

Abstract
Clostridium difficile induces mucosal inflammation via secreted toxins A and B and initial interactions between the toxins and intestinal epithelial cells (which lead to loss of barrier function) are believed to be important in disease pathogenesis. Secreted toxin-specific antibodies may inhibit such interactions. Using the Caco-2 epithelial cell line, we have investigated the use of an anti-toxin A monoclonal antibody (ATAA) in providing protection against toxin A-mediated disruption of epithelial barrier function (assessed by measurement of transepithelial electrical resistance and luminal to basolateral flux of labelled dextran). In contrast to free antibody, ATAA conjugated to sepharose beads was more effective in neutralizing the activity of purified toxin A. Sepharose bead-conjugated ATAA was subsequently used to investigate the contribution of toxin A in epithelial injury mediated by C. difficile supernatant samples (containing toxins A, B and other products). Loss of barrier function mediated by apical application of supernatant samples of reference and epidemic 027 strains of C. difficile was abrogated by neutralization of toxin A. However, this was not the case when the supernatant samples were applied to the basal surface of epithelial monolayers. In conclusion, our studies have shown that (i) sepharose bead-conjugated ATAA is more effective in neutralizing toxin A than free antibody and (ii) when the apical (luminal) surface of epithelial monolayers is exposed to the secretory products of reference and 027 strains of C. difficile, toxin A is required for the initial injury that leads to loss of barrier function.
Keywords: anti-toxin antibody, colitis, paracellular permeability
Introduction
Toxigenic Clostridium difficile is a Gram-positive anaerobic bacillus that is a major cause of diarrhoea and colitis (pseudomembranous colitis) in hospitalized patients. It secretes two toxins, A and B, which are responsible for colonic inflammation and disease. Intestinal epithelial cells are believed to be the first host cells that interact with C. difficile toxins and responses by these mucosal cells may determine the development and nature of the colonic disease. Early effects of C. difficile toxins include loss of epithelial barrier function and expression of proinflammatory cytokines, followed by programmed cell death [1–5]. Inhibition of epithelial-toxin interactions via secreted antibody and agents that bind the toxins [6] are therefore likely to be protective. Indeed, studies suggest that impaired antibody-mediated protection may be responsible for the development of disease and its recurrence [7–9]. Orally administered antibodies to C. difficile toxins may be therapeutically beneficial [10] and their efficacy is likely to be related to their ability to inhibit the toxin-mediated effects on epithelial cells described above.
Both toxins A and B express cytopathic and cytotoxic activities in cultured cells in vitro but studies in animals have shown that toxin A plays an essential role in inducing intestinal disease. Thus, intragastric administration of purified toxin A has been reported to induce intestinal inflammation similar to that seen following infection with toxigenic C. difficile[11]. By contrast, intragastric administration of purified toxin B did not cause any disease unless there was prior mucosal damage to the intestinal mucosa. Moreover, purified toxin A, but not toxin B, induced inflammation when injected into rabbit ileal and colonic loops [12,13]. However, studies using mucosal samples have shown that toxin B is cytotoxic to human colonic epithelial cells [14,15]. Toxin A-negative, toxin B-positive strains of C. difficile have also been reported to be capable of inducing disease [16]. However, the majority of patients with C. difficile-associated disease are infected with bacterial strains that express both toxin A and B. The recent increases in the number of cases of severe disease and also outbreaks have been attributed to a virulent strain (designated PCR ribotype 027/NAP1; [17–19]) that produces greater amounts (compared with other toxigenic strain) of toxin A and B in vitro[20]. The 027 strain also secretes binary toxin, whose role in disease pathogenesis remains to be determined.
To date, in vitro studies to investigate their effects on intestinal epithelial cells have involved the use of purified preparations of C. difficile toxins A and B. Such preparations may not necessarily reflect the relative importance of each toxin in initiating epithelial injury, especially when the contributions of other secreted products (such as binary toxin) have not been fully characterized. We have therefore used supernatant samples of cultured toxigenic C. difficile and a specific monoclonal antibody to investigate the contribution of toxin A in inducing loss of epithelial barrier function. We show that during apical (but not basolateral) exposure to supernatant samples of reference and epidemic strains of C. difficile, toxin A is required to induce loss of epithelial barrier function. Additionally, we show that anti-toxin A antibody has a greater capacity (compared with free antibody) to neutralize toxin A when it is conjugated to sepharose beads.
Materials and methods
Cells
The human intestinal epithelial cell lines Caco-2, T84 and Vero cells (derived from African Green monkey kidney) were obtained from American Type Culture Collection (ATCC, via LGC Promochem, Teddington, UK). T84 cells were maintained in 50% Dulbecco's modified Eagle's medium (DMEM)/50% Ham's F-12 medium (Gibco-Invitrogen, Paisley, UK), supplemented with 10% fetal calf serum (FCS; Gibco-Invitrogen), 2 mmol/l glutamine (Sigma-Aldrich, Poole, UK), 0·1 mg/ml penicillin G (Britannia Pharmaceuticals, Redhill, UK), 0·1 mg/ml streptomycin (Sigma-Aldrich) and 0·1 mg/ml gentamicin (Mayne Pharma Plc, Warwickshire, UK). Caco-2 cells were cultured at 37°C in 5% CO, in DMEM containing 10% FCS, 10 pg/ml holotransferrin (Sigma-Aldrich), 2 mmol/l glutamine, 0·1 mg/ml penicillin G, 0·1 mg/ml streptomycin and 0·1 mg/ml gentamicin. Vero cells were maintained in DMEM supplemented with 10% FCS, 2 mmol/l glutamine, and antibiotics as above.
Clostridium difficile and purification of toxin A
Clostridium difficile toxigenic strain VPI 10463 (obtained from ATCC via LGC Promochem) was used for purification of toxin A, as previously described [21,22]. In brief, C. difficile was grown anaerobically in brain heart–infusion (BHI) broth (Oxoid, Basingstoke, UK) and supernatant samples applied to a bovine thyroglobulin affinity column. Toxin-A-containing fractions (which demonstrated cytotoxicity in Vero cells) were subsequently subjected to two sequential anion exchange chromatographic steps with Q Sepharose FF and Mono Q columns (GE Healthcare, Sweden). Aliquots of the purified toxin A were frozen at −80°C until used.
Supernatant samples were obtained from three strains of C. difficile, VPI 10463 (toxin A+/B+, Toxinotype 0), PCR ribotype 017 (A-/B+, toxinotype VIII) and an epidemic PCR ribotype NAP1/027 (toxin A+/B+, Toxinotype III). After anaerobic culture (at 37°C) of a single colony of C. difficile in BHI broth for 48–72 h, supernatant samples were obtained by centrifugation (×2000 g) for 5 min and passage through a 0·2 micron filter. To study their effects on epithelial barrier function, supernatant samples were applied to upper or lower compartments of transwell inserts (see below) at final concentrations ranging from 1:10 to 1:10 000, depending on the efficiency of bacterial growth and/or cytotoxicity in Vero cells. In each experiment, BHI broth (at the same final concentration as supernatant sample) was used in control transwells.
Anti-toxin A antibody
Hybridoma cells of the anti-toxin A monoclonal antibody (ATAA) PCG-4 were obtained from ATCC (via LGC Promochem) and grown in RPMI (Gibco-Invitrogen, Paisley, UK), supplemented with 10% horse serum (Gibco-Invitrogen), 2 mmol/l glutamine, and antibiotics (as above). The PCG-4 antibody was purified from hybridoma supernatant samples using a protein A affinity column (Perbio Science, Cheshire, UK). Antibody purification was confirmed by SDS polyacrylamide gel electrophoresis and quantified using Coomassie Plus protein assay kit (Perbio Science). The purified antibody was conjugated to protein-A sepharose 4B fast flow beads (Sigma-Aldrich) by incubation at 4°C in Dulbecco's phosphate buffered saline (Sigma-Aldrich) for 4 h, followed by three washes. PCG-4 antibody bound to the beads was calculated from the initial amount applied to beads and the quantity left in the supernatant after incubation (with the beads) and centrifugation. Free (unconjugated) or sepharose bead-conjugated PCG-4 antibody was used in the transwells at a final concentration of 70 µg/ml.
Assessment of epithelial barrier function
Barrier function of monolayers of Caco-2 and T84 cells was assessed by measurement of transepithelial electrical resistance (TER) and permeability to FITC-dextran. Caco-2 and T84 cells were grown in the upper compartment of 12 mm diameter Transwell inserts (Costar, New York, USA) at 37°C, 5% CO2. TER measurements were undertaken with the use of an Epithelial Volt-Ohmmeter (World Precision Instruments Ltd, Stevenage, UK), as previously described [5,23]. Permeability to FITC-labelled dextran (4 kDa, from Sigma-Aldrich) was assessed following its application (final concentration 0·2 mg/ml) to the upper compartment of transwell inserts.
Following the application of toxin A or C. difficile supernatant samples (pre-incubated for 1 h with control buffer, free ATAA or sepharose bead-conjugated ATAA), either to the upper or lower compartments of transwell inserts, TER was measured at 4 h, 24 h and 48 h and expressed as a percentage of electrical resistance at time 0 (immediately after application of samples). Aliquots (from upper and lower chambers) were also collected at 4 h, 24 h and 48 h for assessment of permeability to FITC-dextran. FITC-associated fluorescence was measured by Multiskan Ascent Platereader (Labsystems Affinity Sensors, Cambridge, UK) after excitation at 488nm and detection at 530nm, in aliquots of samples obtained from the upper and lower compartments of transwell inserts. Leakage of FITC-dextran was defined by fluorescence in the bottom compartment and expressed as a percentage of total fluorescence (combined measurements in upper and lower compartments).
Statistical analysis
Data are expressed as mean (±standard error of the mean) and analysed by anova and students t-test.
Results
Sepharose bead-conjugated ATAA provides greater protection than free antibody against toxin A-induced loss of epithelial barrier function
The ATAA PCG-4 has previously been shown to be capable of neutralizing enterotoxic activity of toxin A in hamster and rabbit intestine [24,25]. We tested the ability of this purified antibody to modulate epithelial barrier disrupting effects of toxin A.
In experiments using Caco-2 monolayers, both free and sepharose bead-conjugated ATAA inhibited loss of epithelial barrier function induced by low concentration (10 ng/ml) of toxin A (Fig. 1a and b). At higher concentration (100 ng/ml) of toxin A, sepharose bead-conjugated ATAA, but not free antibody (both used at the same concentration), provided complete protection against loss of TER and increase in permeability to FITC-dextran (Fig. 2a and b). Studies using T84 cells showed largely similar results (Fig. 2c and d).
Fig. 1.
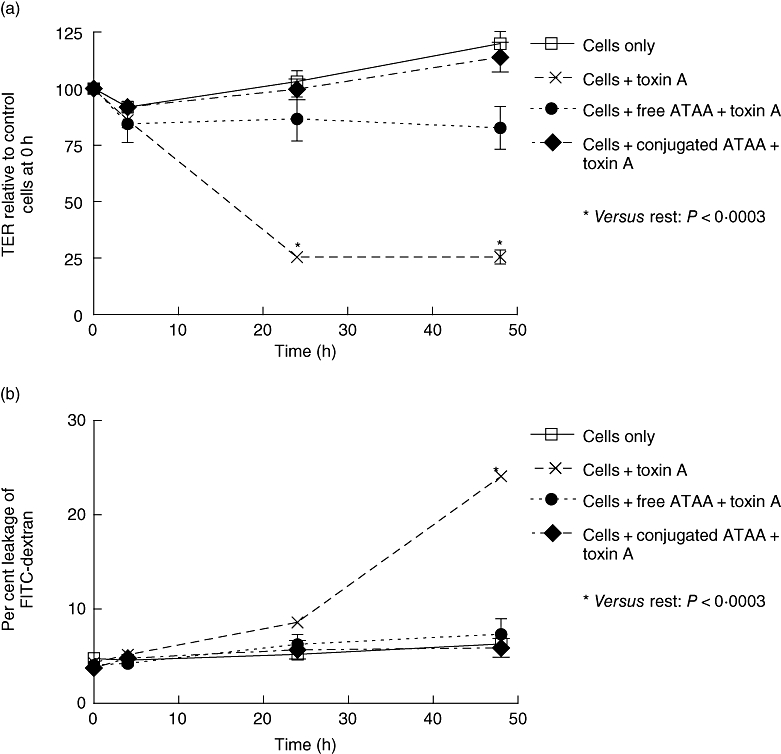
Free and sepharose bead-conjugated anti-toxin A antibody (ATAA) inhibits loss of transepithelial electrical resistance (TER) (a) and increase in permeability to FITC-dextran (b) induced by low concentration of toxin A. Purified toxin A was pre-incubated (for 1 h) with buffer, free ATAA or sepharose bead-conjugated ATAA, before application (final concentration of toxin A – 10 ng/ml) to the apical surface of Caco-2 monolayers in transwell inserts. TER was measured at 4 h, 24 h and 48 h and is expressed as % of electrical resistance at time 0. Leakage of FITC-dextran was defined by fluorescence in the bottom compartment and expressed as a percentage of total fluorescence (combined measurements in upper and lower compartments). *P < 0·0003, versus the rest at relevant time points. The figure represents mean data from three experiments (performed in duplicate).
Fig. 2.
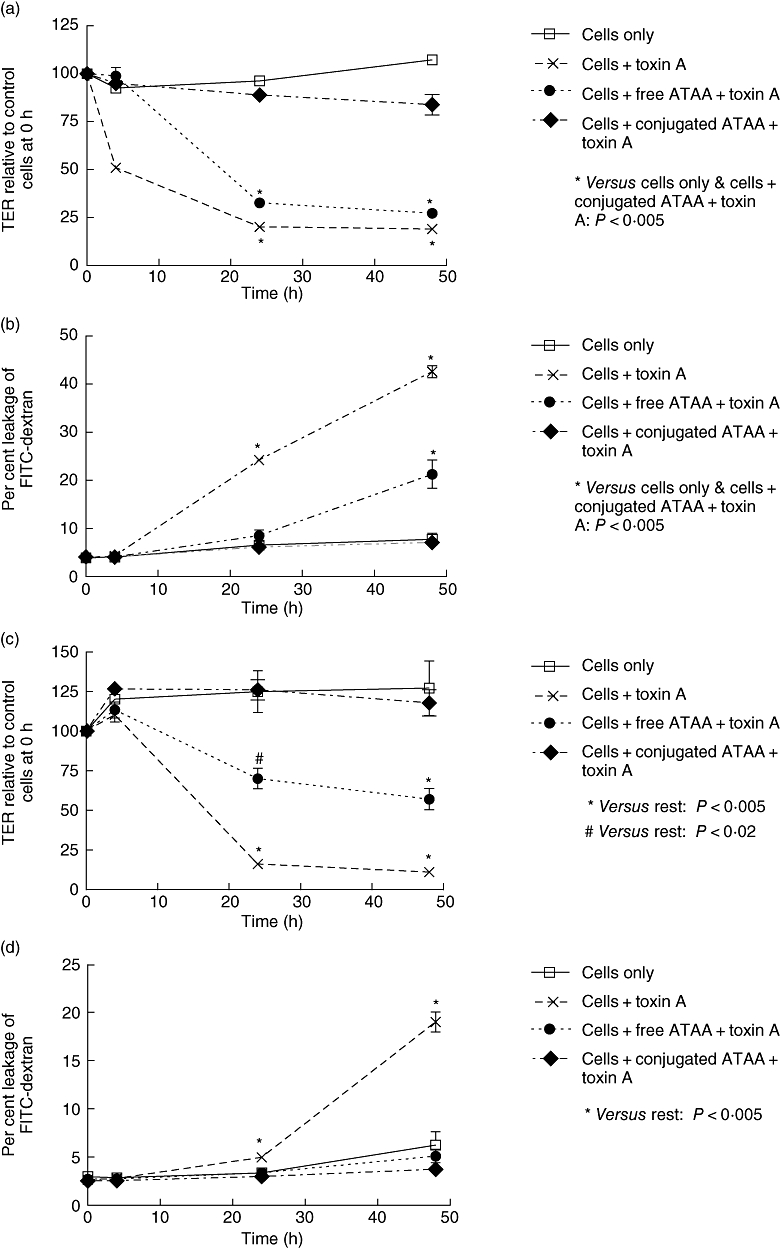
Sepharose bead-conjugated anti-toxin A antibody (ATAA) provides greater protection than free ATAA antibody against loss of transepithelial electrical resistance (TER) (a and c) and increase in permeability to FITC-dextran (b and d) induced by high concentration (100 ng/ml) of toxin A. Purified toxin A was pre-incubated (for 1 h) with buffer, free ATAA or sepharose bead-conjugated ATAA, before application to the apical surface of Caco-2 (a and b) or T84 (c and d) monolayers in transwell inserts (final concentration of toxin A – 100 ng/ml). #P < 0·02 and *P < 0·005, versus the rest at relevant time points. The figure represents mean data from three experiments (performed in duplicate).
Sepharose beads by themselves (without antibody) had no effect on toxin A-induced changes in epithelial barrier function (not shown). Free and sepharose bead-conjugated ATAA, in the absence of toxin A, also did not have any effect on epithelial barrier function (not shown).
Sepharose bead-conjugated ATAA protects against loss of epithelial barrier function induced by apical, but not basal, application of C. difficile supernatant
In experiments performed using Caco-2 monolayers, sepharose bead-conjugated ATAA protected against loss of TER and increase in permeability to FITC-dextran, following apical exposure (application to upper chamber of transwell inserts) to supernatant samples of anaerobically cultured C. difficile strain VPI 10463 (Fig. 3a and b). When the C. difficile supernatant samples were applied to the bottom chamber (basal exposure), the same sepharose bead-conjugated antibody (used at the same concentration as in the upper chamber) did not abrogate the loss of epithelial barrier function. Compared with apical application, basal exposure of Caco-2 monolayers to C. difficile supernatant samples led to significantly greater increase in permeability to FITC-dextran (Fig. 3b).
Fig. 3.
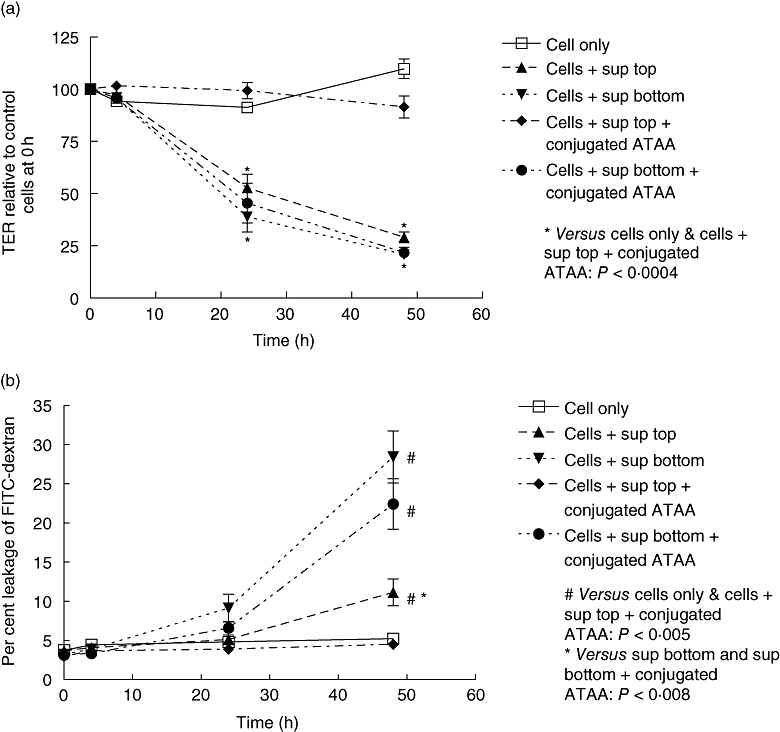
Anti-toxin A antibody (ATAA) protects against loss of epithelial barrier function induced by apical, but not basal, application of supernatant sample of VPI 10463 strain of C. difficile. VPI 10463 strain of C. difficile was cultured anaerobically and diluted supernatant samples were pre-incubated with PBS (for 1 h) or sepharose bead-conjugated ATAA, before application to the upper (apical exposure) or lower (basal exposure) chambers of transwells containing confluent monolayers of Caco-2 cells. Transepithelial electrical resistance (TER; a) and permeability to dextran (b) was assessed at 4 h, 24 h and 48 h. The figure represents mean data from four experiments (performed in duplicate). For TER at 24 h and 48 h, cells only ( ) and cells + supernatant + conjugated ATAA to top chamber (
) and cells + supernatant + conjugated ATAA to top chamber ( ) versus the rest: P < 0·0004 (*). The following P values refer to dextran permeability at 48 h; cells only (
) versus the rest: P < 0·0004 (*). The following P values refer to dextran permeability at 48 h; cells only ( ) and cells + supernatant + conjugated ATAA to top chamber (
) and cells + supernatant + conjugated ATAA to top chamber ( ) versus the rest: P < 0·005 (#). Cells + supernatant to top chamber versus cells + supernatant to bottom chamber, or cells + supernatant + conjugated ATAA to bottom: P < 0·008 (*for both comparisons).
) versus the rest: P < 0·005 (#). Cells + supernatant to top chamber versus cells + supernatant to bottom chamber, or cells + supernatant + conjugated ATAA to bottom: P < 0·008 (*for both comparisons).
When supernatant samples of an epidemic 027 strain of C. difficile were used, the findings were similar to those described for the reference strain, with sepharose bead-conjugated ATAA providing protection against loss of epithelial barrier function induced by apical, but not basal, exposure to supernatant samples (Fig. 4a and b).
Fig. 4.
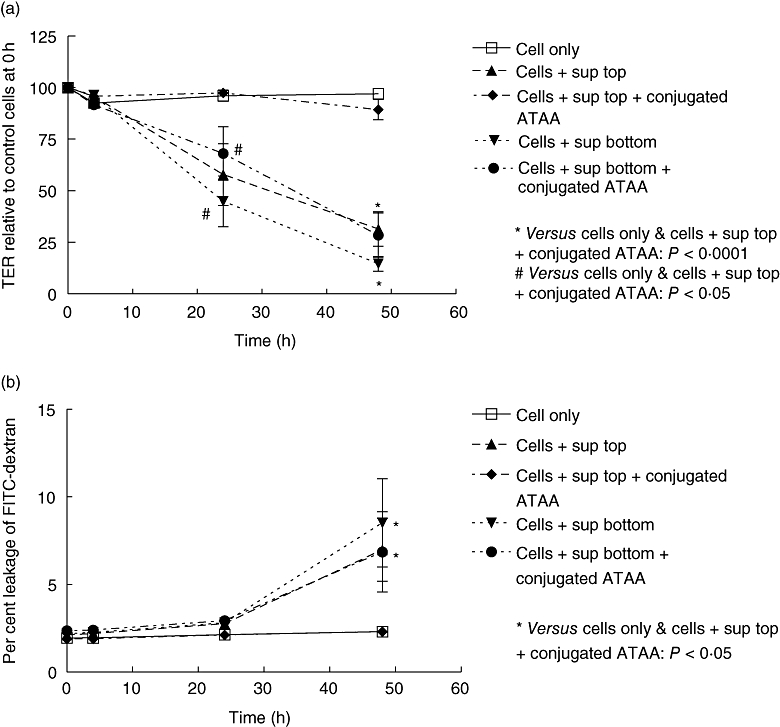
Anti-toxin A antibody (ATAA) protects against loss of epithelial barrier function induced by apical, but not basal, application of supernatant samples of NAP1/027 strain of C. difficile. A NAP1/027 strain of C. difficilewas cultured anaerobically and diluted supernatant samples were pre-incubated with PBS (for 1 h) or sepharose bead-conjugated ATAA, before application to the upper (apical exposure) or lower (basal exposure) chambers of transwells containing confluent monolayers of Caco-2 cells. Transepithelial electrical resistance (TER; a) and permeability to dextran (b) was assessed at 4 h, 24 h and 48 h. The figure represents mean data from three experiments (performed in duplicate). For TER at 24 h and 48 h, cells only ( ) and cells + supernatant + conjugated ATAA to top chamber (
) and cells + supernatant + conjugated ATAA to top chamber ( ) versus the rest: P < 0·05 (#) and P≤ 0·0001 (*) respectively. For dextran permeability at 48 h, cells only (
) versus the rest: P < 0·05 (#) and P≤ 0·0001 (*) respectively. For dextran permeability at 48 h, cells only ( ) and cells + supernatant + conjugated ATAA to top chamber (
) and cells + supernatant + conjugated ATAA to top chamber ( ) versus the rest: P < 0·05 (*).
) versus the rest: P < 0·05 (*).
Basal, but not apical, application of supernatant sample of toxin A-negative, toxin B-positive strain of C. difficile induces loss of epithelial barrier function
Clostridium difficile strain 017 expresses active toxin B, but not toxin A. Following anaerobic culture, supernatant samples of this strain were applied apically or basally to confluent monolayers of Caco-2 cells. Exposure of the supernatant samples to the basal surface of the monolayers led to loss of TER and an increase in permeability to FITC-dextran (Fig. 5a and b). By contrast, apical exposure of the Caco-2 monolayers to the same concentration of the C. difficile 017 supernatant samples did not exhibit significant changes to TER or permeability to FITC-dextran.
Fig. 5.
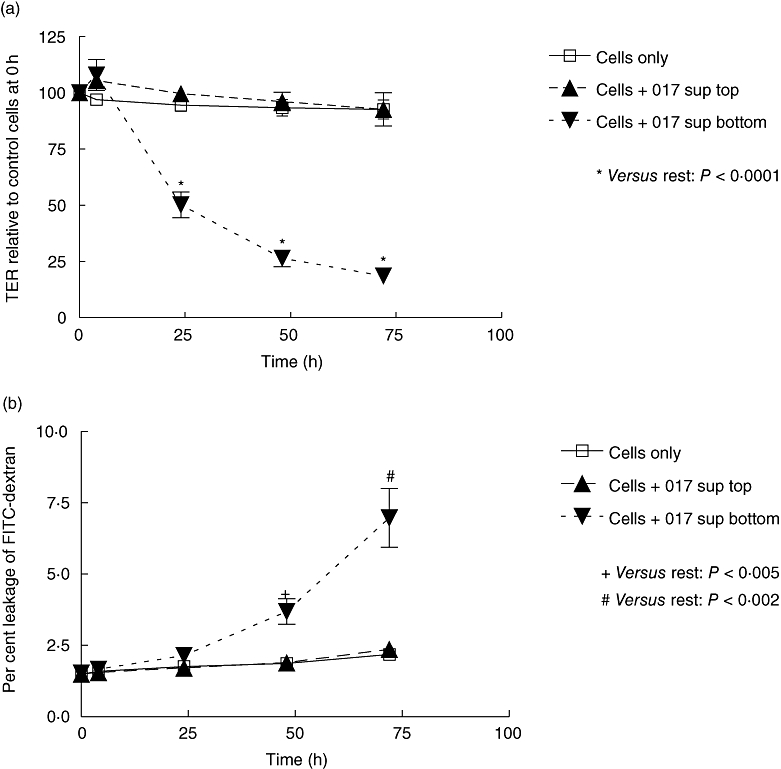
Basal, but not apical, application of supernatant sample of toxin A-negative, toxin B-positive strain of C. difficile induces loss of TER (a) and increase in permeability to FITC-dextran (b). C. difficilewas cultured anaerobically and supernatant samples were subsequently applied to the apical or basal surface of monolayers of Caco-2 cells grown in transwell inserts. +P < 0·005, #P < 0·002 and *P < 0·0001, versus the rest at relevant time points. The figure represents mean data from three experiments (performed in duplicate). TER, transepithelial electrical resistance.
Discussion
Histologically, pseudomembranous colitis because of toxigenic C. difficile is characterized by acute inflammation associated with epithelial ulceration. The focal nature of colonic disease [26] could be due to the production of high concentrations of toxins by bacteria in close vicinity to the surface epithelium, which could lead to the expression of proinflammatory cytokines such as IL-8 followed by cell death [2–5]. Early effects of purified C. difficile toxin A on intestinal epithelial monolayers in vitro include an increase in paracellular permeability, which can be assessed by measurement ofTER and luminal to basolateral flux of labelled dextran. Using this methodology, we show that ATAA PCG-4 neutralizes the barrier-disrupting effect of toxin A. Compared with free antibody, the efficacy of ATAA in neutralizing toxin A was greater when the antibody was conjugated with sepharose beads. Possible reasons for this difference include the ability of a number of antibodies in close proximity on the beads to bind the toxin, leading to more stable bead-antibody-toxin complexes. Another possibility is that the presence of sepharose beads inhibits epithelial uptake of ATAA-toxin complexes.
Using T84 epithelial monolayers, the superiority of polymeric immunoglobulin A (IgA) ATAA (compared with monomeric IgA and IgG antibodies) in protection against toxin A-induced epithelial injury has previously been reported [27]. In the latter study, the efficacy of monomeric (free) IgG ATAA was transient, whereas we have shown that it is capable of neutralizing low concentrations of toxin A over 48 h. As the host antibody response to C. difficile toxins appears to be an important determinant of clinical disease [7,9], the delivery to the colonic lumen of orally-administered anti-toxin antibodies conjugated to an inert support could be considered for therapeutic purposes. Indeed, orally-administered avian antibodies to C. difficile toxins have been reported to prevent morbidity, mortality and relapse in a hamster model of C. difficile-associated disease [10]. Based on our novel findings, we postulate that orally administered anti-C. difficile toxin antibodies conjugated to an inert support (similar to that provided by sepharose beads) will have greater therapeutic efficacy than free (unconjugated) antibodies.
The ability of sepharose bead-conjugated ATAA to effectively neutralize toxin A for a significant period of time was subsequently utilized to investigate the relative contribution of toxin A in the initial disruption of intestinal epithelial barrier function induced by secreted products of toxigenic C. difficile. We show that in both the reference and epidemic 027 strains of toxigenic C. difficile, toxin A is an essential apically-applied secretory component required to induce epithelial injury, which is characterized by an increase in paracellular permeability. We are not aware of such studies undertaken previously in which supernatant samples of toxigenic C. difficile have been used to investigate the contribution of toxin A in the disruption of epithelial barrier function. Our findings using Caco-2 cells are consistent with those previously reported using the T84 epithelial cell line and purified toxins A and B [27]. The inability of supernatant samples of a toxin A-negative, toxin B-positive strain of C. difficile, as shown by us and also reported previously [16], further supports the concept of the lack of a biological effect of apically-applied toxin B in intestinal epithelial monolayers. These findings imply the presence of distinct receptors for toxins A and B, with receptors for the latter only present on the basolateral surface of epithelial cells.
When applied to the surface of human intestinal mucosal samples, purified toxin B has been reported to induce epithelial injury [14,15]. A possible reason for this disparity between studies using human mucosal samples and epithelial cell lines is that toxin B may be taken up by dendritic cells in mucosal samples via processes between enterocytes [28,29], with subsequent loss of barrier function via induction of proinflammatory cytokine secretion [30–32], thereby enabling toxin B access to the basolateral surface of epithelial cells. It is possible that primary human intestinal epithelial cells are able to respond to luminally-applied toxin B because of the presence of specific receptors on the apical surface. The monolayer of primary colonic epithelial cells in vivo consists of stem cells at the crypt base that give rise to two main types of epithelial cells, enterocytes and goblet cells (and also a small number of enteroendocrine cells) that differentiate as they migrate up to the surface. Monolayers of Caco-2 and T84 cells express many phenotypic and functional characteristics of mature enterocytes and colonic crypt cells respectively [33,34]. It is possible therefore that the epithelial cells in whole mucosal samples that respond to luminally-applied toxin B are distinct from mature enterocytes and include immature cells (stem cells and their immediate progeny) and goblet cells. In this case, the increase in permeability via the immature and/or goblet cells would allow toxin B access to the basolateral surface of neighbouring enterocytes.
The occurrence of diarrhoea because of toxin A-negative toxin B-positive strains of C. difficile is now well recognized, although it generally occurs much less frequently than that because of strains that express both toxins A and B. However, there have recently been a number of reports of the isolation of toxin A-negative toxin B-positive strains from patients [16,35,36]. Studies in animals suggest that non-toxin A and toxin B enterotoxic factors may be responsible for fluid accumulation and tissue damage because of these strains [37]. Binary toxin, which is expressed by the epidemic 027 strain of C. difficile[17,18], is one such factor. Although binary toxin has been implicated in fluid accumulation, it has been reported not to cause disease in hamsters [38].
The ability of toxin A-negative, toxin B-positive strains of C. difficile to cause disease in humans could also be explained by its occurrence in those with pre-existing impairment of epithelial barrier function, as has been shown for healthy relatives of patients with Crohn's disease [39], which may represent a genetically determined primary abnormality in epithelial cells or could be due to proinflammatory cytokines secreted by underlying lamina propria cells in the mildly inflamed mucosa [31], such that toxin B is able to get access to the basolateral surface of epithelial cells.
In conclusion, using assessment of changes in barrier function as a sensitive measure of intestinal epithelial injury, we have shown that sepharose bead-conjugated ATAA has a greater capacity to neutralize toxin A than free antibody. The sepharose bead-conjugated ATAA was subsequently used to show that of the apically-applied secreted products of toxigenic C. difficile, toxin A is required to disrupt the barrier function of Caco-2 epithelial monolayers.
References
- 1.Hecht G, Pothoulakis C, LaMont JT, Madara JL. Clostridium difficile toxin A perturbs cytoskeletal structure and tight junction permeability of cultured human intestinal epithelial monolayers. J Clin Invest. 1988;82:1516–24. doi: 10.1172/JCI113760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mahida YR, Makh S, Hyde S, Gray T, Borriello SP. Effect of Clostridium difficile toxin A on human intestinal epithelial cells: induction of interleukin 8 production and apoptosis after cell detachment. Gut. 1996;38:337–47. doi: 10.1136/gut.38.3.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Branka JE, Vallette G, Jarry A, et al. Early functional effects of Clostridium difficile toxin A on human colonocytes. Gastroenterology. 1997;112:1887–94. doi: 10.1053/gast.1997.v112.pm9178681. [DOI] [PubMed] [Google Scholar]
- 4.Brito GA, Fujji J, Carneiro-Filho BA, Lima AA, Obrig T, Guerrant RL. Mechanism of Clostridium difficile toxin A-induced apoptosis in T84 cells. J Infect Dis. 2002;186:1438–47. doi: 10.1086/344729. [DOI] [PubMed] [Google Scholar]
- 5.Johal SS, Solomon K, Dodson S, Borriello SP, Mahida YR. Differential effects of varying concentrations of Clostridium difficile toxin A on epithelial barrier function and expression of cytokines. J Infect Dis. 2004;189:2110–19. doi: 10.1086/386287. [DOI] [PubMed] [Google Scholar]
- 6.Kurtz CB, Cannon EP, Brezzani A, et al. GT160-246, a toxin binding polymer for treatment of Clostridium difficile colitis. Antimicrob Agents Chemother. 2001;45:2340–7. doi: 10.1128/AAC.45.8.2340-2347.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maroo S, Lamont JT. Recurrent Clostridium difficile. Gastroenterology. 2006;130:1311–16. doi: 10.1053/j.gastro.2006.02.044. [DOI] [PubMed] [Google Scholar]
- 8.Kyne L, Warny M, Qamar A, Kelly CP. Asymptomatic carriage of Clostridium difficile and serum levels of IgG antibody against toxin A. N Engl J Med. 2000;342:390–7. doi: 10.1056/NEJM200002103420604. [DOI] [PubMed] [Google Scholar]
- 9.Johal SS, Lambert CP, Hammond J, James PD, Borriello SP, Mahida YR. Colonic IgA producing cells and macrophages are reduced in recurrent and non-recurrent Clostridium difficile associated diarrhoea. J Clin Pathol. 2004;57:973–9. doi: 10.1136/jcp.2003.015875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kink JA, Williams JA. Antibodies to recombinant Clostridium difficile toxins A and B are an effective treatment and prevent relapse of C. difficile-associated disease in a hamster model of infection. Infect Immun. 1998;66:2018–25. doi: 10.1128/iai.66.5.2018-2025.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lyerly DM, Saum KE, MacDonald DK, Wilkins TD. Effects of Clostridium difficile toxins given intragastrically to animals. Infect Immun. 1985;47:349–52. doi: 10.1128/iai.47.2.349-352.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitchell TJ, Ketley JM, Haslam SC, et al. Effect of toxin A and B of Clostridium difficile on rabbit ileum and colon. Gut. 1986;27:78–85. doi: 10.1136/gut.27.1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Triadafilopoulos G, Pothoulakis C, O’Brien MJ, LaMont JT. Differential effects of Clostridium difficile toxins A and B on rabbit ileum. Gastroenterology. 1987;93:273–9. doi: 10.1016/0016-5085(87)91014-6. [DOI] [PubMed] [Google Scholar]
- 14.Riegler M, Sedivy R, Pothoulakis C, et al. Clostridium difficile toxin B is more potent than toxin A in damaging human colonic epithelium in vitro. J Clin Invest. 1995;95:2004–11. doi: 10.1172/JCI117885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Savidge TC, Pan WH, Newman P, O’Brien M, Anton PM, Pothoulakis C. Clostridium difficile toxin B is an inflammatory enterotoxin in human intestine. Gastroenterology. 2003;125:413–20. doi: 10.1016/s0016-5085(03)00902-8. [DOI] [PubMed] [Google Scholar]
- 16.Alfa MJ, Kabani A, Lyerly D, et al. Characterization of a toxin A-negative, toxin B-positive strain of Clostridium difficile responsible for a nosocomial outbreak of Clostridium difficile-associated diarrhea. J Clin Microbiol. 2000;38:2706–14. doi: 10.1128/jcm.38.7.2706-2714.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McDonald LC, Killgore GE, Thompson A, et al. An epidemic, toxin gene-variant strain of Clostridium difficile. N Engl J Med. 2005;353:2433–41. doi: 10.1056/NEJMoa051590. [DOI] [PubMed] [Google Scholar]
- 18.Loo VG, Poirier L, Miller MA, et al. A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality. N Engl J Med. 2005;353:2442–9. doi: 10.1056/NEJMoa051639. [DOI] [PubMed] [Google Scholar]
- 19.Stubbs SL, Brazier JS, O’Neill GL, Duerden BI. PCR targeted to the 16S-23S rRNA gene intergenic spacer region of Clostridium difficile and construction of a library consisting of 116 different PCR ribotypes. J Clin Microbiol. 1999;37:461–3. doi: 10.1128/jcm.37.2.461-463.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Warny M, Pepin J, Fang A, et al. Toxin production by an emerging strain of Clostridium difficile associated with outbreaks of severe disease in North America and Europe. Lancet. 2005;366:1079–84. doi: 10.1016/S0140-6736(05)67420-X. [DOI] [PubMed] [Google Scholar]
- 21.Kamiya S, Reed PJ, Borriello SP. Purification and characterisation of Clostridium difficile toxin A by bovine thyroglobulin affinity chromatography and dissociation in denaturing conditions with or without reduction. J Med Microbiol. 1989;30:69–77. doi: 10.1099/00222615-30-1-69. [DOI] [PubMed] [Google Scholar]
- 22.Solomon K, Webb J, Ali N, Robins RA, Mahida YR. Monocytes are highly sensitive to Clostridium difficile toxin A-induced apoptotic and nonapoptotic cell death. Infect Immun. 2005;73:1625–34. doi: 10.1128/IAI.73.3.1625-1634.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beltinger J, McKaig BC, Makh S, Stack WA, Hawkey CJ, Mahida YR. Human colonic subepithelial myofibroblasts modulate transepithelial resistance and secretory response. Am J Physiol. 1999;277(2) 1 Pt:C271–9. doi: 10.1152/ajpcell.1999.277.2.C271. [DOI] [PubMed] [Google Scholar]
- 24.Lyerly DM, Phelps CJ, Toth J, Wilkins TD. Characterization of toxins A and B of Clostridium difficile with monoclonal antibodies. Infect Immun. 1986;54:70–6. doi: 10.1128/iai.54.1.70-76.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frey SM, Wilkins TD. Localization of two epitopes recognized by monoclonal antibody PCG-4 on Clostridium difficile toxin A. Infect Immun. 1992;60:2488–92. doi: 10.1128/iai.60.6.2488-2492.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Price AB, Davies DR. Pseudomembranous colitis. J Clin Pathol. 1977;30:1–12. doi: 10.1136/jcp.30.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stubbe H, Berdoz J, Kraehenbuhl JP, Corthesy B. Polymeric IgA is superior to monomeric IgA and IgG carrying the same variable domain in preventing Clostridium difficile toxin A damaging of T84 monolayers. J Immunol. 2000;164:1952–60. doi: 10.4049/jimmunol.164.4.1952. [DOI] [PubMed] [Google Scholar]
- 28.Rescigno M, Urbano M, Valzasina B, et al. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol. 2001;2:361–7. doi: 10.1038/86373. [DOI] [PubMed] [Google Scholar]
- 29.Niess JH, Brand S, Gu X, et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science. 2005;307:254–8. doi: 10.1126/science.1102901. [DOI] [PubMed] [Google Scholar]
- 30.Souza MH, Melo-Filho AA, Rocha MF, et al. The involvement of macrophage-derived tumour necrosis factor and lipoxygenase products on the neutrophil recruitment induced by Clostridium difficile toxin B. Immunology. 1997;91:281–8. doi: 10.1046/j.1365-2567.1997.00243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bruewer M, Luegering A, Kucharzik T, et al. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol. 2003;171:6164–72. doi: 10.4049/jimmunol.171.11.6164. [DOI] [PubMed] [Google Scholar]
- 32.Marano CW, Lewis SA, Garulacan LA, Soler AP, Mullin JM. Tumor necrosis factor-alpha increases sodium and chloride conductance across the tight junction of CACO-2 BBE, a human intestinal epithelial cell line. J Membr Biol. 1998;161:263–74. doi: 10.1007/s002329900333. [DOI] [PubMed] [Google Scholar]
- 33.Zweibaum A, Laburthe M, Grasset E, Louvard D. Use of cultured cell lines in studies of intestinal cell differntiation and function. In: Schultz SG, Field M, Rauner BB, editors. Handbook of physiology. Bethesda, MD: American Physiological Society; 1991. pp. 223–55. [Google Scholar]
- 34.Madara JL, Dharmsathaphorn K. Occluding junction structure-function relationships in a cultured epithelial monolayer. J Cell Biol. 1985;101:2124–33. doi: 10.1083/jcb.101.6.2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van den Berg RJ, Claas EC, Oyib DH, et al. Characterization of toxin A-negative, toxin B-positive Clostridium difficile isolates from outbreaks in different countries by amplified fragment length polymorphism and PCR ribotyping. J Clin Microbiol. 2004;42:1035–41. doi: 10.1128/JCM.42.3.1035-1041.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pituch H, Brazier JS, Obuch-Woszczatynski P, Wultanska D, Meisel-Mikolajczyk F, Luczak M. Prevalence and association of PCR ribotypes of Clostridium difficile isolated from symptomatic patients from Warsaw with macrolide-lincosamide-streptogramin B (MLSB) type resistance. J Med Microbiol. 2006;55:207–13. doi: 10.1099/jmm.0.46213-0. [DOI] [PubMed] [Google Scholar]
- 37.Borriello SP, Wren BW, Hyde S, et al. Molecular, immunological, and biological characterization of a toxin A-negative, toxin B-positive strain of Clostridium difficile. Infect Immun. 1992;60:4192–9. doi: 10.1128/iai.60.10.4192-4199.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geric B, Carman RJ, Rupnik M, et al. Binary toxin-producing, large clostridial toxin-negative Clostridium difficile strains are enterotoxic but do not cause disease in hamsters. J Infect Dis. 2006;193:1143–50. doi: 10.1086/501368. [DOI] [PubMed] [Google Scholar]
- 39.Hollander D, Vadheim CM, Brettholz E, Petersen GM, Delahunty T, Rotter JI. Increased intestinal permeability in patients with Crohn's disease and their relatives. A possible etiologic factor. Ann Intern Med. 1986;105:883–5. doi: 10.7326/0003-4819-105-6-883. [DOI] [PubMed] [Google Scholar]