

Abstract
Identifying and characterizing small-molecule inhibitors of protein-protein interactions is of high interest for drug discovery and for chemical genetics studies of biological pathways. Very often initial hits or first-generation compounds have low-micromolar dissociation constants and cause line broadening in NMR spectra. It is very important for subsequent structure-based compound optimization to know if this line broadening is caused by intermediate exchange of the dissociation kinetics only, or in addition by multiple binding modes. Here, we present an approach of how to distinguish these two situations and demonstrate its experimental application. Two very similar small-molecule ligands of Bcl-xL are considered that both cause severe line-broadening of interface residues. We show that one compound exhibits single-mode binding and broadening is just due to dissociation kinetics in the intermediate exchange regime, and the line broadening can be overcome by providing excess ligand. In the other case, line broadening is due to dissociation kinetics and exchange between multiple bound conformations, and broadening cannot be overcome by providing excess ligand. The procedures used are very general and can also be applied to characterizing protein-protein and protein-nucleic acid interactions.
Identifying and characterizing small-molecule inhibitors of protein-protein interactions is of high interest for drug discovery and for chemical genetics studies of biological pathways. Such efforts often start with high-throughput screening (HTS) of compound libraries using a variety of assays. Fluorescence polarization methods, for example, have been particularly successful for identification of inhibitors of protein-protein interactions 1. Depending on the design of the assays, initial hits typically have equilibrium dissociation constants in the low micromolar range at best. To be considered serious drug candidates the affinities of such weak hit compounds have to be improved, and structure-based medicinal chemistry efforts have been found particularly powerful.
For a structure-based optimization of hit compounds it is important to know whether a compound binds in a single well-defined conformation, or adopts two or multiple bound states of comparable energy (Figure 1). This is related to specific and nonspecific binding, respectively. However, non-specific binding is often but not always used synonymously with non-saturable binding while the situation depicted at the bottom panel of Fig. 1 applies to both saturable and non-saturable binding. In order to pursue a rational structure-based optimization of screening hits it is important to know the binding mode. Only if a compound binds in a single conformation a meaningful structure of the complex can be determined and used to rationally optimize the affinity; compounds exhibiting multiple binding modes should be avoided or modified to adopt a single binding mode. Here we present a simple approach to distinguish single-mode from multiple-mode binding and show examples for both situations with ligands to the protein Bcl-xL.
Figure 1.
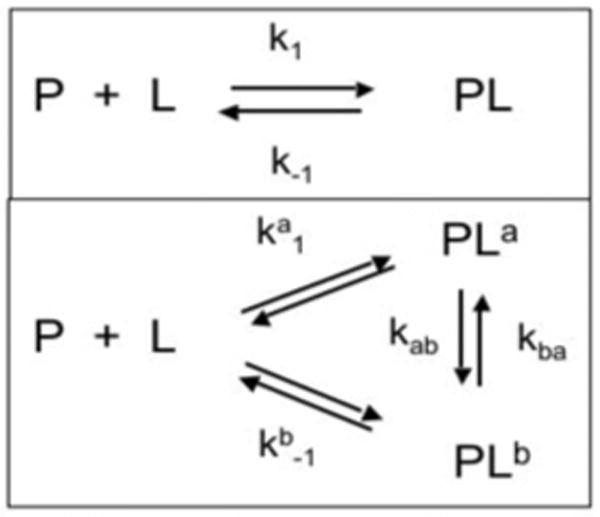
Schemes for single- (top) and multiple-mode (bottom) binding.
Binding of weak initial hits that have low-micromolar dissociation constants typically leads to chemical shift changes and/or broadening of protein resonances in the binding site. For example, Figure 2 shows a ligand titration in the 1H-13C HSQC spectrum of the methyl region of a Bcl-xL sample that has been 13C labeled and protonated at the methyl groups of Ile, Leu and Val but deuterated everywhere else using methods described previously 2-4. Clearly, resonances that are most affected exhibit chemical shift changes and increasing line broadening, such as V91γ1 (identified with a box in Figure 2). We wanted to know whether the line broadening is just due to dissociation kinetics on the intermediate exchange time scale or might also be caused by multiple binding modes.
Figure 2.
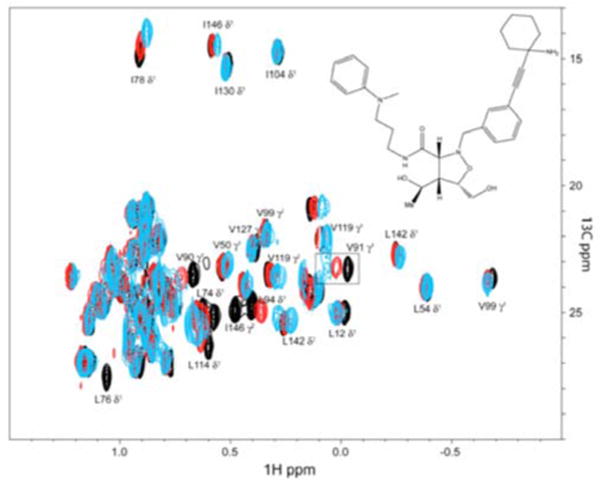
1H-13C HSQC spectrum of a 25 μM sample of Bcl-xL that is 13C labeled only at the methyl groups of Ile, Leu and Val 3, 4. Black: no ligand, red 25 μM and blue 250 μM ligand. The ligand is a first-generation compound from the primary screen, KD 80μM (shown on the insert). The cross peak of V91γ1 is placed in a box and further analyzed with cross sections in Figures 3 and 4.
A distinction of these two cases could be made based on the following consideration. If the line broadening is due to intermediate exchange of the dissociation, excess of ligand should cause all protein to be in a bound state, and all protein resonances should become sharper compared to the stoichiometric ratio. This would not be the case if the broadening were also due to multiple bound conformations because of inherent conformational exchange between different bound states. To further analyze this, we have simulated the spectrum of a protein proton for the two situations outlined in Figure 1. The chemical shift change Δν between free and bound protein was set to 100 Hz, the line width of the protein resonance was set to 20 Hz, and KD values of 20 μM and 80 μM were assumed to match situations for two ligands studied experimentally (see below). Using a procedure described previously5, the spectrum was simulated for protein:ligand concentrations of 1:0, 1:0.1, 1:0.25, 1:0.5,… 1:10. The dynamic exchange between different bound states was modeled assuming two bound conformations and using an exchange relaxation term ΔRa of 80 Hz6. The simulation for the absence of multiple conformations shows the resonance indeed sharpening up upon excess of ligand (Figure 3, bottom); in contrast, if there is chemical exchange between different bound states the line does not sharpen upon providing excess ligand (Figure 3, top).
Figure 3.
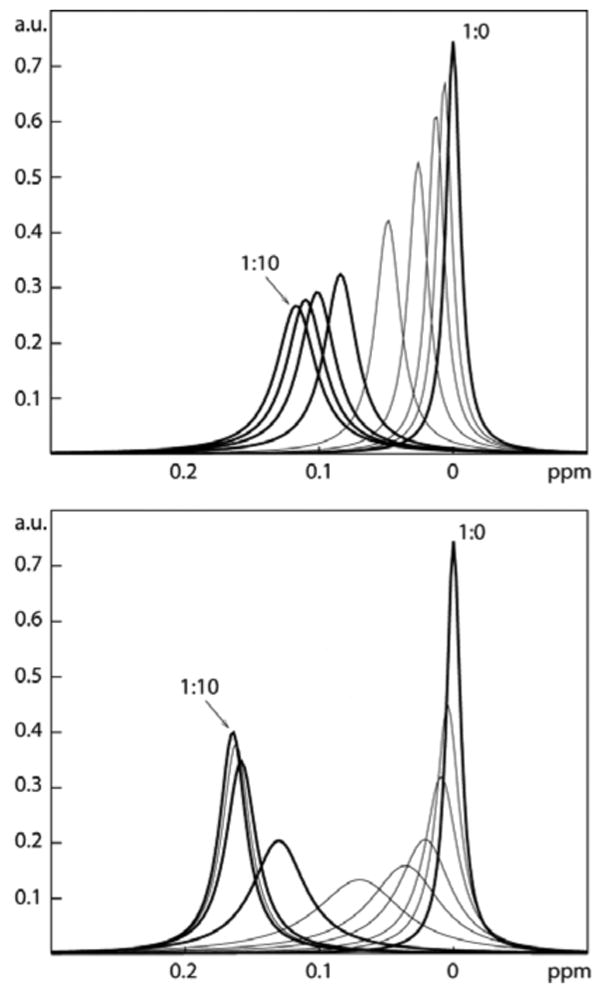
Simulated NMR lineshapes (at 500 MHz) for binding of a compound with two (above, KD 80 μM) and a single bound conformation in the intermediate exchange regime (below, KD 20 μM) at different compound/protein concentration ratios (see text). The same ratios are used for both cases. Thick lines correspond to actual experimental conditions shown in Figure 4. The line width in the free protein is 20 Hz, Δν is 100 Hz, the effective line broadening ΔRa caused by conformational exchange is 80 Hz. The resonance of the free protein is set to 0 ppm,
Indeed, both situations have been found experimentally multiple times in our studies of Bcl-xL ligand interactions. The top trace of Figure 4 shows a cross section along the proton frequency of V91γ1 in the 1H-13C HSQC of Bcl-xL at different ligand concentrations of a compound that was found to bind with an apparent KD (KDapp) of 80 μM as measured with fluorescence spectroscopy. Clearly, as much as ten-fold excess of this first-generation compound does not lead to sharpening of the bound resonance indicating that there is an exchange between two or more different bound states. However, a slightly modified compound that binds with a KDapp of 20 μM (replacing the left-hand aromatic group shown in Figure 2 with a substituted amino(bicycloheptane)) shows a different behavior (Figure 4 bottom). Here excess of ligand leads to sharpening of the bound resonance, which indicates that the compound binds in a single conformation.
Figure 4.
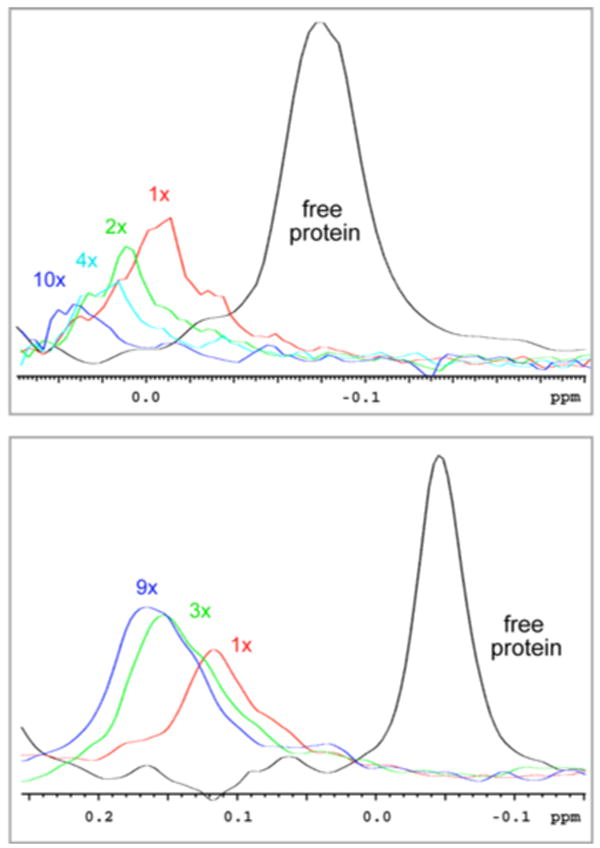
Cross sections of HSQC spectra of 13C ILV labeled Bcl-xL corresponding to V91γ1 resonance at different compound to protein concentration ratios. Above: first-generation compound (KDapp 80μM, causes conformational exchange in the bound state of the protein); below: modified compound (KDapp 20μM, clean intermediate chemical exchange).
Thus, lineshape analysis in a simple titration with excess ligand can clarify whether a compound binds with a single or with multiple conformation(s). In our experience with Bcl-xL and translation initiation factors, small-molecule inhibitors of protein-protein interactions identified with high-throughput screening of compound libraries often cause broadening of protein resonances, and it is desirable to identify the cause. Line broadening is also commonly observed for protein-protein and protein-nucleic acid interactions, where distinguishing between single- and multiple-mode binding is very important.
The titration experiments described above are often difficult to perform since small molecule inhibitors are typically not soluble at high concentrations, and thus the binding mode cannot easily be tested at excess ligand conditions7. The Bcl-xL inhibitors studied here, for example, are soluble to at most 250 μM. Thus, providing a 10-fold excess of compound means that the protein concentration has to be as low as 25 μM. Fortunately, modern cryogenic probes allow recording spectra as shown in Figure 2 at such low concentrations and enable the elucidation of the binding mechanism. The use of 1H-13C HSQC spectra of methyl labeled proteins is beneficial compared to 1H-15N labeling: besides providing a 3-fold higher signal intensity, the experiments can be performed in D2O, and methyl resonances appear to be less susceptible to line broadening compared to backbone amides8. The effect is probably most easily detectable as a gradual loss of peak intensities. The same procedures are applicable to characterize the interaction mode of proteins with other proteins or nucleic acids7.
Acknowledgments
This research was supported by the National Institute of Health (grant GM47467).
References
- 1.Roehrl MH, Wang JY, Wagner G. Biochemistry. 2004;43:16056–66. doi: 10.1021/bi048233g. [DOI] [PubMed] [Google Scholar]
- 2.Rosen MK, Gardner KH, Willis RC, Parris WE, Pawson T, Kay LE. J Mol Biol. 1996;263:627–36. doi: 10.1006/jmbi.1996.0603. [DOI] [PubMed] [Google Scholar]
- 3.Medek A, Olejniczak ET, Meadows RP, Fesik SW. J Biomol NMR. 2000;18:229–38. doi: 10.1023/a:1026544801001. [DOI] [PubMed] [Google Scholar]
- 4.Gross JD, Gelev VM, Wagner G. J Biomol NMR. 2003;25:235–42. doi: 10.1023/a:1022890112109. [DOI] [PubMed] [Google Scholar]
- 5.Matsuo H, Walters KJ, Teruya K, Tanaka T, Gasser GT, Lippard SJ, Kyogoku Y, Wagner G. J Am Chem Soc. 1999;121:9903–9904. [Google Scholar]
- 6.Sandstrom J. Dynamic NMR Spectroscopy. Academic Press; New York: 1982. [Google Scholar]
- 7.Feigon J, Denny WA, Leupin W, Kearns DR. J Med Chem. 1984;27(4):450–65. doi: 10.1021/jm00370a007. [DOI] [PubMed] [Google Scholar]
- 8.Tugarinov V, Kay LE. Chembiochem. 2005;6(9):1567–77. doi: 10.1002/cbic.200500110. [DOI] [PubMed] [Google Scholar]