

Abstract
Excessive airway obstruction is the cause of symptoms and abnormal lung function in asthma.
As airway smooth muscle (ASM) is the effecter controlling airway calibre, it is suspected that dysfunction of ASM contributes to the pathophysiology of asthma. However, the precise role of ASM in the series of events leading to asthmatic symptoms is not clear. It is not certain whether, in asthma, there is a change in the intrinsic properties of ASM, a change in the structure and mechanical properties of the noncontractile components of the airway wall, or a change in the interdependence of the airway wall with the surrounding lung parenchyma. All these potential changes could result from acute or chronic airway inflammation and associated tissue repair and remodelling.
Anti-inflammatory therapy, however, does not “cure” asthma, and airway hyperresponsiveness can persist in asthmatics, even in the absence of airway inflammation. This is perhaps because the therapy does not directly address a fundamental abnormality of asthma, that of exaggerated airway narrowing due to excessive shortening of ASM.
In the present study, a central role for airway smooth muscle in the pathogenesis of airway hyperresponsiveness in asthma is explored.
Keywords: Airway mechanics, interdependence, lung function, muscle adaptation, muscle contraction, parenchyma
Airway hyperresponsiveness (AHR) to nonspecific irritants or pharmacological agonists is a characteristic feature of asthma. The hyperresponsiveness is defined by exaggerated airway narrowing, which can usually be reversed by bronchodilators that relax airway smooth muscle (ASM). Although an important role of ASM in asthma has long been recognised, the precise nature of its involvement in the pathogenesis of AHR is not clear. The observations that the basic features of asthma (intermittent and excessive airway narrowing) are associated with airway inflammation and changes in airway structure (remodelling) have led to the predominant view in the past decade that smooth muscle was primarily an effecter, whereas airway inflammation was thought to be the causal pathophysiological mechanism underlying AHR. This concept has recently been questioned by studies showing dissociation between AHR and airway inflammation [1, 2]. Furthermore, not only does ASM appear to be a very active player in producing pro-inflammatory cytokines and growth factors [3], more importantly, AHR and airway inflammation can manifest independently following specific interventions at the cytokine level. For instance, anti-immunoglobulin E [4] or anti-interleukin (IL)-5 [5] therapy greatly diminish multiple features of inflammation without altering hyperresponsiveness in asthma. Conversely, anti-tumour necrosis factor (TNF)-α treatment was shown to be associated with reduction of AHR without changing the markers of airway inflammation (except for a decrease in histamine concentrations in sputum supernatant), suggesting that the benefit of the treatment is derived primarily by an effect on ASM and mast cells [6]. These findings indicate that the primary functional abnormality in asthma cannot simply be explained by specific inflammatory pathways without an involvement of ASM.
In parallel with these developments, physiological studies have improved current understanding of ASM in terms of its structural plasticity, functional dynamics and interaction with its local environment both within and outside the airway wall. In addition to revealing numerous novel candidate mechanisms for AHR, these studies have also reminded us about the complexity of the role of ASM in AHR in asthma. In the present study, a central role for ASM in the pathogenesis of AHR in asthma is explored.
EVIDENCE OF ASM INVOLVEMENT IN ASTHMA
It is known that lung volume is a key determinant of airway resistance (Raw) [7–14] and that changes in lung volume associated with tidal breathing and deep inspiration differentially modulate airway conductance (Gaw) in normal and asthmatic subjects [15–21]. Although it is speculated that the target of modulation by lung volume change is the smooth muscle embedded in the airways [21–30], a direct relationship between lung volume change and alteration in ASM contractility is yet to be established. Investigations into the differences in the effects of deep inspiration on Raw in normal and asthmatic subjects have provided important insights into the disease mechanism of asthma and have implicated the involvement of ASM [31, 32]. However, even if ASM is involved in the pathophysiology of asthma, it does not necessarily mean that the muscle itself is abnormal. It is possible that AHR results from normal smooth muscle operating in an abnormal airway/lung environment [33, 34].
Difference in response to deep inspiration implicates smooth muscle involvement
Fish et al. [16] were the first to show that a difference in response to deep inspiration distinguishes asthmatic from nonasthmatic subjects. They compared the effect of deep inspiration on Gaw and forced expiratory volume in one second (FEV1) before and after inhalation of methacholine in asthmatic and nonasthmatic allergic subjects (i.e. rhinitis sufferers). Subjects with allergic rhinitis responded to deep inspirations in a similar way to healthy subjects [15, 16]. That is, prior to bronchoprovocation, deep inspiration induced no change in the Gaw, but, after methacholine-induced bronchoconstriction, deep inspiration caused a marked increase in Gaw. Conversely, the Gaw of asthmatic subjects decreased transiently after a deep inspiration in the baseline state, and deep inspiration after methacholine-induced bronchoconstriction caused no significant improvement in Gaw. Subsequent studies of induced bronchial obstruction have shown that timing of the measurements after a deep inspiration is important. Pellegrino et al. [19] showed that with methacholine-induced obstruction in asthmatics, a deep inspiration caused consistent, but transient, bronchodilation when lung resistance was measured on a breath-by-breath basis. The rate of restitution of pre-deep inspiration Raw was much more rapid in asthmatics than in nonasthmatics. Jensen et al. [21] confirmed that after airway provocation at the peak of a deep inspiration, healthy subjects could still reduce their Raw (i.e. dilate their airways) to levels similar to that achievable at baseline. In striking contrast, at baseline, asthmatics dilated their airways less with a deep inspiration compared with healthy subjects, and their dilating ability was subsequently diminished after provocation. Moreover, the same study suggests that airways re-constrict faster in asthmatics than in healthy subjects. Jackson et al. [35] showed consistent findings and found an exponential return to pre-deep inspiration levels, a finding compatible with a first order process; whereas, Thorpe et al. [36] reported a time course following a power law, which is inconsistent with a first order process. Nevertheless, these findings are all consistent with the greater velocity of contraction of asthmatic smooth muscle, as has been shown by Ma et al. [37] in isolated human asthmatic ASM cells and by Mitchell et al. [38] in passively sensitised human bronchial smooth muscle. In the studies by Pellegrino et al. [19] and Jackson et al. [35], the relative magnitude of the reversal was less in asthmatic subjects. Thus the relative magnitude of the reversal, rather than the reversal itself, appears to differentiate the asthmatic response to induced obstruction. The constrictor response to a deep inspiration with spontaneous obstruction, however, clearly distinguishes the asthmatic subjects.
Possible roles of ASM in mediating effects of deep inspiration
What makes asthmatic airways uniquely different in their response to deep inspiration? At least three possible answers are presented in the literature. The first is provided by Burns et al. [17], Pichurko et al. [39], Lim and co-workers [40, 41] and Pliss et al. [42], which is based on the concept of relative hysteresis of airway versus lung and first developed by Froeb and Mead [43]. They found, as did Fish et al. [16], that in asthmatics, not only did deep inspiration fail to reverse spontaneous obstruction, it sometimes resulted in worsening of airway narrowing. Furthermore, Lim and co-workers [40, 41] showed that the degree of bronchoconstriction induced by deep inspiration was related to the severity of spontaneous obstruction at the time of testing. That is, asthmatic subjects with severe spontaneous airway obstruction responded to deep inspiration with a further decrement in maximal expiratory flow (on maximal as opposed to partial expiratory flow–volume curves). The deep inspiration-induced bronchoconstriction waned during the course of intensive anti-inflammatory treatment, and when the same subjects were subsequently challenged to reduce their maximal expiratory flow to a level comparable to that during the spontaneous exacerbation of their disease, deep inspiration became an effective bronchodilating manoeuvre. These effects are illustrated in figure 1. Using the concept of relative parenchymal airway hysteresis, Lim and co-workers [40, 41] postulated that during a spontaneous asthma attack, peripheral parenchymal hysteresis was much greater than that of the airways. An increase in lung pressure–volume hysteresis is characterised by greatly increased elastic recoil pressures during inflation and much lower recoil pressures during deflation at any given volume. As the forces acting on the airways are proportional to the recoil pressures, diminished radial forces acting on airways during deflation will lead to smaller airways than before a deep inhalation, with a concomitant increase in resistance. Although bronchial provocation does not explicitly alter parenchymal tissue properties [44], it is likely that ASM participates in changes in both parenchymal and airway hysteresis. One important factor determining the magnitude of airway hysteresis is ASM tone and the response of that tone to the stretch induced by deep inspiration; this will be discussed later in the current study. Therefore, the analysis of Burns et al. [17], Pichurko et al. [39], Lim and co-workers [40, 41] and Pliss et al. [42] highlights the importance of ASM in mediating the deep inspiration-induced changes in airway calibre.
FIGURE 1.
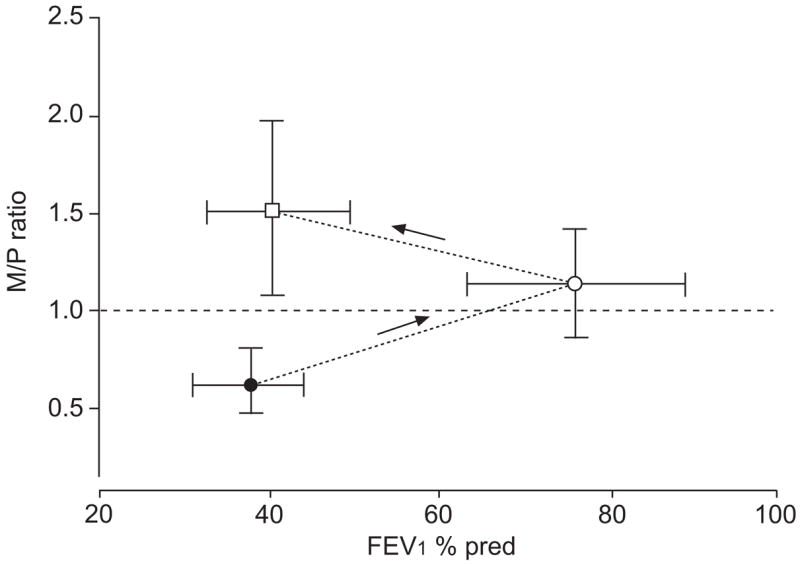
Maximal (M)/partial (P) ratio measured during spontaneous asthma (●), following treatment (○) and following acutely induced bronchoconstriction (□; induced by either dry air or histamine). To calculate the M/P ratio, the maximal mid-expiratory flow on a P and complete M expiratory flow volume curve are compared. An M/P ratio of 1 indicates that the deep inspiration accompanying the complete flow–volume manoeuvre causes neither an increase nor a decrease in mid-expiratory flow. An M/P ratio of <1 means that the deep inspiration decreased mid-expiratory flow; while an M/P ratio of >1 indicates that the deep inspiration increased mid-expiratory flow. During spontaneous asthma, a deep inspiration causes further airway narrowing. This paradoxical bronchoconstriction declines with effective treatment and, when comparable airway narrowing is induced by inhalation of histamine or dry air, a bronchodilating effect of deep inspiration is apparent. FEV1: forced expiratory volume in one second; % pred: % predicted. Modified from [41] with permission.
The second possible answer takes a complementary view to that of the first [17, 39, 40–42] and is based on a distinctly different mechanism [22, 23, 45–51]. Both Gunst and co-workers [22, 23] and Fredberg et al. [52] focused not so much on the parenchyma and changes of parenchymal hysteresis, but rather called attention to the changes in ASM itself. They subjected isolated bronchial segments and isolated ASM tissues in vitro to tidal oscillations and observed that the oscillatory perturbations caused a marked reduction in the contractile responsiveness of the tissues. These observations suggested that volume or load oscillations imposed on the airways during normal breathing might be an important physiological mechanism for reducing airway responsiveness in vivo. Gunst [22] initially suggested that the reduction in airway muscle stiffness and contractility caused by tidal volume oscillations might result from a slow rate of cross-bridge cycling relative to the oscillation rate of the muscle. However, Gunst and co-workers [53–55] subsequently hypothesised that the mechanical stretch and oscillation of ASM might trigger changes in the organisation of the cytoskeletal and contractile filaments that decreased the stiffness and contractility of the muscle.
Fredberg et al. [52] noted that the increase of muscle hysteresis referred to by Lim and co-workers [40, 41] and Sasaki and Hoppin [56] and upon which their argument hinges, happens if, and only if, the muscle is activated and subject to large ongoing tidal stretches; the opposite response occurs to smaller tidal stretches. That is, the hysteresis decreases and does so rather dramatically. Moreover, this progressive decrease of muscle hysteresis with activation has been linked directly, quantitatively, and at the molecular level to progressive decreases of shortening velocity, downregulation of cross-bridge cycling rates, decreases of myosin adenosine triphosphatase activity and the conversion of rapidly cycling cross-bridges to slowly cycling latch-bridges, all of which are now understood to be characteristics of the different phases of smooth muscle activation [57–59]. In the process of activation and tension generation, the muscle can become so stiff that in response to a deep inspiration, it stretches little; it can then become frozen, and virtually stuck in the high-stiffness, low-hysteresis latch state. Such behaviour is consistent with observations from studies in isolated nonasthmatic ASM or in situ in human asthmatics [21, 27, 58, 60, 61]. Such an outcome becomes all the more likely when the ASM mass is increased, when the muscle becomes uncoupled from the lung parenchyma, when expansion of the chest wall is restricted, or when large lung inflations are prohibited during a bronchial challenge [18, 62, 63], all of which are factors that reduce the stretch experienced by the smooth muscle and circumstances relevant to AHR [49, 64]. That being the case, this muscle-based molecular mechanism explains not only how the airways can become refractory to the effects of a deep inspiration, but also how it can exhibit a bronchoconstrictor response.
The third possible answer stems from studies in which the protective effect of deep inspirations taken before experimentally induced bronchoconstriction is examined [18, 27, 60, 65–68]. Malmberg et al. [65] were the first to notice that if a deep inspiration was taken shortly before (<6 min) acutely induced bronchoconstriction, the extent of airway narrowing during the induced constriction was reduced. Subsequently, Scichilone et al. [68] found that deep inspiration exhibited its bronchoprotective effect only in healthy subjects. Using a somewhat different protocol, King et al. [60] found that deep inspiration also had a limited bronchoprotective effect in mild asthmatics. This cannot be easily explained by the mechanical perturbation of activated smooth muscle described immediately above. However, length oscillation applied to resting ASM was observed to diminish the muscle’s ability to generate force in the subsequent contractions [69]. It was postulated by King et al. [26] that this post-oscillation refractoriness of resting ASM was responsible for the bronchoprotective effect of deep inspiration taken before bronchoprovocation seen in nonasthmatics, and that in asthmatics, this refractoriness might be absent. Crimi et al. [70], however, found that this effect was only evident when lung function parameters preceded by a full inflation were used, such as FEV1 or forced vital capacity. Using parameters not preceded by full inflation, (e.g. iso-volume partial forced expiratory flow, residual volume after partial expiration, or specific Gaw), multiple deep inspirations taken prior to methacholine inhalation were found to be associated with increased rather than depressed response, especially in asthmatics. Whether the response to deep inspiration taken before bronchoconstriction is associated with ASM is still a matter of debate.
Morphological evidence of ASM involvement in asthma
There is general agreement that the total amount of smooth muscle is increased in asthma [71–73], and increased muscle mass, even without a change in contractile phenotype, has the potential to enhance airway responsiveness, thus contributing to asthma symptoms [74]. The increased area of the layer of smooth muscle involves small and large airways and may be related to the clinical severity [75] and duration [76] of asthma. Modelling studies predict amplified responsiveness consequent to increased smooth muscle mass. It is important to recognise that this prediction relies on the assumption that the maximal tension that the muscle can generate increases in proportion to its mass. Thickening of the muscle layer alone without such an assumption will only mildly enhance reactivity [74, 77]. Measurements of the area of ASM have usually been made on transverse sections of airways obtained from autopsies. Results from these measurements will depend on the number and size of smooth muscle cells, as well as the extracellular matrix (ECM) between cells [78]. There have been three studies showing more cells (hyperplasia), two in cases of fatal asthma [72, 79] and one in a case of asthma of mild-to-moderate severity [80]. Although Ebina et al. [72] also reported hypertrophy of ASM cells, their measurements included ECM and therefore may have over-estimated cell size.
Benayoun et al. [81] excluded the ECM elements from their measurements and reported hypertrophy of ASM based on cell thickness in patients with severe persistent asthma, based upon thickness of the smooth muscle cells. However, the effects of cell shortening on cell thickness may have confounded these measurements. Bai et al. [76] showed both increased smooth muscle and ECM within the smooth muscle layer in cases of asthma. This study was undertaken on “thick” (relative to submicron-thin sheets of ECM) sections and may have overestimated the true volume fraction of the ECM due to an overlap effect. To date, there are few published quantitative studies regarding the type of ECM within the smooth muscle bundle in asthmatic airways. Recently, Pini et al. [82] have shown that the area fractions of the proteoglycans lumican and biglycan, but not decorin or versican, are increased within the smooth muscle layer in cases of moderate asthma, compared with both nonasthmatic and severe asthma cases. Changes in the matrix components of ASM may alter the mechanical properties of the muscle.
There are no published studies showing increased proliferation of smooth muscle in situ in asthma. Data presented in abstract form [83–85] have not shown differences between cases of asthma and nonasthmatic controls using either proliferating cell nuclear antigen or cyclin D1 to label proliferating smooth muscle cells. Conversely, increased expression of proliferation markers has been observed in the epithelium of patients with asthma [86, 87]. The increased expression of proliferation markers in ASM in animal models of asthma [88] suggests that, in humans, proliferation may not be an important mechanism for increased ASM in asthma. Alternate explanations may be that current techniques do not detect subtle increases in the rate of smooth muscle proliferation in the chronically inflamed airway, or that increased proliferation occurs in “bursts”, in relation to airway development, early at the “onset” of asthma or during episodes of acute inflammation. Since different signals may be involved in smooth muscle hypertrophy, hyperplasia or increased deposition of ECM, sorting out the contribution of these elements to the increased thickness of the smooth muscle layer in asthma is an important research goal.
Apoptosis and increased ASM mass
Reduced apoptosis could in part explain increased ASM mass in asthmatics and account for limited evidence of increased proliferation of ASM cells in situ [89]. Peptide growth factors, which are elevated in asthmatic airways, play pivotal roles in modulating cell survival, proliferation and migration in the lung [90]. For example, fibroblast growth factor, which is elevated in premature and neonatal lungs exposed to hyperoxia, suppresses apoptosis required for normal alveolo-genesis, thus underpinning the development of chronic neonatal lung injury [91, 92]. To date, only a few studies have assessed the impact of asthma-associated biomolecules on ASM apoptosis.
Human ASM cell apoptosis is potentiated by TNF-α, which concomitantly upregulates myocyte-expressed Fas, a transmembrane component of the TNF-α receptor [93]. Binding of inflammatory cell-derived Fas-Ligand (FasL) greatly induces human ASM apoptosis [93]. However, the pro-apoptotic role of TNF-α on ASM in vitro is concentration dependent [94], and appears to be modulated by other mediators, since pro-mitogenic effects of TNF-α have also been reported [94, 95]. Notably, cardiotrophin-1, a member of the gp130/IL-6 cytokine family that is synthesised by ASM cells, inhibits, via p42/p44 mitogen-activated protein kinase (MAPK), human ASM cell apoptosis induced by serum deprivation or FasL/TNF-α exposure [96]. A subsequent study revealed that endothelin-1, which is elevated in asthmatic airways [97], may also promote increased ASM mass via its effects on cell growth and apoptosis [98]. Indeed, endothelin-1 markedly reduced the apoptosis of human ASM cells induced by serum withdrawal, and, in combination with cardiotrophin-1, further promoted ASM hypertrophy and accumulation of contractile apparatus-associated proteins [96, 98]. Freyer et al. [99] reported that ECM components, such as fibronectin, laminin-1 and collagens I and IV, provide strong survival signals to cultured human ASM cells, an effect that is mediated, in part, via integrin α5β1. Interestingly, proteinases derived from inflammatory cells, such as neutrophils, degrade ECM proteins, including fibronectin, to promote human ASM apoptosis [100]. Collectively, these observations suggest that inflammation and changes in ECM associated with airway remodelling in asthma could directly modulate apoptotic responses of ASM cells.
The few studies of apoptosis of ASM in animal models of airway remodelling support the notion that the rate of apoptosis could be a contributor to the regulation of ASM mass. Using an adoptive transfer model of CD4+ T-cell-driven remodelling, Ramos-Barbon et al. [101] observed a reduction in ASM apoptosis at the same time that there was an increase in proliferation of ASM. The airway remodelling was triggered by three consecutive allergen exposures. Interestingly, a change in matrix proteins was also induced by allergen challenge in this model. In horses with heaves, a form of equine asthma, increase in ASM proliferation was also confirmed by proliferating cell nuclear antigen immunoreactivity. However, there was an increase in rates of apoptosis [88]. In the steady state, it seems more likely that an increase in apoptosis will occur to adjust tissue mass. Therapeutic approaches to alter rates of apoptosis might well be useful to diminishing airway remodelling. Peroxisome proliferator-activated receptor (PPAR)-γ ligands have also been shown to reduce ASM proliferation in response to thrombin, basic fibroblast growth factor and foetal bovine serum [102, 103]. The PPAR-γ ligand ciglitazone induces ASM cell apoptosis [102] at concentrations exceeding that required to inhibit proliferation [103]. Future studies are needed in this important area to assess the therapeutic potential of pro-apoptotic agents in reducing or reversing ASM tissue thickening.
ASM growth responses
The controversies over the relative importance of hyperplastic and hypertrophic growth in asthma appear to have had little influence on the extent to which smooth muscle proliferation or hypertrophy have been explored in culture as potential mechanisms for the increase in smooth muscle volume. Proliferation has been extensively explored in smooth muscle cultures from post mortem tissue, resections, lung transplants and biopsies. There are a number of recent studies that identify the diversity of growth stimuli that range from growth factors (fibroblast growth factor (FGF)-β), spasmogens (cysteinyl leukotrienes), cytokines (TNF-α), and the ECM components (monomeric type I collagen) through to physical influences, such as stretch (supra-tidal breathing volumes) and stasis, when smooth muscle is cultured on flexible supports that can be subjected to pressure cycles that simulate the impact of different breathing patterns on the forces acting on smooth muscle [104–106]. Furthermore, signal transduction pathways have been extensively examined, leading to identification of roles for extracellular signal-related kinase 1 and 2, phosphoinositide-3 kinase and p38MAPK in the initial signalling followed by activation of cyclin-dependent kinases and diminished levels of cyclin-dependent kinase inhibitors, including p27kip1 andp21cip1, which culminate in retinoblastoma phosphorylation to allow cells to continue through the final stages of G1 and onto S-phase.
The range of inhibitors of proliferation that have been identified include a number of endogenous regulators including 2-methoxyestradiol [107], cortisol, adrenaline, natriuretic peptides and interferon-β [108]. There has been an even greater level of interest in the impact of existing and potential anti-asthma agents. Both short- and long-acting β2-adrenoceptor agonists reduce proliferation, as do synthetic glucocorticoids, including budesonide and fluticasone propionate, which are used clinically in combination with the long-acting β2-agonists (LABA). Combinations of LABA and glucocorticoids show some synergy in regulating smooth muscle proliferation [109]. However, there are differences amongst mitogens in their susceptibility to regulation, with thrombin responses appearing to be more readily regulated than those to stronger mitogens, such as FGF-β. Although stimulus-dependency of inhibition by glucocorticoids is also observed for cytokine production, this response is more readily regulated when thrombin, rather than IL-1α, is the stimulus [110].
The ECM may also modulate drug-responsiveness of remodelling responses, such as proliferation and migration [111, 112], as cells cultured on monomeric collagen show steroid-resistance whereas cells cultured directly on plastic or on laminin are highly steroid-responsive [113]. Interest in migration of smooth muscle is increasing in light of the possibility that smooth muscle proliferation does not appear to take place in the muscle bundle, but rather in the subepithelial tissue, where the source of the proliferating cells may be stem cells recruited from the bone marrow [114], fibroblasts that have undergone differentiation [115] or smooth muscle cells that have migrated off the potentially antiproliferative ECM that is rich in proteoglycans and laminins [89]. Migration is required for each of these alternative mechanisms for smooth muscle hyperplasia. Migration of ASM can be induced by platelet-derived growth factor [116, 117], enhanced by cys-leukotrienes [118] modulated by β2-adrenoceptor agonists and glucocorticoids [119], with the latter agents losing activity when cells are seeded onto a matrix containing monomeric type-1 collagen [120, 121].
Hypertrophic growth of cultured ASM has been largely ignored, possibly because it is more difficult to establish. Work in human ASM identified the peptide cardiotrophin as an hypertrophic (and anti-apoptotic) influence [96], and in guinea pig ASM culture, IL-1 has reported hypertrophic actions. The most widely investigated hypertrophic factor for vascular smooth muscle, transforming growth factor-β, has no detectable effect on the size of cultured human ASM (T. Harris and A. G. Stewart, Dept of Pharmacology, University of Melbourne, Parkville, Australia; unpublished communication).
Removal of smooth muscle from asthmatic airways
Recently, another line of evidence supporting a role of ASM in asthma has emerged from studies in which ASM mass of asthmatic subjects was reduced partially and permanently through a procedure called bronchial thermoplasty [122]. Rationale for the procedure is perhaps based on the fact that there is no clearly demonstrated physiological benefit of ASM contraction, and that only negative consequences accompany enhanced ASM tone [28, 123]. It should be pointed out that there is evidence to suggest various roles of ASM contraction, from controlling the ventilation/perfusion ratio [124] to stiffening of airways [12, 125, 126]. A review by Seow and Fredberg [28] gives more details. Although a complete ablation of ASM is not feasible with current technology, a recent report by Cox et al. [127], with comments by Bel [128], has shown that partial removal of ASM in asthmatic lungs alleviates asthma symptoms without introducing severe or persistent undesirable side-effects. By reducing the mass of smooth muscle in the walls of conducting airways (intralobar bronchi with diameters >3 mm), Cox et al. [127] showed in a group of mild-to-moderate asthmatics, that the treatment was effective in reducing AHR and asthma exacerbation, and that the reduction persisted throughout the 2 yrs following the procedure, during which the subjects were monitored. The direct effect of bronchial thermoplasty on airway calibre during methacholine challenge was assessed by Brown et al. [129, 130] and Cox et al. [127], who found that the decrease in ASM resulting from thermoplasty led to an increased airway calibre at any dose of inhaled methacholine, compared with the untreated airways. One cautionary note here is that airway calibre data relies on imaging that can detect only a limited number of airways per subject, with these being primarily larger airways. Based on imaging and oscillatory mechanical data [131], it is difficult to imagine that asthmatics do not experience substantial constriction in airways smaller in size than those treated with thermoplasty. Nevertheless, while the thermoplasty studies do not provide much insight into the pathogenesis of the disease, their results argue strongly that ASM is involved in the exacerbation of asthma. If the disease could be cured by eliminating the smooth muscle, there might be a shift in emphasis from understanding the pathogenic role of muscle in the disease to this novel therapy. However, the risk and cost associated with the procedure will probably warrant a continued search for a better therapy, of lower invasiveness and expense, based on a thorough understanding of the disease mechanism.
LUNG VOLUME, AIRWAY DIAMETER AND ASM: MECHANICAL COUPLINGS THAT DEFINE THEIR RELATIONSHIPS
A common explanation for the in vivo observations described in the previous section is that the contractility of ASM is somehow affected by changes in lung volume and the resulting strain of ASM associated with deep inspirations. The validity of this explanation rests on the assumption that there is a tight coupling between lung volume and airway calibre, as well as between airway calibre and ASM length. This important and yet unconfirmed assumption is discussed in the present section, which is mostly confined to in situ studies, predominantly involving human subjects. In the subsequent sections, in vitro studies of airway mechanics and ASM function will be discussed in terms of their relevance to human asthma. The current section focuses on the following two aspects of lung function related to volume change. 1) The nature of coupling between ASM, the airway wall and the rest of the lung; and how this coupling may modulate ASM function, which in turn may alter lung function. 2) The entire airway tree as a system in which large and small airways interact in the context of changing lung volume to either promote or attenuate AHR.
Coupling between the airway and the lung parenchyma in situ
First, the term “coupling” must be clarified. As lung volume increases, two forces act to potentially dilate the airways and hence stretch the ASM. One relates to the bulk modulus and is associated with the pressure difference acting directly across the airway wall, transmitted from the pleural space through the parenchyma and relative to the intraluminal pressure for each airway. This is sometimes referred to as airway–parenchymal interdependence. The other force relates to the shear modulus of the lung parenchyma and occurs because of local tethering, or pulling, of the surrounding parenchyma as it expands with lung volume [132, 133]. The forces of interdependence have a potent inhibitory effect on airway narrowing and can prevent airway closure at high transpulmonary pressures [9, 12]. In the context of lung inflation, the former force is likely to be more important than the latter [133, 134]; however, shear forces have been thought to be important in the context of airway narrowing produced by bronchial provocation, where they may contribute to forces opposing airway narrowing and limit the capacity of the airways to close off or to reach high levels of resistance [8, 9, 12, 135, 136]. Hence, what is of primary concern in the context of the present review is the coupling between the length of ASM and increases in the recoil pressure of the lung during a deep inspiration, rather than the coupling of the length of ASM and actual change in lung volume itself. This is rather important because, if there is a deficiency in the ability to stretch ASM with a deep breath, one needs to distinguish whether it arises simply from a reduced ability to generate an ~2.9 kPa transmural pressure at total lung capacity (TLC), or whether it arises because of a real stiffening in the local airway wall, perhaps due to alterations in ASM properties or other wall constituents. A recent study by Fredberg and Kamm [137] gives a thorough description of force transmission in the lung.
Two recent in situ approaches show the best promise for assessing potential coupling. One method involves direct imaging of the airways. High resolution computed tomography (HRCT) has been used to show that airways dilate during lung inflation from functional residual capacity (FRC) to TLC [138–140]. The studies by Brown et al. [138, 139] suggest that the degree of dilation is similar in milder asthmatics and healthy subjects, even after provocation. However, in the studies by Brown et al. [138, 139], only relatively few large airways were imaged in each subject and changes in airway dimension (during bronchial provocation and during a deep inspiration) occurring in the huge number of small airways were not accessible with the HRCT. Also, closure of these small airways will probably contribute to changes in lung volume (“air trapping”) and could affect coupling between the length of ASM and increases in recoil pressure. Subsequent imaging studies, reviewed by Brown and Mitzner [140], by the same group suggest that the constriction of airways is highly heterogeneous, that they may be capable of closing, and that airways in more severe asthmatics are likely to dilate less during deep inspiration than in those with healthy lungs. Of course, the imaging approach is inherently static, that is, each image is acquired during zero-flow conditions at a fixed lung volume. Thus, one cannot distil the dynamic force–length behaviour of the ASM in situ as it responds to deep inspiration. Another less explicit approach, but one that might permit inference of dynamic force–length behaviour, is to track Raw (or conductance) during a deep inspiration using a forced oscillation method. The Raw obtained from this method is used as a surrogate for airway diameter [21]. An advantage of this approach is that it explicitly captures the functional impact of the deep inspiration on the net change in airway diameters averaged over the entire airway tree. Also, one can track the dynamics of airway reconstriction [21, 63, 141], which may be sensitive to altered ASM function in situ. A disadvantage is that one cannot distinguish airway dilation from either airway or alveolar recruitment. Generally, this approach shows that at TLC, despite achieving a normal recoil pressure, asthmatics cannot reach the same degree of reduction in Raw at baseline as normal subjects [21]. This problem is amplified by bronchoprovocation with inhaled methacholine (or similar bronchoconstrictors). Even if baseline Raw increases in healthy subjects to that of symptomatic asthmatics, the asthmatics have a lesser decrease in Raw with a deep inspiration [63]. The implication is that the tracheobronchial tree is inherently stiffer in asthma [125]. There is also evidence from animal models that allergen exposure increases airway stiffness [142].
An inherent limitation of these approaches is that one cannot determine whether the reduced ability to dilate airways, and hence to stretch ASM with a deep inspiration, lies at the level of ASM. For example, the reduced response to deep inspiration could be due to a change in airway distensibility due to remodelling in those portions of the wall distinct from the ASM, such as the adventitia, the lamina propria or the reticular layer under the airway epithelium, where deposition of connective tissue proteins can alter mechanical properties of the whole airway. Mauad et al. [143] have shown that the content of elastic fibres is decreased in the adventitial wall of the small airways in patients who died of asthma. This could contribute to the increased airway wall stiffness associated with asthma. A role for local connective tissue elements in the airway wall in the dynamics of bronchoconstriction has been shown in nondiseased isolated airways [144, 145]. Airway remodelling is a well-documented phenomenon associated with asthma development. Thickening of some airway wall components seen in asthmatic airways may contribute to excessive airway narrowing [146]; thickening of other components may have a protective effect against excessive narrowing [147–151]. Many of these predictions were based on computer simulations of geometrical changes in airway components and, therefore, were limited by the assumptions associated with the simulation. As highlighted by Pare [152], “geometry is not everything”; analysis of the effect of airway remodelling has to be based not only on altered geometry, but also on the mechanical properties of the altered airway components. For example, deposition of connective tissue in the adventitia could attenuate the cyclically varying strain from the surrounding parenchyma to smooth muscle if the material deposited in this layer was stiff. Conversely, if the deposited material was highly compliant, the parenchymal strain could not be effectively transmitted to the smooth muscle either. It appears, therefore, that there should exist an optimal coupling stiffness that allows maximal transmission of undulating strain from parenchyma to smooth muscle in order for the airways to receive the maximal benefit of the bronchodilating effect of tidal breathing and deep inspirations.
In situ HRCT imaging of canine airways of sizes ranging 1.8–19.1 mm by Brown and Mitzner [153] showed that the diameters of fully relaxed airways increased with transpulmonary pressures up to 0.5–0.7 kPa, where they reached a plateau, suggesting that in these relatively large airways, the diameter does not relate linearly to the cube root of lung volume, at least not at volumes near TLC. This is in contrast to studies [154–156], which concluded that relaxed airways expanded isotropically with the lung. The discrepancy appears to be in the sizes of airways examined. Klingele and Staub [154] based their conclusion on the measurements of diameters of terminal bronchioles. Hahn et al. [156] showed that in airways <2 mm in diameter, isotropic expansion with lung volume was observed throughout the entire range of lung volumes in both relaxed and activated airways. The larger airways deviated from the isotropic behaviour when smooth muscle tone was introduced; they expanded significantly less with lung volume in the high volume range [156]. The effect of smooth muscle tone was also observed by Brown and Mitzner [153]. They showed that with a moderate tone, the airways were less distensible in the low-pressure range, but their diameters continued to increase with increasing transpulmonary pressure at high pressures.
In summary, there is accumulating in situ evidence suggesting that airways (especially small airways) in the lung are sufficiently compliant to be stretched during a deep inspiration, although the extent of stretch is still debatable. In other words, ASM in healthy lungs is likely to be stretched periodically during tidal breathing and deep inspirations, and responds to that stretch by relaxation, even after pharmacological stimulation. Modulation of the mechanical properties of ASM by oscillatory strains, either in the resting or activated state, is discussed elsewhere in the present study. In asthma, it appears that ASM is either refractory to the modulating effect of oscillatory strain, or that the oscillatory strain is not transmitted to the ASM.
AHR and lung volume
Although ASM contraction may not be the only factor that contributes to AHR, it is the final common pathway leading to excessive airway narrowing. Other components, such as inflammatory changes and cellular infiltrates, and structural changes, such as increased or decreased elastin and collagen, as well as proteoglycans, can modify the ASM responses and thus contribute to AHR. These latter changes are commonly referred to as airway remodelling, which has been defined as changes in the composition, quantity and organisation of the cellular and molecular constituents of the airway wall. The topic of airway remodelling is discussed in more detail elsewhere in the present study. Certain aspects of airway remodelling may affect the relationship between AHR and lung volume, while other types may not. For example, airway wall oedema may thicken the wall, but may have little effect on the airway luminal diameter. Brown et al. [157] have shown in a sheep model that airway oedema has a minimal effect on Raw in general, and on the relationship between lung volume and extent of airway constriction, specifically. They found that a 50% increase in airway wall oedema caused a 13% decrease in airway luminal area; and at high lung volume, luminal area was not affected by the oedema. These observations are consistent with another study in which the effects of lung volume on oedematous airways were measured using HRCT [158]. In that study, airway narrowing caused by wall oedema at low lung volume was completely reversed by lung expansion. These findings in sheep are analogous to the changes in airway dimensions observed using computed tomography scanning in patients with asthma. Nakano et al. [159] showed that although patients with asthma had increased thickness of the airway wall, their lumen area was not significantly reduced, compared with nonasthmatic control cases.
In contrast, Brown et al. [138, 139] found that the airway wall thickness was increased and the lumen diameter was decreased in patients with mild asthma. In that same study, it was shown that the airway dilatation caused by lung inflation was comparable in asthmatics and healthy individuals. That is, both at baseline and after methacholine challenge, deep inspiration dilated airways to a similar extent. However, after the lung inflation manoeuvre, when the lung volumes returned to FRC, there was a difference in response. The airways of the healthy individuals remained somewhat dilated, while the airways of the subjects with mild asthma constricted further. This visual evidence of differential hysteresis confirms the early studies of Fish et al. [16], and suggests that the difference between mild asthmatics and nonasthmatics is not in the transmission of the strain to the airway. It remains to be determined whether a comparable dilatation occurs following deep inspiration in moderate or severe asthmatics in whom airway remodelling may cause baseline airway narrowing and/or an increase in airway wall thickness.
IN VITRO BEHAVIOUR OF ASM
ASM in situ is only one component of an extraordinarily complex system, which ranges from single cells within airways to an extensively branched bifurcating tree embedded in a distensible parenchyma. Thus, any explanation of in vivo phenomena, such as deep inspiration-induced bronchodilation and bronchoprotection based on evidence gathered from studies of isolated ASM, has to have caveats attached, and an explanation based on the observations from isolated tissue or cultured cells in vitro can only be regarded as provisional until all interactions between ASM and other lung components are worked out.
Response of activated ASM to oscillatory stress or strain
As discussed briefly above, it has been known for a long time that length or force oscillations at physiologically relevant frequencies and amplitudes applied to activated ASM result in a reduction in the ability of the muscle to contract [22, 23, 45, 56, 58, 160–164]. Evidence that tidal volume oscillations had similar effects on airway calibre and Raw in experimental animals in vivo led Warner et al. [24], Tepper et al. [25] and Shen et al. [160] to suggest that the oscillation of the airways that occurs during tidal breathing might be a physiologically important mechanism for decreasing airway responsiveness.
A theoretical explanation for this muscle behaviour was provided by Fredberg et al. [58] based on predictions by Mijailovich et al. [59] of a four-state cross-bridge model of smooth muscle contraction. Fredberg et al. [58] and Mijailovich et al. [59] were able to describe the relationship between oscillation amplitude and the extent of shortening in the muscle, and these studies [58, 59] suggest that if tidal breathing or deep inspiration causes mechanical perturbation to contracted ASM in situ, bronchodilation could result. The relatively small amplitude of length oscillation (comparable to that associated with tidal breathing) required to produce a substantial decrease in active force in ASM prompted Gump et al. [163] to suggest that tidal breathing is a potent bronchodilator.
Structural rearrangement of cytoskeleton and contractile apparatus during active contraction may also be responsible for the bronchodilatory effect. Fredberg et al. [58] observed that after large-amplitude oscillations, a muscle temporarily lost some of its ability to shorten, and this loss of ability was not related to perturbed actomyosin interaction, but was likely to be related to plastic rearrangement of cell structure. It has been theorised that actin filament length may be a factor determining the extent of muscle relaxation (lengthening) due to oscillatory stress [165]. Abnormally long actin filaments may increase the range of sliding of contractile filaments without diminishing the overlap between myosin and actin filaments, and thus render the muscle more resistant to the relaxing effect of oscillatory stress. This was proposed as one of the possible defects of asthmatic ASM [165]. Computer simulation of this scenario [166, 167] also demonstrated that longer actin filaments were associated with greater resistance of the simulated muscle to relax in the presence of oscillatory strain.
Response of resting ASM to mechanical perturbation
It was mentioned briefly previously that length oscillation applied to ASM in the relaxed state has been found to have an inhibitory effect on the muscle’s ability to generate force in the subsequent contractions [69]. The same study also revealed that the inhibitory effect was linearly correlated to the amplitude and duration of oscillation, but was relatively independent of oscillation frequency. This finding suggests that if deep inspirations are taken just before a bronchial provocation (e.g. with methacholine inhalation), the induced bronchoconstriction may be attenuated due to the inability of the muscle to generate maximal force. This interpretation, of course, is subject to the caveats mentioned above regarding mechanical coupling between changes in lung volume and ASM length. This interesting response of resting muscle to mechanical strain is transient and appears to be part of the process of the muscle’s adaptation to length change. Kuo et al. [168] have shown that the length-oscillation-induced decrease in force is accompanied by a similar amount of reduction in myosin filament density in the muscle. Transmission of force, therefore, can occur throughout the whole lung structure right down to the molecules [137]. Length adaptation appears to be responsible for many aspects of the unique behaviour of ASM and may be an integral part of normal airway function. A failure of length adaptation, due to either changes within the smooth muscle or external strain coupling to the smooth muscle, could explain some abnormalities seen in asthma.
Length adaptation in ASM
As in striated muscle, a sufficiently large change in length (from one that corresponds to optimal overlap of contractile filaments) in ASM can reduce force production [47, 169, 170]. What is different in the smooth muscle response is that the force decrease is often transient. Different extents of force recovery have been observed after a length change if the muscle is allowed to relax and is re-activated repeatedly at the new length [47, 170–172]. The dynamic process of force recovery is termed length adaptation [173]. Adaptation of ASM to a new length leads to a shift in the length–force relationship along the length axis, as illustrated in figure 2, and consequently alters the muscle’s ability to regulate airway diameter. Although the mechanism for length adaptation is still largely unknown, the ability of smooth muscle to adapt to length change is likely to stem from structural malleability in the muscle’s cytoskeleton and contractile apparatus. This novel concept deviates from the static model of fixed myofilament lattice that describes striated muscle. The molecular events associated with structural reorganisation in smooth muscle probably involve rearrangement of actin and myosin filaments, attachment of actin filaments to dense bodies and plaques, and polymerisation and depolymerisation of these filaments. The reorganisation is thought to serve the purpose of facilitating the cell in maintaining optimal contractile filament overlap and orientation at different cell lengths [174].
FIGURE 2.
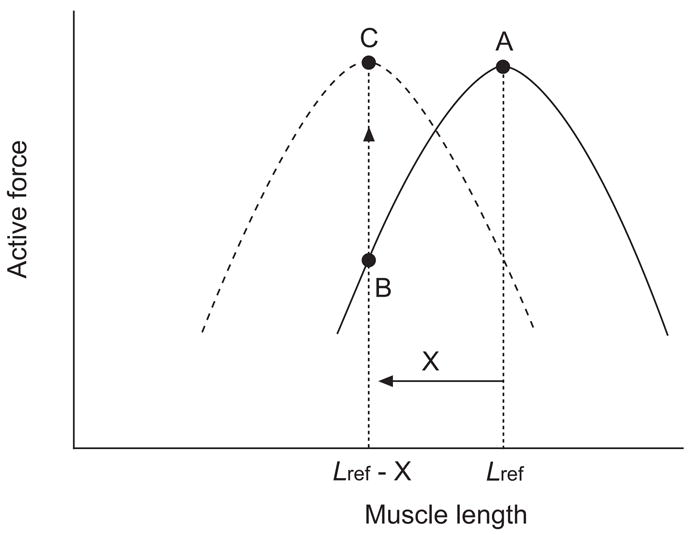
Shift in the length–force relationship of airway smooth muscle due to length adaptation. The solid curve (———) represents the length–force relationship of a muscle adapted at an arbitrarily chosen reference length (Lref). Upon shortening by an amount (X), the actively developed force decreases from A to B. After adaptation at the new length (Lref - X), usually through repeated activation and relaxation, the maximal active force for the muscle at the new length recovers to the level before the length change (C), and the muscle now possesses a new length–force relationship (- - - - -).
Gunst and co-workers [53, 169] suggest that actin filaments may not attach firmly to dense plaques in the relaxed state; they do so only when the muscle is activated. Thus, it was proposed that the reorganisation of the attachment of actin filaments to membrane adhesion sites might be accompanied by changes in the organisation of the actin filament lattice resulting in reorientation of the contractile apparatus [53, 169]. This would allow ASM in the relaxed state to change length without altering contractile-filament overlap and enable the muscle to optimise the organisation of the contractile filaments to maximise the contractility of the muscle when length changes are imposed on it by the external environment. The details on how the attachment of thin filaments to adhesion proteins is regulated, especially in partially activated states, remain to be established. However, there is evidence that contractile stimulation of ASM activates a complex of cytoskeletal proteins that associate with transmembrane integrins at sites of cell adhesion [175, 176], and that the activation of these proteins is sensitive to mechanical strain [177, 178]. The cytoskeletal protein complex that forms at cell adhesion sites may transduce mechanical signals sensed by transmembrane integrin proteins, and initiate signalling pathways that regulate cytoskeletal events, such as the polymerisation of actin, the anchoring of the actin cytoskeleton to adhesion proteins, and the strain-sensitive activation of contractile proteins [55, 179–181].
The “fluidity” of the cell structure is not limited to the anchorage sites where actin filaments attach to dense plaques. In fact, the whole cell structure appears to be in a constant state of restructuring and adaptation to external stress and strain [182–184]. Depression of force and stiffness due to length change can be explained by a disruption or weakening of structures along the force-generating and -transmitting pathway. The actomyosin cross-bridge is only one of many such structures. Other structures include myosin filaments, actin filaments, cytoskeletal cross-linker molecules, cytosolic dense bodies, membrane adhesion plaques and cell–cell connections [55, 185]. There is ample evidence for each of these candidates to be somehow involved in the response to external perturbation [168, 174, 186–191].
The overall interaction of these subcellular components can be described as weakly interacting discrete elements in a “crowded”, out-of-equilibrium micro-environment. In other words, the cell behaves like soft glass [182, 183]. According to the theory of soft glass, as applied to smooth muscle, the ability to deform, to flow and to remodel is governed by nonthermal agitation (motion) energy of the cytoskeletal elements relative to the height of bond energies that constrain their motion [182, 183]. A mechanical strain can act as an energy source that helps individual elements to jump out of their local energy well, so the cytoskeleton essentially “heats up” and “melts”. Over time, however, the cytoskeletal elements evolve into configurations that are more and more stable and stiffer [183]. Slowly evolving dynamics of this kind are called “ageing”, in the terminology of material sciences. External forces can reverse the ageing process, and the system can be “rejuvenated” by shear stress, as found by Bursac et al. [183] in ASM. The theory explains many aspects of time-dependent behaviour observed in ASM [29].
By examining the details of length-dependent properties of ASM, including adapted and nonadapted isometric force, shortening velocity, power output, rate of adenosine tiphosphate utilisation and change in myosin filament density (or filament mass), Kuo et al. [188], Qi et al. [192] and Herrera and co-workers [192–195] have provided evidence that length adaptation in ASM involves changes in the number of contractile units in series and in parallel. They also showed that externally applied strain on ASM in the relaxed state led directly to partial disassembly of the myosin filaments [168], thus providing an explanation for the strain-induced inhibition of muscle force, which could underlie the phenomenon of deep-inspiration-induced bronchoprotection, as discussed in the previous section.
The mechanisms described are not mutually exclusive, as suggested by simulations of dynamic networks mimicking intracellular structure of smooth muscle [166].
EFFECTS OF CHRONIC LUNG-VOLUME ALTERATION AND SMOOTH MUSCLE LENGTH
As a consequence of its effect on airway calibre and ASM length, lung volume as a determinant of lung function has recently received more attention [30, 196]. McClean et al. [13] produced a chronic decrease in lung volume (25% below FRC) in sheep by restricting their chests with a corset, and examined the subsequent in vitro contractility of ASM from these animals after 4 weeks. They found an increased rate of force generation in bronchial rings of adult and adolescent sheep. In humans, Torchio et al. [14] found that chest wall strapping, sufficient to reduce FRC by ~1 L, enhanced airway narrowing when applied during, but not after, the inhalation of methacholine. This suggests that the length of ASM at the time of stimulation is more important than the geometric effect of lung volume on airway calibre. Moreover, Xue et al. [197] examined the effect of lung volume in tracheotomised rabbits breathing at a chronically elevated lung volume using positive end-expiratory pressure (PEEP). They found that chronic PEEP reduced airway responsiveness. In addition, bronchial segments isolated from these animals were larger and generated reduced maximal pressures in response to acetylcholine when compared with controls. Although the exact mechanism is not clear, these studies implicate changes in ASM function in response to alterations in resting lung volume.
In cultured explants of ASM bundles [198], chronic shortening of the tissue produced shifts in the passive and active length–force relationships, similar to those found in acute [34, 170] and subacute [171] length adaptation. However, this longer term (>3 days) adaptation of the tissue at short lengths was found to be associated with an increased passive stiffness and partial loss of the ability to re-adapt when the muscle was returned to its in situ length [198]. Such a change in ASM properties could explain the ineffectiveness of deep inspiration in relaxing the airways in asthmatics [18], i.e. the ASM in asthmatics may be chronically adapted to a shortened state. In cultured explants of bronchial segments [199], prolonged distension of the segment produced a larger airway lumen size and a decrease in the stiffness of the segment, consistent with the “right” shift in the passive length–force relationship of ASM adapted at long lengths observed by Naghshin et al. [198]. The reduced responsiveness of distended airways in terms of their ability to narrow [199] also suggests that the active length–force relationship has shifted with the passive relationship.
It is thus plausible that chronic changes in ASM length could contribute to alterations in bronchial responsiveness in humans. This raises the question of how such changes in length could arise. The length of the ASM in situ is determined by the balance between forces that tend to expand the airway wall and those that tend to collapse it. Among the expansive forces, the most important and easily modulated is that due to the outward recoil of the parenchymal attachments. These attachments transmit the transpulmonary pressure across the airway wall and so they are greatly affected by lung volume [136], which is a strong modulator of bronchial responsiveness [8, 200]. Consequently, any condition that causes a chronic alteration in lung volume would also be expected to affect mean muscle length. For example, obesity and the supine position associated with bed rest both reduce FRC and so should reduce ASM length, while the hyperinflation associated with obstructive lung disease would have the opposite effect. Interestingly, it appears that patients with fatal asthma have damaged alveolar attachments, which may lead to irreversible uncoupling between expansive forces and the airways, including the smooth muscle [143]. Factors that interfere with the ability of the parenchyma to distend the airway wall should also affect wall circumference and hence ASM length. In particular, it has been suggested that peribronchial inflammation may uncouple the parenchyma from the airways [201], thereby allowing the airways to narrow more easily under the influence of airway elastic recoil or active ASM tone. Indeed, elevations in airway tone resulting from elevated levels of contractile mediators could lead to a chronically shortened ASM [34]. Such conditions might be secondary to the chronic inflammatory state that characterises asthma. In this context, the actions of the endogenous muscle relaxants, such as prostaglandin E2 and adrenaline are also relevant. Moreover, by extension, the long-acting β2-adrenoceptor agonists, which are presumed to cause chronic lengthening of ASM, may have, as a consequence, a self-reinforcing impact on airway calibre. The intermittent and abbreviated nature of the muscle lengthening induced by short-acting β2-adrenoceptor agonists, even when used regularly, may not share this amplifier effect.
Another possible mechanism for chronically altering ASM length, as briefly mentioned above, is by changing the mechanical properties of the airway wall itself. Alterations in the connective tissue structure and composition of the airway wall, a process termed remodelling, has been documented in human asthmatic airways [71, 73, 202] and in animal models [203, 204]. Such changes might result in alterations in the passive mechanical properties of the airway wall, leading to a change in the wall diameter at which its inward recoil is balanced by the outward pull of the parenchymal attachments. If any of these changes were associated with a reduction in either airway lumen or wall compliance, then increased bronchial responsiveness might be a consequence. This analysis, however, does not take into account the possibility that a stiffened airway may be more resistant to forces that tend to collapse the airway.
Age and maturity may also be important in determining the effects of changes in ASM length on contractility, particularly in view of the changes in airway reactivity that occur with lung growth and development [25, 205–207]. For example, the mechanical interdependence between the airways and parenchyma is weaker in young compared with mature animals [208, 209], and the airway wall of immature animals is more compliant than that of mature animals [210]. Also, the more compliant chest wall of the infant compared with the adult results in FRC being lower relative to TLC in the infant than in the adult lung [211]. These mechanical factors may explain why ASM contractility is affected by chronic corseting in adolescent and adult sheep but not in neonates [13]. In addition, in vitro evidence suggests that the contractility of ASM may undergo significant changes in the transition from infancy to adulthood. ASM becomes progressively less compliant but more sensitive to acetylcholine with maturation in sheep [212], while the shortening velocity and passive stiffness of guinea pig trachealis both increase in the first 3 weeks of life and then decline [213, 214]. More significantly, the force-generating ability of ASM is potentiated by stretch in infant guinea pigs but not in adult animals [215], and appears to be related to maturational changes in the release of prostanoids [216]. These findings may have relevance to the observations of Weist et al. [206], who showed that airway reactivity in healthy infants is not only greater than that in healthy adults, but is also insensitive to deep inspiration, similar to the situation in asthmatic adults [18] and in passively sensitised human bronchi [217]. Thus, there is reason to suspect that immature and asthmatic ASM may adapt differently to chronic length change.
There is therefore abundant evidence that ASM responds to changes in operating length by adjusting its internal structure to optimise force generation. Such effects can occur rapidly, in a matter of minutes; but they can be reversed just as quickly. There is now increasing evidence both in vitro and in vivo that chronic changes in ASM length also lead to persistent adjustments in force-generating capacity, and that these changes are less reversible than those in acute length adaptation. These subacute and chronic changes in ASM length may be important contributors to the AHR characteristic of asthma.
CHRONIC OSCILLATORY LENGTH CHANGE IN CULTURED ASM CELLS
The use of cultured cells to investigate specific components of cell function and its control is routine in cell biology and physiology research, and has been extensively employed in the study of ASM. The approach affords the opportunity to manipulate conditions, such as soluble biomarkers and ECM composition, to assess their effects on a defined cell population, thus eliminating many difficult-to-control variables associated with in situ and in vivo experimental models. It must be kept in mind, however, that cells in culture undergo phenotype modulation such that their contractile protein expression is quite different from that in vivo, and this could significantly alter cell mechanics [218–226]. Nevertheless, as a way to study the effects of mechanical strain on ASM contractility and mechanical plasticity, in vitro models offer some advantages including the ability to apply acute or chronic mechanical strain varying in magnitude, frequency and direction. Furthermore, real-time and time-lapse light and fluorescence microscopy can be used effectively both to measure contraction, stiffening and receptor-mediated signalling, and to assess the effects of mechanical strain on cytoskeletal dynamics and organisation, cell–cell and cell–matrix interactions, and mechanotransduction. Conversely, videomicrometry of enzymatically dissociated cells can be used to examine the dynamics of repeated ASM cell shortening and re-elongation in the absence of any contributions from the ECM [227]; this approach perhaps best reveals the internal force(s) within the smooth muscle cell that assist the external tethering force(s) to oppose/reverse shortening. For studies in which oscillatory mechanical strain is applied for days or weeks, the most widely used approach is to grow cells on flexible membranes that can be stretched either uniaxially or biaxially with a vacuum system [228, 229]. This can be adapted for acute studies to enable simultaneous assessment of cell morphology and signal transduction events, such as intracellular Ca2+ flux. Another widely used approach for acute studies is to use magnetic twisting cytometry to apply force and measure stiffness of the underlying cytoskeleton [230, 231]. Twisting cytometry, atomic force microscopy and videomicroscopy are currently the most effective approaches with which to assess cell stiffness and shortening in cultured cells.
Changes in morphology and cell attachment in culture
It is now well established that mature ASM cells with contractile phenotype undergo spontaneous, reversible phenotype switching when placed in cell culture [221, 226]. This has promoted understanding of the functional properties of ASM cells that contribute to airway remodelling, including myocyte proliferation, secretion of pro-inflammatory bio-molecules, and synthesis of ECM components [232–234]. Nonetheless, key ultrastructural changes are evident when smooth muscle cells are plated as a monolayer in culture dishes, and this underpins a need for careful assessment of the effects of external mechanical strain on myocyte function. In tissues, the contractile apparatus of smooth muscle cells is dotted with a more-or-less regular pattern of dense bodies to which thin filaments are anchored [185, 235]. Furthermore, dense plaques, arranged in discrete linear plasma membrane domains between caveolae-rich regions, anchor the contractile apparatus to the cell periphery and serve as junctions between adjacent cells, thereby creating a contractile filament syncytium that is sensitive to externally applied mechanical strain [185, 236]. In contrast, cultured myocytes characteristically possess multiple, longitudinally oriented cables of actin and myosin called “stress fibres”. In specialised, long-term, serum-deficient culture conditions, the protein content of contractile cables in cultured cells can be induced to a higher level resembling the contractile apparatus of smooth muscle cells in intact tissue, and dense bodies are visible within contractile cables [237, 238]. Moreover, as seen in tissue, some ASM cells in these cultures exhibit discrete longitudinal caveolae-rich plasma membrane domains, and these appear to be in register with underlying contractile cables [238].
In cultured ASM cells, stress fibres are anchored at either end to the extracellular substratum via focal adhesion complexes [179]. These junctions, like dense plaques in tissue-situated myocytes, transmit strain due to deformation of the substratum, and are composed of integrins and associated proteins, such as talin, α-actinin and vinculin. However, a notable difference between myocytes in tissue and in culture is that there is little or no connection between the cytoskeleton of adjacent cells in the latter environment. Since this difference is likely to have direct effects on the response of myocytes to external mechanical strain, it needs to be considered in analysis of data obtained using in vitro systems. It seems likely that by placing myocytes in monolayer culture on inflexible plastic plates, structural constraints are imposed so as to preclude cytoskeletal organisation mimicking the in situ situation. A potential solution may lie in recent advances in developing cultures in which cells are cast in three-dimensional matrices of biopolymers [239–241]. Though a systematic appraisal of the ultrastructure and phenotype of myocytes in such cultures has not yet been reported, future directions using this approach should provide valuable insight, and may significantly extend the utility of in vitro systems to study the molecular signalling, cytoskeletal remodelling and functional consequences of oscillatory mechanical strain on airway myocytes.
Changes in mechanical properties after chronic strain
Application of chronic mechanical strain in cultured ASM cells leads to several changes in cell structure and function. These changes depend on strain magnitude and orientation, as discussed in sections below. In several studies, Smith and coworkers [230, 242–244] have demonstrated that a predominantly uniaxial (stretch in one direction) strain of ~10% applied for several days in culture increases proliferation, force generation, expression of contractile proteins, calcium sensitivity and cytoskeletal stiffness with contractile activation. Perhaps the most dramatic changes they have been shown are a doubling in shortening capacity and shortening velocity [245]. Furthermore, while the ability to relax and decrease cell stiffness following acute stretches in cultured cells (whether they have been exposed to strain or not) is unaltered and the response is comparable to that of intact tissues [52, 246], the ability to recover following such stretches is significantly altered by previous exposure to chronic strain. In single cells lifted from culture dishes and then subjected to large acute stretches of 12% (simulating stretches caused by sighs) followed by smaller stretches of 2% (simulating tidal breathing), cells that had been grown under conditions of chronic cyclic strain for several days recovered their stiffness rapidly within tens of seconds, compared with cells that were not grown under chronic cyclic strain, which recovered after >100 s [246]. This more rapid recovery following an acute stretch may be important as it is reminiscent of the more rapid re-narrowing (and thus lack of bronchodilatory effect) observed following a deep inspiration in asthma [20, 35, 138, 139]. Together, these changes suggest that chronic strain may play an important role in promoting the contractile function of ASM, and possibly in the pathogenesis of asthma.
Cell alignment and dependence on strain profile and underlying matrix
The changes described for ASM cells cultured in the presence of oscillatory mechanical strain appear to depend on the magnitude, nature and orientation of the strain that is applied. In the studies described in the preceding paragraph, cyclical negative pressure applied below the circular flexible membranes strained the cultured cells. This method results in uneven strain across the membranes with biaxial strain (i.e. the stretch is the same in every direction in a two-dimensional plane) of ~10% in the central region, changing gradually to uniaxial stretch of 10% in the radial direction near the membrane edge [246, 247]. After 2–4 days, the cells in the region of predominantly uniaxial strain re-orient, forming a ring of cells aligned circumferentially, while cells in the central region do not show any particular preferred direction. Thus, over most of the dish, stretch is primarily across the “waists” of the cells, with very little stretch occurring along the cell’s long axis [242, 243]. This is illustrated in figure 3.
FIGURE 3.
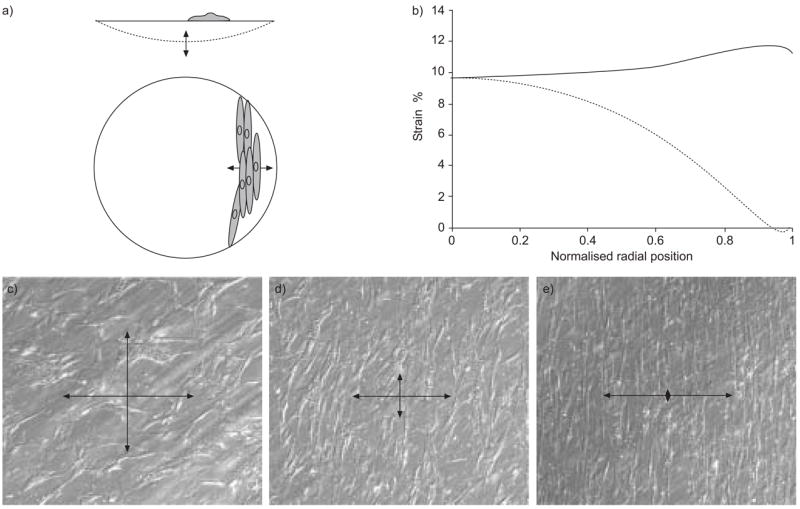
a) Airway smooth muscle cells being stretched in circular dishes by oscillating negative pressure below the flexible membrane. b) The strain field is anisotropic with nearly constant strain in the radial strain (–––––––) but gradually decreasing stretch in the circumferential strain (-------), which results in nearly uniaxial strain near the edge where the membrane is held. c, d, e) Cells are shown in Hoffmann contrast microscopy after 7 days of continuous cyclic stretching in culture. Cells do not align in the biaxial strain region, where strain is the same in every direction in the plane (arrows, as shown in c), but near the edge become almost completely aligned transverse to the applied strain direction (e). c, d, e) Microscopy shows the strain profile near radial positions 0, 0.5. and 1.0, respectively. As indicated by the normalised radial position in (b). Reproduced from [246] with permission.
It has been observed for many cell types that cells align in response to anisotropic or uniaxial strain, including fibroblasts [248] and vascular endothelial cells [249, 250]. Interestingly, the cellular response to strain also depends on the orientation of the applied strain relative to the cells. For example, in response to uniaxial strain, fibroblasts that are forced to align in grooves with the applied strain increase their expression of α-actin more than cells aligned perpendicular to the strain direction, although α-actin is increased in both cases. Interestingly, fibroblasts forced to align in grooves perpendicular to the applied strain increase secretory behaviour compared with those aligned parallel to the strain [248]. In contrast, in response to biaxial strain, no orientation behaviour is observed in fibroblast or ASM cells [229, 248].
It is unknown why almost all cells re-orient in response to mostly uniaxial or anisotropic strain; however, there is recent evidence that a difference in strain profile leads to quite different changes in cell structure and function in ASM cells. Most studies of mechanically strained ASM cells including the results described above were primarily from cells in the outer area of the membranes on which they were grown, and thus the results represent the response to a primarily uniaxial strain. In contrast to these results, Wang et al. [229] found that when subjected to uniform biaxial strain, there was no cell alignment, no increase in proliferation, a ~50% decrease in smooth muscle protein 22 and smooth muscle myosin heavy chain promoter activity, and decreased filamentous to globular-actin ratio, compared with cells not exposed to any strain. These observations are consistent with decreased Rho activation, which is in contrast to the increased Rho-A activation observed in response to predominantly uniaxial strain [231] and would appear to represent anticontractile effects of strain rather than pro-contractile effects of strain as occurring with primarily uniaxial strain. Furthermore, increases in amount of myosin light chain kinase after 11 days of strain seem to occur primarily in uniaxially strained cells [243, 246]. Interestingly, proliferation is reduced in cells grown on laminin and subjected to lower strains of 4% compared with cells grown on collagen with either 0 or 4% strain. Conversely, proliferation is enhanced in cells grown on collagen subjected to 10% strain, which indicates that the response to strain is sensitive to both strain magnitude and ECM [242, 251]. While these results point to the importance to cell function of strain profile and magnitude, how relevant are the strain conditions experienced by cells in culture to strain experienced in vivo, and do strain conditions change in disease?
Strain in culture and strain in vivo
In vivo, the strain produced by lung expansion is three-dimensional, and thus applied to all cell surfaces, in contrast to most in vitro strain systems. In vivo, the airways are constantly being stretched circumferentially and longitudinally. The magnitude of the strain depends on airway size as discussed previously, but typically is ~2–4% during tidal breathing, ~12% during sighs, with a maximal of ~25% during inflation to TLC [52, 154–156]. Thus in healthy lungs, smooth muscle strain more closely matches the biaxial strain that does not promote contractile changes in cultured cells. However, the strain profile must be altered in the presence of elevated ASM tone and with airway narrowing, such as that occurs in asthma. As ASM is predominantly oriented circumferentially about the airways with an average pitch close to 13° [252], increased tone would increase circumferential stiffness relative to the stiffness along the length of the airway. Thus, the stretch proffered by breathing would tend to be more axial than circumferential, leading to development of strain anisotropy in vivo. Indeed, while this has not yet been examined directly, if true, more stretch would be conferred across the waists of the ASM cells, matching conditions leading to pro-contractile changes in ASM in vitro.
Thus, increased tone may lead to enhanced contractile function by enhancing strain anisotropy in vivo. Interestingly, elevated tone appears to be required to elicit stretch-induced changes in cultured ASM cells. For example, cytoskeletal remodelling and stiffening in response to mechanical stress are inhibited during β-agonist-induced reduction of tone in culture [253]. While it has yet to be demonstrated that reduced tone would inhibit the pro-contractile changes previously demonstrated in uniaxially strained cells, this suggests that in addition to promoting shortened ASM as discussed previously [34], tone may play a harmful role in the pathogenesis of asthma by also leading to strain anisotropy and potentiation of structural remodelling of the ASM cells. However, while changes in cell structure and function with strain are well documented in vivo, the acquisition of a more contractile phenotype due to mechanical strain in vivo has not been established.
THE INFLUENCE OF CA2+ ON THE MECHANICS OF ASM
As mentioned previously, a major determinant of the mechanical properties of smooth muscle cells is the extent of the actomyosin cross-bridge interactions with a high rate or sustained level of cross-bridge formation leading to muscle contraction or stiffness. The rate of cross-bridge activity depends on the phosphorylation state of the regulatory myosin light chain (rMLC) which, in turn, is determined by the antagonistic activities of myosin light chain kinase (MLCK) and myosin light chain phosphatase (MLCP). With respect to changes in smooth muscle cell length, the potential for cross-bridge formation is also increased by filament elongation or formation of additional contractile units. While MLCK activity is generally a function of the intracellular Ca2+ concentration ([Ca2+]i), MLCP activity is a complex function of the activity of a variety of kinases and phosphatases and, in most cases, independent of [Ca2+]i. This ability of the smooth muscle cell to modulate MLCP activity independently of the Ca2+-dependent MLCK activity is referred to as Ca2+ sensitivity. As the Ca2+ signalling and sensitivity of smooth muscle cells are often receptor-mediated processes, muscle mechanics may be altered in the inflammatory environment with additional cytokines or growth factors associated with asthma.
While ASM contraction usually requires an increase in [Ca2+]i, this agonist-induced stimulus (i.e. acetylcholine, 5-hydroxytryptamine, endothelin) usually consists of Ca2+ oscillations that propagate along the muscle cells as Ca2+ waves [254–258]. The frequency of these Ca2+ oscillations correlate with the muscle’s contractile state and this has led to the consensus that ASM cells frequency encode their Ca2+ signals [259–261]. These Ca2+ oscillations are primarily mediated by repetitive Ca2+ release from internal stores and require refilling by Ca2+ influx [255, 256, 260, 261].
The mechanism responsible for the release of Ca2+ from the internal store is currently under discussion. There are two basic ideas centred on either the inositol 1,4,5-triphosphate (IP3) receptor (IP3R) or ryanodine receptor (RyR). In one hypothesis, the Ca2+ oscillations are mediated by calcium-induced calcium-release (CICR) via the IP3R sensitised by IP3 [254, 260]. Alternatively, Ca2+ oscillations may be mediated by CICR via the RyR [261–264], although the initiation of the Ca2+ oscillations may require an IP3-induced Ca2+ release [265]. The release of Ca2+ via the RyR may also require sensitisation with cADP-ribose. Although it would be convenient to have a unifying mechanism, these data indicate that the mechanism of CICR varies with smooth muscle species and airway location.
As refilling of the Ca2+ store is essential for Ca2+ oscillations, the mechanisms of Ca2+ influx are vital. Although voltage-dependent Ca2+ channels are commonly implicated in Ca2+ influx, agonist stimulation does not sufficiently depolarise smooth muscle cells to fully activate such channels [266]. Similarly, L-type Ca2+ channel blockers, such as nifedipine, have variable effects [258, 260, 261, 267, 268]. These data indicate that there is a range of involvement of voltage-gated channels in refilling internal stores. The variability may arise from the weak depolarisation induced by the inward, Ca2+-dependent, Cl− current and nonspecific cation currents that occur in conjunction with Ca2+ oscillations [269–271]. However, changes in Cl− may have an additional electrogenic action to facilitate Ca2+ release and uptake across the sarcoplasmic reticulum membrane [272]. Ca2+ entry may also occur as the result of the emptying of the internal stores via store-operated Ca2+ channels [273]. The mechanism linking store-depletion to influx is unclear, but recent studies have identified a potential Ca2+ sensor (STIM1) within the sarcoplasmic reticulum that communicates with a plasma membrane protein or channel (Orai 1) [274, 275]. Ca2+ influx can also be influenced by the contractile agonist indicating either activation of receptor-operated channels or a second messenger-operated channel. In addition, the occurrence of Ca2+-dependent nonspecific cation currents can lead to increased intracellular Na+ and the reversal of Na+/Ca2+ exchanger to increase Ca2+ entry [276]. Clearly, ASM cells are well equipped with multiple Ca2+ import pathways to ensure store refilling.
The propagating Ca2+ waves within smooth muscle cells arise from the sequential release or CICR of Ca2+ via individual receptor channels. However, the independent activity of these receptors, or elemental Ca2+ signalling, has been implicated in smooth muscle contraction. Elemental Ca2+ signals are termed Ca2+ sparks or puffs if predominantly mediated by RyR or IP3Rs, respectively. The correlation of Ca2+ sparks with spontaneous transient outward currents mediated by Ca2+-activated K+ channels has led to the idea, mainly in vascular smooth muscle, that smooth muscle cells are relaxed by Ca2+ sparks that hyperpolarise the membrane potential to reduce Ca2+ entry into the cell via voltage-dependent channels.
Although Ca2+ sparks and associated spontaneous transient outward currents have been observed in isolated tracheal smooth muscle cells [277, 278], ASM cells do not appear to rely on membrane potential to develop tone. Consequently, Ca2+ sparks may not be required for muscle relaxation. In ASM, Ca2+ sparks have been observed in isolated cells or in bronchial smooth muscle cells under conditions of stress [260] where the Ca2+ store may be overfilled. It is possible that Ca2+ sparks occur to regulate the Ca2+ concentration of the internal store. Ca2+ uptake into the internal stores would counter Ca2+ leaking into the cell, but once these stores reach their capacity, the release of Ca2+ via Ca2+ sparks would serve to minimise further influx. However, if the stores become overloaded, elemental Ca2+ events can lead to propagating Ca2+ waves [260]. This activity is similar to the observation that Ca2+ waves in isolated smooth muscle cells are initiated from areas of high Ca2+ spark activity, although this activity is thought to be the result of the sensitisation of the RyRs by cADP-ribose [256, 278].
While Ca2+ initiates contraction, the Ca2+ sensitivity of the smooth muscle cells (mediated by decreased MLCP activity) strongly influences the effect of contraction [279]. There are two major pathways that lead to decreased MLCP activity; these are the phosphorylation and inhibition of the regulator unit of MLCP by Rho kinase and the phosphorylation of the inhibitory protein CPI-17 by protein kinase C. A second form of sensitisation can also occur by altering the phosphorylation of rMLC in a Ca2+-independent manner by kinases, such as luceine zipper kinase, or by actin-binding proteins, such as caldesmon. Most importantly, these kinases can be activated in a variety of ways and this provides a broad spectrum of possibilities by which smooth muscle contractility can be modulated.
In view of these mechanisms of Ca2+-induced contraction, a key question here is, does mechanical stretch of smooth muscle cells (by inspiration) alter the contractile response of the muscle to Ca2+? Stretch-induced responses can be mediated by the opening of ion channels to either alter membrane potential or directly induce Ca2+ influx. However, an increase in Ca2+ would seem counter-productive for relaxation, unless these Ca2+ increases occur locally to alter membrane potential to decease the driving force for Ca2+ entry, which in turn will slow Ca2+ oscillations. Smooth muscle relaxation usually results from either decreased Ca2+ or Ca2+ desensitisation, and a major relaxant pathway is mediated via increases in cyclic adenosine monophosphatase (cAMP) usually following activation by β-adrenergic agonists. The effect of cAMP appears to reduce the influx of Ca2+ that correlates with its ability to reduce the frequency of Ca2+ oscillations [280, 281]. A similar slowing of Ca2+ oscillations is induced by nitric oxide, which acts via guanylate cyclase and cyclic guanosine monophosphate (cGMP) [268]. However, cAMP also inhibits Ca2+ release from the IP3R [282]. Consequently, stretch may act by stimulating cAMP or cGMP increases. Similarly, stretch may act by reducing the Ca2+ sensitivity of the smooth muscle cells. In this case, membrane potential or cyclic nucleotide-dependent kinases may alter the activity of Rho kinase or protein kinase C, or act directly on the regulatory proteins. In the mouse, sustained high Ca2+ also decreases Ca2+ sensitivity [283]. In addition to a direct effect on the smooth muscle cells, an additional source for these cAMP and cGMP stimuli, as mentioned below, may be the release of products by epithelial cells.
In addition to the range of processes outlined above, the tone or stiffness of the ASM cells is also a function of the antagonistic activity of agonist-dependent kinases and phosphates. Consequently, it must be considered how these enzymes are influenced by airway mechanics and inflammation, since airway hyperactivity can result from multiple defects working independently or in parallel. It is an interesting concept that smooth muscle tone could be set by the balance between the contractile response to the rhythm of intracellular Ca2+ oscillations and relaxation response mediated by the rhythm of breathing.
OTHER MODULATORS OF ASM
Within the airway wall environment, there are cells known to modulate the function of smooth muscle. The most notable one is perhaps the epithelium. The major function of the respiratory epithelium was once thought to be primarily that of a physical barrier. It constitutes the interface between the internal milieu and the external environment. It is the first point of contact for inhaled substances, respiratory viruses and airborne allergens, as well as being a primary target for inhaled respiratory drugs [284]. In this context, the mucociliary layer provides an efficient barrier. Tight junctions located between the apices of adjacent cells restrict paracellular diffusion of electrolytes and other molecules [285]. Desmosomes, integrins and intermediate- and gap-junctions also maintain the structural integrity of the epithelium.
In addition to its barrier function, it is now generally accepted that the epithelium also plays an active role in controlling many airway functions, including ASM contractility. The epithelium produces a diverse array of lipid mediators, growth factors, chemokines and cytokines, as well as both bronchoprotective and bronchoconstricting peptides [286]. However, it is beyond the scope of the present article to study comprehensively the critical role the epithelium plays in regulating airway function. The reader is referred to several articles specifically dealing with this topic [287, 288]. Little is known about the effect of oscillatory strain due to tidal breathing on epithelium and the subsequent epithelial modulation of ASM contractility. To understand the regulation of airway patency in the mechanically dynamic in vivo environment, this topic needs to be explored.
EVIDENCE NOT CONSISTENT WITH TIGHT COUPLING AMONG PARENCHYMA, AIRWAY AND ASM
As discussed at the beginning of the present study, the presumption that parenchyma, airway and ASM are tightly linked mechanically, at least in healthy lungs, comes from studies involving lung function tests and in vivo imaging in human subjects. The mechanical properties of these anatomical linkages, however, have never been directly examined in human subjects or human lung tissue. In a recent study, Noble et al. [289] examined pig airway narrowing either with or without parenchyma and concluded that under some physiological conditions, parenchymal tethering played no role in altering airway calibre. Their findings showed that while lung inflation did increase the diameter of relaxed airways and reduce the extent of narrowing produced by ASM stimulation, expected effects of parenchymal tethering from shear forces on airway diameter or airway narrowing were absent. This is somewhat surprising and contradictory to findings suggesting that parenchymal tethering is an important determinant of airway reactivity [8, 9, 134–136, 200]. Although the reason for the discrepancy is not clear, the mid-sized (10th generation) airways studied by Noble et al. [289] are more cartilaginous than those of comparable size and generation in humans. In fact, the seventh generation human bronchi were found to be cartilage-free [290], but the pig airways appear not to be stiffer than human bronchi [291, 292]. However, measures of airway wall compliance may not be a good predictor of structural or mechanical coupling between the ASM and parenchyma, because the airway wall is multilayered and anisotropic. For example, airways from pig and dog show that muscle contraction stretches connective tissue elements between the ASM and cartilage, expanding the wall area [144, 145, 293]. There is earlier, qualitative evidence that some human airways may behave similarly [294]. For this to happen in vivo implies an accompanying fluid flux within the airway wall or shortening of the airways longitudinally. The elastic properties of the different stress-bearing structures that lead to coupling of the parenchyma to the ASM are relatively unexplored, and, as mentioned elsewhere in the current study, these structures could allow uncoupling of ASM from the adventitia and account for the reported dissociation of the parenchyma and ASM described above [289]. It is also possible that differences in airway and parenchymal hysteresis could reduce the effect of parenchymal tethering on airway diameter. Under conditions following a deep inspiration, if lung hysteresis exceeds airway hysteresis, it is predicted that parenchymal tethering would have a minimal effect on airway calibre. In the study by Noble et al. [289], pressure–strain curves showed minimal hysteresis in the airways in contrast to the much greater hysteresis in the lung lobe, which is consistent with the reported lack of tethering effect reported. However, in that study, airway narrowing was recorded under pressure-controlled conditions, over a range of inspiratory lung pressures and therefore without the complicating effects of hysteresis caused by a deep inspiration.
In vitro studies on the behaviour of isolated pig airway segments also produced results not predicted by the behaviour of isolated ASM [295, 296]. Wang et al. [69] found that periodic stretching of isolated porcine ASM in the relaxed state resulted in a decrease of force production with subsequent contractions. However, pressure oscillation in isolated airway segments that periodically strained the ASM embedded within the segment led to a slight increase of the pressure generated in the subsequent isovolumic “constriction” [295, 296]. Airways were subjected to either radial [295] or axial [296] distension, with similar findings. Radial strain increased ASM length, whereas under the experimental conditions applied, axial distension reduced ASM length. These in vitro findings are, however, in agreement with those reported in vivo by Crimi et al. [70], showing that in both asthmatic and healthy humans, multiple deep inspirations taken prior to inhaling a constrictor agent led to airway narrowing rather than dilatation, thus arguing against the existence of an in vivo bronchoprotective mechanism. Disparate findings like these serve to re-emphasise the caution required in the interpretation of data from isolated ASM in terms of in vivo airway function. There are still many gaps in the present knowledge regarding how ASM exerts its influence on airway calibre and lung function. The same gaps also prevent us from gaining a firm grasp on how lung volume regulates ASM contractility.
UNANSWERED QUESTIONS
Although there is strong in vivo evidence to suggest a connection between ASM and AHR in asthma, details of the connection in terms of mechanical coupling and/or chemical pathways linking events in the lung parenchyma to the function of ASM, and how the connection is altered in asthma, are vague. The tendency for investigators to concentrate on well-defined systems, such as the whole lung, the airway segment or the ASM, and to pay less attention to the details of how these separate systems are interrelated, has contributed to the current state of knowledge: one that is full of gaps. It will not be easy to fill these gaps, because to do so, “longitudinal” experiments are needed that examine in vivo lung function, parenchymal-airway interdependence, airway segment mechafonics, couplings among components within the airway wall and ASM cell properties, all in a single species (preferably human, or a species with which human-like asthma can be induced). Obviously some of these gaps are impossible to fill in the near future; however, there are relevant and important questions that can be readily addressed now.
Parenchyma-airway interdependence
As mentioned previously, there appears to be a lack of parenchymal-airway interdependence in the pig lung, at least as far as mid-sized airways are concerned [289]. If this is also found in the human lung, it will drastically change current interpretation of many previous studies, and indeed, it will change the view of how the lung works. Unanswered questions remain from the studies of Noble et al. [289], an obvious one being “does human lung behave in the same way as pig lung?”. More exact determination of the interdependence of airway size on parenchymal mechanics also needs to be derived for the small airways at the periphery of the lung. The static conditions under which the interdependence was assessed by Noble et al. [289] do not represent the real environfment of the lung [297]. The viscoelastic lung parenchyma under dynamic conditions interacts with the airways very differently [298, 299], and this should be kept in mind if the experiments of Noble et al. [289] are revisited.
Stretch-induced release of transmitters in the parenchyma and airways
The influence of mechanical stretch on ASM contractility has so far been interpreted as strictly a “physical” phenomenon: 1) perturbation of actomyosin cross-bridge interaction [58]; 2) disruption of myosin filament assembly [168]; or 3) changes in cytoskeletal organisation [55]. It is entirely possible that a mechanical signal can be transduced into other signals via some kind of stretch receptor coupled to chemical signalling pathways [231, 300]. It has been speculated [68] that stretch-induced bronchodilation acts through inhibition of cholinergic tone or activation of the nonadrenergic noncholinergic system and results in the release of nitric oxide [140]. Stretch is also known to increase prostaglandin production, and certain ion channels may be directly influenced. Surfactant release after a deep inspiration could also affect airway impedance, especially that of the small airways [301]. It is crucial to establish the relative importance of the various contributing factors involved in stretch-induced smooth muscle response and their relevance to asthma.
Discrepancies in findings from airway segments and ASM
So far, there is no clear explanation as to why periodic stretches applied, by pressure oscillation, to ASM embedded in intact airways increase muscle contractility, and why stretches applied to isolated ASM bundles have an opposite effect [295, 296]. This question needs to be answered before any interpretation of stretch-induced alteration in ASM properties in terms of lung function can be taken seriously. Perhaps more studies need to be performed using airway segments from different animal species and from different generations of the airway tree, so that the generality of this observation can be confirmed. It will be informative to perform similar experiments using the whole lung, which is one-step closer to the in vivo lung function test, compared with the isolated airway experiment. In such experiments, Raw could be measured in a lung lobe before and after a period of volume oscillation. Airway diameter should be monitored throughout the experiment, during volume oscillation and during broncho-challenge under static conditions. Experimentally, there are at least two ways by which to oscillate lung volume. One is through changes in airway pressure (i.e. in a system where a closed airway tree in the lung is connected to a pressure source); the other is by changing the volume of pleural cavity or an experimental device serving the same function. The latter is more physiologically relevant. Different modes of volume oscillation may produce different experimental outcomes. If this is found to be the case, then the reason for the disparity may provide insights into the mechanism responsible for the altered ASM properties.
Differences between normal and asthmatic ASM
Excessive shortening of ASM leading to excessive narrowing of the airways could occur under at least four scenarios. Each scenario is associated with a different mechanism. One scenario is that the local airway environment allows the muscle to adapt at abnormally short lengths and, when the muscle is subsequently activated, it shortens further than it would do normally. Although it is not clear whether it happens in vivo, this behaviour has been reproduced in vitro in healthy ASM [34, 195]. Hence, excessive shortening of airways does not have to be associated with altered intrinsic muscle properties [33]. Therefore, any experiment designed to illustrate the relationship between airway narrowing and ASM contractility needs to establish what the normal in situ length is for the muscle cells. Without that reference, comparison of the amounts of shortening in two muscle preparations would be meaningless, because the amount of shortening is partially determined by the initial length of the muscle [195]. For the same reason, comparison of shortening velocities needs to be carried out at appropriate lengths of reference.
The second scenario involves changes in the mechanical properties or the amount of the ECM surrounding ASM. Bramley et al. [302] have shown that treatment of human bronchial smooth muscle with collagenase augments the muscle’s ability to shorten. Assuming that the collagenase has no direct effect on muscle contractility, the results indicate that collagen fibres (and probably other components of the ECM) constrain muscle shortening, perhaps through restricting lateral expansion of the muscle cells, as elegantly demonstrated by Meiss [303] in an in vitro model. Changes in the ECM composition could also affect ASM phenotype by increasing its proliferative state and altering its secretory properties [234].
The third scenario is that the smooth muscle mass within the airway wall has increased, due to hypertrophy and/or hyperplasia, without a change in the intrinsic contractile properties of individual muscle cells. This could lead to excessive airway narrowing simply because of the increased constricting force associated with the greater muscle mass, if other factors influencing airway narrowing remain the same. This emphasises the importance of proper normalisation of muscle properties by appropriate parameters, such as muscle cross-section and length, and local airway geometry and mechanical properties. More quantitative information is needed about smooth muscle cell number and size, and the amount and type of ECM between the muscle cells in cases of asthma of varying duration and severity.
The fourth scenario is that the intrinsic contractile properties of asthmatic ASM are indeed different from normal. A change in intrinsic properties does not automatically mean that there will be a change in the degree of airway narrowing. Other changes in the airway property may negate or enhance the influence of smooth muscle. If an increase in contractility of ASM is a contributing factor in the excessive airway narrowing in asthma, then further questions should be asked. Can over-expression of MLCK [37, 304] be responsible for the exaggerated airway narrowing [305]? Can a change in myosin or actin filament length and integrity alter the response of ASM to length fluctuation associated with lung volume change [164]? Can a change in the composition of myosin heavy chain isoforms in asthmatics [306] lead to excessive airway narrowing and through what mechanism? Studies on a rodent model of innate AHR suggest that the abnormalities in the pathways of biochemical signalling and the isoform mix of myosin molecules may underlie the hyperresponsiveness. Tracheal smooth muscle from the hyperresponsive Fisher rat generates more force [307, 308] and shortens faster [309] compared with the Lewis rat (control). Experiments on airway explants from Fisher rats also demonstrate a more rapid and excessive airway narrowing in response to agonist stimulation, compared with explants from the Lewis rats [310, 311]. In cultured ASM cells from Fisher rats, the observed excessive calcium response can be attributed to higher levels of IP3, which in turn could be caused by a slower rate of IP3 degradation in the cells from the Fisher rats [312]. Increased activation of the contractile pathway is also shown by the increased phosphorylation of the regulatory myosin light chain [308]. The Fisher ASM cells are also characterised by a greater complement of the fast myosin isoform (SM-B) [308]. Preliminary results from bronchial biopsies of asthmatic subjects show that the fast myosin isoform is also expressed to a greater extent than that found in normal subjects [306]. Examination of the differences in biochemical pathways and myosin isoforms in airway tissues from animal models of hyperresponsiveness may point to mechanisms pertinent to human asthma.
Effects of steroids on smooth muscle length adaptation
Inhaled steroids are the most effective drugs in reducing AHR in asthma [313]. The working hypothesis is that this is due to a reduction in airway inflammation [314], which indirectly changes the stimulation and the micro-environment of ASM, leading to reduced airway narrowing at a given dose of inhaled agonist. However, smooth muscle itself responds to steroids by changing its expression of a host of genes that are involved in cell adhesion, proliferation, ECM formation, cell signalling, cytokine production and signal-transduction molecules regulating smooth muscle responsiveness [315–318]. It can be postulated that this also influences the structure of the cytoskeleton and the arrangement of contractile proteins, and thus affects the muscle’s ability to adapt to length or force perturbation [319]. Hence, it needs to be investigated whether the benefits of steroids in asthma are based on changing the dynamics in structure and function of ASM rather than suppressing the surrounding airway inflammation alone. As suggested by the recent reports regarding the lack of antiproliferative activity of steroids on smooth muscle cells from asthmatics [320] and the impairment of glucocorticoid action by type I monomeric collagen (presumably present in the micro-environment where ASM cells reside in asthmatic airways) [251, 321], the efficacy of steroids in modulating smooth muscle gene transcription may be diminished in asthmatics. This indicates a need to look for alternative mechanisms for steroid action that are beneficial to (at least some) asthmatics. Given the interaction of β-agonists with steroids [109], as well as the relaxing effect of mechanical stretch in smooth muscle [163], examination is required as to whether there is a three-party interaction functioning in normal ASM and whether this interaction is impaired in ASM obtained from patients with asthma.
Possible abnormal relaxation of ASM in asthma
The role of impaired relaxation needs to be dealt with in consideration of airway hyperreactivity, since it could contribute to bronchospasm. Several investigators have reported alterations in relaxation in ASM from animal models of asthma [322–325]. Unfortunately, most of them dealt with isometric relaxation (relaxation of tension in muscle maintained at a constant length after cessation of stimulation), which provided information relating to stiffening of the airway wall and not to the relevant parameter of airway lumen narrowing [322–324]. In vivo measurements of FEV1 and specific Gaw are better in this regard, but suffer from the drawback that multiple confounding variables are operating simultaneously. Jiang and Stephens [325] have studied isotonic relaxation (lengthening of muscle under a constant load after active shortening) and have shown that its half-relaxation time is prolonged in sensitised ASM. The magnitude of the residual failure to relax could account for the magnitude of bronchospasm seen in the sensitised ASM. No such experiment has been carried out using human ASM. The bottom line, therefore, is that in studies of ASM dynamics in asthma, relaxation must be included and that for it to be relevant the studies should be in isotonic mode.
FUTURE DIRECTIONS
The recent burst of knowledge of the dynamic behaviour of airway smooth muscle has provided new and constructive concepts for understanding the mechanisms of episodic excessive airway narrowing in asthma. To move the field forward, the unresolved discrepancies discussed previously must be addressed. The weakest link in the ability to acquire information regarding the contribution of the various lung components to total lung function appears to lie at the boundaries separating the components: between airways smooth muscle and the rest of the airway wall components, and between the airway wall and lung parenchyma. Interpretation of results from studies of isolated or cultured airway smooth muscle in terms of lung function will only be meaningful when these “boundary” problems are solved. Creative solutions are needed to bridge this impasse. One interesting example is the use of videomicrometric recordings of airway narrowing in thin lung slices (~100–200 μm thick), which leaves parenchymal airway connections intact and yet retains many of the advantages of in vitro experimentation. Meanwhile, the opportunity should be taken to translate the newly gained knowledge and novel concepts into possible interventions aimed at preventing airway impairment in asthma and/or returning airway function towards normal.
Acknowledgments
The present study originated from a workshop supported by the American Thoracic Society.
Footnotes
STATEMENT OF INTEREST
Statements of interest for M.J. Sanderson, A.G. Stewart, J. Solway, N.L. Stephens, W. Mitzner, T.M. Murphy, P.D. Paré, G.G. King, V. Brusasco and T.R. Bai can be found at www.erj.ersjournals.com/misc/statements.shtml
References
- 1.Brusasco V, Crimi E, Pellegrino R. Airway hyperresponsiveness in asthma: not just a matter of airway inflammation. Thorax. 1998;53:992–998. doi: 10.1136/thx.53.11.992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crimi E, Spanevello A, Neri M, Ind PW, Rossi GA, Brusasco V. Dissociation between airway inflammation and airway hyperresponsiveness in allergic asthma. Am J Respir Crit Care Med. 1998;157:4–9. doi: 10.1164/ajrccm.157.1.9703002. [DOI] [PubMed] [Google Scholar]
- 3.Howarth PH, Knox AJ, Amrani Y, Tliba O, Panettieri RA, Johnson M. Synthetic responses in airway smooth muscle. J Allergy Clin Immunol. 2004;114(Suppl 2):S32–50. doi: 10.1016/j.jaci.2004.04.041. [DOI] [PubMed] [Google Scholar]
- 4.Djukanovic R, Wilson SJ, Kraft M, et al. Effect of treatment with anti-immunoglobulin E antibody omalizumab on airway inflammation in allergic asthma. Am J Respir Crit Care Med. 2004;170:583–593. doi: 10.1164/rccm.200312-1651OC. [DOI] [PubMed] [Google Scholar]
- 5.Leckie MJ, ten Brinke A, Khan J, et al. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyperresponsiveness, and the late asthmatic response. Lancet. 2000;356:2144–2148. doi: 10.1016/s0140-6736(00)03496-6. [DOI] [PubMed] [Google Scholar]
- 6.Berry MA, Hargadon B, Shelley M, et al. Evidence of a role of tumor necrosis factor alpha in refractory asthma. N Engl J Med. 2006;354:697–708. doi: 10.1056/NEJMoa050580. [DOI] [PubMed] [Google Scholar]
- 7.Macklem PT, Mead J. Resistance of central and peripheral airways measured by a retrograde catheter. J Appl Physiol. 1967;22:395–401. doi: 10.1152/jappl.1967.22.3.395. [DOI] [PubMed] [Google Scholar]
- 8.Ding DJ, Martin JG, Macklem PT. Effects of lung volume on maximal methacholine-induced bronchoconstriction in normal humans. J Appl Physiol. 1987;62:1324–1330. doi: 10.1152/jappl.1987.62.3.1324. [DOI] [PubMed] [Google Scholar]
- 9.Gunst SJ, Warner DO, Wilson TA, Hyatt RE. Parenchymal interdependence and airway response to methacholine in excised dog lobes. J Appl Physiol. 1988;65:2490–2497. doi: 10.1152/jappl.1988.65.6.2490. [DOI] [PubMed] [Google Scholar]
- 10.Nagase T, Martin JG, Ludwig MS. Comparative study of mechanical interdependence: effect of lung volume on Raw during induced constriction. J Appl Physiol. 1993;75:2500–2505. doi: 10.1152/jappl.1993.75.6.2500. [DOI] [PubMed] [Google Scholar]
- 11.Bates JH, Schuessler TF, Dolman C, Eidelman DH. Temporal dynamics of acute isovolume bronchoconstriction in the rat. J Appl Physiol. 1997;82:55–62. doi: 10.1152/jappl.1997.82.1.55. [DOI] [PubMed] [Google Scholar]
- 12.Irvin CG, Pak J, Martin RJ. Airway-parenchyma uncoupling in nocturnal asthma. Am J Respir Crit Care Med. 2000;161:50–56. doi: 10.1164/ajrccm.161.1.9804053. [DOI] [PubMed] [Google Scholar]
- 13.McClean MA, Matheson MJ, McKay K, et al. Low lung volume alters contractile properties of airway smooth muscle in sheep. Eur Respir J. 2003;22:50–56. doi: 10.1183/09031936.03.00099802. [DOI] [PubMed] [Google Scholar]
- 14.Torchio R, Gulotta C, Ciacco C, et al. Effects of chest wall strapping on mechanical response to methacholine in humans. J Appl Physiol. 2006;101:430–438. doi: 10.1152/japplphysiol.00379.2005. [DOI] [PubMed] [Google Scholar]
- 15.Nadel JA, Tierney DF. Effect of a previous deep inspiration on airway resistance in man. J Appl Physiol. 1961;16:717–719. doi: 10.1152/jappl.1961.16.4.717. [DOI] [PubMed] [Google Scholar]
- 16.Fish JE, Peterman VI, Cugell DW. Effect of deep inspiration on airway conductance in subjects with allergic rhinitis and allergic asthma. J Allergy Clin Immunol. 1977;60:41–46. doi: 10.1016/0091-6749(77)90081-1. [DOI] [PubMed] [Google Scholar]
- 17.Burns CB, Taylor WR, Ingram RH., Jr Effects of deep inhalation in asthma: relative airway and parenchymal hysteresis. J Appl Physiol. 1985;59:1590–1596. doi: 10.1152/jappl.1985.59.5.1590. [DOI] [PubMed] [Google Scholar]
- 18.Skloot G, Permutt S, Togias A. Airway hyperresponsiveness in asthma: a problem of limited smooth muscle relaxation and inspiration. J Clin Invest. 1995;96:2393–2403. doi: 10.1172/JCI118296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pellegrino R, Wilson O, Jenouri G, Rodarte JR. Lung mechanics during induced bronchoconstriction. J Appl Physiol. 1996;81:964–975. doi: 10.1152/jappl.1996.81.2.964. [DOI] [PubMed] [Google Scholar]
- 20.Pellegrino R, Violante B, Brusasco V. Bronchoconstriction in humans. Relationship with the effects of deep inhalation and airway sensitivity. Am J Respir Crit Care Med. 1996;153:115–121. doi: 10.1164/ajrccm.153.1.8542103. [DOI] [PubMed] [Google Scholar]
- 21.Jensen A, Atilah H, Suki B, Ingenito E, Lutchen KR. Airway caliber in healthy subjects and asthmatics: effects of deep inspirations and bronchial challenge. Highlighted topics: signal transduction in smooth muscle. J Appl Physiol. 2001;91:506–515. doi: 10.1152/jappl.2001.91.1.506. [DOI] [PubMed] [Google Scholar]
- 22.Gunst SJ. Contractile force of canine airway smooth muscle during cyclical length changes. J Appl Physiol. 1983;55:759–769. doi: 10.1152/jappl.1983.55.3.759. [DOI] [PubMed] [Google Scholar]
- 23.Gunst SJ, Stropp JQ, Service J. Mechanical modulation of pressure-volume characteristics of contracted canine airways in vitro. J Appl Physiol. 1990;68:2223–2229. doi: 10.1152/jappl.1990.68.5.2223. [DOI] [PubMed] [Google Scholar]
- 24.Warner DO, Gunst SJ. Limitation of maximal bronchoconstriction in living dogs. Am Rev Respir Dis. 1992;145:553–560. doi: 10.1164/ajrccm/145.3.553. [DOI] [PubMed] [Google Scholar]
- 25.Tepper RS, Shen X, Bakan E, Gunst SJ. Maximal airway response in mature and immature rabbits during tidal ventilation. J Appl Physiol. 1995;79:1190–1198. doi: 10.1152/jappl.1995.79.4.1190. [DOI] [PubMed] [Google Scholar]
- 26.King GG, Pare PD, Seow CY. The mechanics of exaggerated airway narrowing in asthma: the role of smooth muscle. Respir Physiol. 1999;118:1–13. doi: 10.1016/s0034-5687(99)00076-6. [DOI] [PubMed] [Google Scholar]
- 27.King GG, Moore BJ, Seow CY, Pare PD. Time course of increased airway narrowing caused by inhibition of deep inspiration during methacholine challenge. Am J Respir Crit Care Med. 1999;160:454–457. doi: 10.1164/ajrccm.160.2.9804012. [DOI] [PubMed] [Google Scholar]
- 28.Seow CY, Fredberg JJ. Historical perspective on airway smooth muscle: the saga of a frustrated cell. J Appl Physiol. 2001;91:938–952. doi: 10.1152/jappl.2001.91.2.938. [DOI] [PubMed] [Google Scholar]
- 29.Gunst SJ, Fredberg JJ. The first three minutes: smooth muscle contraction, cytoskeletal events, and soft glasses. J Appl Physiol. 2003;95:413–425. doi: 10.1152/japplphysiol.00277.2003. [DOI] [PubMed] [Google Scholar]
- 30.Irvin CG. Lung volume: a principle determinant of airway smooth muscle function. Eur Respir J. 2003;22:3–5. doi: 10.1183/09031936.03.00035703. [DOI] [PubMed] [Google Scholar]
- 31.Ingram RH., Jr Relationships among airway-parenchymal interactions, lung responsiveness, and inflammation in asthma. Giles F. Filley Lecture. Chest. 1995;107(Suppl 3):148S–152S. doi: 10.1378/chest.107.3_supplement.148s. [DOI] [PubMed] [Google Scholar]
- 32.Pellegrino R, Sterk PJ, Sont JK, Brusasco V. Assessing the effect of deep inhalation on airway calibre: a novel approach to lung function in bronchial asthma and COPD. Eur Respir J. 1998;12:1219–1227. doi: 10.1183/0903.1936.98.12051219. [DOI] [PubMed] [Google Scholar]
- 33.Wagers S, Lundblad LK, Ekman M, Irvin CG, Bates JH. The allergic mouse model of asthma: normal smooth muscle in an abnormal lung. J Appl Physiol. 2004;96:2019–2027. doi: 10.1152/japplphysiol.00924.2003. [DOI] [PubMed] [Google Scholar]
- 34.McParland BE, Tait RR, Pare PD, Seow CY. The role of airway smooth muscle during an attack of asthma simulated in vitro. Am J Respir Cell Mol Biol. 2005;33:500–504. doi: 10.1165/rcmb.2005-0183OC. [DOI] [PubMed] [Google Scholar]
- 35.Jackson AC, Murphy MM, Rassulo J, Celli BR, Ingram RH., Jr Deep breath reversal and exponential return of methacholine-induced obstruction in asthmatic and non-asthmatic subjects. J Appl Physiol. 2004;96:137–142. doi: 10.1152/japplphysiol.00504.2003. [DOI] [PubMed] [Google Scholar]
- 36.Thorpe CW, Salome CM, Berend N, King GG. Modeling airway resistance dynamics after tidal and deep inspirations. J Appl Physiol. 2004;97:1643–1653. doi: 10.1152/japplphysiol.01300.2003. [DOI] [PubMed] [Google Scholar]
- 37.Ma X, Cheng Z, Kong H, et al. Changes in biophysical and biochemical properties of single bronchial smooth muscle cells from asthmatic subjects. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1181–1189. doi: 10.1152/ajplung.00389.2001. [DOI] [PubMed] [Google Scholar]
- 38.Mitchell RW, Rühlmann E, Magnussen H, Leff AR, Rabe KF. Passive sensitization of human bronchi augments smooth muscle shortening velocity and capacity. Am J Physiol Lung Cell Mol Physiol. 1994;11:L218–222. doi: 10.1152/ajplung.1994.267.2.L218. [DOI] [PubMed] [Google Scholar]
- 39.Pichurko BM, Ingram RH., Jr Effects of airway tone and volume history on maximal expiratory flow in asthma. J Appl Physiol. 1987;62:1133–1140. doi: 10.1152/jappl.1987.62.3.1133. [DOI] [PubMed] [Google Scholar]
- 40.Lim TK, Pride NB, Ingram RH., Jr Effects of volume history during spontaneous and acutely induced air-flow obstruction in asthma. Am Rev Respir Dis. 1987;135:591–596. doi: 10.1164/arrd.1987.135.3.591. [DOI] [PubMed] [Google Scholar]
- 41.Lim TK, Ang SM, Rossing TH, Ingenito EP, Ingram RH., Jr The effects of deep inhalation on maximal expiratory flow during intensive treatment of spontaneous asthmatic episodes. Am Rev Respir Dis. 1989;140:340–343. doi: 10.1164/ajrccm/140.2.340. [DOI] [PubMed] [Google Scholar]
- 42.Pliss LB, Ingenito BP, Ingram RH., Jr Responsiveness, inflammation, and effects of deep breaths on obstruction in mild asthma. J Appl Physiol. 1989;66:2298–2304. doi: 10.1152/jappl.1989.66.5.2298. [DOI] [PubMed] [Google Scholar]
- 43.Froeb HF, Mead J. Relative hysteresis of the dead space and lung in vivo. J Appl Physiol. 1968;25:244–248. doi: 10.1152/jappl.1968.25.3.244. [DOI] [PubMed] [Google Scholar]
- 44.Lutchen KR, Hantos Z, Petak F, Adamicza A, Suki B. Airway inhomogeneities contribute to apparent lung tissue mechanics during constriction. J Appl Physiol. 1996;80:1841–1849. doi: 10.1152/jappl.1996.80.5.1841. [DOI] [PubMed] [Google Scholar]
- 45.Gunst SJ, Mitzner W. Mechanical properties of contracted canine bronchial segments in vitro. J Appl Physiol. 1981;50:1236–1247. doi: 10.1152/jappl.1981.50.6.1236. [DOI] [PubMed] [Google Scholar]
- 46.Gunst SJ, Russell JA. Contractile force of canine tracheal smooth muscle during continuous stretch. J Appl Physiol. 1982;52:655–663. doi: 10.1152/jappl.1982.52.3.655. [DOI] [PubMed] [Google Scholar]
- 47.Gunst SJ. Effect of length history on contractile behavior of canine tracheal smooth muscle. Am J Physiol. 1986;250:C146–154. doi: 10.1152/ajpcell.1986.250.1.C146. [DOI] [PubMed] [Google Scholar]
- 48.Fredberg JJ. Airway smooth muscle in asthma: flirting with disaster. Eur Respir J. 1998;12:1252–1256. doi: 10.1183/09031936.98.12061252. [DOI] [PubMed] [Google Scholar]
- 49.Fredberg JJ. Frozen objects: small airways, big breaths, and asthma. J Allergy Clin Immunol. 2000;106:615–624. doi: 10.1067/mai.2000.109429. [DOI] [PubMed] [Google Scholar]
- 50.Fredberg JJ. Airway narrowing in asthma: does speed kill. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1179–1180. doi: 10.1152/ajplung.00190.2002. [DOI] [PubMed] [Google Scholar]
- 51.Fredberg JJ. Bronchospasm and its biophysical basis in airway smooth muscle. Respir Res. 2004;5:2. doi: 10.1186/1465-9921-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fredberg JJ, Inouye D, Miller B, et al. Airway smooth muscle, tidal stretch, and dynamically determined contractile state. Am J Respir Critical Care Med. 1997;156:1752–1759. doi: 10.1164/ajrccm.156.6.9611016. [DOI] [PubMed] [Google Scholar]
- 53.Gunst SJ, Wu MF, Smith DD. Contraction history modulates isotonic shortening velocity in smooth muscle. Am J Physiol. 1993;265:C467–476. doi: 10.1152/ajpcell.1993.265.2.C467. [DOI] [PubMed] [Google Scholar]
- 54.Gunst SJ, Wu MF. Plasticity of airway smooth muscle stiffness and extensibility: role of length-adaptive mechanisms. J Appl Physiol. 2001;90:741–749. doi: 10.1152/jappl.2001.90.2.741. [DOI] [PubMed] [Google Scholar]
- 55.Gunst SJ, Tang DD, Opazo Saez A. Cytoskeletal remodeling of the airway smooth muscle cell: a mechanism for adaptation to mechanical forces in the lung. Respir Physiol Neurobiol. 2003;137:151–168. doi: 10.1016/s1569-9048(03)00144-7. [DOI] [PubMed] [Google Scholar]
- 56.Sasaki H, Hoppin FG., Jr Hysteresis of contracted airway smooth muscle. J Appl Physiol. 1979;47:1251–1262. doi: 10.1152/jappl.1979.47.6.1251. [DOI] [PubMed] [Google Scholar]
- 57.Fredberg JJ, Jones KA, Nathan M, et al. Friction in airway smooth muscle: mechanism, latch, and implications in asthma. J Appl Physiol. 1996;81:2703–2712. doi: 10.1152/jappl.1996.81.6.2703. [DOI] [PubMed] [Google Scholar]
- 58.Fredberg JJ, Inouye DS, Mijailovich SM, Butler JP. Perturbed equilibrium of myosin binding in airway smooth muscle and its implications in bronchospasm. Am J Respir Crit Care Med. 1999;159:959–967. doi: 10.1164/ajrccm.159.3.9804060. [DOI] [PubMed] [Google Scholar]
- 59.Mijailovich SM, Butler JP, Fredberg JJ. Perturbed equilibria of myosin binding airway smooth muscle: bond-length distributions, mechanics and ATP metabolism. Biophys J. 2000;79:2667–2681. doi: 10.1016/S0006-3495(00)76505-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.King GG, Moore BJ, Seow CY, Pare PD. Airway narrowing associated with inhibition of deep inspiration during methacholine inhalation in asthmatics. Am J Respir Crit Care Med. 2001;164:216–218. doi: 10.1164/ajrccm.164.2.2006101. [DOI] [PubMed] [Google Scholar]
- 61.Latourelle J, Fabry B, Fredberg JJ. Dynamic equilibration of airway smooth muscle contraction during physiological loading. J Appl Physiol. 2002;92:771–779. doi: 10.1152/japplphysiol.01090.2000. [DOI] [PubMed] [Google Scholar]
- 62.Brusasco V, Crimi E, Barisione G, Spanevello A, Rodarte JR, Pellegrino R. Airway responsiveness to methacholine: effects of deep inhalations and airway inflammation. J Appl Physiol. 1999;87:567–573. doi: 10.1152/jappl.1999.87.2.567. [DOI] [PubMed] [Google Scholar]
- 63.Black L, Henderson AC, Atileh H, Israel E, Ingenito EP, Lutchen KR. Relating maximum airway dilation and subsequent reconstruction to reactivity in human lungs. J Appl Physiol. 2004;96:1808–1814. doi: 10.1152/japplphysiol.01170.2003. [DOI] [PubMed] [Google Scholar]
- 64.Shore SA, Fredberg JJ. Obesity, smooth muscle, and airway hyperresponsiveness. J Allergy Clin Immunol. 2005;115:925–927. doi: 10.1016/j.jaci.2005.01.064. [DOI] [PubMed] [Google Scholar]
- 65.Malmberg P, Larsson K, Sundblad BM, Zhiping W. Importance of the time interval between FEV1 measurements in a methacholine provocation test. Eur Respir J. 1993;6:680–686. doi: 10.1016/s0012-3692(16)47463-5. [DOI] [PubMed] [Google Scholar]
- 66.Scichilone N, Kapsali T, Permutt S, Togias A. Deep inspiration-induced bronchoprotection is stronger than bronchodilation. Am J Respir Crit Care Med. 2000;162:910–916. doi: 10.1164/ajrccm.162.3.9907048. [DOI] [PubMed] [Google Scholar]
- 67.Kapsali T, Permutt S, Laube B, Scichilone N, Togias A. Potent bronchoprotective effect of deep inspiration and its absence in asthma. J Appl Physiol. 2000;89:711–720. doi: 10.1152/jappl.2000.89.2.711. [DOI] [PubMed] [Google Scholar]
- 68.Scichilone N, Permutt S, Togias A. The lack of the bronchoprotective and not the bronchodilatory ability of deep inspiration is associated with airway hyperrespon-siveness. Am J Respir Crit Care Med. 2001;163:413–419. doi: 10.1164/ajrccm.163.2.2003119. [DOI] [PubMed] [Google Scholar]
- 69.Wang L, Pare PD, Seow CY. Effects of length oscillation on the subsequent force development in swine tracheal smooth muscle. J Appl Physiol. 2000;88:2246–2250. doi: 10.1152/jappl.2000.88.6.2246. [DOI] [PubMed] [Google Scholar]
- 70.Crimi E, Pellegrino R, Milanese M, Brusasco V. Deep breaths, methacholine, and airway narrowing in healthy and asthmatic subjects. J Appl Physiol. 2002;93:1384–1390. doi: 10.1152/japplphysiol.00209.2002. [DOI] [PubMed] [Google Scholar]
- 71.Carroll N, Elliot J, Morton A, James A. The structure of large and small airways in nonfatal and fatal asthma. Am Rev Respir Dis. 1993;147:405–410. doi: 10.1164/ajrccm/147.2.405. [DOI] [PubMed] [Google Scholar]
- 72.Ebina M, Takahashi T, Chiba T, Motomiya M. Cellular hypertrophy and hyperplasia of airway smooth muscles underlying bronchial asthma. A 3-D morphometric study. Am Rev Respir Dis. 1993;148:720–726. doi: 10.1164/ajrccm/148.3.720. [DOI] [PubMed] [Google Scholar]
- 73.Kuwano K, Bosken CH, Pare PD, Bai TR, Wiggs BR, Hogg JC. Small airways dimensions in asthma and in chronic obstructive pulmonary disease. Am Rev Respir Dis. 1993;148:1220–1225. doi: 10.1164/ajrccm/148.5.1220. [DOI] [PubMed] [Google Scholar]
- 74.Lambert RK, Wiggs BR, Kuwano K, Hogg JC, Pare PD. Functional significance of increased airway smooth muscle in asthma and COPD. J Appl Physiol. 1993;74:2771–2781. doi: 10.1152/jappl.1993.74.6.2771. [DOI] [PubMed] [Google Scholar]
- 75.James AL. Relationship between airway wall thickness and airway hyperresponsiveness. In: Stewart AG, editor. Airway Wall Remodeling in Asthma, Pharmacology and Toxicology: Basic and Clinical Aspects. New York: CRC Press Inc; 1997. pp. 1–28. [Google Scholar]
- 76.Bai TR, Cooper J, Koelmeyer T, Pare PD, Weir TD. The effect of age and duration of disease on airway structure in fatal asthma. Am J Respir Crit Care Med. 2000;162:663–669. doi: 10.1164/ajrccm.162.2.9907151. [DOI] [PubMed] [Google Scholar]
- 77.Affonce DA, Lutchen KR. New perspectives on the mechanical basis for airway hyperreactivity and hypersensitivity in asthma. J Appl Physiol. 2006;101:1710–1719. doi: 10.1152/japplphysiol.00344.2006. [DOI] [PubMed] [Google Scholar]
- 78.Thomson RJ, Bramley AM, Schellenberg RR. Airway muscle stereology: implications for increased shortening in asthma. Am J Respir Crit Care Med. 1996;154:749–757. doi: 10.1164/ajrccm.154.3.8810615. [DOI] [PubMed] [Google Scholar]
- 79.Heard BE, Hossain S. Hyperplasia of bronchial muscle in asthma. J Pathol. 1973;110:319–331. doi: 10.1002/path.1711010212. [DOI] [PubMed] [Google Scholar]
- 80.Woodruff PG, Dolganov GM, Ferrando RE, et al. Hyperplasia of smooth muscle in mild to moderate asthma without changes in cell size or gene expression. Am J Respir Crit Care Med. 2004;169:1001–1006. doi: 10.1164/rccm.200311-1529OC. [DOI] [PubMed] [Google Scholar]
- 81.Benayoun L, Druilhe A, Dombret MC, Aubier M, Pretolani M. Airway structural alterations selectively associated with severe asthma. Am J Respir Crit Care Med. 2003;167:1360–1368. doi: 10.1164/rccm.200209-1030OC. [DOI] [PubMed] [Google Scholar]
- 82.Pini L, Hamid Q, Shannon J, et al. Differences in proteoglycan deposition in the airway of moderate and severe asthmatics. Eur Respir J. 2007;29:71–77. doi: 10.1183/09031936.00047905. [DOI] [PubMed] [Google Scholar]
- 83.Bamford TL, Mast AE, Wilson JW, et al. Relationship between asthma severity, airway smooth muscle and PCNA expression in bronchial biopsies. Respirology. 2003;8:A4. [Google Scholar]
- 84.James AL, Carroll M, Dromey J, et al. In-situ proliferation of inflammatory cells and smooth muscle cells in patients with and without asthma. Respirology. 2002;7:A11. [Google Scholar]
- 85.van Kan CI, Wu J, Willems LNA, et al. Airway smooth muscle proliferation in bronchial biopsy specimens of patients with asthma. Am J Respir Crit Care Med. 1996;153:A743. [Google Scholar]
- 86.Druilhe A, Wallaert B, Tsicopoulos A, et al. Apoptosis, proliferation, and expression of Bcl-2, Fas, and Fas ligand in bronchial biopsies from asthmatics. Am J Respir Cell Mol Biol. 1998;19:747–757. doi: 10.1165/ajrcmb.19.5.3166. [DOI] [PubMed] [Google Scholar]
- 87.Bamford TL, Mast AE, Wilson JW, et al. Relationship between asthma severity, airway smooth muscle and PCNA expression in bronchial biopsies. Respirology. 2003;8:A4. [Google Scholar]
- 88.Herszberg B, Ramos-Barbon D, Tamaoka M, Martin JG, Lavoie JP. Heaves, an asthma-like equine disease, involves airway smooth muscle remodeling. J Allergy Clin Immunol. 2006;118:382–388. doi: 10.1016/j.jaci.2006.03.044. [DOI] [PubMed] [Google Scholar]
- 89.Stewart AG. Emigration and immigration of mesenchymal cells: a multicultural airway wall. Eur Respir J. 2004;24:515–517. doi: 10.1183/09031936.04.00067404. [DOI] [PubMed] [Google Scholar]
- 90.Vantler M, Huntgeburth M, Caglayan E, Ten Freyhaus H, Schnabel P, Rosenkranz S. PI3-kinase/Akt-dependent antiapoptotic signaling by the PDGF alpha receptor is negatively regulated by Src family kinases. FEBS Lett. 2006;580:6769–6776. doi: 10.1016/j.febslet.2006.11.034. [DOI] [PubMed] [Google Scholar]
- 91.Jankov RP, Luo X, Campbell A, et al. Fibroblast growth factor receptor-1 and neonatal compensatory lung growth after exposure to 95% oxygen. Am J Respir Crit Care Med. 2003;167:1554–1561. doi: 10.1164/rccm.200207-662OC. [DOI] [PubMed] [Google Scholar]
- 92.Yi M, Belcastro R, Shek S, Luo D, Post M, Tanswell AK. Fibroblast growth factor-2 and receptor-1alpha(IIIc) regulate postnatal rat lung cell apoptosis. Am J Respir Crit Care Med. 2006;174:581–589. doi: 10.1164/rccm.200511-1718OC. [DOI] [PubMed] [Google Scholar]
- 93.Hamann KJ, Vieira JE, Halayko AJ, et al. Fas cross-linking induces apoptosis in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2000;278:L618–624. doi: 10.1152/ajplung.2000.278.3.L618. [DOI] [PubMed] [Google Scholar]
- 94.Stewart AG, Tomlinson PR, Fernandes DJ, Wilson JW, Harris T. Tumor necrosis factor alpha modulates mitogenic responses of human cultured airway smooth muscle. Am J Respir Cell Mol Biol. 1995;12:110–119. doi: 10.1165/ajrcmb.12.1.7529028. [DOI] [PubMed] [Google Scholar]
- 95.Amrani Y, Panettieri RA, Jr, Frossard N, Bronner C. Activation of the TNF alpha-p55 receptor induces myocyte proliferation and modulates agonist-evoked calcium transients in cultured human tracheal smooth muscle cells. Am J Respir Cell Mol Biol. 1996;15:55–63. doi: 10.1165/ajrcmb.15.1.8679222. [DOI] [PubMed] [Google Scholar]
- 96.Zhou D, Zheng X, Wang L, et al. Expression and effects of cardiotrophin-1 (CT-1) in human airway smooth muscle cells. Br J Pharmacol. 2003;140:1237–1244. doi: 10.1038/sj.bjp.0705562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Redington AE, Springall DR, Ghatei MA, et al. Airway endothelin levels in asthma: influence of endobronchial allergen challenge and maintenance corticosteroid therapy. Eur Respir J. 1997;10:1026–1032. doi: 10.1183/09031936.97.10051026. [DOI] [PubMed] [Google Scholar]
- 98.McWhinnie R, Pechkovsky DV, Zhou D, et al. Endothelin-1 induces hypertrophy and inhibits apoptosis in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2007;292:L278–286. doi: 10.1152/ajplung.00111.2006. [DOI] [PubMed] [Google Scholar]
- 99.Freyer AM, Johnson SR, Hall IP. Effects of growth factors and extracellular matrix on survival of human airway smooth muscle cells. Am J Respir Cell Mol Biol. 2001;25:569–576. doi: 10.1165/ajrcmb.25.5.4605. [DOI] [PubMed] [Google Scholar]
- 100.Oltmanns U, Sukkar MB, Xie S, John M, Chung KF. Induction of human airway smooth muscle apoptosis by neutrophils and neutrophil elastase. Am J Respir Cell Mol Biol. 2005;32:334–341. doi: 10.1165/rcmb.2004-0321OC. [DOI] [PubMed] [Google Scholar]
- 101.Ramos-Barbon D, Presley JF, Hamid QA, Fixman ED, Martin JG. Antigen-specific CD4+ T cells drive airway smooth muscle remodeling in experimental asthma. J Clin Invest. 2005;115:1580–1589. doi: 10.1172/JCI19711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Patel HJ, Belvisi MG, Bishop-Bailey D, Yacoub MH, Mitchell JA. Activation of peroxisome proliferator-activated receptors in human airway smooth muscle cells has a superior anti-inflammatory profile to corticosteroids: relevance for chronic obstructive pulmonary disease therapy. J Immunol. 2003;170:2663–2669. doi: 10.4049/jimmunol.170.5.2663. [DOI] [PubMed] [Google Scholar]
- 103.Ward JE, Gould H, Harris T, Bonacci JV, Stewart AG. PPAR gamma ligands, 15-deoxy-delta12,14-prostaglandin J2 and rosiglitazone regulate human cultured airway smooth muscle proliferation through different mechanisms. Br J Pharmacol. 2004;141:517–525. doi: 10.1038/sj.bjp.0705630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hirst SJ, Martin JG, Bonacci JV, et al. Proliferative aspects of airway smooth muscle. J Allergy Clin Immunol. 2004;114(Suppl 2):S2–17. doi: 10.1016/j.jaci.2004.04.039. [DOI] [PubMed] [Google Scholar]
- 105.Stewart AG, Bonacci JV, Quan L. Factors controlling airway smooth muscle proliferation in asthma. Curr Allergy Asthma Rep. 2004;4:109–115. doi: 10.1007/s11882-004-0055-8. [DOI] [PubMed] [Google Scholar]
- 106.Tang ML, Wilson JW, Stewart AG, Royce SG. Airway wall remodelling in asthma: current understanding and implications for future therapies. Pharmacol Therapeut. 2006;112:474–488. doi: 10.1016/j.pharmthera.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 107.Hughes RA, Harris T, Altmann E, et al. 2-Methoxyes-tradiol and analogs as novel antiproliferative agents: analysis of three-dimensional quantitative structure-activity relationships for DNA synthesis inhibition and estrogen receptor binding. Mol Pharmacol. 2002;61:1053–1069. doi: 10.1124/mol.61.5.1053. [DOI] [PubMed] [Google Scholar]
- 108.Tliba O, Tliba S, Da Huang C, et al. Tumor necrosis factor alpha modulates airway smooth muscle function via the autocrine action of interferon beta. J Biol Chem. 2003;278:50615–50623. doi: 10.1074/jbc.M303680200. [DOI] [PubMed] [Google Scholar]
- 109.Roth J, Johnson PRA, Rudiger JJ, et al. Interaction between glucocorticoids and beta-2-agonists on bronchial airway smooth muscle cells through synchronised cellular signalling. Lancet. 2002;360:1293–1299. doi: 10.1016/S0140-6736(02)11319-5. [DOI] [PubMed] [Google Scholar]
- 110.Tran T, Fernandes DJ, Schuliga M, Harris T, Landells L, Stewart AG. Stimulus-dependent glucocorticoid-resistance of GM-CSF production in human cultured airway smooth muscle. Br J Pharmacol. 2005;145:123–131. doi: 10.1038/sj.bjp.0706174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hirst SJ, Twort CH, Lee TH. Differential effects of extracellular matrix proteins on human airway smooth muscle cell proliferation and phenotype. Am J Respir Cell Mol Biol. 2000;23:335–344. doi: 10.1165/ajrcmb.23.3.3990. [DOI] [PubMed] [Google Scholar]
- 112.Johnson PR, Burgess JK, Underwood PA, et al. Extracellular matrix proteins modulate asthmatic airway smooth muscle cell proliferation via an autocrine mechanism. J Allergy Clin Immunol. 2004;113:690–696. doi: 10.1016/j.jaci.2003.12.312. [DOI] [PubMed] [Google Scholar]
- 113.Bonacci JV, Harris T, Wilson JW, Stewart AG. Collagen-induced resistance to glucocorticoid anti-mitogenic actions: a potential explanation of smooth muscle hyperplasia in the asthmatic remodelled airway. Br J Pharmacol. 2003;138:1203–1206. doi: 10.1038/sj.bjp.0705135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schmidt M, Sun G, Stacey MA, Mori L, Mattoli S. Identification of circulating fibrocytes as precursors of bronchial myofibroblasts in asthma. J Immunol. 2003;171:380–389. doi: 10.4049/jimmunol.171.1.380. [DOI] [PubMed] [Google Scholar]
- 115.Gizycki MJ, Adelroth E, Rogers AV, O’Byrne PM, Jeffery PK. Myofibroblast involvement in the allergen-induced late response in mild atopic asthma. Am J Respir Cell Mol Biol. 1997;16:664–673. doi: 10.1165/ajrcmb.16.6.9191468. [DOI] [PubMed] [Google Scholar]
- 116.Goncharova EA, Vorotnikov AV, Gracheva EO, et al. Activation of p38 MAP-kinase and caldesmon phosphorylation are essential for urokinase-induced human smooth muscle cell migration. Biol Chem. 2002;383:115–126. doi: 10.1515/BC.2002.012. [DOI] [PubMed] [Google Scholar]
- 117.Irani C, Goncharova EA, Hunter DS, Walker CL, Panettieri RA, Krymskaya VP. Phosphatidylinositol 3-kinase but not tuberin is required for PDGF-induced cell migration. Am J Physiol Lung Cell Mol Physiol. 2002;282:L854–862. doi: 10.1152/ajplung.00291.2001. [DOI] [PubMed] [Google Scholar]
- 118.Parameswaran K, Cox G, Radford K, Janssen LJ, Sehmi R, O’Byrne PM. Cysteinyl leukotrienes promote human airway smooth muscle migration. Am J Respir Crit Care Med. 2002;166:738–742. doi: 10.1164/rccm.200204-291OC. [DOI] [PubMed] [Google Scholar]
- 119.Goncharova EA, Billington CK, Irani C, et al. Cyclic AMP-mobilizing agents and glucocorticoids modulate human smooth muscle cell migration. Am J Respir Cell Mol Biol. 2003;29:19–27. doi: 10.1165/rcmb.2002-0254OC. [DOI] [PubMed] [Google Scholar]
- 120.Bonacci JV, Schuliga M, Harris T, Stewart AG. Collagen impairs glucocorticoid actions in airway smooth muscle through integrin signalling. Br J Pharmacol. 2006;149:365–373. doi: 10.1038/sj.bjp.0706881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bonacci JV, Stewart AG. Regulation of human airway mesenchymal cell proliferation by glucocorticoids and beta(2)-adrenoceptor agonists. Pulm Pharmacol Ther. 2006;19:32–38. doi: 10.1016/j.pupt.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 122.Cox PG, Miller J, Mitzner W, Leff AR. Radiofrequency ablation of airway smooth muscle for sustained treatment of asthma: preliminary investigations. Eur Respir J. 2004;24:659–663. doi: 10.1183/09031936.04.00054604. [DOI] [PubMed] [Google Scholar]
- 123.Mitzner W. Airway smooth muscle: the appendix of the lung. Am J Respir Crit Care Med. 2004;169:787–790. doi: 10.1164/rccm.200312-1636PP. [DOI] [PubMed] [Google Scholar]
- 124.Kuriyama T, Latham LP, Horwitz LD, Reeves JT, Wagner WW., Jr Role of collateral ventilation in ventilation-perfusion balance. J Appl Physiol. 1984;56:1500–1506. doi: 10.1152/jappl.1984.56.6.1500. [DOI] [PubMed] [Google Scholar]
- 125.Brackel HJ, Pedersen OF, Mulder PG, Overbeek SE, Kerrebijn KF, Bogaard JM. Central airways behave more stiffly during forced expiration in patients with asthma. Am J Respir Crit Care Med. 2000;162:896–904. doi: 10.1164/ajrccm.162.3.9905034. [DOI] [PubMed] [Google Scholar]
- 126.Olsen CR, Stevens AE, McIlroy MB. Rigidity of tracheae and bronchi during muscular constriction. J Appl Physiol. 1967;23:27–34. doi: 10.1152/jappl.1967.23.1.27. [DOI] [PubMed] [Google Scholar]
- 127.Cox G, Miller JD, McWilliams A, Fitzgerald JM, Lam S. Bronchial Thermoplasty for asthma. Am J Respir Crit Care Med. 2006;173:965–969. doi: 10.1164/rccm.200507-1162OC. [DOI] [PubMed] [Google Scholar]
- 128.Bel EH. “Hot stuff”: bronchial thermoplasty for asthma. Am J Respir Crit Care Med. 2006;173:941–942. doi: 10.1164/rccm.2602002. [DOI] [PubMed] [Google Scholar]
- 129.Brown RH, Wizeman W, Danek C, Mitzner W. In vivo evaluation of the effectiveness of bronchial thermoplasty with computed tomography. J Appl Physiol. 2005;98:1603–1606. doi: 10.1152/japplphysiol.01210.2004. [DOI] [PubMed] [Google Scholar]
- 130.Brown RH, Wizeman W, Danek C, Mitzner W. Effect of bronchial thermoplasty on airway distensibility. Eur Respir J. 2005;26:277–282. doi: 10.1183/09031936.05.00006605. [DOI] [PubMed] [Google Scholar]
- 131.Tgavalekos NT, Tawhai M, Harris SR, et al. Identifying airways responsible for heterogeneous ventilation and mechanical dysfunction in asthma: an image-functional modeling approach. J Appl Physiol. 2005;99:2388–2397. doi: 10.1152/japplphysiol.00391.2005. [DOI] [PubMed] [Google Scholar]
- 132.Lai-Fook SJ, Hyatt RE, Rodarte JR, Wilson TA. Behavior of artificially produced holes in lung parenchyma. J Appl Physiol. 1977;43:648–655. doi: 10.1152/jappl.1977.43.4.648. [DOI] [PubMed] [Google Scholar]
- 133.Lai-Fook SJ, Hyatt RE, Rodarte JR. Effect of parenchymal shear modulus and lung volume on bronchial pressure-diameter behavior. J Appl Physiol. 1978;44:859–868. doi: 10.1152/jappl.1978.44.6.859. [DOI] [PubMed] [Google Scholar]
- 134.Lambert RK, Pare PD. Lung parenchymal shear modulus, airway wall remodeling, and bronchial hyperresponsiveness. J Appl Physiol. 1997;83:140–147. doi: 10.1152/jappl.1997.83.1.140. [DOI] [PubMed] [Google Scholar]
- 135.Hughes JM, Jones HA, Wilson AG, Grant BJ, Pride NB. Stability of intrapulmonary bronchial dimensions during expiratory flow in excised lungs. J Appl Physiol. 1974;37:684–694. doi: 10.1152/jappl.1974.37.5.684. [DOI] [PubMed] [Google Scholar]
- 136.Adler A, Bates JH. A micromechanical model of airway–parenchymal interdependence. Ann Biomed Eng. 2000;28:309–317. doi: 10.1114/1.270. [DOI] [PubMed] [Google Scholar]
- 137.Fredberg JJ, Kamm RD. Stress transmission in the lung: pathways from organ to molecule. Annu Rev Physiol. 2006;68:507–541. doi: 10.1146/annurev.physiol.68.072304.114110. [DOI] [PubMed] [Google Scholar]
- 138.Brown RH, Scichilone N, Mudge B, Diemer FB, Permutt S, Togias A. HRCT evaluation of airways distensibility and the effects of lung inflation on airway caliber in healthy and asthmatic individuals. Am J Respir Crit Care Med. 2001;163:989–993. doi: 10.1164/ajrccm.163.4.2007119. [DOI] [PubMed] [Google Scholar]
- 139.Brown RH, Scichilone N, Mudge B, Diemer FB, Permutt S, Togias A. High-resolution computed tomographic evaluation of airway distensibility and the effects of lung inflation on airway caliber in healthy subjects and individuals with asthma. Am J Respir Crit Care Med. 2001;163:994–1001. doi: 10.1164/ajrccm.163.4.2007119. [DOI] [PubMed] [Google Scholar]
- 140.Brown RH, Mitzner W. Airway response to deep inspiration: role of nitric oxide. Eur Respir J. 2003;22:57–61. doi: 10.1183/09031936.03.00090403. [DOI] [PubMed] [Google Scholar]
- 141.Salome CM, Thorpe CW, Diba C, Brown NJ, Berend N, King GG. Airway re-narrowing following deep inspiration in asthmatic and nonasthmatic subjects. Eur Respir J. 2003;22:62–68. doi: 10.1183/09031936.03.00117502. [DOI] [PubMed] [Google Scholar]
- 142.Mitchell HW, Turner DJ, Gray PR, McFawn PK. Compliance and stability of the bronchial wall in a porcine model of allergen-induced lung inflammation. J Appl Physiol. 1999;86:932–937. doi: 10.1152/jappl.1999.86.3.932. [DOI] [PubMed] [Google Scholar]
- 143.Mauad T, Silva FF, Santos MA, et al. Abnormal alveolar attachments with decreased elastic fiber content in distal lung in fatal asthma. Am J Respir Crit Care Med. 2004;170:857–862. doi: 10.1164/rccm.200403-305OC. [DOI] [PubMed] [Google Scholar]
- 144.Mitchell HW, Gray PR. Uncoupling in the wall of the cartilaginous bronchus of the pig produced by smooth muscle contraction. Pulm Pharmacol. 1996;9:29–34. doi: 10.1006/pulp.1996.0003. [DOI] [PubMed] [Google Scholar]
- 145.Mitchell HW, Gray PR. Assessment of the dynamic relationship between external diameter and lumen flow in isolated bronchi. Respir Physiol. 1999;116:67–76. doi: 10.1016/s0034-5687(99)00034-1. [DOI] [PubMed] [Google Scholar]
- 146.Wiggs BR, Moreno R, Hogg JC, Hilliam C, Pare PD. A model of the mechanics of airway narrowing. J Appl Physiol. 1990;69:849–860. doi: 10.1152/jappl.1990.69.3.849. [DOI] [PubMed] [Google Scholar]
- 147.Lambert RK, Codd SL, Alley MR, Pack RJ. Physical determinants of bronchial mucosal folding. J Appl Physiol. 1994;77:1206–1216. doi: 10.1152/jappl.1994.77.3.1206. [DOI] [PubMed] [Google Scholar]
- 148.Okazawa M, Vedal S, Verburgt L, Lambert RK, Pare PD. Determinants of airway smooth muscle shortening in excised canine lobes. J Appl Physiol. 1995;78:608–614. doi: 10.1152/jappl.1995.78.2.608. [DOI] [PubMed] [Google Scholar]
- 149.Seow CY, Wang L, Pare PD. Airway narrowing and internal structural constraints. J Appl Physiol. 2000;88:527–533. doi: 10.1152/jappl.2000.88.2.527. [DOI] [PubMed] [Google Scholar]
- 150.Milanese M, Crimi E, Scordamaglia A, et al. On the functional consequences of reticular basement membrane thickening in the airways. J Appl Physiol. 2001;91:1035–1040. doi: 10.1152/jappl.2001.91.3.1035. [DOI] [PubMed] [Google Scholar]
- 151.Niimi A, Matsumoto H, Takemura M, Ueda T, Chin K, Mishima M. Relationship of airway wall thickness to airway sensitivity and airway reactivity in asthma. Am J Respir Crit Care Med. 2003;168:983–988. doi: 10.1164/rccm.200211-1268OC. [DOI] [PubMed] [Google Scholar]
- 152.Pare PD. Airway hyperresponsiveness in asthma: geometry is not everything! Am J Respir Crit Care Med. 2003;168:913–914. doi: 10.1164/rccm.2307005. [DOI] [PubMed] [Google Scholar]
- 153.Brown RH, Mitzner W. Effect of lung inflation and airway muscle tone on airway diameter in vivo. J Appl Physiol. 1996;80:1581–1588. doi: 10.1152/jappl.1996.80.5.1581. [DOI] [PubMed] [Google Scholar]
- 154.Klingele TG, Staub NC. Terminal bronchiole diameter changes with volume in isolated, air-filled lobes of cat lung. J Appl Physiol. 1971;30:224–227. doi: 10.1152/jappl.1971.30.2.224. [DOI] [PubMed] [Google Scholar]
- 155.Hughes JM, Hoppin FG, Jr, Mead J. Effect of lung inflation on bronchial length and diameter in excised lungs. J Appl Physiol. 1972;32:25–35. doi: 10.1152/jappl.1972.32.1.25. [DOI] [PubMed] [Google Scholar]
- 156.Hahn HL, Watson A, Wilson AG, Pride NB. Influence of bronchomotor tone on airway dimensions and resistance in excised dog lungs. J Appl Physiol. 1980;49:270–278. doi: 10.1152/jappl.1980.49.2.270. [DOI] [PubMed] [Google Scholar]
- 157.Brown RH, Mitzner W, Wagner EM. Interaction between airway edema and lung inflation on responsiveness of individual airways in vivo. J Appl Physiol. 1997;83:366–370. doi: 10.1152/jappl.1997.83.2.366. [DOI] [PubMed] [Google Scholar]
- 158.Brown RH, Mitzner W, Bulut Y, Wagner EM. Effects of lung inflation in vivo on airways with smooth muscle tone or edema. J Appl Physiol. 1997;82:491–499. doi: 10.1152/jappl.1997.82.2.491. [DOI] [PubMed] [Google Scholar]
- 159.Nakano Y, Muller NL, King GG, et al. Quantitative assessment of airway remodeling using high-resolution CT. Chest. 2002;122(Suppl 6):271S–275S. [PubMed] [Google Scholar]
- 160.Shen X, Gunst SJ, Tepper RS. Effect of tidal volume and frequency on airway responsiveness in mechanically ventilated rabbits. J Appl Physiol. 1997;83:1202–1208. doi: 10.1152/jappl.1997.83.4.1202. [DOI] [PubMed] [Google Scholar]
- 161.Shen X, Wu MF, Tepper RS, Gunst SJ. Mechanisms for the mechanical response of airway smooth muscle to length oscillation. J Appl Physiol. 1997;83:731–738. doi: 10.1152/jappl.1997.83.3.731. [DOI] [PubMed] [Google Scholar]
- 162.Shen X, Wu MF, Tepper RS, Gunst SJ. Pharmacological modulation of the mechanical response of airway smooth muscle to length oscillation. J Appl Physiol. 1997;83:739–745. doi: 10.1152/jappl.1997.83.3.739. [DOI] [PubMed] [Google Scholar]
- 163.Gump A, Haughney L, Fredberg JJ. Relaxation of activated airway smooth muscle: relative potency of isoproterenol vs. tidal stretch. J Appl Physiol. 2001;90:2306–2310. doi: 10.1152/jappl.2001.90.6.2306. [DOI] [PubMed] [Google Scholar]
- 164.Dowell ML, Lakser OJ, Gerthoffer WT, et al. Latrunculin B increases force fluctuation-induced relengthening of ACh-contracted, isotonically shortened canine tracheal smooth muscle. J Appl Physiol. 2005;98:489–497. doi: 10.1152/japplphysiol.01378.2003. [DOI] [PubMed] [Google Scholar]
- 165.Solway J, Bellam S, Dowell M, et al. Actin dynamics: a potential integrator of smooth muscle (dys-)function and contractile apparatus gene expression in asthma. Parker B. Francis lecture. Chest. 2003;123(Suppl 3):392S–398S. doi: 10.1378/chest.123.3_suppl.392s. [DOI] [PubMed] [Google Scholar]
- 166.Silveira PS, Butler JP, Fredberg JJ. Length adaptation of airway smooth muscle: a stochastic model of cytoskeletal dynamics. J Appl Physiol. 2005;99:2087–2098. doi: 10.1152/japplphysiol.00159.2005. [DOI] [PubMed] [Google Scholar]
- 167.Silveira PSP, Fredberg JJ. Smooth muscle length adaptation and actin filament length: a network model of the cytoskeletal dysregulation. Can J Physiol Pharmacol. 2005;83:923–931. doi: 10.1139/y05-092. [DOI] [PubMed] [Google Scholar]
- 168.Kuo KH, Wang L, Pare PD, Ford LE, Seow CY. Myosin thick filament lability induced by mechanical strain in airway smooth muscle. J Appl Physiol. 2001;90:1811–1816. doi: 10.1152/jappl.2001.90.5.1811. [DOI] [PubMed] [Google Scholar]
- 169.Gunst SJ, Meiss RA, Wu MF, Rowe M. Mechanisms for the mechanical plasticity of tracheal smooth muscle. Am J Physiol. 1995;268:C1267–1276. doi: 10.1152/ajpcell.1995.268.5.C1267. [DOI] [PubMed] [Google Scholar]
- 170.Pratusevich VR, Seow CY, Ford LE. Plasticity in canine airway smooth muscle. J Gen Physiol. 1995;105:73–94. doi: 10.1085/jgp.105.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wang L, Pare PD, Seow CY. Selected contribution: effect of chronic passive length change on airway smooth muscle length–tension relationship. J Appl Physiol. 2001;90:734–740. doi: 10.1152/jappl.2001.90.2.734. [DOI] [PubMed] [Google Scholar]
- 172.Silberstein J, Hai CM. Dynamics of length–force relations in airway smooth muscle. Respir Physiol Neurobiol. 2002;132:205–221. doi: 10.1016/s1569-9048(02)00112-x. [DOI] [PubMed] [Google Scholar]
- 173.Bai TR, Bates JH, Brusasco V, et al. On the terminology for describing the length-force relationship and its changes in airway smooth muscle. J Appl Physiol. 2004;97:2029–2034. doi: 10.1152/japplphysiol.00884.2004. [DOI] [PubMed] [Google Scholar]
- 174.Seow CY. Myosin filament assembly in an ever-changing myofilament lattice of smooth muscle. Am J Physiol Cell Physiol. 2005;289:C1363–1368. doi: 10.1152/ajpcell.00329.2005. [DOI] [PubMed] [Google Scholar]
- 175.Pavalko FM, Adam LP, Wu MF, Walker TL, Gunst SJ. Phosphorylation of dense-plaque proteins talin and paxillin during tracheal smooth muscle contraction. Am J Physiol. 1995;268:C563–571. doi: 10.1152/ajpcell.1995.268.3.C563. [DOI] [PubMed] [Google Scholar]
- 176.Wang Z, Pavalko FM, Gunst SJ. Tyrosine phosphorylation of the dense plaque protein paxillin is regulated during smooth muscle contraction. Am J Physiol. 1996;271:C1594–602. doi: 10.1152/ajpcell.1996.271.5.C1594. [DOI] [PubMed] [Google Scholar]
- 177.Tang DD, Mehta D, Gunst SJ. Mechanosensitive tyrosine phosphorylation of paxillin and focal adhesion kinase in tracheal smooth muscle. Am J Physiol. 1999;276:C250–258. doi: 10.1152/ajpcell.1999.276.1.C250. [DOI] [PubMed] [Google Scholar]
- 178.Tang DD, Gunst SJ. Roles of focal adhesion kinase and paxillin in the mechanosensitive regulation of myosin phosphorylation in smooth muscle. J Appl Physiol. 2001;91:1452–1459. doi: 10.1152/jappl.2001.91.3.1452. [DOI] [PubMed] [Google Scholar]
- 179.Gerthoffer WT, Gunst SJ. Focal adhesion and small heat shock proteins in the regulation of actin remodeling and contractility in smooth muscle. J Appl Physiol. 2001;91:963–972. doi: 10.1152/jappl.2001.91.2.963. [DOI] [PubMed] [Google Scholar]
- 180.Tang DD, Turner CE, Gunst SJ. Expression of non-phosphorylatable paxillin mutants in canine tracheal smooth muscle inhibits tension development. J Physiol. 2003;553:21–35. doi: 10.1113/jphysiol.2003.045047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Zhang W, Wu Y, Du L, Tang DD, Gunst SJ. Activation of the Arp2/3 Complex by N-WASp is required for actin polymerization and contraction in canine tracheal smooth muscle. Am J Physiol Cell Physiol. 2005;288:C1145–1160. doi: 10.1152/ajpcell.00387.2004. [DOI] [PubMed] [Google Scholar]
- 182.Fabry B, Maksym GN, Butler JP, et al. Time scale and other invariants of integrative mechanical behavior in living cells. Phys Rev E Stat Nonlin Soft Matter Phys. 2003;68:041914. doi: 10.1103/PhysRevE.68.041914. [DOI] [PubMed] [Google Scholar]
- 183.Bursac P, Lenormand G, Fabry B, et al. Cytoskeletal remodelling and slow dynamics in the living cell. Nat Mater. 2005;4:557–561. doi: 10.1038/nmat1404. [DOI] [PubMed] [Google Scholar]
- 184.Seow CY. Biophysics: fashionable cells. Nature. 2005;435:1172–1173. doi: 10.1038/4351172a. [DOI] [PubMed] [Google Scholar]
- 185.Kuo KH, Seow CY. Contractile filament architecture and force transmission in swine airway smooth muscle. J Cell Sci. 2004;117:1503–1511. doi: 10.1242/jcs.00996. [DOI] [PubMed] [Google Scholar]
- 186.An SS, Fabry B, Mellema M, et al. Role of heat shock protein 27 in cytoskeletal remodeling of the airway smooth muscle cell. J Appl Physiol. 2004;96:1701–1713. doi: 10.1152/japplphysiol.01129.2003. [DOI] [PubMed] [Google Scholar]
- 187.Gunst SJ, Tang DD. The contractile apparatus and mechanical properties of airway smooth muscle. Eur Respir J. 2000;15:600–616. doi: 10.1034/j.1399-3003.2000.15.29.x. [DOI] [PubMed] [Google Scholar]
- 188.Kuo KH, Herrera AM, Wang L, et al. Structure–function correlation in airway smooth muscle adapted to different lengths. Am J Physiol Cell Physiol. 2003;285:C384–390. doi: 10.1152/ajpcell.00095.2003. [DOI] [PubMed] [Google Scholar]
- 189.Mehta D, Gunst SJ. Actin polymerization stimulated by contractile activation regulates force development in canine tracheal smooth muscle. J Physiol. 1999;519:829–840. doi: 10.1111/j.1469-7793.1999.0829n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Opazo Saez A, Zhang W, Wu Y, Turner CE, Tang DD, Gunst SJ. Tension development during contractile stimulation of smooth muscle requires recruitment of paxillin and vinculin to the membrane. Am J Physiol Cell Physiol. 2004;286:C433–447. doi: 10.1152/ajpcell.00030.2003. [DOI] [PubMed] [Google Scholar]
- 191.Smolensky AV, Ragozzino J, Gilbert SH, Seow CY, Ford LE. Length-dependent filament formation assessed from birefringence increases during activation of porcine tracheal muscle. J Physiol. 2005;563:517–527. doi: 10.1113/jphysiol.2004.079822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Qi D, Mitchell RW, Burdyga T, Ford LE, Kuo KH, Seow CY. ssMyosin light chain phosphorylation facilitates in vivo myosin filament reassembly after mechanical perturbation. Am J Physiol Cell Physiol. 2002;282:C1298–1305. doi: 10.1152/ajpcell.00554.2001. [DOI] [PubMed] [Google Scholar]
- 193.Herrera AM, Kuo KH, Seow CY. Influence of calcium on myosin thick filament formation in intact airway smooth muscle. Am J Physiol Cell Physiol. 2002;282:C310–316. doi: 10.1152/ajpcell.00390.2001. [DOI] [PubMed] [Google Scholar]
- 194.Herrera AM, Martinez EC, Seow CY. Electron microscopic study of actin polymerization in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1161–1168. doi: 10.1152/ajplung.00298.2003. [DOI] [PubMed] [Google Scholar]
- 195.Herrera AM, McParland BE, Bienkowska A, Tait R, Pare PD, Seow CY. “Sarcomeres” of smooth muscle: functional characteristics and ultrastructural evidence. J Cell Sci. 2005;118:2381–2392. doi: 10.1242/jcs.02368. [DOI] [PubMed] [Google Scholar]
- 196.Tschumperlin DJ, Drazen JM. Chronic effects of mechanical force on airways. Annu Rev Physiol. 2006;68:563–583. doi: 10.1146/annurev.physiol.68.072304.113102. [DOI] [PubMed] [Google Scholar]
- 197.Xue Z, Zhang L, Ramchandani R, et al. Respiratory system responsiveness in rabbits in vivo is reduced by prolonged continuous positive airway pressure. J Appl Physiol. 2005;99:677–682. doi: 10.1152/japplphysiol.00165.2005. [DOI] [PubMed] [Google Scholar]
- 198.Naghshin J, Wang L, Pare PD, Seow CY. Adaptation to chronic length change in explanted airway smooth muscle. J Appl Physiol. 2003;95:448–453. doi: 10.1152/japplphysiol.01180.2002. [DOI] [PubMed] [Google Scholar]
- 199.Tepper RS, Ramchandani R, Argay E, et al. Chronic strain alters the passive and contractile properties of rabbit airways. J Appl Physiol. 2005;98:1949–1954. doi: 10.1152/japplphysiol.00952.2004. [DOI] [PubMed] [Google Scholar]
- 200.Bates JH, Lauzon AM, Dechman GS, Maksym GN, Schuessler TF. Temporal dynamics of pulmonary response to intravenous histamine in dogs: effects of dose and lung volume. J Appl Physiol. 1994;76:616–626. doi: 10.1152/jappl.1994.76.2.616. [DOI] [PubMed] [Google Scholar]
- 201.Hoppin FG., Jr Parenchymal mechanics and asthma. Chest. 1995;107(Suppl 3):140S–144S. doi: 10.1378/chest.107.3_supplement.140s. [DOI] [PubMed] [Google Scholar]
- 202.Pepe C, Foley S, Shannon J, et al. Differences in airway remodeling between subjects with severe and moderate asthma. J Allergy Clin Immunol. 2005;116:544–549. doi: 10.1016/j.jaci.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 203.Pini L, Torregiani C, Martin JG, Hamid Q, Ludwig MS. Airway remodeling in allergen challenged brown Norway rats: distribution of proteoglycans. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1052–1058. doi: 10.1152/ajplung.00122.2005. [DOI] [PubMed] [Google Scholar]
- 204.Martin JG, Tamaoka M. Rat models of asthma and chronic obstructive lung disease. Pulm Pharmacol Ther. 2006;19:377–385. doi: 10.1016/j.pupt.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 205.Sparrow MP, Mitchell HW. Contraction of smooth muscle of pig airway tissues from before birth to maturity. J Appl Physiol. 1990;68:468–477. doi: 10.1152/jappl.1990.68.2.468. [DOI] [PubMed] [Google Scholar]
- 206.Weist A, Williams T, Kisling J, Clem C, Tepper RS. Volume history and effect on airway reactivity in infants and adults. J Appl Physiol. 2002;93:1069–1074. doi: 10.1152/japplphysiol.00986.2001. [DOI] [PubMed] [Google Scholar]
- 207.Ramchandani R, Shen X, Gunst SJ, Tepper RS. Comparison of elastic properties and contractile responses of isolated airway segments from mature and immature rabbits. J Appl Physiol. 2003;95:265–271. doi: 10.1152/japplphysiol.00362.2002. [DOI] [PubMed] [Google Scholar]
- 208.Tepper RS, Wiggs B, Gunst SJ, Pare PD. Lower shear modulus of the lung for immature than mature rabbits. Am J Respir Crit Care Med. 1999;159:A406. [Google Scholar]
- 209.Gomes RF, Shardonofsky F, Eidelman DH, Bates JH. Respiratory mechanics and lung development in the rat from early age to adulthood. J Appl Physiol. 2001;90:1631–1638. doi: 10.1152/jappl.2001.90.5.1631. [DOI] [PubMed] [Google Scholar]
- 210.Shen X, Ramchandani R, Dunn B, Lambert R, Gunst SJ, Tepper RS. Effect of transpulmonary pressure on airway diameter and responsiveness of immature and mature rabbits. J Appl Physiol. 2000;89:1584–1590. doi: 10.1152/jappl.2000.89.4.1584. [DOI] [PubMed] [Google Scholar]
- 211.Bryan AC, Wohl MEB. Respiratory mechanics in children. In: Macklem PT, Mead J, editors. American Physiological Society Handbook of Physiology: Respiratory System. V3: Mechanics of Breathing Section 3. Philadelphia: Lippincott Williams and Wilkins; 1986. pp. 179–191. [Google Scholar]
- 212.Panitch HB, Deoras KS, Wolfson MR, Shaffer TH. Maturational changes in airway smooth muscle structure–function relationships. Pediatr Res. 1992;31:151–156. doi: 10.1203/00006450-199202000-00012. [DOI] [PubMed] [Google Scholar]
- 213.Chitano P, Wang J, Cox CM, Stephens NL, Murphy TM. Different ontogeny of rate of force generation and shortening velocity in guinea pig trachealis. J Appl Physiol. 2000;88:1338–1345. doi: 10.1152/jappl.2000.88.4.1338. [DOI] [PubMed] [Google Scholar]
- 214.Wang L, Chitano P, Murphy TM. Maturation of guinea pig tracheal strip stiffness. Am J Physiol Lung Cell Mol Physiol. 2005;289:L902–908. doi: 10.1152/ajplung.00005.2005. [DOI] [PubMed] [Google Scholar]
- 215.Wang L, Chitano P, Murphy TM. Length oscillation induces force potentiation in infant guinea pig airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2005;289:L909–915. doi: 10.1152/ajplung.00128.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Chitano P, Turato G, Murphy TM. The release of prostaglandin E2 by sustained electrical field stimulation increases with age in guinea pig trachea. Am J Respir Crit Care Med. 2000;161:A693. [Google Scholar]
- 217.Mitchell RW, Magnussen H, Rabe KF, Leff AR. Passive sensitization of human airways induces myogenic contractile responses in vitro. J Appl Physiol. 1997;83:1276–1281. doi: 10.1152/jappl.1997.83.4.1276. [DOI] [PubMed] [Google Scholar]
- 218.Kawamoto S, Adelstein RS. Characterization of myosin heavy chains in cultured aorta smooth muscle cells. A comparative study. J Biol Chem. 1987;262:7282–7288. [PubMed] [Google Scholar]
- 219.Babij P, Kawamoto S, White S, Adelstein RS, Periasamy M. Differential expression of SM1 and SM2 myosin isoforms in cultured vascular smooth muscle. Am J Physiol. 1992;262:C607–613. doi: 10.1152/ajpcell.1992.262.3.C607. [DOI] [PubMed] [Google Scholar]
- 220.Thyberg J. Differentiated properties and proliferation of arterial smooth muscle cells in culture. Int Rev Cytol. 1996;169:183–265. doi: 10.1016/s0074-7696(08)61987-7. [DOI] [PubMed] [Google Scholar]
- 221.Halayko AJ, Salari H, Ma X, Stephens NL. Markers of airway smooth muscle phenotype. Am J Physiol. 1996;270:L1040–1051. doi: 10.1152/ajplung.1996.270.6.L1040. [DOI] [PubMed] [Google Scholar]
- 222.Wong JZ, Woodcock-Mitchell J, Mitchell J, et al. Smooth muscle actin and myosin expression in cultured airway smooth muscle cells. Am J Physiol. 1998;274:L786–792. doi: 10.1152/ajplung.1998.274.5.L786. [DOI] [PubMed] [Google Scholar]
- 223.Ma X, Wang Y, Stephens NL. Serum deprivation induces a unique hypercontractile phenotype of cultured smooth muscle cells. Am J Physiol. 1998;274:C1206–1214. doi: 10.1152/ajpcell.1998.274.5.C1206. [DOI] [PubMed] [Google Scholar]
- 224.Lin VK, Robertson JB, Lee IL, Zimmern PE, McConnell JD. Smooth muscle myosin heavy chains are developmentally regulated in the rabbit bladder. J Urol. 2000;164:1376–1380. [PubMed] [Google Scholar]
- 225.Arafat HA, Kim GS, DiSanto ME, Wein AJ, Chacko S. Heterogeneity of bladder myocytes in vitro: modulation of myosin isoform expression. Tissue Cell. 2001;33:219–232. doi: 10.1054/tice.2001.0171. [DOI] [PubMed] [Google Scholar]
- 226.Halayko AJ, Solway J. Molecular mechanisms of phenotypic plasticity in smooth muscle cells. J Appl Physiol. 2001;90:358–368. doi: 10.1152/jappl.2001.90.1.358. [DOI] [PubMed] [Google Scholar]
- 227.Janssen LJ, Sims SM. Emptying and refilling of Ca2+ store in tracheal myocytes as indicated by Ach-evoked currents and contraction. Am J Physiol. 1993;265:C877–886. doi: 10.1152/ajpcell.1993.265.4.C877. [DOI] [PubMed] [Google Scholar]
- 228.Smith PG, Garcia R, Kogerman L. Strain reorganizes focal adhesions and cytoskeleton in cultured airway smooth muscle cells. Exp Cell Res. 1997;232:127–136. doi: 10.1006/excr.1997.3492. [DOI] [PubMed] [Google Scholar]
- 229.Wang L, Liu HW, McNeill KD, Stelmack GL, Scott JE, Halayko AJ. Mechanical strain inhibits airway smooth muscle gene transcription via protein kinase C signaling. Am J Respir Cell Mol Biol. 2004;31:54–61. doi: 10.1165/rcmb.2003-0240OC. [DOI] [PubMed] [Google Scholar]
- 230.Smith PG, Deng L, Fredberg JJ, Maksym GN. Mechanical strain increases cell stiffness through cytoskeletal filament reorganization. Am J Physiol. 2003;285:L456–463. doi: 10.1152/ajplung.00329.2002. [DOI] [PubMed] [Google Scholar]
- 231.Smith PG, Roy C, Zhang YN, Chauduri S. Mechanical stress increases RhoA Activation in airway smooth muscle cells. Am J Respir Cell Mol Biol. 2003;28:436–442. doi: 10.1165/rcmb.4754. [DOI] [PubMed] [Google Scholar]
- 232.Halayko AJ, Amrani Y. Mechanisms of inflammation-mediated airway smooth muscle plasticity and airways remodeling in asthma. Respir Physiol Neurobiol. 2003;137:209–222. doi: 10.1016/s1569-9048(03)00148-4. [DOI] [PubMed] [Google Scholar]
- 233.Black JL, Burgess JK, Johnson PR. Airway smooth muscle--its relationship to the extracellular matrix. Respir Physiol Neurobiol. 2003;137:339–346. doi: 10.1016/s1569-9048(03)00157-5. [DOI] [PubMed] [Google Scholar]
- 234.Hirst SJ, Martin JG, Bonacci JV, et al. Proliferative aspects of airway smooth muscle. J Allergy Clin Immunol. 2004;114(Suppl 2):S2–17. doi: 10.1016/j.jaci.2004.04.039. [DOI] [PubMed] [Google Scholar]
- 235.Small JV, Gimona M. The cytoskeleton of the vertebrate smooth muscle cell. Acta Physiol Scand. 1998;164:341–348. doi: 10.1046/j.1365-201X.1998.00441.x. [DOI] [PubMed] [Google Scholar]
- 236.North AJ, Galazkiewicz B, Byers TJ, Glenney JR, Small JV. Complementary distributions of vinculin and dystrophin define two distinct sarcolemma domains in smooth muscle. J Cell Biol. 1993;120:1159–1167. doi: 10.1083/jcb.120.5.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Halayko AJ, Camoretti-Mercado B, Forsythe SM, et al. Divergent differentiation paths in airway smooth muscle culture: induction of functionally contractile myocytes. Am J Physiol. 1999;276:L197–206. doi: 10.1152/ajplung.1999.276.1.L197. [DOI] [PubMed] [Google Scholar]
- 238.Halayko AJ, Stelmack GL. The association of caveolae with the dystrophin-glycoprotein complex: a role in smooth muscle phenotype and function. Can J Physiol Pharmacol. 2005;83:877–891. doi: 10.1139/y05-107. [DOI] [PubMed] [Google Scholar]
- 239.Jeong SI, Kwon JH, Lim JI, et al. Mechano-active tissue engineering of vascular smooth muscle using pulsatile perfusion bioreactors and elastic PLCL scaffolds. Biomaterials. 2005;26:1405–1411. doi: 10.1016/j.biomaterials.2004.04.036. [DOI] [PubMed] [Google Scholar]
- 240.Cukierman E, Pankov R, Stevens DR, Yamada KM. Taking cell-matrix adhesions to the third dimension. Science. 2001;294:1708–1712. doi: 10.1126/science.1064829. [DOI] [PubMed] [Google Scholar]
- 241.Choe MM, Sporn PH, Swartz MA. Extracellular matrix remodeling by dynamic strain in a 3D tissue engineered human airway wall model. Am J Respir Cell Mol Biol. 2006;35:306–313. doi: 10.1165/rcmb.2005-0443OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Smith PG, Janiga K, Bruce MC. Strain increases airway smooth muscle cell proliferation. Am J Respir Cell Mol Biol. 1994;10:85–90. doi: 10.1165/ajrcmb.10.1.8292386. [DOI] [PubMed] [Google Scholar]
- 243.Smith PG, Moreno R, Ikebe M. Strain increases airway smooth muscle contractile and cytoskeletal proteins in vitro. Am J Physiol. 1997;272:L20–27. doi: 10.1152/ajplung.1997.272.1.L20. [DOI] [PubMed] [Google Scholar]
- 244.Smith PG, Roy C, Fisher S, Huang QQ, Brozovich F. Mechanical strain increases force production and calcium sensitivity in cultured airway smooth muscle cells. J Appl Physiol. 2000;89:2092–2098. doi: 10.1152/jappl.2000.89.5.2092. [DOI] [PubMed] [Google Scholar]
- 245.Smith PG, Roy C, Dreger J, Brozovich F. Mechanical strain increases velocity and extent of shortening in cultured airway smooth muscle cells. Am J Physiol. 1999;277:L343–348. doi: 10.1152/ajplung.1999.277.2.L343. [DOI] [PubMed] [Google Scholar]
- 246.Maksym GN, Deng L, Fairbank NJ, Connolly SC. Beneficial and harmful effects of oscillatory mechanical strain on airway smooth muscle. Can J Physiol Pharmacol. 2005;83:913–922. doi: 10.1139/y05-091. [DOI] [PubMed] [Google Scholar]
- 247.Williams JL, Chen JH, Belloli DM. Strain fields on cell stressing devices employing clamped circular elastic diaphragms as substrates. J Biomech Eng. 1992;114:377–384. doi: 10.1115/1.2891398. [DOI] [PubMed] [Google Scholar]
- 248.Wang JH, Yang G, Li Z, Shen W. Fibroblast responses to cyclic mechanical stretching depend on cell orientation to the stretching direction. J Biomech. 2004;37:573–576. doi: 10.1016/j.jbiomech.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 249.Wang JH, Goldschmidt-Clermont P, Wille J, Yin FC. Specificity of endothelial cell reorientation in response to cyclic mechanical stretching. J Biomech. 2001;34:1563–1572. doi: 10.1016/s0021-9290(01)00150-6. [DOI] [PubMed] [Google Scholar]
- 250.Yamada T, Naruse K, Sokabe M. Stretch-induced morphological changes of human endothelial cells depend on the intracellular level of Ca2+ rather than of cAMP. Life Sci. 2000;67:2605–2613. doi: 10.1016/s0024-3205(00)00844-4. [DOI] [PubMed] [Google Scholar]
- 251.Bonacci JV, Harris T, Stewart AG. Collagen-induced resistance to glucocorticoid anti-mitogenic actions: a potential explanation of smooth muscle hyperplasia in the asthmatic remodelled airway. Br J Pharmacol. 2003;138:1203–1206. doi: 10.1038/sj.bjp.0705135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Lei M, Ghezzo H, Chen MF, Eidelman DH. Airway smooth muscle orientation in intraparenchymal airways. J Appl Physiol. 1997;82:70–77. doi: 10.1152/jappl.1997.82.1.70. [DOI] [PubMed] [Google Scholar]
- 253.Deng L, Fairbank NJ, Cole DJ, Fredberg JJ, Maksym GN. Airway smooth muscle tone modulates mechanically induced cytoskeletal stiffening and remodelling. J Appl Physiol. 2005;99:634–641. doi: 10.1152/japplphysiol.00025.2005. [DOI] [PubMed] [Google Scholar]
- 254.Liu X, Farley JM. Acetylcholine-induced Ca++-dependent chloride current oscillations are mediated by inositol 1,4,5-trisphosphate in tracheal myocytes. J Pharmacol Exp Ther. 1996;277:796–804. [PubMed] [Google Scholar]
- 255.Prakash YS, Kannan MS, Sieck GC. Regulation of intracellular calcium oscillations in porcine tracheal smooth muscle cells. Am J Physiol. 1997;272:C966–975. doi: 10.1152/ajpcell.1997.272.3.C966. [DOI] [PubMed] [Google Scholar]
- 256.Pabelick CM, Sieck GC, Prakash YS. Invited review: significance of spatial and temporal heterogeneity of calcium transients in smooth muscle. J Appl Physiol. 2001;91:488–496. doi: 10.1152/jappl.2001.91.1.488. [DOI] [PubMed] [Google Scholar]
- 257.Perez JF, Sanderson MJ. The frequency of calcium oscillations induced by 5-HT, ACH, and KCl determine the contraction of smooth muscle cells of intrapulmonary bronchioles. J Gen Physiol. 2005;125:535–553. doi: 10.1085/jgp.200409216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Dai JM, Kuo KH, Leo JM, Pare PD, van Breemen C, Lee CH. Acetylcholine-induced asynchronous calcium waves in intact human bronchial muscle bundle. Am J Respir Cell Mol Biol. 2007 doi: 10.1165/rcmb.2006-0096OC. [Epub ahead of print, PMID: 17170384] [DOI] [PubMed] [Google Scholar]
- 259.Prakash YS, Pabelick CM, Kannan MS, Sieck GC. Spatial and temporal aspects of ACh-induced [Ca2+]i oscillations in porcine tracheal smooth muscle. Cell Calcium. 2000;27:153–162. doi: 10.1054/ceca.1999.0106. [DOI] [PubMed] [Google Scholar]
- 260.Perez JF, Sanderson MJ. The frequency of calcium oscillations induced by 5-HT, ACH, and KCl determine the contraction of smooth muscle cells of intrapulmonary bronchioles. J Gen Physiol. 2005;125:535–553. doi: 10.1085/jgp.200409216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Dai JM, Kuo KH, Leo JM, van Breemen C, Lee CH. Mechanism of ACh-induced asynchronous calcium waves and tonic contraction in porcine tracheal muscle bundle. Am J Physiol Lung Cell Mol Physiol. 2006;290:L459–469. doi: 10.1152/ajplung.00092.2005. [DOI] [PubMed] [Google Scholar]
- 262.Prakash YS, Kannan MS, Walseth TF, Sieck GC. Role of cyclic ADP-ribose in the regulation of [Ca2+]i in porcine tracheal smooth muscle. Am J Physiol. 1998;274:C1653–1660. doi: 10.1152/ajpcell.1998.274.6.C1653. [DOI] [PubMed] [Google Scholar]
- 263.Deshpande DA, White TA, Dogan S, et al. CD38/cyclic ADP-ribose signaling: role in the regulation of calcium homeostasis in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2005;288:L773–788. doi: 10.1152/ajplung.00217.2004. [DOI] [PubMed] [Google Scholar]
- 264.Kip SN, Smelter M, Iyanoye A, et al. Agonist-induced cyclic ADP ribose production in airway smooth muscle. Arch Biochem Biophys. 2006;452:102–107. doi: 10.1016/j.abb.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 265.Kannan MS, Prakash YS, Brenner T, Mickelson JR, Sieck GC. Role of ryanodine receptor channels in Ca2+ oscillations of porcine tracheal smooth muscle. Am J Physiol. 1997;272:L659–664. doi: 10.1152/ajplung.1997.272.4.L659. [DOI] [PubMed] [Google Scholar]
- 266.Janssen LJ. Ionic mechanisms and Ca(2+) regulation in airway smooth muscle contraction: do the data contradict dogma. Am J Physiol Lung Cell Mol Physiol. 2002;282:L1161–1178. doi: 10.1152/ajplung.00452.2001. [DOI] [PubMed] [Google Scholar]
- 267.Kannan MS, Prakash YS, Johnson DE, Sieck GC. Nitric oxide inhibits calcium release from sarcoplasmic reticulum of porcine tracheal smooth muscle cells. Am J Physiol. 1997;272:L1–7. doi: 10.1152/ajplung.1997.272.1.L1. [DOI] [PubMed] [Google Scholar]
- 268.Prakash YS, Kannan MS, Sieck GC. Nitric oxide inhibits ACh-induced intracellular calcium oscillations in porcine tracheal smooth muscle. Am J Physiol. 1997;272:L588–596. doi: 10.1152/ajplung.1997.272.4.L588. [DOI] [PubMed] [Google Scholar]
- 269.Janssen LJ, Sims SM. Acetylcholine activates non-selective cation and chloride conductances in canine and guinea-pig tracheal myocytes. J Physiol. 1992;453:197–218. doi: 10.1113/jphysiol.1992.sp019224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Janssen LJ, Sims SM. Spontaneous transient inward currents and rhythmicity in canine and guinea-pig tracheal smooth muscle cells. Pflugers Arch. 1994;427:473–480. doi: 10.1007/BF00374263. [DOI] [PubMed] [Google Scholar]
- 271.Liu X, Farley JM. Acetylcholine-induced chloride current oscillations in swine tracheal smooth muscle cells. J Pharmacol Exp Ther. 1996;276:178–186. [PubMed] [Google Scholar]
- 272.Hirota S, Trimble N, Pertens E, Janssen LJ. Intracellular Cl− fluxes play a novel role in Ca2+ handling in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1146–1153. doi: 10.1152/ajplung.00393.2005. [DOI] [PubMed] [Google Scholar]
- 273.Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 274.Parekh AB. A CRAC current tango. Nat Cell Biol. 2006;8:655–656. doi: 10.1038/ncb0706-655. [DOI] [PubMed] [Google Scholar]
- 275.Smyth JT, Dehaven WI, Jones BF, et al. Emerging perspectives in store-operated Ca2+ entry: roles of Orai, Stim and TRP. Biochim Biophys Acta. 2006;1763:1147–1160. doi: 10.1016/j.bbamcr.2006.08.050. [DOI] [PubMed] [Google Scholar]
- 276.Hirota S, Pertens E, Janssen LJ. The reverse mode of the Na(+)/Ca(2+) exchanger provides a source of Ca(2+) for store refilling following agonist-induced Ca(2+) mobilization. Am J Physiol Lung Cell Mol Physiol. 2007;292:L438–447. doi: 10.1152/ajplung.00222.2006. [DOI] [PubMed] [Google Scholar]
- 277.ZhuGe R, Sims SM, Tuft RA, Fogarty KE, Walsh JV., Jr Ca2+ sparks activate K+ and Cl− channels, resulting in spontaneous transient currents in guinea-pig tracheal myocytes. J Physiol. 1998;513:711–718. doi: 10.1111/j.1469-7793.1998.711ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Pabelick CM, Prakash YS, Kannan MS, Sieck GC. Spatial and temporal aspects of calcium sparks in porcine tracheal smooth muscle cells. Am J Physiol. 1999;277:L1018–1025. doi: 10.1152/ajplung.1999.277.5.L1018. [DOI] [PubMed] [Google Scholar]
- 279.Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83:1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- 280.Nuttle LC, Farley JM. Frequency modulation of acetylcholine-induced oscillations in Ca++ and Ca(++)-activated Cl− current by cAMP in tracheal smooth muscle. J Pharmacol Exp Ther. 1996;277:753–760. [PubMed] [Google Scholar]
- 281.Prakash YS, van der Heijden HF, Kannan MS, Sieck GC. Effects of salbutamol on intracellular calcium oscillations in porcine airway smooth muscle. J Appl Physiol. 1997;82:1836–1843. doi: 10.1152/jappl.1997.82.6.1836. [DOI] [PubMed] [Google Scholar]
- 282.Bai Y, Sanderson MJ. Airway smooth muscle relaxation results from a reduction in the frequency of Ca2+ oscillations induced by a cAMP-mediated inhibition of the IP3 receptor. Respir Res. 2006;7:34. doi: 10.1186/1465-9921-7-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Bai Y, Sanderson MJ. Modulation of the Ca2+ sensitivity of airway smooth muscle cells in murine lung slices. Am J Physiol Lung Cell Mol Physiol. 2006;291:L208–221. doi: 10.1152/ajplung.00494.2005. [DOI] [PubMed] [Google Scholar]
- 284.Knight DA, Holgate ST. The airway epithelium: structural and functional properties in health and disease. Respirology. 2003;8:432–446. doi: 10.1046/j.1440-1843.2003.00493.x. [DOI] [PubMed] [Google Scholar]
- 285.Wan H, Winton HL, Soeller C, et al. Der p 1 facilitates transepithelial allergen delivery by disruption of tight junctions. J Clin Invest. 1999;104:123–133. doi: 10.1172/JCI5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.McGrogan I, Janssen LJ, Wattie J, O’Byrne PM, Daniel EE. Release of epithelium-derived PGE2 from canine trachea after antigen inhalation. Am J Physiol. 1998;274:L220–225. doi: 10.1152/ajplung.1998.274.2.L220. [DOI] [PubMed] [Google Scholar]
- 287.Holgate ST. Epithelial damage and response. Clin Exp Allergy. 2000;30(Suppl 1):37–41. doi: 10.1046/j.1365-2222.2000.00095.x. [DOI] [PubMed] [Google Scholar]
- 288.Knight DA, Stewart GA, Thompson PJ. Histamine tachyphylaxis in human airway smooth muscle. The role of H2- receptors and the bronchial epithelium. Am Rev Respir Dis. 1992;146:137–140. doi: 10.1164/ajrccm/146.1.137. [DOI] [PubMed] [Google Scholar]
- 289.Noble PB, Sharma A, McFawn PK, Mitchell HW. Airway narrowing in porcine bronchi with and without lung parenchyma. Eur Respir J. 2005;26:804–811. doi: 10.1183/09031936.05.00065405. [DOI] [PubMed] [Google Scholar]
- 290.Mitchell RW, Rühlmann E, Magnussen H, Munoz NM, Leff AR, Rabe KF. Conservation of bronchiolar wall area during constriction and dilation of human airways. J Appl Physiol. 1997;82:954–958. doi: 10.1152/jappl.1997.82.3.954. [DOI] [PubMed] [Google Scholar]
- 291.McFawn PK, Mitchell HW. Bronchial compliance and wall structure during development of the immature human and pig lung. Eur Respir J. 1997;10:27–34. doi: 10.1183/09031936.97.10010027. [DOI] [PubMed] [Google Scholar]
- 292.Tiddens HA, Hofhuis W, Bogaard JM, et al. Compliance, hysteresis, and collapsibility of human small airways. Am J Respir Crit Care Med. 1999;160:1110–1118. doi: 10.1164/ajrccm.160.4.9709004. [DOI] [PubMed] [Google Scholar]
- 293.Stephens NL, Jiang H. Basic physiology of airway smooth muscle. In: Busse WW, Holgate ST, editors. Asthma and Rhinitis. London: Blackwell Scientific Publications; 1995. pp. 1087–1115. [Google Scholar]
- 294.Von Hayek H. The Human Lung. New York: Hafner; 1960. [Google Scholar]
- 295.Noble PB, McFawn PK, Mitchell HW. Intraluminal pressure oscillation enhances subsequent airway contraction in isolated bronchial segments. J Appl Physiol. 2004;96:1161–1165. doi: 10.1152/japplphysiol.01082.2003. [DOI] [PubMed] [Google Scholar]
- 296.Khangure SR, Noble PB, Sharma A, Chia PY, McFawn PK, Mitchell HW. Cyclical elongation regulates contractile responses of isolated airways. J Appl Physiol. 2004;97:913–919. doi: 10.1152/japplphysiol.00262.2004. [DOI] [PubMed] [Google Scholar]
- 297.Seow CY. Are you pulling my airway. Eur Respir J. 2005;26:759–761. doi: 10.1183/09031936.05.00099905. [DOI] [PubMed] [Google Scholar]
- 298.Butler J, Caro CG, Alcala R, Dubois AB. Physiological factors affecting airway resistance in normal subjects and in patients with obstructive respiratory disease. J Clin Invest. 1960;39:584–591. doi: 10.1172/JCI104071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Caro CG, Butler J, Dubois AB. Some effects of restriction of chest cage expansion on pulmonary function in man: an experimental study. J Clin Invest. 1960;39:573–583. doi: 10.1172/JCI104070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Smith PG, Garcia R, Kogerman L. Mechanical strain increases protein tyrosine phosphorylation in airway smooth muscle cells. Exp Cell Res. 1998;239:353–360. doi: 10.1006/excr.1997.3905. [DOI] [PubMed] [Google Scholar]
- 301.Dietl P, Frick M, Mair N, Bertocchi C, Haller T. Pulmonary consequences of a deep breath revisited. Biol Neonate. 2004;85:299–304. doi: 10.1159/000078176. [DOI] [PubMed] [Google Scholar]
- 302.Bramley AM, Roberts CR, Schellenberg RR. Collagenase increases shortening of human bronchial smooth muscle in vitro. Am J Respir Crit Care Med. 1995;152:1513–1517. doi: 10.1164/ajrccm.152.5.7582286. [DOI] [PubMed] [Google Scholar]
- 303.Meiss RA. Influence of intercellular tissue connections on airway muscle mechanics. J Appl Physiol. 1999;86:5–15. doi: 10.1152/jappl.1999.86.1.5. [DOI] [PubMed] [Google Scholar]
- 304.Ammit AJ, Armour CL, Black JL. Smooth-muscle myosin light-chain kinase content is increased in human sensitized airways. Am J Respir Crit Care Med. 2000;161:257–263. doi: 10.1164/ajrccm.161.1.9901005. [DOI] [PubMed] [Google Scholar]
- 305.Solway J, Fredberg JJ. Perhaps airway smooth muscle dysfunction contributes to asthmatic bronchial hyperresponsiveness after all. Am J Respir Cell Mol Biol. 1997;17:144–146. doi: 10.1165/ajrcmb.17.2.f137. [DOI] [PubMed] [Google Scholar]
- 306.Leguillette R, Laviolette M, Lauzon AM. Quantitative smooth muscle RNA analysis of asthmatic human bronchial biopsies. Proc Am Thor Soc. 2005;2:A338. [Google Scholar]
- 307.Florio C, Styhler A, Heisler S, Martin JG. Mechanical responses of tracheal tissue in vitro: dependence on the tissue preparation employed and relationship to smooth muscle content. Pulmon Pharmacol. 1996;9:157–166. doi: 10.1006/pulp.1996.0018. [DOI] [PubMed] [Google Scholar]
- 308.Gil FR, Zitouni NB, Azoulay E, Maghni K, Lauzon AM. Smooth muscle myosin isoforms and LC20 phosphorylation in innate rat airway hyperresponsiveness. Am J Physiol Lung Cell Mol Physiol. 2006;291:L932–940. doi: 10.1152/ajplung.00339.2004. [DOI] [PubMed] [Google Scholar]
- 309.Blanc FX, Coirault C, Salmeron S, Chemla D, Lecarpentier Y. Mechanics and crossbridge kinetics of tracheal smooth muscle in two inbred rat strains. Eur Respir J. 2003;22:227–234. doi: 10.1183/09031936.03.00064203. [DOI] [PubMed] [Google Scholar]
- 310.Wang CG, Almirall JJ, Dolman CS, Dandurand RJ, Eidelman DH. In vitro bronchial responsiveness in two highly inbred rat strains. J Appl Physiol. 1997;82:1445–1452. doi: 10.1152/jappl.1997.82.5.1445. [DOI] [PubMed] [Google Scholar]
- 311.Tao FC, Tolloczko B, Eidelman DH, Martin JG. Enhanced Ca(2+) mobilization in airway smooth muscle contributes to airway hyperresponsiveness in an inbred strain of rat. Am J Respir Crit Care Med. 1999;160:446–453. doi: 10.1164/ajrccm.160.2.9811098. [DOI] [PubMed] [Google Scholar]
- 312.Tao FC, Tolloczko B, Mitchell CA, Powell WS, Martin JG. Inositol (1,4,5)trisphosphate metabolism and enhanced calcium mobilization in airway smooth muscle of hyperresponsive rats. Am J Respir Cell Mol Biol. 2002;23:514–520. doi: 10.1165/ajrcmb.23.4.3966. [DOI] [PubMed] [Google Scholar]
- 313.Currie GP, Fowler SJ, Lipworth BJ. Dose response of inhaled corticosteroids on bronchial hyperresponsiveness: a meta-analysis. Chest. 2003;90:194–198. doi: 10.1016/S1081-1206(10)62140-0. [DOI] [PubMed] [Google Scholar]
- 314.Leung DYM, Bloom JW. Update on glucocorticoid action and resistance. J Allergy Clin Immunol. 2003;111:3–22. doi: 10.1067/mai.2003.97. [DOI] [PubMed] [Google Scholar]
- 315.Fernandes DJ, Guida E, Kalafatis V, Harris T, Wilson J, Stewart AG. Glucocorticoids inhibit proliferation, cyclin D1 expression and retinoblastoma protein phosphorylation, but not mitogen-activated protein kinase activity in human cultured airway smooth muscle. Am J Respir Cell Mol Biol. 1999;21:77–88. doi: 10.1165/ajrcmb.21.1.3396. [DOI] [PubMed] [Google Scholar]
- 316.Hakonarson H, Halapi E, Whelan R, Gulcher J, Stefansson K, Grunstein MM. Association between IL-1β/TNF-a induced glucocorticoid-sensitive changes in multiple gene expression and altered responsiveness in airway smooth muscle. Am J Respir Cell Mol Biol. 2001;25:761–771. doi: 10.1165/ajrcmb.25.6.4628. [DOI] [PubMed] [Google Scholar]
- 317.Tran T, Fernandes DJ, Schuliga M, Harris T, Landells L, Stewart AG. Stimulus-dependent glucocorticoid-resistance of GM-CSF production in human cultured airway smooth muscle. Br J Pharmacol. 2004;145:123–131. doi: 10.1038/sj.bjp.0706174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Borger P, Tamm M, Black JL, Roth M. Asthma? Is it due to an abnormal airway smooth muscle cell. Am J Respir Crit Care Med. 2006;174:367–372. doi: 10.1164/rccm.200501-082PP. [DOI] [PubMed] [Google Scholar]
- 319.Lakser OJ, Dowell ML, Mitchell RW, Solway J. Dexamethasone enhances fluctuation-induced relengthening of canine tracheal smooth muscle (TSM) Proc Am Thor Soc. 2006;3:A771. [Google Scholar]
- 320.Roth M, Johnson PRA, Borger P, et al. Dysfunctional interaction of C/EBPa and the glucocorticoid receptor in asthmatic bronchial smooth muscle cells. N Engl J Med. 2004;351:560–574. doi: 10.1056/NEJMoa021660. [DOI] [PubMed] [Google Scholar]
- 321.Bonacci JV, Harris T, Wilson JW, Stewart AG. Impact of extracellular matrix and strain on proliferation of bovine airway smooth muscle. Clin Exp Pharmacol Physiol. 2003;30:324–328. doi: 10.1046/j.1440-1681.2003.03838.x. [DOI] [PubMed] [Google Scholar]
- 322.Boyle JP, Davies JM, Foster RW, Morgan GP, Small RC. Inhibitory responses to nicotine and transmural stimulation in hyoscine-treated guinea-pig isolated trachealis: an electrical and mechanical study. Br J Pharmacol. 1987;90:733–744. doi: 10.1111/j.1476-5381.1987.tb11227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Farmer SG, Hay DW, Raeburn D, Fedan JS. Relaxation of guinea-pig tracheal smooth muscle to arachidonate is converted to contraction following epithelium removal. Br J Pharmacol. 1987;92:231–236. doi: 10.1111/j.1476-5381.1987.tb11316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Small RC, Foster RW. Electrophysiology of airway smooth muscle. In: Barnes PJ, Roger IW, Thomson NC, editors. Asthma: Basic Mechanisms and Clinical Management. Academic Press; New York: 1988. p. 47. [Google Scholar]
- 325.Jiang H, Stephens NL. Isotonic relaxation of sensitized bronchial smooth muscle. Am J Physiol. 1992;262:L344–350. doi: 10.1152/ajplung.1992.262.3.L344. [DOI] [PubMed] [Google Scholar]