

Abstract
Both vanadium and molybdenum cofactor clusters are found in nitrogenase. In biomimetic research, many fewer heterometal MFe3S4 cubane-type clusters have been synthesized with M = V than with M = Mo because of the well-established structural relationship of the latter to the molybdenum coordination unit in the enzyme. In this work, a series of single cubane and edge-bridged double cubane clusters containing the cores [VFe3(μ3-S)4]2+ and [V2Fe6(μ3-S)6(μ4-S)2]2+ have been prepared by ligand substitution of the phosphine clusters [(Tp)VFe3S4(PEt3)3]1+ and [(Tp)2V2Fe6S8(PEt3)4]. The single cubanes [(Tp)VFe3S4L3]2− and double cubanes [(Tp)2V2Fe6S8L4]4− (L = F−, N3−, CN−, PhS−) are shown by X-ray structures to have trigonal symmetry and centrosymmetry, respectively. Single cubanes form the three-member electron transfer series [(Tp)VFe3S4L3]3−,2−,1−. The ligand dependence of redox potentials and electron distribution in cluster cores as sensed by 57Fe isomer shifts (δ) have been determined. Comparison of these results with those previously determined for the analogous molybdenum clusters (Pesavento, Berlinguette, and Holm Inorg. Chem. 2007, 46, 510) allows detection of the influence of heterometal M on the properties. At constant M and variable L, redox potentials are lowest for π-donor ligands and largest for cyanide and relate approximately with decreasing ferrous character in clusters with constant charge z = 2−. At constant L and z and variable M, EV > EMo and , demonstrating that M = Mo clusters are more readily oxidized and suggesting a qualitative relation between lower potentials (greater ease of oxidation) and ferrous character.
Introduction
Heterometal cubane-type clusters containing the [MFe3S4] unit are essential building blocks in our exploration by synthesis of clusters relevant to those of the enzymes nitrogenase (M = V, Mo)1–4 and carbon monoxide dehydrogenase (M = Ni).5–7 The clusters of interest in this context are of two types: single cubanes (SC)8 containing the [MFe3(μ3-S)4] core of idealized trigonal symmetry and edge-bridged double cubanes (EBDC) with the core [M2Fe6S(μ3-S)6(μ4-S)2] of idealized centrosymmetry. The large majority of such clusters have been prepared with M = Mo because the immediate core coordination sphere of the molybdenum atom (MoFe3S3) is structurally closely related to that in the nitrogenase MoFe protein.1,2,9,10 An X-ray crystal structure of a vanadium nitrogenase is not available; however, a similar structural relationship appears to exist between VFe3S4 clusters and the vanadium site in VFe proteins.11–14 Far fewer vanadium than molybdenum SC clusters have been prepared since their inception in 1986–1987,15–17 and the first and only examples of vanadium EBDCs were obtained in 2002. The two cluster types may be generalized as [L′MFe3S4L3]z and [(L′)2M2Fe6S8L4]z of variable oxidation level z with diverse ligands L′ bound to the octahedral heterometal site and ligands L = phosphine, thiolate, or halide at the tetrahedral iron sites.
The existence of the foregoing SC and EBDC clusters with two different heterometals raises several basic issues. These include the scope of electron transfer series and values of redox potentials at constant M, L, and L′ and the effect of controlled variation of these constituents on these properties. Of particular interest are the influence of M and L varied separately on potentials and core–electron delocalization. These matters are best pursued with clusters in which L′ is constant, facilitating the isolation of clusters of either heterometal with the same charge z. For this purpose, we employ the tris(pyrazolyl)borate (Tp) ligand, which conforms to the trigonal symmetry of the SC core and in synthesis generally affords SCs with z = 2− and EBDCs with z = 3− or 4− in species carrying monoanionic ligands L.3,4,18–21 We have recently completed an investigation of the clusters [(Tp)MoFe3S4L3]z and [(Tp)2Mo2Fe6S8L4]z.20,21 Among the leading findings are the existence of three-member SC and four-member EBDC electron transfer series, isolation of different oxidation states within the series, and the isolation of the first SC in the all-ferrous oxidation state. We report here the results of a complementary study of vanadium-containing clusters.
Experimental Section
Preparation of Compounds
All reactions and manipulations were performed under a pure dinitrogen atmosphere using either Schlenk techniques or an inert atmosphere box. Solvents were passed through an Innovative Technology or MBraun solvent purification system and degassed prior to use. Volume reduction steps were carried out in vacuo. The compounds [(Tp)2V2-Fe6S8(PEt3)4], (Et4N)4[(Tp)2V2Fe6S8Cl4],19 and (Et4N)4[(Tp)2V2-Fe6S8(SPh)4]4 were prepared by published methods. Because of the small scales of some preparations, not all compounds were subjected to elemental analysis. The six compounds in Table 1 were identified by X-ray diffraction. Like all previously reported MFe3S4-type clusters, not all proton resonances were detectable due to paramagnetic broadening. 1H NMR spectra were recorded in CD3CN solutions and display distinctive isotropically shifted spectra, fully consistent with solid-state structures and satisfactory purity of the isolated compounds. Chemical shifts of anions only are given below. Double cubane clusters proved somewhat unstable in solution and were characterized by X-ray structure determinations and electrospray mass spectroscopy. Ions with isotope distributions consistent with the formulations given were observed in electrospray mass spectra (M = cluster). In the following preparations, yields are based on the unsolvated formula weights of the cluster compounds.
Table 1.
Crystallographic Data for Single and Double Cubane Cluster Compounds
| (Et4N)2[3] ·MeCN | (Et4N)2[4] | (Bu4N)2[6] | (Et4N)4[9] ·8MeCN ·3H2O | (Et4N)4[10] ·2MeCN | (Bu4N)4[12] ·6MeCN | |
|---|---|---|---|---|---|---|
| formula | C27H53BF3Fe3N9S4V | C25H50BFe3N17S4V | C44H82BFe3N11S4V | C66H130B2F4Fe6N24O3S8V2 | C54H106B2Fe6N30S8V2 | C98H182B2Fe6N26S8V2 |
| fw | 918.08 | 946.09 | 1122.32 | 2098.36 | 1890.21 | 2438.79 |
| crystal system | monoclinic | monoclinic | monoclinic | orthorhombic | monoclinic | monoclinic |
| space group | P21/c | P21/c | P21/n | Pnnm | P21/c | P21/n |
| Z | 4 | 12 | 4 | 4 | 2 | 2 |
| a, Å | 10.8425(6) | 23.671(8) | 11.224(2) | 14.743(2) | 11.867(10) | 18.617(3) |
| b, Å | 16.8430(10) | 34.548(12) | 40.248(7) | 29.397(4) | 20.779(19) | 16.165(3) |
| c, Å | 21.8996(13) | 15.700(5) | 12.602(2) | 11.0978(15) | 17.148(15) | 23.299(4) |
| β, deg | 96.1270(10) | 108.308(7) | 97.759(3) | 90.00 | 100.134(14) | 110.141(2) |
| V, Å3 | 3976.5(4) | 12189(7) | 5640.8(17) | 4809.7(11) | 4162(6) | 6583(2) |
| dcalcd g/cm3 | 1.534 | 1.547 | 1.322 | 1.955 | 1.509 | 1.189 |
| μ, 1/mm | 1.557 | 1.522 | 1.105 | 2.539 | 1.484 | 0.950 |
| 2θ range, deg | 1.53 → 27.49 | 0.91 → 28.35 | 1.01 → 27.89 | 1.39 → 25.21 | 1.55 → 25.09 | 1.57 → 25.02 |
| GOF (F2) | 0.994 | 1.058 | 1.027 | 1.005 | 1.028 | 0.993 |
| R1a/wR2b | 0.0381/0.0847 | 0.0795/0.1328 | 0.0663/0.1740 | 0.0608/0.1755 | 0.0585/0.1593 | 0.0866/0.1711 |
Single Cubanes. (Et4N)2[(Tp)VFe3S4F3]
A solution of 66 mg (0.36 mmol) of (Et4N)F ·2H2O in 2 mL of acetonitrile was added to a solution of 125 mg (0.12 mmol) of [(Tp)VFe3S4(PEt3)3](PF6) 19 in 1.5 mL of acetonitrile generating a black solution. The solution was stirred for 1.5 h and filtered through Celite. Vapor diffusion of ether into the black-red filtrate afforded the product as 95 mg (89%) of black plate-like crystals. 1H NMR (CD3CN, anion): δ 19.20 (1) 5.73 (1). Anal. Calcd. for C25H50BF3Fe3N8S4V: C, 34.21; H, 5.75; N, 12.77. Found: C, 34.38; H, 5.70; N, 12.88.
(Et4N)2[(Tp)VFe3S4(N3)3]
A solution of 62 mg (0.36 mmol) of (Et4N)(N3) in 2 mL of acetonitrile was added to a solution of 125 mg (0.12 mmol) of [(Tp)VFe3S4(PEt3)3](PF6) in 1.5 mL of acetonitrile generating a black solution. The solution was stirred for 1.5 h and filtered through Celite. Vapor diffusion of ether into the black-red filtrate afforded the product as 89 mg (79%) of black plate-like crystals. 1H NMR: δ 17.56 (1) 6.23 (1). Anal. Calcd. for C25H50BFe3N17S4V: Calcd. C, 31.73; H, 5.33; N, 25.16. Found: C, 31.78; H, 5.37; N, 25.14.
(Bu4N)2[(Tp)VFe3S4(CN)3]
A solution of 96 mg (0.36 mmol) of (Bu4N)(CN) in 2 mL of acetonitrile was added to a solution of 125 mg (0.12 mmol) of [(Tp)VFe3S4(PEt3)3](PF6) in 1.5 mL of acetonitrile generating a black solution. The solution was stirred for 1.5 h and filtered through Celite. Vapor diffusion of ether into the black-red filtrate afforded the product as 97 mg (73%) of black platelike crystals. 1H NMR: δ 15.63 (1) 6.29 (1). Anal. Calcd. for C44H82BFe3N11S4V: Calcd: C, 47.07; H, 7.36; N, 13.72. Found: C, 46.95; H, 7.42; N, 13.67.
(Me4N)2[(Tp)VFe3S4(SPh)3]
A solution of 58 mg (0.44 mmol) of NaSPh in 2 mL of acetonitrile was added to a solution of 125 mg (0.15 mmol) of (Me4N)2[(Tp)VFe3S4Cl3]22 in 1.5 mL of acetonitrile generating a black solution. The solution was stirred for 1.5 h and filtered trough Celite, removing a pale solid. Vapor diffusion of ether into the black-red filtrate afforded the product as 67 mg (45%) of black platelike crystals. The 1H NMR spectrum of the cluster was identical to that of the Et4N+ salt.4
Double Cubanes. (Et4N)4[(Tp)2V2Fe6S8F4]
A solution of 24 mg (0.12 mmol) of (Et4N)F ·2(H2O) in 2 mL of acetonitrile was added to 50 mg (0.03 mmol) of [(Tp)2V2Fe6S8(PEt3)4]19 suspended in 0.5 mL of acetonitrile. After about 15 min, the solid dissolved, forming a solution that was stirred for 1.5 h and filtered through Celite. Vapor diffusion of ether into the black-red filtrate afforded the product as 60 mg (77%) of black platelike crystals. 1H NMR: δ 23.0 (1), 19.74 (2), 5.91 (2). ES-MS: m/z 1698.5 ({M − F + 4Et4N + H}+ 1698.1), 1717.5 ({M + 4Et4N + H} 1717.2), 1847.8 ({M + 5Et4N + H}+ 1847.3).
(Et4N)4[(Tp)2V2Fe6S8(N3)4]
A solution of 22 mg (0.12 mmol) of (Et4N)(N3) in 2 mL of acetonitrile was added to 50 mg (0.03 mmol) of [(Tp)2V2Fe6S8(PEt3)4] suspended in 0.5 mL of acetonitrile. After about 15 min, the solid dissolved to form a solution that was stirred for 1.5 h and filtered through Celite. Vapor diffusion of ether into the black-red filtrate afforded the product as 45 mg (78%) of black blocklike crystals. ES-MS: m/z 1768.0 ({M − 3N + 4Et4N + 2H}+ 1768.2), 1809.0 ({M + 4Et4N + H}+ 1809.2), 1939.3 ({M + 5Et4N + H}+ 1939.3).
(Bu4N)4[(Tp)2V2Fe6S8(CN)4]
A solution of 34 mg (0.12 mmol) of (Bu4N)(CN) in 2 mL of acetonitrile was added to 50 mg (0.030 mmol) of [(Tp)2V2Fe6S8(PEt3)4] suspended in 0.5 mL of acetonitrile. After about 15 min, the solid dissolved forming a solution that was stirred for 1.5 h and filtered through Celite. Vapor diffusion of ether into the black-red filtrate at −35 °C afforded the product as 47 mg (71%) black blocklike crystals. ES-MS: m/z 2194.6 ({M + 4Bu4N + 2H}+ 2194.7), 2168.6 ({M − CN + 4Bu4N + 2H}+ 2168.7), 2438.7 ({M + 5Bu4N + 4H}+ 2438.9).
In the sections that follow, clusters are numerically designated according to Chart 1.
Chart 1.

Designation of Clustersa
X-ray Structure Determinations
The structures of the six compounds in Table 1 were determined. Suitable crystals of compounds containing clusters 3, 4, 6, 10, and 12 were obtained by ether vapor diffusion into acetonitrile solutions; crystals of the compound containing 9 were produced by layering a saturated acetonitrile solution onto THF. In the case of double cubane cluster salts, crystals were obtained from reaction mixture filtrates. Crystals were mounted with paratone oil. Diffraction data were collected with a Bruker Apex D8 CCD diffractometer at 193 K using Mo Kα radiation (λ= 0.71073 Å). Cell parameters were retrieved using SMART software and refined using SAINT on all observed reflections. Data were collected in 0.3° intervals in Φ and ω for 30–60 s/frame such that a hemisphere of data was collected. A total of 1271 frames were collected with a maximum resolution of 0.75 Å. The first 50 frames were recollected at the end of the data collection to monitor for decay; none was found. The highly redundant data sets were reduced using SAINT and corrected for Lorentz and polarization effects. Absorption corrections were applied using SADABS. Structures were solved by direct methods with use of SHELXL-97. Positions of metal atoms and their first coordination sphere atoms were located from direct-methods E-maps; other nonhydrogen atoms were found in alternating difference-Fourier syntheses and least-squares refinement cycles and were refined anisotropically during the final cycles. Hydrogen atoms were placed in calculated positions in the final refinements. Crystallographic parameters and agreement factors are contained in Table 1.23
Other Physical Measurements
All measurements were performed under anaerobic conditions. 1H NMR spectra were obtained with a Varian AM-400 spectrometer. Electrochemical measurements were made with a Princeton Applied Research model 263 potentiostat/galvanostat or a BAS Epsilon EZ system using acetonitrile solutions, a glassy carbon working electrode, and 0.1 M (Bu4N)(PF6) supporting electrolyte. Potentials are referenced to a standard calomel electrode (SCE). 57Fe Mossbauer spectra were collected with a constant acceleration spectrometer. Data were analyzed using WMOSS software (WEB Research Corp., Edina, MN); isomer shifts are referenced to iron metal at room temperature.
Results and Discussion
Cluster Synthesis
Single and double cubane-type vanadium–iron–sulfur clusters in the [VFe3S4]2+ and [V2Fe6S8]2+ oxidation states are accessible by the subsitution reactions 1 and 2, respectively, which are summarized in Figure 1. All clusters were prepared in acetonitrile and isolated as black, dioxygen-sensitive R4N+ salts. These reactions follow from earlier
Figure 1.

Summary scheme for the synthesis of single and double cubane VFe3S4 clusters in acetonitrile solutions, including present and previous results. Clusters 3, 4, 6, 9, 10, and 12 were prepared in this investigation.
| (1) |
| (2) |
demonstrations of the substitutional lability of the molybdenum clusters [(Tp)MoFe3S4(PEt3)3]1+ and [(Tp)2Mo2Fe6-S8(PEt3)4].3,20,21 While MFe3S4 clusters with labile ligands at the M = V and Mo sites are readily substituted at those sites,17,24–27 Tp ligation directs substitution to the iron sites exclusively in the formation of both SCs and EBDCs. Chloride cluster 1 is readily converted to precursor phosphine cluster 2.19 Reaction of 2 with 3 equiv of fluoride, azide, thiolate, or cyanide leads to clusters 3–6 in ca. 70–90% yield. SCs are most easily identified by their characteristic isotropically shifted 1H NMR spectra shown in Figure 2. Two of the three pyrazolyl proton signals expected under trigonal symmetry are clearly observed. The third signal (3-H) is broadened by cluster paramagnetism and appears as a barely detectable broad feature just upfield of the resonances in 15–19 ppm region. The spectral patterns are similar to those of [(Tp)MoFe3S4L3]2− clusters with the same ligands whose signals occur in the 3.5–21 ppm interval.20,21 Cluster 1, for example, shows resonances at 6.21 and 17.76 ppm. The clusters are paramagnetic but with different ground states, S = 3/2 for [VFe3S4]2+ 19,28 and S = 2 for [MoFe3S4]2+ 29clusters.
Figure 2.
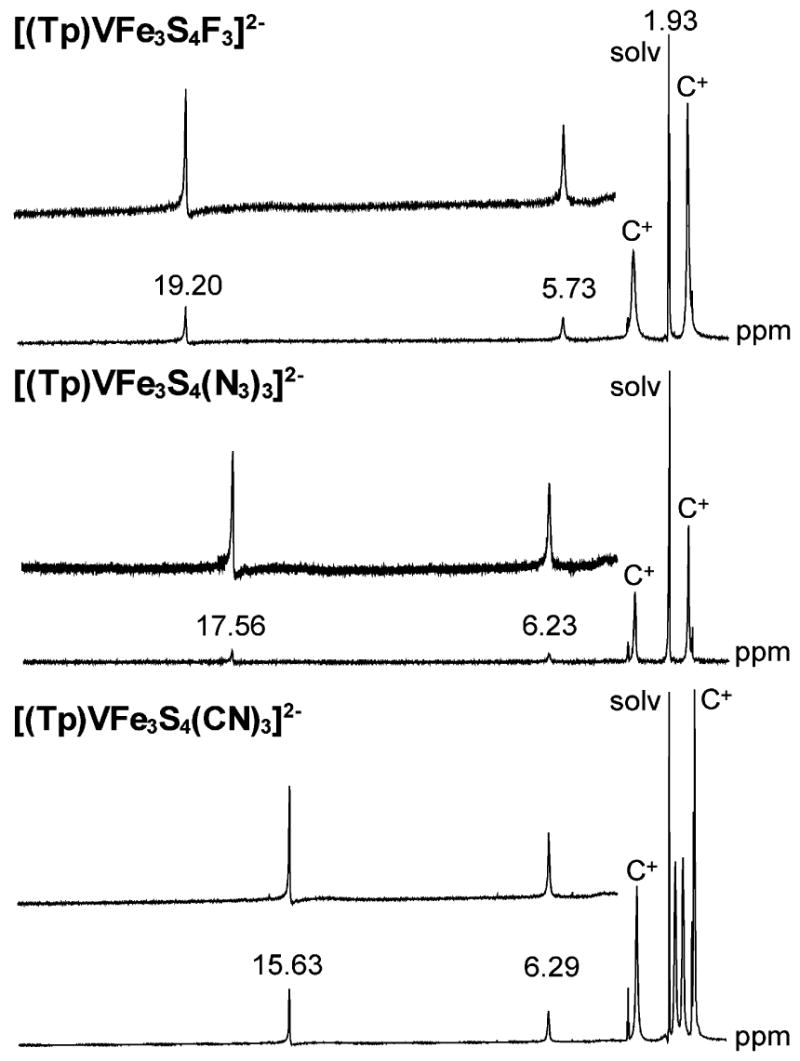
1H NMR spectra of [(Tp)VFe3S4L3]2−, L = F− (top), N3−(middle), and CN− (bottom) in CD3CN solutions (solv = solvent, C+ = Et4N+ or Bu4N+).
Similarly, the phosphine EBDC cubane 7, readily obtained by one-electron reduction of SC 2,19 forms chloride cluster 8,19 and with same set of ligands used with 2 affords clusters 9–12 isolated in ca. 70–80% yield. Single crystals of cluster salts were acquired by crystallization directly from reaction mixtures. Clusters in acetonitrile solutions tended to decompose partially upon dissolution, such that 1H NMR spectra and voltammograms contained unidentified features. Consequently, reliable solution data were not obtained. These clusters were authenticated by X-ray structure determinations and electrospray mass spectra of reaction solutions.
Cluster Structures
Structures of SCs 3, 4, and 6 and EBDCs 9, 10, and 12 are provided in Figures 3 and 4, respectively. Inasmuch as the structures of Tp-ligated SCs4,19,22 and EBDCs4,19 presented earlier are similar to those of the present clusters, detailed descriptions of the latter are not necessary. We summarize essential features; full structural information is available elsewhere.23 Metric data for cyanide clusters 6 and 12 in Table 2 suffice for this purpose. SC clusters closely approach idealized trigonal symmetry with distorted tetrahedral iron and octahedral vanadium sites. The majority of metric data are averaged under this symmetry. Dimensions conform well with Tp-ligated and other trigonal17,30 and nontrigonal17,31 [VFe3S4]2+ clusters. The solid state structures are obviously consistent with the equivalence of pyrazolyl rings in the 1H NMR spectra (Figure 2).
Figure 3.
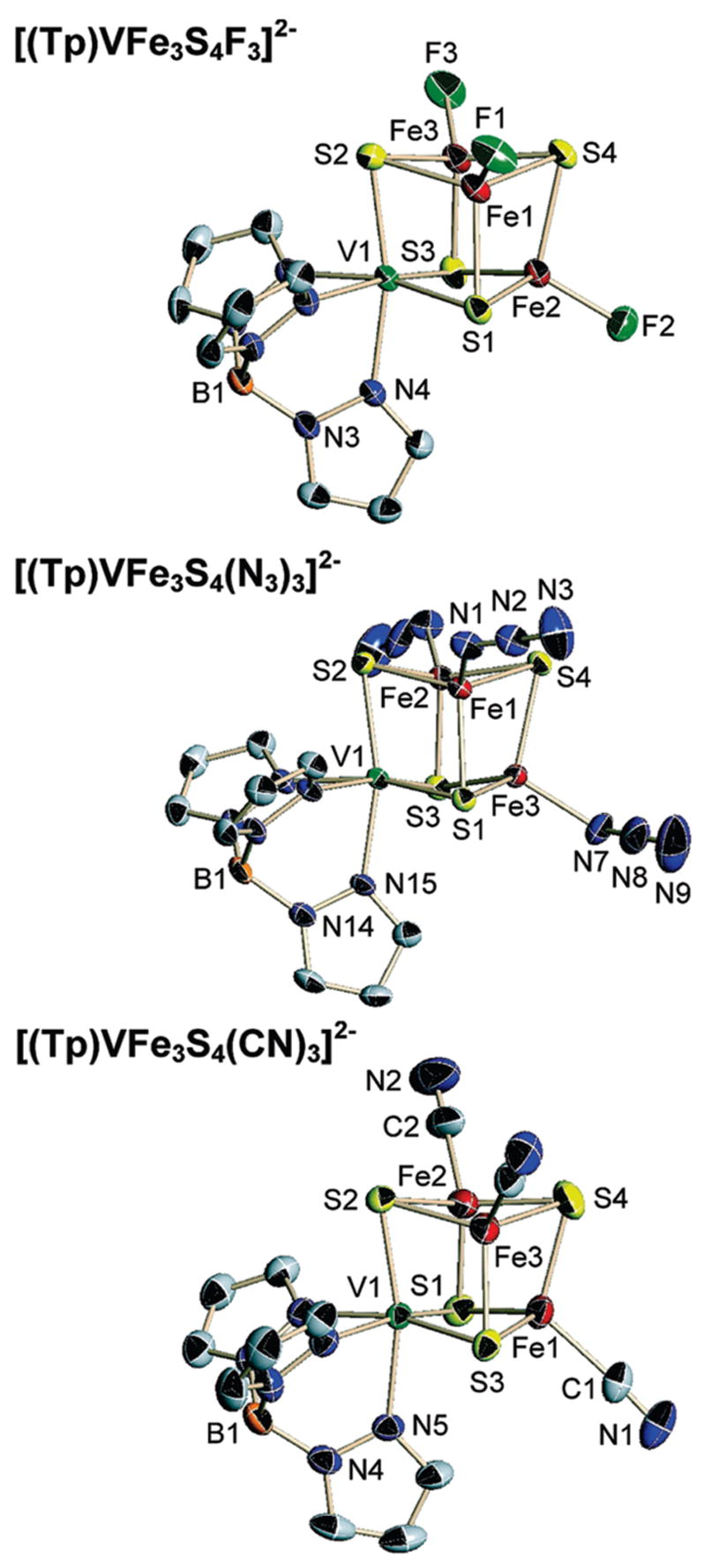
Structures of the SC clusters [(Tp)VFe3S4L3]2−, L = F− (top), N3− (middle), and CN− (bottom) showing 50% probability level ellipsoids and partial atom labeling schemes.
Figure 4.
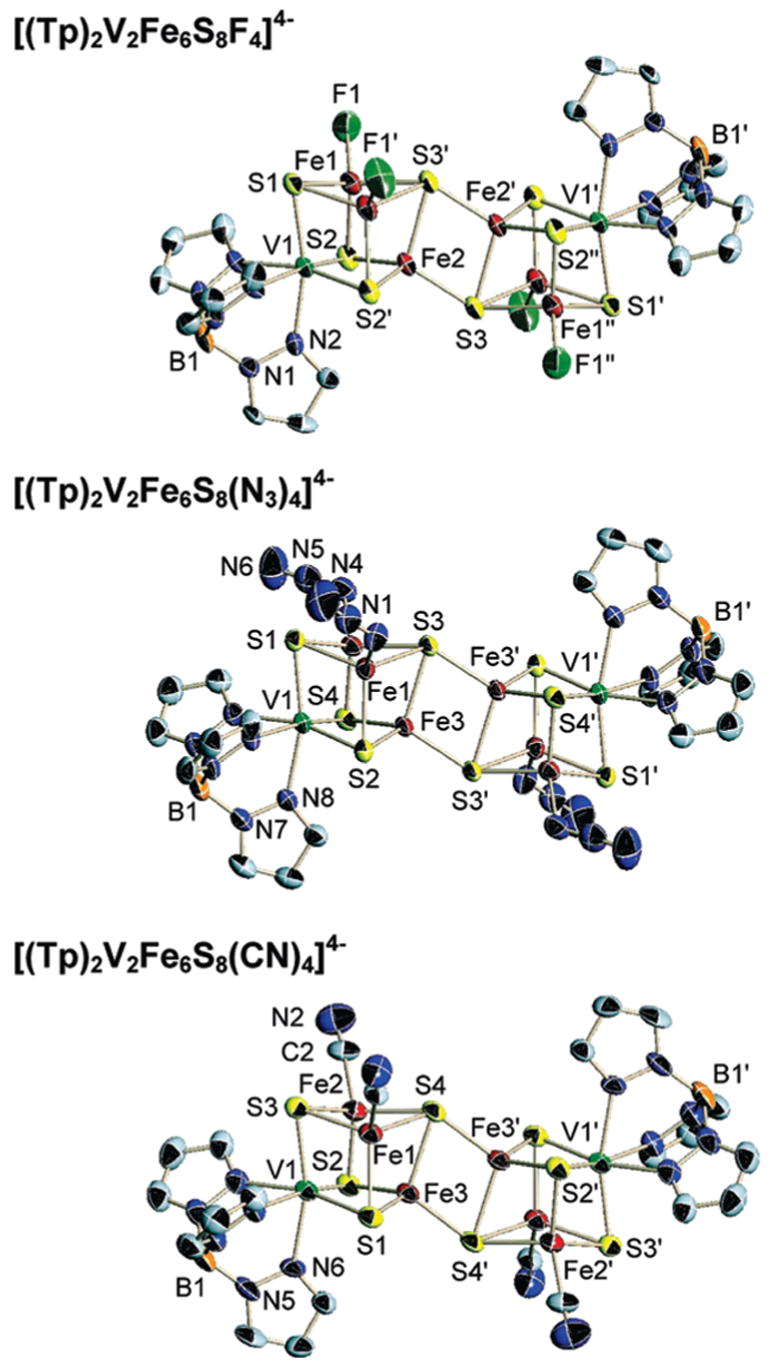
Structures of the EBDC clusters [(Tp)2V2Fe6S8L4]4−, L = F− (top), N3− (middle), and CN− (bottom) showing 50% probability level ellipsoids and partial atom labeling schemes. Primed and unprimed atoms are related by a symmetry center.
Table 2.
Selected Mean Values or Ranges of Bond Distances (Å) and Angles (deg) of Cyanide-Ligated Clusters
| [(Tp)VFe3S4(CN)3]2−a | |||
| V–N | 2.188(4) | N–V–N | 81.4(3) |
| V–S | 2.34(1) | N–V–S | 87.1(7)c |
| Fe–S4 | 2.27(3) | S–V–S | 102.2(6) |
| Fe(n)–S(n) | 2.26(1)e | V–S–Fe | 73.2(4)c |
| Fe(n)–S(n′) | 2.26(2)b | Fe–S4–Fe | 70.55(5)–72.27(5) |
| V–Fe | 2.74(1) | Fe–S–Fe | 71.31(4)–72.73(4) |
| Fe–Fe | 2.650(7) | S–Fe–S | 103.5(1)–108.3(1)d |
| Fe–C | 2.04(1) | C–Fe–S | 103.1(1)–115.7(1) |
| [(Tp)2V2Fe6S8(CN)4]4− | |||
| core | |||
| V–N | 2.192(4) | N–V–N | 80.8(2) |
| V–S | 2.35(2) | N–V–S | 85.8(2)–88.7(2) |
| Fe(1,2)–S3 | 2.271(2) | S–V–S | 101.0(1)–103.0(1) |
| Fe(n)–S(n) | 2.252(4)f | V–S–Fe | 72.13(8)–74.32(8) |
| Fe3–S(1,2) | 2.252(1) | Fe–S–Fe | 69.24(8)–73.82(8) |
| Fe(1,2)–S4 | 2.356(4) | S–Fe–S | 104.2(1)–109.4(1) |
| V–Fe | 2.74(4) | C–Fe–S | 102.7(3)–121.7(3) |
| Fe1–Fe2 | 2.580(2) | ||
| Fe(1,2)–Fe3 | 2.701(1) | ||
| Fe–C | 2.061(3) | ||
| bridge | |||
| Fe3–S4 | 2.352(2) | S4–Fe3–S4 | 111.17(8) |
| Fe3–S4 | 2.280(3) | Fe3–S4–Fe3′ | 68.83(8) |
| Fe3–Fe3 | 2.618(2) | S1/S2–Fe–S4′ | 114.8(1)/113.5(1) |
Averaged under trigonal symmetry unless otherwise indicated.
n ≠ n′.
Mean of six.
Range of nine.
n = 1, 2, 3.
n = 1, 2.
EBDCs consist of two subclusters connected by two Fe–(μ4-S) bridges to form a planar rhomb (Fe2(3,3′)S2(4,4′) in 12, Figure 4) in structures with imposed centrosymmetry. Individual cubane portions are approximately isostructural with SCs but metric features of bridge atoms depart owing to the presence of two μ4-S atoms. Most notable is the Fe3–S4 bond length which is ca. 0.08–0.10 Å longer than other subcluster values. Also, it is 0.07 Å longer than the intersubcluster Fe3–S4′ bond. Shorter Fe–S bridge bonds than subcluster bonds is a characteristic structural feature of all M2Fe6S8 EBDCs with M = V,4,19 Mo,3,20,21 and Fe.32
When examined at constant oxidation state with variant ligands at the iron sites, core structures of vanadium or molybdenum SCs and EBDCs are practically congruent and while differing by one or two valence electrons (e.g., [VFe3S4]2+ vs [MoFe3S4]2+,3+) are nearly isometric.
Isomer Shifts and Oxidation States
Heterometal cubane-type clusters provide a unique opportunity to examine separately the effects of variable terminal ligands and heterometal atom on two fundamental properties, scope and potentials of electron transfer series and core–electron distribution, in molecules of essentially constant structure. With the generalized clusters [(Tp)MFe3S4L3]z (M = V, Mo), potentials can be determined at constant M and z and variable L and at constant L and z and variable M. Comparisons at constant z and variable M involve clusters differing by one electron but with the same (mean) iron oxidation state based on 57Fe isomer shifts.18,19,21,33 In the case of z = 2−, for example, the [MFe3S4]2+ core is formalized as M3+ + 3Fe2.33+ (2Fe2+ + Fe3+). For z = 2− and 1−, iron atoms are electronically delocalized and redox changes are effectively localized in the Fe3portion of the core. Clusters with z = 3− have an all-ferrous core and are difficult to isolate because of their instability toward oxidation. Redox potentials, isomer shifts, and data comparison with the corresponding molybdenum clusters are collected in Table 3.
Table 3.
Redox Potentials and 57Fe Mössbauer Parameters for the Clusters [(Tp)VFe3S4L3]2− and [(Tp)2V2Fe6S8L4]4−
|
E1/2 (V)a |
EV − EMob |
|||||||
|---|---|---|---|---|---|---|---|---|
| L | 3−/2− | 2−/1− | 3−/2− | 2−/1− | δ (mm/s)c | ΔEQ (mm/s) | δavV − δavMob | |
| SC | F− | −1.57 | −0.25 | 0.30 | 0.48 | 0.47(1), 0.57(2) | 0.86(1), 1.22(2) | −0.09 |
| Cl− | −1.36 | −0.10 | 0.28 | 0.47 | 0.65(1), 0.50(2) | 1.15(1), 1.10(2) | −0.06 | |
| N3- | −1.29 | −0.11 | 0.25 | 0.42 | 0.68(1), 0.50(2) | 1.56(1), 1.14(2) | −0.02 | |
| PhS− | −1.50 | −0.42 | 0.26 | 0.27 | 0.48(1), 0.46(2) | 1.07(1), 0.67(2) | −0.05 | |
| CN− | −1.18 | (−0.04)d | 0.31 | (0.19)e | 0.42(1), 0.42(2) | 1.01(1), 1.15(2) | −0.05 | |
| PEt3 | 0.38 | 1.02 | +0.04 | |||||
| EBDC | F− | 0.63 | 0.85 | −0.08 | ||||
| Cl− | 0.64 | 0.83 | −0.07 | |||||
| N3− | f | 0.60 | 0.76 | −0.08 | ||||
| PhS− | 0.60(1), 0.56(2) | 0.73(1), 0.96(2) | g | |||||
| CN− | 0.56 | 0.95 | −0.05 | |||||
| PEt3 | 0.49(1), 0.51(2) | 1.32(1), 0.90 | +0.08 | |||||
Vanadium SCs, like their molybdenum counterparts, exhibit three-member electron transfer series as demonstrated by the voltammograms of 1, 3, and 5 in Figure 5. The 3−/2− couple is chemically reversible (ipc/ipa ≈ 1) for all clusters and the 2−/1− couple behaves similarly but is irreversible for 6. Redox potential order 3 for variable L holds for the 3−/2− couple, with the result that ease of oxidation decreases from fluoride to cyanide. The order for the 2−/1− couple inverts fluoride and thiolate but retains cyanide as the anionic ligand most effective in stabilizing the lower oxidation states. As one example, cyanide stabilizes [(Tp)VFe3S4(CN)3]3− by 320 mV compared to [(Tp)VFe3S4(SPh)3]3−. These features are the same as those displayed by molybdenum SCs.20,21 However, for either redox couple the potential difference EV −EMo at constant L is substantial, being ca. 200–500 mV.
Figure 5.
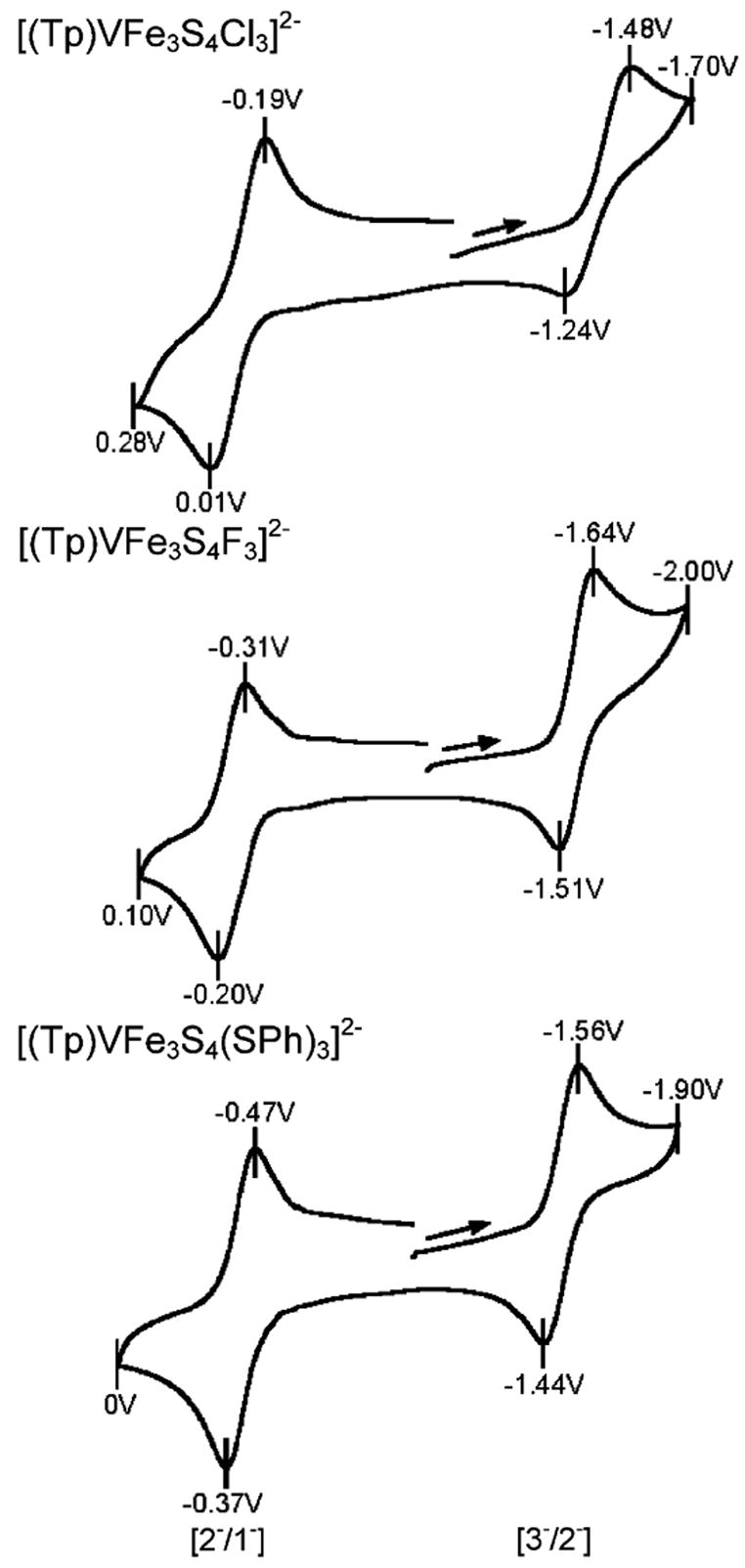
Cyclic voltammograms of SC clusters [(Tp)VFe3S4L3]2− with L = Cl− (top), F− (middle), and PhS− (bottom) in acetonitrile demonstrating the three-member electron transfer series. The top voltammogram was recorded at 500 mV/s, and the others, at 100 mV/s; peak potentials and redox couples are indicated.
| (3) |
| (4) |
Mössbauer spectra of both SCs and EBDCs consist of a single sometimes broadened quadrupole doublet and resemble those recently reported.20 As previously, the spectra are analyzed as a single quadrupole doublet or two overlapping doublets. No distinct Fe2+ or Fe3+ sites are observed. Weighted mean isomer shifts for SC clusters with variable L and z = 2− follow the series 4 with the π-donor ligands effecting more ferrous character as represented by the larger shifts. The order is about the same for EBDCs in that cyanide and phosphine induce less ferrous character. In these clusters six iron atoms are potentially influenced by four ligands as opposed to the same number of ligands and iron atoms in SCs, so the effect is reduced. Several observations of a qualitative nature follow. An increase in ferrous character is crudely associated with more negative redox potentials at constant z. The difference in isomer shifts at constant L and z is a measure of the intrinsic effect of heterometal M. Although the effect is small (<0.10 mm/s), the larger average isomer shifts of the molybdenum clusters tend to correlate with more negative redox potentials; i.e., the more reduced the iron portion, the easier the oxidation.
From the preceding trends in potentials and isomer shifts, we offer several additional observations. The molybdenum clusters are more susceptible to oxidation, an effect that operates in higher nuclearity clusters as well. Two practical consequences are the extremely facile oxidation of the all-ferrous EBDCs [(Tp)2Mo2Fe6S8L4]4− in solution to 3− species,20 and the ready isolation of the all-ferrous cluster [(Tp)2V2Fe6S9(SH)2]4− 4 whereas [(Tp)2Mo2Fe6S9(SH)2]3− (E1/24−/3− = −1.80 V) is the most reduced molybdenum cluster of this structural type that has been isolated.34 The influence of cyanide in stabilizing low cluster oxidation states is made additionally apparent by potentials of the [Fe4S4L4]4−/3− couples which for L = CN− is 310 mV less negative than for L = PhS−. Further, the 4− cyanide cluster is the only synthetic [Fe4S4]0 cluster with this charge that has been isolated.35 Likewise, [(Tp)MoFe3S4(CN)3]3− is the only all-ferrous heterometal SC to be isolated.21 There is scant information on heterometal influence on redox potentials at parity of cluster structure and ligation. In the only extensive study, which utilized the isostructural thiolate-bridged double cubanes [M2Fe6S8(SEt)9]3−, EV − EMo = 0.27 and 0.28 V for the 5−/4− and 4−/3−, respectively, and .36 While examples are limited, all current data on trends in redox potentials and isomer shifts are consistent, suggesting possibly general empirical relationships.
This investigation provides basic information on accessible core oxidation states, redox stability, and electron distribution as a function of terminal ligation and heterometal in vanadium and molybdenum cubane-type clusters. Cognizance of these properties, both absolute and relative, will continue to be of consequence in the synthetic manipulation of cubane clusters directed toward new structures and their reactivity.
Supplemental Materials
Acknowledgments
This research was supported by NIH Grant GM 28856.
Footnotes
Supporting Information Available: X-ray crystallographic files in CIF format for the compounds in Table 1. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lee SC, Holm RH. Proc Natl Acad Sci USA. 2003;100:3595–3600. doi: 10.1073/pnas.0630028100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee SC, Holm RH. Chem Rev. 2004;104:1135–1157. doi: 10.1021/cr0206216. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y, Holm RH. J Am Chem Soc. 2003;125:3910–3920. doi: 10.1021/ja0214633. [DOI] [PubMed] [Google Scholar]
- 4.Zuo JL, Zhou HC, Holm RH. Inorg Chem. 2003:4624–4631. doi: 10.1021/ic0301369. [DOI] [PubMed] [Google Scholar]
- 5.Panda R, Zhang Y, McLauchlan CC, Rao PV, Tiago de Oliveira FA, Münck E, Holm RH. J Am Chem Soc. 2004;126:6448–6459. doi: 10.1021/ja030627s. [DOI] [PubMed] [Google Scholar]
- 6.Panda R, Berlinguette CP, Zhang Y, Holm RH. J Am Chem Soc. 2005;127:11092–11101. doi: 10.1021/ja052381s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun J, Tessier C, Holm RH. Inorg Chem. 2007;46:2691–2699. doi: 10.1021/ic062362z. [DOI] [PubMed] [Google Scholar]
- 8.Abbreviations are given in Chart 1.
- 9.Peters JW, Stowell MHB, Soltis SM, Finnegan MG, Johnson MK, Rees DC. Biochemistry. 1997;36:1181–1187. doi: 10.1021/bi9626665. [DOI] [PubMed] [Google Scholar]
- 10.Mayer SM, Lawson DM, Gormal CA, Roe SM, Smith BE. J Mol Biol. 1999;292:871–891. doi: 10.1006/jmbi.1999.3107. [DOI] [PubMed] [Google Scholar]
- 11.Eady RR. Chem Rev. 1996;96:3013–3030. doi: 10.1021/cr950057h. [DOI] [PubMed] [Google Scholar]
- 12.Smith BE. Adv Inorg Chem. 1999;47:160–218. [Google Scholar]
- 13.Eady RR. Coord Chem Rev. 2003;237:23–30. [Google Scholar]
- 14.Strange RW, Eady RR, Lawson DM, Hasnain SS. J Synchrotron Rad. 2003;10:71–75. doi: 10.1107/s0909049502017272. [DOI] [PubMed] [Google Scholar]
- 15.Kovacs JA, Holm RH. J Am Chem Soc. 1986;108:340–341. [Google Scholar]
- 16.Kovacs JA, Holm RH. Inorg Chem. 1987;26:702–711. [Google Scholar]
- 17.Kovacs JA, Holm RH. Inorg Chem. 1987;26:711–718. [Google Scholar]
- 18.Fomitchev DV, McLauchlan CC, Holm RH. Inorg Chem. 2002;41:958–966. doi: 10.1021/ic011106d. [DOI] [PubMed] [Google Scholar]
- 19.Hauser C, Bill E, Holm RH. Inorg Chem. 2002;41:1615–1624. doi: 10.1021/ic011011b. [DOI] [PubMed] [Google Scholar]
- 20.Berlinguette CP, Miyaji T, Zhang Y, Holm RH. Inorg Chem. 2006;45:1997–2007. doi: 10.1021/ic051770k. [DOI] [PubMed] [Google Scholar]
- 21.Pesavento RP, Berlinguette CP, Holm RH. Inorg Chem. 2007;46:510–516. doi: 10.1021/ic061704y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malinak SM, Demadis KD, Coucouvanis D. J Am Chem Soc. 1995;117:3126–3133. [Google Scholar]
- 23.See the Supporting Information.
- 24.Palermo RE, Holm RH. J Am Chem Soc. 1983;105:4310–4318. [Google Scholar]
- 25.Palermo RE, Singh R, Bashkin JK, Holm RH. J Am Chem Soc. 1984;106:2600–2612. [Google Scholar]
- 26.Demadis KD, Coucouvanis D. Inorg Chem. 1995;34:436–448. [Google Scholar]
- 27.Malinak SM, Coucouvanis D. Prog Inorg Chem. 2001;49:599–662. [Google Scholar]
- 28.Carney MJ, Kovacs JA, Zhang YP, Papaefthymiou GC, Spartalian K, Frankel RB, Holm RH. Inorg Chem. 1987;26:719–724. [Google Scholar]
- 29.Mascharak PK, Papaefthymiou GC, Armstrong WH, Foner S, Frankel RB, Holm RH. Inorg Chem. 1983;22:2851–2858. [Google Scholar]
- 30.Zhu HP, Liu QT, Chen CN. Chin J Struct Chem. 2001;20:19–23. [Google Scholar]
- 31.Huang J, Mukerjee S, Segal BM, Akashi H, Zhou J, Holm RH. J Am Chem Soc. 1997;119:8662–8674. [Google Scholar]
- 32.Zhou HC, Holm RH. Inorg Chem. 2003;42:11–21. doi: 10.1021/ic020464t. [DOI] [PubMed] [Google Scholar]
- 33.Osterloh F, Segal BM, Achim C, Holm RH. Inorg Chem. 2000;39:980–989. doi: 10.1021/ic991016x. [DOI] [PubMed] [Google Scholar]
- 34.Berlinguette CP, Holm RH. J Am Chem Soc. 2006;128:11993–12000. doi: 10.1021/ja063604x. [DOI] [PubMed] [Google Scholar]
- 35.Scott TA, Berlinguette CP, Holm RH, Zhou HC. Proc Natl Acad Sci USA. 2005;102:9741–9744. doi: 10.1073/pnas.0504258102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cen W, Lee SC, Li J, MacDonnell FM, Holm RH. J Am Chem Soc. 1993;115:9515–9523. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.