recombinant XAGE-1b was purified using Ni-NTA agarose (Novagen, Madison, WI, USA) and an anion exchange column (Mono-Q; Novagen) by fast protein liquid chromatography (AKTA-FPLC; Amersham Biosciences, Piscataway, NJ, USA). The purity of the proteins was analysed with Bandscan software (http://www.glyco.com).
Identification of recombinant XAGE-1b
The molecular mass of the recombinant protein was determined by 18% SDS-PAGE and the protein was identified by Western blotting. A mouse anti-His-tag monoclonal antibody (Novagen) and rabbit anti-XAGE-1b polyclonal antibody (Novus Biologicals, Littleton, CO, USA) were used as the primary antibodies, respectively. Peroxidase-conjugated anti-mouse rabbit IgG (Sigma) was used as the secondary antibody.
Generation of monocyte-derived DCs and pulsing with XAGE-1b
Based on the RT–PCR results, five patients with XAGE-1b-positive tumour cells and five patients with XAGE-1b-negative tumour cells were chosen for further analysis. Peripheral blood (20 ml) was collected from each patient and the peripheral blood mononuclear cells (PBMCs) were isolated. Monocyte-derived DCs were then generated as described previously [21]. On day 6, XAGE-1b was added to the immature DCs at a final concentration of 0·2 µg/ml. After 24 h (on day 7), the DCs were matured by the addition of rhIL-1β (10 ng/ml), prostaglandin E2 (PGE2) (1 µg/ml), recombinant human interleukin (rhIL)-6 (1000 U/ml) and recombinant human tumour necrosis factor (rhTNF)-α (50 ng/ml). The mature DCs were harvested on day 9. The culture supernatants were collected on days 2, 5 and 9 and assayed for IL-12 secretion by enzyme-linked immunosorbent assay (ELISA) (R&D Systems, Minneapolis, MN, USA). All non-adherent mononuclear cells were cryopreserved for future use.
Immunofluorescence and flow cytometric analysis of the XAGE-1b-pulsed DCs
DCs pulsed with XAGE-1b were collected on slides coated with l-polylysine and fixed at 4°C in acetone. Phosphate-buffered saline (PBS) containing 1% Triton X-100 was used to increase the membrane permeability of the DCs. The slides were incubated first with rabbit anti-XAGE-1b polyclonal antibodies at 37°C for 2 h then with goat anti-rabbit IgG CY3 fluorescent antibodies (Sigma) at 37°C for 30 min. Finally, the slides were analysed under a fluorescence microscope (TE2000-S; Nikon, Tokyo, Japan). The XAGE-1b-pulsed DCs were stained with fluorescein isothiocyanate (FITC)- or phycoerythrin (PE)-conjugated monoclonal antibodies (BD Biosciences Pharmingen, San Diego, CA, USA) against human leucocyte antigen D-related (HLA-DR), CD83, CD40, CD80 and CD86. The stained DCs were analysed on a flow cytometer (Bio-Rad, Hercules, CA, USA).
Induction of XAGE-1b-specific CTLs
The cryopreserved non-adherent mononuclear cells isolated from peripheral blood were reanimated and co-cultured with XAGE-1b protein-pulsed mature DCs at a ratio of 10:1 in AIM-V® (Gibco–BRL) containing IL-2 (10 U/ml) and IL-7 (5 ng/ml). Fresh medium containing the same cytokines was added every other day. On day 7, the responder cells were restimulated using XAGE-1b protein-pulsed mature DCs. On day 14, the responder cells (XAGE-1b-specific CTLs) were harvested. Non-adherent mononuclear cells co-cultured with mock-pulsed DCs were used as controls in all subsequent experiments.
Cytokine measurement
Enzyme-linked immunospot (ELISPOT) assays were carried out to detect the interferon (IFN)-γ secreted by the T lymphocytes using a human IFN-γ ELISPOT Kit (U-Cy Tech Biosciences, Utrecht, the Netherlands), according to the instruction manual. In total, 2 × 105 T lymphocytes and 1 × 104 XAGE-1b protein-pulsed DCs were plated per well in six-well plates and incubated for 24 h at 37°C under 5% CO2. In separate plates, 2 × 105 T lymphocytes stimulated by XAGE-1b protein-pulsed DCs for 7 days were reincubated with 1 × 104 XAGE-1b protein-pulsed DCs. Additional T lymphocytes (2 × 105) were incubated with 4 µg/ml phytohaemagglutinin (PHA) as a positive control or with nothing as a negative control. Six sets of controls were created. The spots were counted using a Bioreader-4000 (Biosys, Karben, Germany).
Cytotoxicity assay
The cytotoxicity of the XAGE-1b-specific CTLs was assessed as described previously [22] using an adherent target detachment (ATD) assay. Briefly, the target cells were seeded in 384-well cell culture trays at a concentration of 2500–3000 cells/well and incubated overnight to allow adhesion to the surface of the wells. The effector cells were then added at effector : target (E : T) ratios of 10:1, 20:1, 40:1 or 80:1 and incubated with the target cells for 24 h. After incubation, the dead target cells became non-adherent and were removed by washing together with the added effector cells. The remaining adherent target cells were stained with acridine orange (Invitrogen) at a final concentration of 25 µM for 30 min and then washed three times with PBS. Finally, images of the stained target cells were collected under a fluorescence microscope and analysed with micro-cosmos MiVnt Biologic Image Analytical Software (Weiyu, Zhuhai, China). Those wells in which target cells were seeded without effector cells were used as negative controls. The number of target cells in each image was determined using the above software. The percentage of specific lysis was calculated using the following formula: % specific lysis = [(mean number of target cells in the negative control wells − mean number of target cells in the experimental wells)/mean number of target cells in the negative control wells] × 100%.
For the cytotoxicity assay, we selected five patients with XAGE-1b-positive tumour cells and five patients with XAGE-1b-negative tumour cells. Autologous monocyte-derived DCs were pulsed with XAGE-1b and used to induce XAGE-1b-specific CTLs from autologous T lymphocytes; the CTLs were used subsequently as effector cells. Autologous or allogenous cancer cells and normal lung epithelial cells were used as the target cells. T lymphocytes co-cultured with mock-pulsed DCs were used as control effector cells.
Statistical analysis
Fisher's exact test was applied to compare differences in the XAGE-1b expression rate between adenocarcinoma and other histological types. Student's t-test was used to analyse the secretion of IFN-γ and IL-12. Statistical significance was defined as P < 0·05.
Results
XAGE-1b mRNA expression in 30 cases of NSCLC
XAGE-1b mRNA expression was detected in 12 of 30 (40%) tumour tissues (Fig. 1) and in none of the adjacent normal lung tissues. Eleven of the 12 XAGE-1b mRNA-positive patients had adenocarcinoma. XAGE-1b mRNA expression was observed in 11 of 18 (61·1%) cases of adenocarcinoma and in one of 12 (8·3%) cases of lung cancer with other histological types (P = 0·015). The relationship between gene expression and the clinical characteristics of the patients has been analysed and published [20].
Fig. 1.
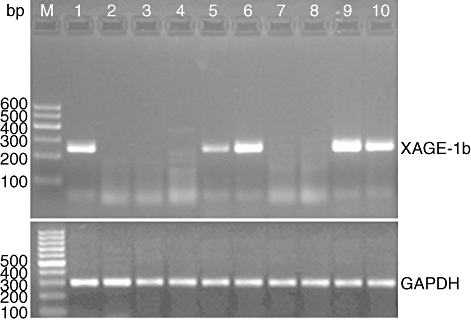
XAGE-1b mRNA expression status in tumour tissues of 10 patients with non-small cell lung cancer. M, DNA marker; lanes 1, 5, 6, 9, 10, XAGE-1b mRNA expression positive; lanes 2, 3, 4, 7, 8, XAGE-1b mRNA expression negative.
Expression vector construction and identification
Agarose gel electrophoresis indicated that B01/XAGE fell behind pReceiver-B01a; the cDNA fragment excised from B01/XAGE using XmnI and XhoI was 246 bp long while the product amplified from transformed BL21 (DE3) cells or the B01/XAGE plasmid was also 246 bp (Fig. 2). Correct insertion of the cDNA was confirmed by sequencing.
Fig. 2.
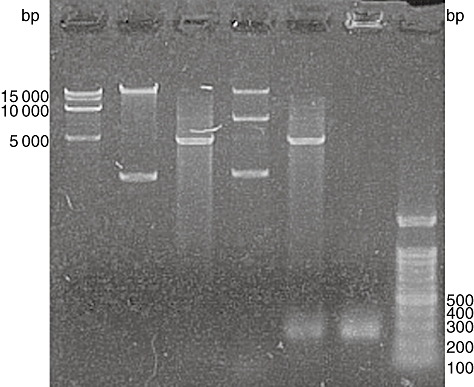
Restriction enzyme analysis and polymerase chain reaction (PCR) identification of recombinant vector B01/XAGE. M, DNA markers; lane 1, pReceiver-B01a expression vector; lane 2, restriction digest analysis of pReceiver-B01a using XmnI and XhoI; lane 3, recombinant vector B01/XAGE; lane 4, restriction digest analysis of recombinant vector B01/XAGE using XmnI and XhoI; lane 5, amplification of XAGE-1b by PCR using recombinant vector B01/XAGE as templates.
Expression, purification and identification of recombinant XAGE-1b
After expression of the protein, the BL21 (DE3) cells were lysed by sonication and centrifuged. Soluble XAGE-1b was identified in the supernatants by SDS-PAGE. The molecular weight was 11 kDa. AKTA-FPLC was performed using nickel-charged resin (Ni-NTA) and an anion exchange column (Mono-Q) to purify XAGE-1b (Fig. 3). Bandscan software analysis indicated that the purity of the protein was 93%. XAGE-1b was confirmed further by Western blot analysis using anti-His-tag and anti-XAGE-1b antibodies, respectively.
Fig. 3.
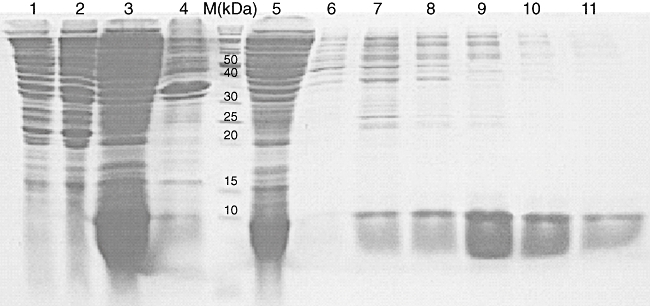
Purification profile of recombinant XAGE-1b analysis by 18% sodium dodecyl sulphate–polyacrylamide gel electrophoresis before and after purification. Lane 1, supernatants of cell lysates in non-induced culture; lane 2, pellets of cell lysates in non-induced culture; lane 3, supernatants of cell lysates in induced culture; lane 4, pellets of cell lysates in induced culture; M, protein marker; lane 5, flow; lane 6, 20 mmol/l imidazole wash; lane 7, 50 mmol/l imidazole eluate; lane 8, 100 mmol/l imidazole eluate; lane 9, 200 mmol/l imidazole eluate; lane 10, 300 mmol/l imidazole eluate; lane 11, 500 mmol/l imidazole eluate.
Identification of the XAGE-1b-pulsed DCs
On day 9, mature DCs pulsed with XAGE-1b were harvested and observed under an inverted phase contrast microscopy, which revealed typical fully differentiated DCs. Immunofluorescence analysis showed intense fluorescence in the cytoplasm and membranes of the mature XAGE-1b-pulsed DCs (Fig. 4). No fluorescence was present in the mock-pulsed DCs. Flow cytometric analysis showed that high levels of HLA-DR, CD83, CD40, CD80 and CD86 were expressed on the mature XAGE-1b-pulsed DCs (Fig. 5). An ELISA-based assay indicated that IL-12 secretion (73·00 ± 9·53 pg/ml) from the mature XAGE-1b-pulsed DCs was higher than that from the immature DCs (54·96 ± 7·73 pg/ml; P = 0·011) or PBMCs (14·72 ± 5·02 pg/ml; P < 0·0001).
Fig. 4.
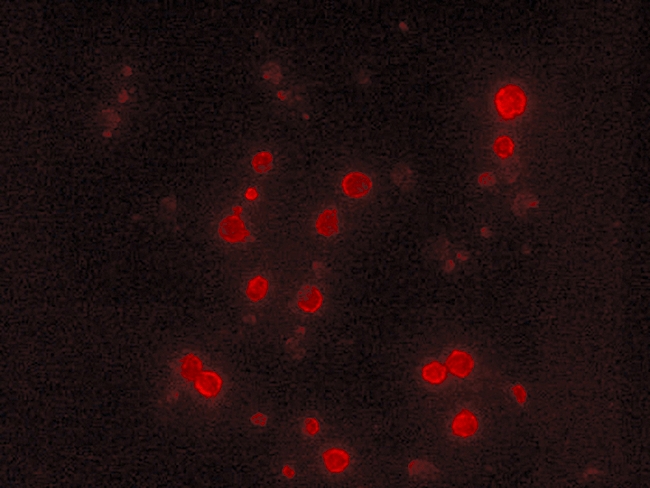
Immunofluorescence analysis of the XAGE-1b-pulsed dendritic cells (DCs). Intense fluorescence in the cytoplasm and membranes of mature XAGE-1b-pulsed DCs were observed under fluorescence microscope.
Fig. 5.
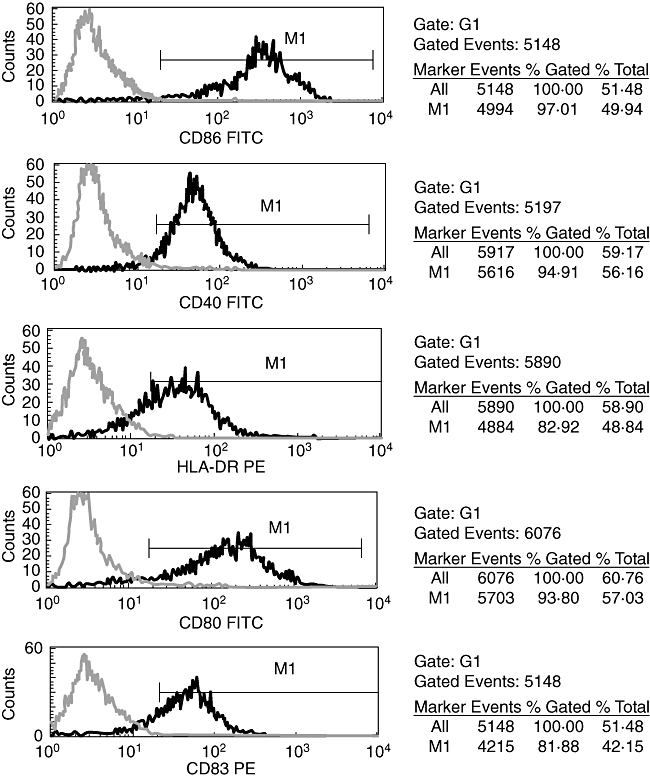
Flow cytometric analysis of mature XAGE-1b-pulsed dendritic cells (DCs). High levels of CD86, CD40, human leucocyte antigen D-related, CD80 and CD83 were expressed on the mature XAGE-1b-pulsed DCs.
Production of XAGE-1b-specific CTLs
XAGE-1b-specific CTLs were generated from non-adherent mononuclear cells by repeated stimulation of XAGE-1b-pulsed mature DCs in the presence of IL-2 and IL-7 for 14 days. An ELISPOT-based IFN-γ secretion assay revealed that the spots number of non-adherent mononuclear cells before stimulation was 3·83 ± 2·21. In comparison, the number of lymphocytes stimulated once and twice with XAGE-1b-pulsed DCs was 71·00 ± 27·42 and 163·58 ± 33·21, respectively. The level of IFN-γ increased significantly (P < 0·0001).
Cytotoxicity of the XAGE-1b-specific CTLs
When XAGE-1b mRNA-positive autologous cancer cells were used as target cells, the XAGE-1b-specific CTLs lysed the cancer cells effectively in a dose-dependent manner, whereas the cancer cells were lysed only minimally by T lymphocytes (Fig. 6a). As for the XAGE-1b mRNA-negative autologous cancer cells, the lytic activity of the XAGE-1b-specific CTLs notably decreased but was still stronger than that of the T lymphocytes (Fig. 6b). Neither the XAGE-1b-specific CTLs nor the T lymphocytes exhibited any lytic activity against normal lung epithelial cells (Fig. 6c). We also used XAGE-1b mRNA-positive allogenous cancer cells as target cells with the XAGE-1b-specific CTLs. The CTLs had some lytic activity against the allogenous cancer cells, but the effect was smaller than that observed against the autologous cancer cells (Fig. 6d).
Fig. 6.
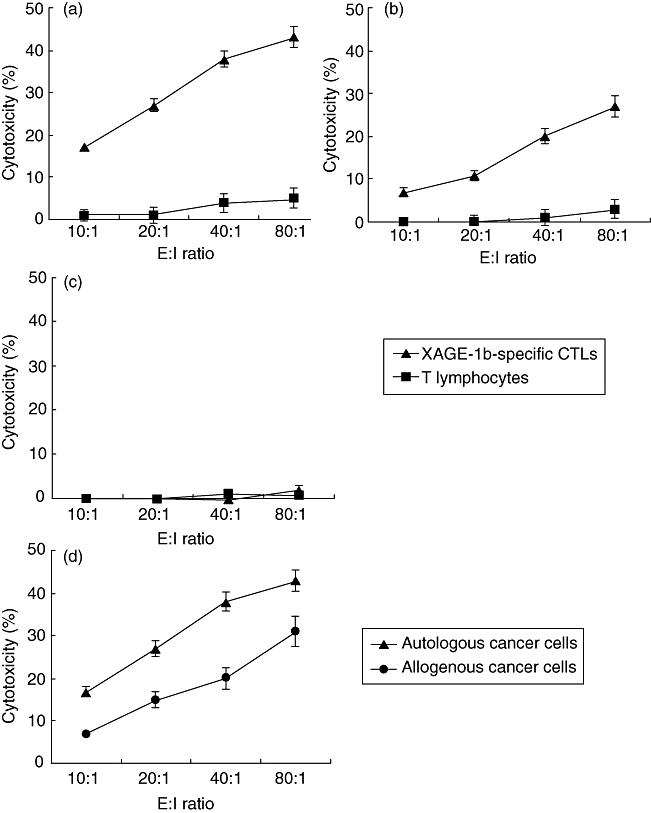
Cytotoxicity of XAGE-1b-specific cytotoxic T lymphocytes (CTLs) against cancer cells and normal lung epithelial cells. Mean ± standard deviation from five patients. T lymphocytes co-cultured with mock-pulsed dendritic cells (DCs) were used as control effector cells. (a) XAGE-1b mRNA-positive autologous cancer cells were used as target cells; (b) XAGE-1b mRNA-negative autologous cancer cells were used as target cells; (c) normal lung epithelial cells were used as target cells; D, XAGE-1b mRNA-positive allogenous cancer cells were used as target cells with the XAGE-1b-specific CTLs.
Discussion
Previous studies on XAGE-1b identified XAGE-1b as an active candidate for lung adenocarcinoma immunotherapy. In this study, we induced XAGE-1b-specific CTLs against lung cancer cells using DCs pulsed with full-length XAGE-1b. An important feature of this study was the use of purified full-length XAGE-1b as antigenic material. In DC-based vaccines, the efficient entry of tumour-associated antigens into DCs and sufficient presentation of the antigens on the DC surface are critical for inducing a potent CTL response. Nikitina et al.[23,24] reported that DCs transduced with full-length wild-type p53 using an adenoviral vector were able to generate a specific anti-tumour immune response against p53-over-expressing tumour cells. This strategy may be effective, but the use of adenoviral vectors in a clinical setting remains controversial. In contrast, pulsing DCs with antigenic peptides or proteins does not pose a safety concern. Compared to antigenic peptides, full-length antigens offer several advantages. After uptake, DCs can process proteins into many different peptides and present them to different major histocompatibility complex (MHC) molecules on the DC surface, which means that the vaccine can be administered to a broad range of individuals. Some of the peptides presented on the DC surface are bound to MHC class II molecules and stimulate CD4+ T helper lymphocytes. In addition to this classic mechanism of antigen presentation, DCs can also process exogenous proteins and present peptides on MHC class I molecules by cross-presentation [25]. Tokunaga [25] reported that the p53-specific CTLs induced by full-length p53 protein-pulsed DCs had similar lytic activity to that induced by stimulation with Ad-p53-transduced DCs in vitro. Using flow cytometric analysis, cytokines measurement and cytotoxicity assay in our study, we demonstrated again that the method used to pulse the DCs with protein (i.e. by incubating them together) was simple and effective. In order to introduce more antigenic proteins into DCs and construct more powerful vaccines, a new method called protein transduction, a process by which proteins are fused with a protein transduction domain (PTD), is developing. Fusion proteins could traverse membrane barriers efficiently, enter the cytosol and have immediate access to the MHC class I pathway for highly efficient anti-tumour CTL induction [26].
The activity of the DCs was an important part of the endocytosis and processing of the proteins and presentation of the peptides. We previously optimized a method for generating active DCs from the PBMCs of patients with lung cancer [21,27]. To obtain DCs suitable for clinical applications, we changed the DC medium from one containing 10% FCS to one containing 5% human antibody serum, and then switched to serum-free DC medium (CellGro serum-free DC medium from CellGenix, Freiburg, Germany). We also adjusted the cytokines used to promote DC maturation by adding rhIL-1β, PGE2, rhIL-6 and rhTNF-α together and by increasing the amount of rhTNF-α to 50 ng/ml. After making these changes, we consistently obtained active DCs of clinical grade.
Another important feature of this study was the use of lung cancer cells and normal lung epithelial cells obtained directly from patients as the target cells in our cytotoxicity assay. Most in vitro studies rely on cell lines as the source of target cells to examine the cytotoxicity of CTLs. Cell lines do have some unique characteristics, but they are very different from cancer cells obtained directly from patients. Our experiments were designed with a future clinical application in mind; thus, we sought to stimulate the process in vivo as much as possible. All the cells in each section, including the DCs from the autologous PBMCs, the CTLs from the autologous lymphocytes and the target cells from the autologous cancer cells, were obtained from the same patient. The overwhelming advantage of this design is that it ensured MHC conformity during the course of presentation and recognition of the antigenic information. It was also unnecessary to identify the MHC types of the patients. Vaccines constructed in this way are similar to the status in vivo; therefore, the results of an in vitro cytotoxicity assay can be used to predict the effects of a vaccine on patients in the future.
The results of our cytotoxicity assay revealed that the XAGE-1b-specific CTLs had specific lytic activity against autologous XAGE-1b mRNA-positive tumour cells. In the XAGE-1b mRNA-negative tumour cells some other CTA might be positive, and cross-reacting antigens between XAGE-1b and the other CTA might lead to the lysis of XAGE-1b-specific CTLs. When we used allogenous XAGE-1b mRNA-positive cancer cells as targets, the lytic activity of the XAGE-1b-specific CTLs was weaker than that against autologous cancer cells. This phenomenon may have occurred because the MHC molecules of the two individuals did not match completely. This result verifies the MHC restriction and specificity of CTL lytic activity. Because different individuals share some MHC molecules which may present parts of antigenic peptides, the allogenous cancer cells were also be lysed to some extent. The results of our cytotoxicity assays show that vaccines constructed by pulsing full-length XAGE-1b with autologous DCs may induce XAGE-1b-specific CTLs against autologous XAGE-1b mRNA-positive tumour cells and that such vaccines are more suitable for patients with XAGE-1b-positive lung cancer.
In conclusion, this study offers a simple and effective method for constructing CTA-based vaccinations against lung cancer. This is the first report to show the induction of XAGE-1b-specific CTLs against lung cancer cells obtained directly from patients with NSCLC using DCs pulsed with full-length XAGE-1b. For more extensive clinical applications, we are now exploring polyvalent vaccines by combining two or more CTAs highly expressed on lung cancer cells, such as XAGE-1b and NY-ESO-1. Polyvalent vaccines may benefit a greater number of patients and theoretically may be more powerful. Further studies in animals are also ongoing.
Acknowledgments
This research was supported by National Nature Science Foundation of China (30100218), foundation of Guangzhou Science and Technology Bureau (2001-Z-044-01) and Key Research Project for Medical Science of Guangdong Province (200321). We thank GeneCopoeia & FulenGen, Inc. for kindly providing pReceiver-B01a expression vector freely in this study.
References
- 1.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–52. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 2.Van der Bruggen P, Traversari C, Chomez P, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254:1643–7. doi: 10.1126/science.1840703. [DOI] [PubMed] [Google Scholar]
- 3.Van den Eynde BJ, van der Bruggen P. T cell defined tumor antigens. Curr Opin Immunol. 1997;9:684–93. doi: 10.1016/s0952-7915(97)80050-7. [DOI] [PubMed] [Google Scholar]
- 4.Brass N, Heckel D, Sahin U, Pfreundschuh M, Sybrecht GW, Meese E. Translation initiation factor eIF-4gamma is encoded by an amplified gene and induces an immune response in squamous cell lung carcinoma. Hum Mol Genet. 1997;6:33–9. doi: 10.1093/hmg/6.1.33. [DOI] [PubMed] [Google Scholar]
- 5.Sahin U, Türeci O, Schmitt H, et al. Human neoplasms elicit multiple specific immune responses in the autologous host. Proc Natl Acad Sci USA. 1995;92:11810–13. doi: 10.1073/pnas.92.25.11810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen YT, Güre AO, Tsang S, et al. Identification of multiple cancer/testis antigens by allogeneic antibody screening of a melanoma cell line library. Proc Natl Acad Sci USA. 1998;95:6919–23. doi: 10.1073/pnas.95.12.6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scanlan MJ, Gure AO, Jungbluth AA, Old LJ, Chen YT. Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol Rev. 2002;188:22–32. doi: 10.1034/j.1600-065x.2002.18803.x. [DOI] [PubMed] [Google Scholar]
- 8.Scanlan MJ, Simpson AJ, Old LJ. The cancer/testis genes: review, standardization, and commentary. Cancer Immun. 2004;4:1. [PubMed] [Google Scholar]
- 9.Old LJ. Cancer/testis (CT) antigens – a new link between gametogenesis and cancer. Cancer Immun. 2001;1:1. [PubMed] [Google Scholar]
- 10.Davis ID, Chen W, Jackson H, et al. Recombinant NY-ESO-1 protein with ISCOMATRIX adjuvant induces broad integrated antibody and CD4(+) and CD8(+) T cell responses in humans. Proc Natl Acad Sci USA. 2004;101:10697–702. doi: 10.1073/pnas.0403572101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y, Sun Z, Nicolay H, et al. Monitoring of anti-vaccine CD4 T cell frequencies in melanoma patients vaccinated with a MAGE-3 protein. J Immunol. 2005;174:2404–11. doi: 10.4049/jimmunol.174.4.2404. [DOI] [PubMed] [Google Scholar]
- 12.Nakagawa K, Noguchi Y, Uenaka A, et al. XAGE-1 expression in non-small cell lung cancer and antibody response in patients. Clin Cancer Res. 2005;11:5496–503. doi: 10.1158/1078-0432.CCR-05-0216. [DOI] [PubMed] [Google Scholar]
- 13.Gure AO, Chua R, Williamson B, et al. Cancer-testis genes are coordinately expressed and are markers of poor outcome in non-small cell lung cancer. Clin Cancer Res. 2005;11:8055–62. doi: 10.1158/1078-0432.CCR-05-1203. [DOI] [PubMed] [Google Scholar]
- 14.Brinkmann U, Vasmatzis G, Lee B, Pastan I. Novel genes in the PAGE and GAGE family of tumor antigens found by homology walking in the dbEST database. Cancer Res. 1999;59:1445–8. [PubMed] [Google Scholar]
- 15.Liu XF, Helman LJ, Yeung C, Bera TK, Lee B, Pastan I. XAGE-1, a new gene that is frequently expressed in Ewing's sarcoma. Cancer Res. 2000;60:4752–5. [PubMed] [Google Scholar]
- 16.Zendman AJ, Van Kraats AA, Weidle UH, Ruiter DJ, Van Muijen GN. The XAGE family of cancer/testis-associated genes: alignment and expression profile in normal tissues, melanoma lesions and Ewing's sarcoma. Int J Cancer. 2002;99:361–9. doi: 10.1002/ijc.10371. [DOI] [PubMed] [Google Scholar]
- 17.Egland KA, Kumar V, Duray P, Pastan I. Characterization of overlapping XAGE-1 transcripts encoding a cancer testis antigen expressed in lung, breast, and other types of cancers. Mol Cancer Ther. 2002;1:441–50. [PubMed] [Google Scholar]
- 18.Ali Eldib AM, Ono T, Shimono M, et al. Immunoscreening of a cDNA library from a lung cancer cell line using autologous patient serum: identification of XAGE-1b as a dominant antigen and its immunogenicity in lung adenocarcinoma. Int J Cancer. 2004;108:558–63. doi: 10.1002/ijc.11587. [DOI] [PubMed] [Google Scholar]
- 19.Sato S, Noguchi Y, Ohara N, et al. Identification of XAGE-1 isoforms: predominant expression of XAGE-1b in testis and tumors. Cancer Immun. 2007;7:5–13. [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou Q, Guo AL, An SJ, Xu CR, Yang SQ, Wu YL. Association of XAGE-1b gene expression with clinical characteristics of non-small cell lung cancer. Nan Fang Yi Ke Da Xue Xue Bao. 2007;27:966–8. in Chinese. [PubMed] [Google Scholar]
- 21.Zhou Q, Wu YL, Guo AL, Wang K, Xu CR, Yang XN. Rapid in vitro generation of clinical grade dendritic cells in serum-free medium. Zhong Guo Zhong Liu Lin Chuang. 2006;33:605–7. in Chinese. [Google Scholar]
- 22.Wang X, Cai J, Zhong H, Denham SA, Terasaki PI. Screening of high cytotoxic tumor killer cells using a sensitive adherent target detachment assay. J Immunol Methods. 2004;295:57–65. doi: 10.1016/j.jim.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 23.Nikitina EY, Clark JI, Van Beynen J, et al. Dendritic cells transduced with full-length wild-type p53 generate antitumor cytotoxic T lymphocytes from peripheral blood of cancer patients. Clin Cancer Res. 2001;7:127–35. [PubMed] [Google Scholar]
- 24.Nikitina EY, Chada S, Muro-Cacho C, et al. An effective immunization and cancer treatment with activated dendritic cells transduced with full-length wild-type p53. Gene Ther. 2002;9:345–52. doi: 10.1038/sj.gt.3301670. [DOI] [PubMed] [Google Scholar]
- 25.Tokunaga N, Murakami T, Endo Y, et al. Human monocyte-derived dendritic cells pulsed with wild-type p53 protein efficiently induce CTLs against p53 overexpressing human cancer cells. Clin Cancer Res. 2005;11:1312–18. [PubMed] [Google Scholar]
- 26.Batchu RB, Moreno AM, Szmania SM, et al. Protein transduction of dendritic cells for NY-ESO-1-based immunotherapy of myeloma. Cancer Res. 2005;65:10041–9. doi: 10.1158/0008-5472.CAN-05-1383. [DOI] [PubMed] [Google Scholar]
- 27.Wang K, Wu YL, Zhou Q, Lin JY, Xu CR, Yang XN. Large-scale generation of mature clinical grade dendritic cells. Zhong Liu Fang Zhi Yan Jiu. 2004;31:458–60. in Chinese. [Google Scholar]