

Abstract
Surface immobilized poly(N-isopropyl acrylamide) (pNIPAM) is currently used for a wide variety of biosensor and biomaterial applications. A thorough characterization of the surface properties of pNIPAM thin films will benefit those applications. In this work, we present analysis of a plasma polymerized NIPAM (ppNIPAM) coating by multiple surface analytical techniques, including time-of-flight secondary ion mass spectrometry (ToF-SIMS), contact angle measurement, atomic force microscopy (AFM) and sum frequency generation (SFG) vibrational spectroscopy. ToF-SIMS data show that the plasma-deposited NIPAM polymer on the substrate is crosslinked with a good retention of the monomer integrity. Contact angle results confirm the thermoresponse of the film as observed by a change of surface wettability as a function of temperature. Topographic and force distance curve measurements by AFM further demonstrate that the grafted film shrinks or swells depending on the temperature of the aqueous environment. A clear transition of the elastic modulus is observed at 31-32°C. The change of the surface wettability and mechanical properties vs. temperature are attributed to different conformations taken by the polymer, which is reflected on the outmost surface as distinct side chain groups orienting outwards at different temperatures as measured by SFG. The results suggest that a ppNIPAM thin film on a substrate experiences similar mechanical and chemical changes to pNIPAM bulk polymers in solution. The SFG result provides evidence supporting the current theory of the lower critical solution temperature (LCST) behavior of pNIPAM.
Keywords: N-isopropylacrylamide, plasma polymerized NIPAM (ppNIPAM), thermoresponsive polymer, surface analysis, elastic modulus, ToF-SIMS, AFM, SFG
Introduction
Poly(N-isopropyl acrylamide) (pNIPAM) has been studied extensively for decades for its reversible phase transition behavior: the swollen polymer matrix in an aqueous solution collapses as the temperature is increased above the lower critical solution temperature (LCST). This transition around physiological temperature has recently found numerous applications1 including drug release,2,3 chromatography for bioseparation,4-7 biosensors,8,9 catalytic reaction control,10,11 gene delivery,12,13 protein folding14 and microactuators.15,16 When immobilized onto a flat substrate, the LCST behavior of pNIPAM leads to a temperature controlled wettability change on the surface and has been used for control of protein adsorption17,18 and cell adhesion.19-22 The recent work by Okano and colleagues to recover cell sheets from pNIPAM-grafted surfaces23,24 brings exciting applications of the grafted polymer to 2-D and 3-D tissue engineering.
In spite of the great interest in thermoresponsive polymer coatings based on pNIPAM, characterization of pNIPAM properties such as chemical,25,26 thermal,27,28 structural,29-31 mechanical,32,33 and optical properties34-36 have mostly been performed with bulk polymer in an aqueous solution. Recent studies have examined the physical and chemical properties of immobilized pNIPAM using various techniques such as IR, contact angle, SIMS and ellipsometry.19,37-40 However, a comprehensive study of the grafted pNIPAM thin film—including its mechanical properties and surface transition details—has yet to be undertaken, and will enable further surface applications of pNIPAM..
In this paper, we studied a pNIPAM-grafted surface with multiple complementary surface analytical techniques. The pNIPAM thin film is produced using plasma polymerization (ppNIPAM),41 which is a one-step, solvent free, vapor-phase technique to produce conformal, sterile, tightly adhering and ultrathin coatings. Previously, we demonstrated that ppNIPAM stimulates different cell responses depending on the surface temperature and explored its application for proteomic and cellomic chips.42,43 Here, we characterized the mechanical and chemical properties of the thin film to better understand the behavior of the polymer itself. We used time-of-flight secondary-ion mass spectroscopy (ToF-SIMS) and principal component analysis (PCA) of the ToF-SIMS data to compare the surface chemistry of ppNIPAM to conventionally synthesized pNIPAM by free radical polymerization. The temperature induced wettability change of the polymer coating was studied by contact angle measurements. To correlate the surface wettability with the polymer mechanical properties, atomic force microscopy (AFM) was applied to study the topography and modulus of the thin film under ambient conditions in water. 44,45 Finally we used sum frequency generation (SFG) vibrational microscopy to probe the species of the surface functional groups. Our study demonstrates that temperature affects the wettability, mechanical properties and conformation of the ppNIPAM thin film. These observations are consistent with those previous reported for bulk pNIPAM in solution.
Experimental Methods
Substrate Preparation
Silicon wafers were obtained from Silicon Valley Microelectronics (San Jose, CA) and diced into 1cm × 1cm squares. Calcium fluoride (CaF2) crystals were purchased from International Scientific Products (Irvington, NY). The wafers and CaF2 samples were cleaned by sonication in methylene chloride, acetone, methanol and 18 mΩ de-ionized (DI) water (Millipore, Billerica, MA) twice each for 10 minutes. The wafers and CaF2 crystals were then subjected to a 30 minutes UV/ozone oxidation treatment to remove trace organic contaminants before further use.
Plasma Polymerization
Plasma polymerization of NIPAM was carried out in a custom-built reactor using the protocol described earlier.41 In brief, the powered electrode is connected to a 13.56 MHz radio frequency power source and a manual impedance matching network. The deposition process included an 80W methane plasma deposition, followed by NIPAM plasma deposition with stepwise decreasing powers from 80W to 1W for 30 minutes with a processing pressure of 140 mTorr. The ppNIPAM-grafted surfaces were rinsed three times with cold deionized water to remove uncrosslinked molecules before use.
Preparation for Film Thickness Measurement
To measure ppNIPAM film thickness, a mask was applied on the center of a silicon chip to block deposition in a small region on the substrate.46 The mask solution was prepared by dissolving poly(lactic acid) (PLA) (average M.W. 75,000-120,000, Fisher Scientific, Fairlawn, NJ) in acetone to make a 10% (w/v) solution. Five microliters of this solution were pipetted onto silicon samples and air dried for a minimum of 5 minutes immediately preceding plasma deposition. After plasma deposition, the PLA mask was carefully removed using tweezers and the film edge was imaged by AFM.
Preparation of pNIPAM by Free Radical Polymerization
The control bulk pNIPAM was synthesized by free radical polymerization using protocols adapted from a previous publication by Schild et al.47 and was generously donated by Thomas Robey of the Bioengineering Department at the University of Washington. The reaction was carried out in benzene using azobisisobutyronitrile (AIBN, C8H12N4) as the free radical initiator to obtain the polymer. One gram of polymer was dissolved in 6 mL deionized water at room temperature to make a pNIPAM solution. Twenty microliters of solution were pipetted on a piece of silicon (8 × 8mm) and spin cast at 4000 rpm for 20 seconds to form the control surface for the ToF-SIMS study.
ToF-SIMS and Principal Component Analysis (PCA)
ToF-SIMS data acquisition were performed using a PHI Model 7200 Physical Electronics instrument (PHI, Eden Prairie, MN) equipped with an 8 keV Cs+ primary ion source, a reflectron time-of-flight mass analyzer, chevron type multichannel plate (MCP), a time-to-digital converter (TDC), and a pulsed flood gun for charge neutralization. Both positive and negative secondary ion mass spectra were collected over a mass range from m/z = 0 to 400 and analyzed with PHI Tofpak software. As both the positive and negative ion mass spectra showed similar trends, only positive ion ToF-SIMS spectra were presented in this study. The area of analysis for each spectrum was 100 μm × 100 μm, and the total ion dose used to acquire each spectrum was less than 2 × 1012 ions/cm2. The mass resolution of the secondary ion peaks in the positive ion spectra was typically between 4000 and 6000. Positive ion spectra were calibrated using CH3+, C2H3+, C3H5+, C7H7+ peaks before further analysis. Three samples were prepared for each sample type, with three spectra acquired from different locations on each sample.
ToF-SIMS spectra from different sample types (ppNIPAM or pNIPAM) were compared using principal component analysis (PCA), which captures the linear combination of peaks that describe the majority of variation in a dataset (the principle components, PCs). A “complete” peak set using all of the peaks with intensities > 3 times the background in the 0-400 m/z region was generated from all of the spectra. The peaks were then normalized to the total ion intensity to account for fluctuations in secondary ion yield between different spectra and all the datasets were mean-centered. PCA was then used to analyze the positive ToF-SIMS spectra, and was performed using PLS Toolbox v. 2.0 (Eigenvector Research, Manson, WA) for MATLAB (MathWorks, Inc., Natick, MA).
Although a detailed description of PCA is not warranted here, the interested reader is referred to a more complete discussion of PCA by Jackson48 or Wold.49 Briefly, the input to PCA is the “complete” peak set constructed using all of the major peaks from each sample type (ppNIPAM or pNIPAM). From this input, an output of both “scores” and “loadings” plots were obtained to compare the main differences of the samples and contribution of the chemistries to the difference. The scores describe the relationship or spread of the samples. They represent the amount of the PC in each sample and are the projection of the samples onto the PC axes. Loadings plots describe which variables are responsible for the differences seen within the dataset.
Contact Angle Measurement
Surface wettability was evaluated by a captive air bubble method. This was done by measuring static contact angles of an air bubble in contact with the samples submerged in pure water using a contact angle goniometer (Ramé-Hart, Mountain Lakes, NJ). The samples were allowed to equilibrate for at least an hour in the water bath at the appropriate temperature before any measurements were taken.
AFM Measurements
All AFM measurements were performed using a PicoScan™ microscope (Molecular Imaging (MI), Phoenix, AZ) on ppNIPAM deposited onto silicon chips. Samples were hydrated in DI-water for an hour and then quickly transferred to a Teflon cell containing deionized water for measurement. The cell was mounted on a MI thermal stage controlled by a temperature controller (Model 321, Lake Shore Cryotronics, Westerville, OH) and data were collected in the range of 25°C to 37°C. The samples were allowed to equilibrate for at least half an hour at each temperature before the measurements were taken.
Topography images were obtained in the acoustic AC mode using rectangular silicon cantilevers with integrated sharp tips, having resonance frequencies in the range of 260 –410 kHz (PPP-NCH type, Nanosensors, Neuchatel, Switzerland). The boundary between the plasma polymer coated and uncoated regions were aligned directly below the AFM tip using an add-on CCD camera. The images were analyzed using Image J software (http://rsb.info.nih.gov/ij/) to get height histograms, average step height and roughness information.
The force displacement curves were taken with standard V-shaped silicon nitride cantilevers (Nanoprobe™ AFM tips, Type NP-S, Digital Instruments, Santa Barbara, CA). The tip spring constant was 0.06 N/m according to the manufacturer's specifications. The tip radius was measured to be 50 – 60 nm by scanning an Ultrasharp calibration grating that contains an array of sharp tips (TGT01 model, Silicon-MDT, Moscow, Russia). These blunt tip cantilevers were selected to satisfy conditions for elastic mechanical contact.50 Five to ten force displacement curves were acquired at randomly selected regions at each temperature using an approach-retract frequency of 0.5Hz.
To calibrate the detector signal (in volts), measurements of the force displacement curves were taken on a reference surface, which is assumed to have infinitely high stiffness and no deformation from pressing. In our study, a piece of clean silicon wafer was used as the reference surface, whose modulus is 160GPa and several orders of magnitude higher than the polymer.
The measured force-displacement curves were converted to plots relating indentation depth vs. loading force according to previously described methods.51 The indentation of the cantilever under a given load was fit to the Hertz model for indentation of a sphere on an elastic solid and used to calculate the elastic modulus of the surface thin film.51,52 The indentation depth was controlled to no greater than 20% of the film thickness to minimize the influence of the solid support.50,53,54 To exclude the possibility of polymer plastic deformation, tens of force displacement curves on the same location were recorded and identical curves were obtained, indicating full recovery of surfaces for each indentation cycle.
SFG Measurements
Surface vibrational spectra were obtained by sum frequency generation (SFG) vibrational spectroscopy to identify the functional groups present at the outermost surface. Details of the technique can be found in some recent papers.55-57 The specific laser system used for our study was described by Kim et al.58 Briefly, surface vibrational spectra of ppNIPAM deposited onto CaF2 crystals were collected by overlapping visible and tunable infrared laser pulses on the polymer surface and measuring the induced sum-frequency signal. The visible laser pulse (ωvis) of 532 nm light was generated by frequency doubling the 1064 nm fundamental output from a passive-active mode-locked Nd:YAG laser (Leopard, Continuum, Santa Clara, CA) using an optical parametric generation/optical parametric amplification (OPG/OPA) stage (Laservison, Bellevue, WA). The untreated radiation has a pulse width of 20 ps and a 20Hz repetition rate. Using the fundamental radiation to pump the stage crystals, the infrared laser pulse (ωir) is generated from the OPG/OPA stage and is tunable from 2000 to 4000 cm−1. The sum frequency output signal (ωsum = ωvis + ωir) was collected by a gated integrator and photon counting system. In this study, the spectra were collected using a ssumsvispIR polarization combination. The SFG spectra in air were taken by aligning the polymer face of the sample toward the light source with the angle of incident light set at 45° with respect to the surface normal. For studies in water, the polymer film was immersed in pure water and the incident light was projected through the crystal to the polymer film. Light reflected from the solid-liquid interface was collected after traveling twice through the crystal. The incident light angle and polarization were the same as those used for the air spectra. The collected spectra cover the CH-stretch region from 2800-3050 cm−1 in 5 cm−1 increments. For a given condition, SFG measurements were repeated on at least three different samples with 1-3 spots probed on each sample. For each sample spot 4-5 scans were performed with 100 shots/data point to increase the signal-to-noise ratio. The collected data were averaged to produce the final spectra presented here.
Results and Discussions
ToF-SIMS Analysis of Surface Chemistry
Previously we presented an ESCA analysis on ppNIPAM indicating that its surface chemical composition is close to that predicted by the monomer's stoichiometry.41 To better assess whether the monomer structure is retained after plasma deposition, ToF-SIMS is used as a complementary technique, as it yields information regarding molecular species at interfaces. A representative ToF-SIMS positive ion spectrum of ppNIPAM in the range from m/z = 0 to 200 is shown in Figure 1(a) as very few peaks of interest are beyond this range. The molecular fragment of the NIPAM monomer (C6H12NO+) is clearly evident at m/z 114.093. Fragments at m/z 43.055 and 58.065 are indicative of the monomer fragments C3H7+ and C3H8N+ N respectively. The ToF-SIMS spectra of pNIPAM by free radical polymerization also contain these peaks. Observation of the monomer molecular fragments supports the hypothesis that at least some of the monomer units remain intact on the surface following plasma deposition.
Figure 1.
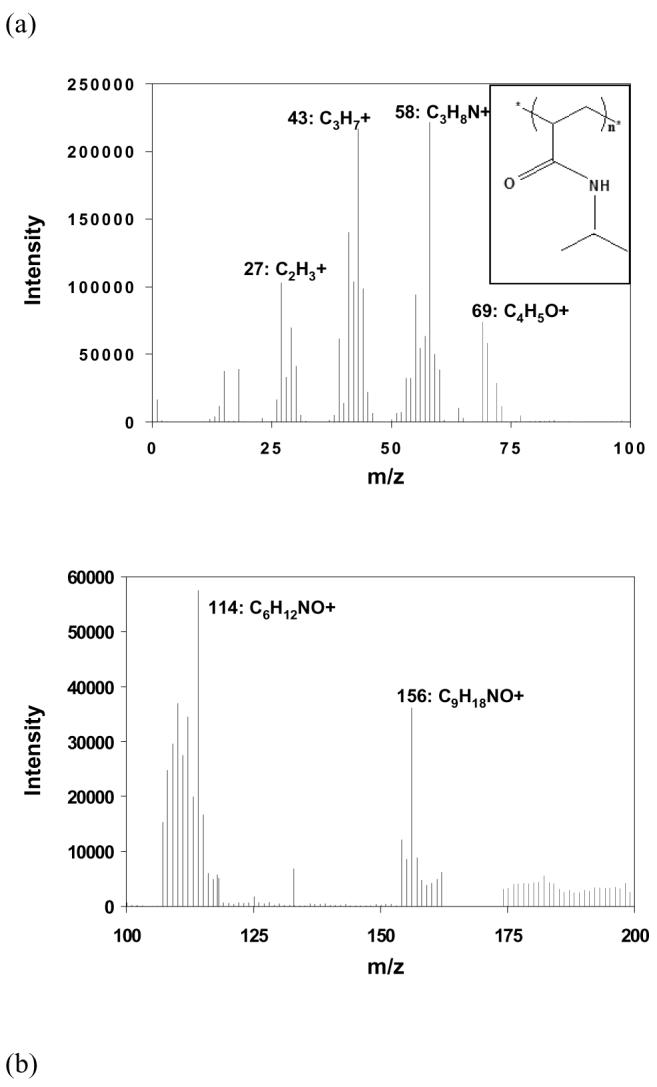

(a) Characteristic ToF-SIMS positive ion spectrum acquired on a ppNIPAM surface. Important peaks corresponding to the monomer fragments are labeled. The theoretical linear polymer structure is shown in the inset. ToF-SIMS spectra of ppNIPAM and conventional pNIPAM prepared by free radical polymerization were subjected to principal component analysis (PCA) to compare their chemistry. The PCA results of positive ion spectra are presented as scores (b) and loadings (c) plots. The differences between the two surfaces are likely produced from crosslinking in the plasma film, as both films show evidence of significant retention of the NIPAM monomer structure.
We next compare the surface chemistry of ppNIPAM to that of conventionally synthesized pNIPAM. ESCA analysis reveals the similarity of ppNIPAM to pNIPAM in both the elemental composition and chemical bonding (data not shown). However, a direct comparison of ppNIPAM and pNIPAM SIMS spectra is difficult, as SIMS spectra from each type contain hundreds of peaks in the 0-400 m/z range. Principal component analysis (PCA) is used to aid in the interpretation of spectra by identifying related variables and focusing on the differences between spectra.48,49 Figure 1(b) shows a scores plot of principal component 1 (PC1), which captures 97% of the variance in the data, vs. the spectrum number. Examination of Figure 1(b) shows that ppNIPAM and pNIPAM samples are distinctly grouped from each other: ppNIPAM samples cluster above the x-axis and pNIPAM samples cluster below the x-axis. To appreciate the reason for these differences, we must inspect the loadings plot for PC 1.
Figure 1(c) represents the loadings from PC1. Peaks are shown only in the mass range from m/z = 0 to 200 as few peaks of interest are observed beyond this range. Each of the positive peaks in the PC1 loadings plot corresponds to samples in the scores plot that load positively. Of key interest in the loadings plot is the absence of molecular fragments arising from the intact NIPAM moiety in the negative loadings corresponding to conventional pNIPAM. This indicates that the differences between ppNIPAM and pNIPAM surfaces are not due to the absence of monomer units. Instead, when comparing the PC1 loadings plot to the PC1 scores, we find that separation of ppNIPAM from pNIPAM is due to peaks loaded positively with ppNIPAM, including 74.062 (C3H8NO+), 97.080 (C5H9N2+) and 156.144 (C9H18NO+). These peaks are likely produced by crosslinking or other reactions occurring during the plasma deposition process as they do not contain the monomer structure. This is expected considering the numerous reactive species created in the energetic plasma process.59 The fact that hydrocarbon peaks at 27.024 (C2H3+) and 43.055 (C3H7+) loaded negatively with pNIPAM indicate the crosslinking may be occurring through the side-chain propyl groups. Therefore, a comparison of the positive ion spectra of the two surfaces indicates that the NIPAM monomer units are immobilized onto the substrate in the plasma film with high retention of the monomer structure in spite of the concern of significant monomer fragmentation in the plasma state.59 Two hypotheses may explain the maintenance of the monomer integrity: 1) low RF power (as used in our process) reduces monomer fragmentation in plasmas;59 and 2) condensation of polymerizable molecules (e.g. molecules with unsaturated backbones) in conjunction with plasma deposition onto the surface helps with the formation of predictable chemical functionalities that resemble the monomer.60-62 That the monomer structure is retained suggests that the phase transition properties of the ppNIPAM thin film will be retained as well. However, as both ESCA and ToF-SIMS are ultrahigh vacuum techniques, they cannot probe the phase transition behavior in situ. Thus we employ several other techniques in the rest of the paper to directly study the structure and behavior of ppNIPAM films below and above the LCST in water.
Change in Surface Wettability by Contact Angles
To directly evaluate the wettability of the ppNIPAM film, a captive air bubble contact angle of the ppNIPAM film surface was measured in water. A summary of the advancing contact angles measured on ppNIPAM coated silicon and bare silicon is found in Table 1. The contact angles on ppNIPAM change from 34 ± 1.0° at 20°C to 40 ± 0.5° at 45°C (n ≥ 11), which are comparable with measurements by Akiyama et al. using a silane grafted pNIPAM thin film.39 Similar measurements taken on untreated bare silicon show no statistical change in the surface contact angles over the same temperature range (41 ± 0.6° at 20°C to 40 ± 0.6° at 45°C). The significantly lower contact angle at room temperature on ppNIPAM indicates that the plasma film becomes more hydrated when the temperature is below the LCST. Thus, the surface wettability change with temperature confirms the thermoresponsive behavior of ppNIPAM film, as expected from the intact monomer structure observed in ToF-SIMS.
Table 1.
Advancing contact angles on ppNIPAM coated silicon and bare silicon measured by trapped air bubbles in water. A significant difference is noticed on ppNIPAM when measurements are taken at the two temperatures, but not on the control silicon substrate.
| PpNIPAM | Silicon | |
|---|---|---|
| 20°C | 34° ± 1.0° | 41° ± 0.6° |
| 40°C | 40° ± 0.5° | 40° ± 0.6° |
Surface Topography by AFM
Bulk pNIPAM polymer demonstrates a transition in volume and modulus through its LCST.32,33 To test whether this is also the case with the plasma deposited thermoresponsive film, we studied the surface topographic and mechanical properties of ppNIPAM using AFM. Figure 2 shows the surface topography of a ppNIPAM film imaged in water by AFM at 25°C and 37°C. The surface is fairly smooth at 37°C with a root-mean-square (RMS) roughness of 3.5 ± 0.5 nm over a 5 μm × 5 μm scan area. When the same size image is taken at 25°C, the RMS roughness increases to 5.3 ± 1.1 nm. It has been previously observed that when ppNIPAM hydrates at room temperature, it creates nano-cavities on the surface,63 that probably account for the slight increase in surface roughness observed in the swollen state. This may suggest an inhomogenous crosslink density over the surface at a nanometer scale causing some areas to swell less than others.
Figure 2.

AFM images of ppNIPAM in water at 25°C (a) and 37°C (b) acquired in the acoustic AC mode. The surfaces are fairly smooth in both cases with a slightly higher root-mean-square roughness at 25°C than at 37°C. Notice the two images have different z scales.
To determine the film thickness in water at different temperatures, ppNIPAM steps are created by plasma deposition over a partially masked sample and then removing the mask afterwards. Two characteristic AFM images of the same area across a ppNIPAM step with the height histograms and section analysis are shown in Figure 3(a) and (d). The height histograms obtained for each image give two characteristic peaks corresponding to the substrate and coating heights (gray areas in Figure 3(b) and (e)). By subtracting the lower peak value from the upper, the film thickness is obtained. To accurately estimate the peak location in the histogram, a Gaussian model is used to fit each peak in the histograms (black curves in Figure 3(b) and (e)). The distances between the two peaks are 73.7 nm and 63.7 nm at 25°C and 37°C respectively, indicating a 10 nm decrease in film thickness above the LCST compared to below the LCST. The section analysis on individual scan lines (Figure 3(c) and (f)) in the two images confirms the values obtained from the histograms. The broadening of the height histogram at 25°C (Figure 3(b)) also indicates higher roughness and scattering of the height distribution. Identical histogram analyses were performed on 4 samples of different deposition batches and 3 spots on each sample. The results are summarized in Figure 3(g). Although the thickness of deposited polymer varies from batch to batch, decreased film thickness is observed in all the samples and areas tested as temperature increases from below to above the LCST, with the error bars in Figure 3(g) resulting from variations in film thickness on individual samples at each temperature. The batch-to-batch variation of the film thickness at the same temperature may arise from variation in the plasma polymerization process.
Figure 3.

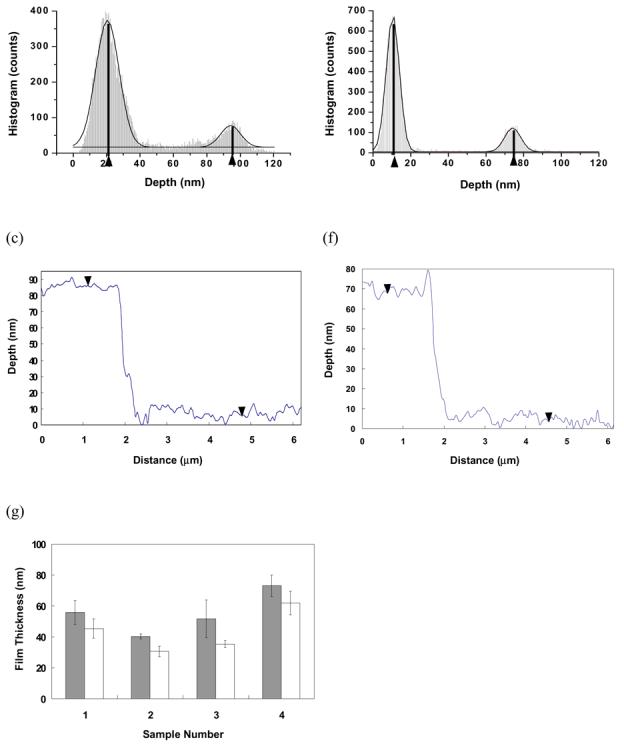
AFM images of a ppNIPAM step on a silicon surface at 25°C (a) and 37°C (d). The corresponding height histograms (gray area) at 25°C (b) and 37°C (e) show two main heights, representing the substrate and plasma polymer surfaces respectively. Each of the peaks is fitted to a Gaussian model (black curve) and the centers of the peaks are denoted by the triangular cursors. The step heights are obtained by subtracting the lower cursor position from the upper, giving a plasma polymer thickness of 73.7 nm at 25°C and 63.7 nm at 37 °C for the scanned region. Section analyses on individual scan lines in each image are shown in (c) for 25°C and (f) for 37°C, which yields step heights of 74.2 nm and 63.1 nm respectively. Film thickness measured on 4 different samples and 3 spots on each samples is summarized in (g) using the histogram analsyis. The gray bar and white bar are film thickness measured at 25°C and 37°C respectively and thicker film is noticed for all measurements at 25°C.
Recently, Akiyama et. al. reported that temperature-controlled cell adhesion and detachment from pNIPAM surface is dependent on the thickness of the thermoresponsive film.39 However, in our cell culture study no obvious difference was observed on different batches of coatings.43 This suggests that the variation in film thickness we observed arise mainly from the thickness variations in the underlying adhesion-promoting layer formed during the high power deposition, not thickness variations in the top functional ppNIPAM coating
Mechanical Property by Force Displacement Curves
Since the ppNIPAM film thickness/volume changes with temperature, it is expected that the polymer mechanical properties also vary.32 Atomic force microscopy has been shown as a unique tool to probe the micromechanical properties of thin film coatings.64-67 Figure 4(a) shows sample AFM force vs. distance curves obtained on bare silicon at room temperature and on ppNIPAM surface at 25°C and 37°C in pure water. In this figure, the cantilever deflection is presented as a function of the z-piezo displacement. It is clearly seen that the cantilever deflects less for the same z-piezo movement at 25°C than at 37°C, indicating the polymer is softer and more swollen at 25°C.
Figure 4.
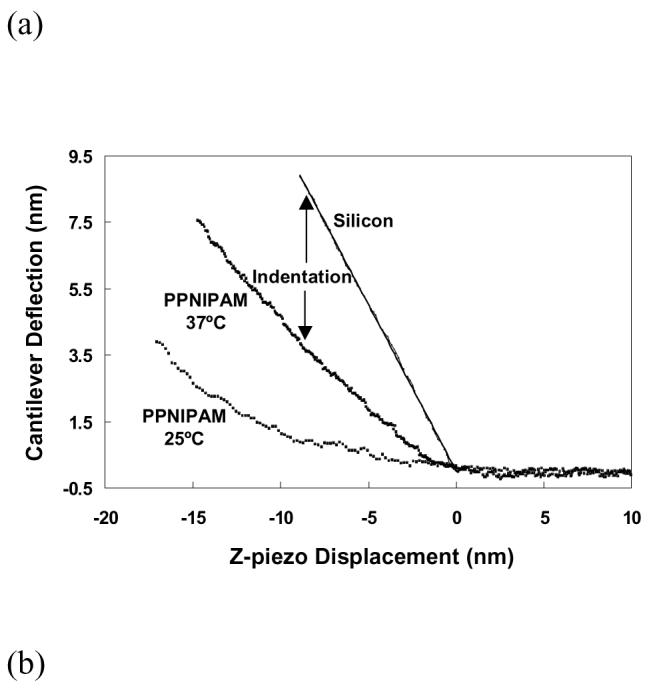

(a) Force displacement curves by AFM on ppNIPAM coatings in pure water at 25°C (bottom line) and 37°C (middle line) and on silicon (top line). The same z-piezo displacement results in a smaller cantilever deflection on the ppNIPAM surface in comparison to the hard silicon surfaces because of elastic indentation. The plasma polymerized NIPAM surface appears to be stiffer at 37°C than at 25°C as there is less deflection from the same z-piezo movement. (b) Plot of indentation vs. loading force obtained on ppNIPAM at 25°C (open circles) and 37°C (solid squares). Each data set has been modeled by the Hertz theory for a sphere indenting a flat surface (solid lines). The optimal fitting parameters from the Hertz theory are used to calculate Young's modulus at each temperature. The corresponding log-log presentation of (b) is given in (c) for easier comparison of the measured data with the theoretical fittings. To determine the transition temperature, Young's modulus of ppNIPAM are calculated from force displacement curves acquired at 25°C to 37°C with 1°C temperature increment. The results are demonstrated in (d) with 5 to 10 replicates for each data point. A clear transition in the surface modulus is noticed at around 31-32°C. The open square at 25°C shows recovery of the film to the swollen state after cooling the sample down from 37°C to 25°C.
To quantitatively compare the thin film mechanical properties, the force-displacement data (Figure 4(a)) are converted to plots relating the indentation depth as a function of the loading force (squares and circles in Figure 4(b)). The elastic moduli are obtained from best fitting to the indentation vs. load data using the Hertz model (thin solid lines in Figure 4(b)). The parameters used are R= 53 nm and ν= 0.5 assuming elastic deformation. The spring constant is 0.06 N/m taken from the manufacturer's specifications. For easier comparison of the fitted curve and experimental data, the same plot is shown in the log-log presentation in Figure 4(c). It is observed that the experimental data are described satisfactorily using a simple Hertz model for the force displacement measurement taken at 37°C (solid squares in Figure 4(b) and 4(c)). However at 25°C, the fitted curve deviates from the experimental measurement when the indentation depth goes beyond 12-13 nm. As the Hertz model assumes that the two contacting surfaces have homogeneous elastic properties in the z direction, this deviation is likely due to the substrate effect that the simple Hertz model cannot explain as reported by Domke and Radmacher.54 Using the optimal fitting parameters, the elastic moduli are calculated to be 185.2 kPa at 25°C and 1.592 MPa at 37°C for the two curves shown in Figure 4(a). Thus, the local stiffness of ppNIPAM is more than 8 times higher in the collapsed state at 37°C than the swollen state at 25°C.
In an aqueous environment, bulk polymer chains of pNIPAM are believed to adopt a random coil configuration below the LCST, and a more compact globular configuration above the LCST. This conformational transition accounts for changes in both polymer volume and mechanical properties.33 The AFM study in our work demonstrates that the ppNIPAM thin film is still able to transition between the collapsed and swollen state as the temperature is changed. However, when comparing the mechanical characteristics of the ppNIPAM thin film produced in our system with those of the pNIPAM gel synthesized by free radical polymerization in the bulk state, some obvious differences are noticed. Matzelle et al. recently reported a swelling ratio of eight and elastic moduli of 2.8 kPa and 183 kPa at 25°C and 35°C respectively for a lightly crosslinked pNIPAM gel (100:1 monomer to crosslinker molar ratio) cast onto mica.63 The surface immobilized ppNIPAM however, has a higher moduli (185kPa and 1.592MPa) and lower swelling ratio (< 1). This would seem to support our conclusion from PCA analysis of the ToF-SIMS data that the ppNIPAM film is more cross-linked, as crosslinks have been shown to restrict the magnitude of the swelling ratio and reinforce the polymer mechanical properties.32,33,68,69 Nevertheless, the moduli below and above the LCST obtained by us still fall in the soft hydrogel range of kilopascals to a few megapscals.70
To determine at which temperature the transition in surface stiffness happens, force-distance curve measurements were taken at 1°C increments from 24°C to 37°C. Five to ten randomly selected spots were probed at each temperature with the average elastic moduli summarized in Figure 4(d). The surface stiffness increases significantly around 31°C −32°C, close to the transition temperature measured by Pan et al. using an AFM modulation measurement.41 To confirm that the stiffness transition behavior is reversible, the surface was cooled down to 25°C again and the measured modulus is shown as an open square in Figure 5(d). It is clearly observed that the film rehydrates and swells after cooling to below its LCST. The relatively larger deviation for measurements taken above the transition temperature may arise from reduced sensitivity to modulus differences in the range of the higher modulus above the LCST.
Figure 5.
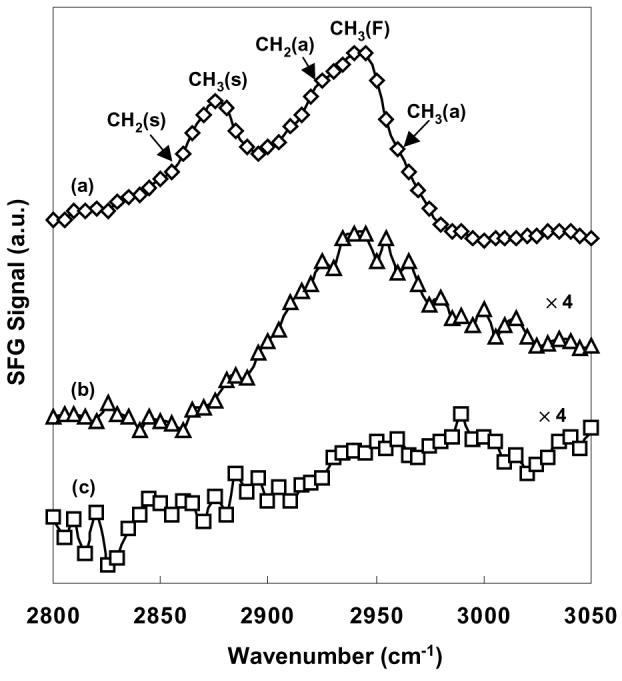
SFG spectra of ppNIPAM in (a) air at room temperature, (b) pure water at 37°C and (c) pure water at room temperature (25°C). The spectra obtained in water are enhanced by a multiple of four to counteract the low signal intensity in the water measurements brought on by the absorption of the IR signal by water. The lines are guides to the eyes and the peak assignments are labeled in the figure.
Surface Group Rearrangement by SFG
To determine whether the surface chemistry of ppNIPAM films changes in situ at different temperatures, we next analyzed the ppNIPAM film with SFG vibrational spectroscopy. SFG selection rules allow the detection of chemical species that are ordered and lack inversion symmetry, thus allowing the top few atomic layers of the polymer surface to be probed directly.55-57,71 Figure 5 shows characteristic SFG spectra for the CH stretching region taken in water at room temperature and 37°C. For comparison, the spectrum of ppNIPAM in air at room temperature is plotted in the same figure. As water absorption tends to attenuate the signal, the spectra in water are multiplied by a factor of four. Figure 5(a) shows the spectrum of fully dehydrated ppNIPAM film in air where two peaks are predominant and centered at 2875 and 2940 cm−1. These two peaks are assigned as the CH3(s) stretch and Fermi resonance between the CH3 stretch and bending mode. There are also three shoulder peaks measured at approximately 2855 cm−1, 2925 cm−1and 2960 cm−1, which are assigned as the CH2(s), CH2(a) and CH3(a) stretches. The strong CH3 stretch peak indicates the presence of ordered isopropyl groups tilting towards the surface normal from the polymer side chain in the dehydrated state. The CH2 stretch in the spectrum may arise from the backbone, or from random crosslinking induced by the plasma deposition process. This is expected given that SFG spectra provide averaged chemical information over a 1mm2 spot size. When the spectrum is acquired in water at 37°C, a broad peak is observed between 2900 and 2980 cm−1, which is associated with the CH2(a) stretch, CH3 Fermi resonance and CH3(a) stretch peaks. This suggests organization of the hydrophobic isopropyl side chain to the aqueous environment above the LCST. In contrast, we find no evidence of these peaks at room temperature. This indicates disordering of the hydrophobic isopropyl groups away from the surface normal, either orienting to the interior of the polymer or in the plane of the surface. It has been hypothesized that below the LCST, the amide groups will extend into the aqueous environment to participate in hydrogen bonding, although we are not able to directly observe this as the characteristic peaks are not detected in this region.
The results from our SFG characterization of ppNIPAM films in situ above and below the LCST support the hypothesis that ppNIPAM films retain the behavior characteristic of their conventionally formed counterparts. The structural rearrangement of the ppNIPAM polymer side chain provides evidence to support the currently proposed mechanism of LCST behavior of thermoresponsive polymers.33,72 It has been proposed, that below the LCST, well-hydrated pNIPAM chains take a random coil configuration in an aqueous environment with amide groups forming hydrogen bonds with water, but a more compact globular configuration and an increased hydrophobic interaction accompanied by a sudden dehydration above its LCST. Associated with the polymer conformational changes, we directly observe the re-orientation of the surface chemical functionalities using SFG. In the dehydrated state, the polymer surface orients the hydrophobic group outwards to maximize hydrogen bonding underneath the surface.70,73 In the aqueous environment at room temperature, on the other hand, the hydrophobic isopropyl groups appear to bend inwards to allow hydrogen bonding of the polar amide groups with water to lower surface energy. Above the LCST in water the surface rearranges to resemble the one in air due to an entropy-driven process to free the bound water molecules and re-form intramolecular hydrogen bonds under the surface.1,72,74 This exposure of surface chemical functionalities of different polarities may also help to explain the change of surface contact angles as a function of temperature. This preliminary examination of the C-H stretch region shows promising results in that we detect rearrangement of the isopropyl groups, although more studies of the amide group transition and water structure on ppNIPAM are needed to provide a more complete picture of the mechanism behind LCST behavior.
Conclusion
We report in this paper characterization of a plasma deposited NIPAM thin film using multiple, complementary techniques. The ppNIPAM film is found to resemble the conventional bulk polymer in surface wettability, structural and elasticity transition through the LCST near 31-32°C. The surface wettability as well as polymer thickness decreases while the film elastic modulus increases when water temperature goes from 25 to 35°C. However, the magnitude of moduli and swelling ratio differs between ppNIPAM and pNIPAM. This may arise from crosslinking in the plasma polymer as suggested by the ToF-SIMS. Associated with the wettability and mechanical property transition, re-orientation of the side-chain groups is directly probed by SFG measurements. Disappearance of the features attributed to methyl groups at room temperature suggests different hydrogen bonding and water structure at the surfaces. The value of SFG to observe transitions in polymers at the nanometer scale is confirmed in this study. Direct molecular measurement of subtle but significant surface changes as a function of temperature in a NIPAM polymer were made for the first time here. Future experiments will further investigate this hypothesis by detecting signals in the hydroxyl and amide stretch region to further understand the orientation of those groups.
Acknowledgements
This research was supported by NSF-Engineering Research Center program grant # ERC-9529161 to the University of Washington Engineered Biomaterials group (UWEB) and NIBIB grant EB-002027 to the National ESCA and Surface Analysis Center for Biomedical Problems (NESAC/BIO). The SFG study was supported by the Director, Office of Science, Office of Basic Energy Sciences, Divisions of Materials Science and Chemical Science of the U.S. Department of Energy under Contract No. DE-AC03-76SF00098. JRH was supported by NIH Biomaterials grant. The authors thank Rosnita Talib and Sally McArthur at the Department of Engineering Materials at University of Sheffield as well as Winston Ciridon and Dr. Vickie Pan for expertise.
References
- 1.Roy I, Gupta MN. Chem. Biol. 2003;10:1161–1171. doi: 10.1016/j.chembiol.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 2.Coughlan DC, Quilty FP, Corrigan OI. J. Control. Release. 2004;98:97–114. doi: 10.1016/j.jconrel.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 3.Kost J, Langer R. Adv. Drug Deliv. Rev. 2001;46:125–148. doi: 10.1016/s0169-409x(00)00136-8. [DOI] [PubMed] [Google Scholar]
- 4.Kikuchi A, Okano T. Macromol. Symp. 2004;207:217–227. [Google Scholar]
- 5.Kobayashi J, Kikuchi A, Sakai K, Okano T. Anal. Chem. 2003;75:3244–3249. doi: 10.1021/ac026364m. [DOI] [PubMed] [Google Scholar]
- 6.Kobayashi J, Kikuchi A, Sakai K, Okano T. Anal. Chem. 2001;73:2027–2033. doi: 10.1021/ac0013507. [DOI] [PubMed] [Google Scholar]
- 7.Fong RB, Ding ZL, Hoffman AS, Stayton PS. Biotechnol. Bioeng. 2002;79:271–276. doi: 10.1002/bit.10315. [DOI] [PubMed] [Google Scholar]
- 8.Malmstadt N, Yager P, Hoffman AS, Stayton PS. Anal. Chem. 2003;75:2943–2949. doi: 10.1021/ac034274r. [DOI] [PubMed] [Google Scholar]
- 9.Zhu QZ, Yang HH, Li DH, Chen QY, Xu JG. Analyst. 2000;125:2260–2263. doi: 10.1039/b005748g. [DOI] [PubMed] [Google Scholar]
- 10.Nagayama H, Maeda Y, Shimasaki C, Kitano H. Macromol. Chem. Phys. 1995;196:611–620. [Google Scholar]
- 11.Stayton PS, Shimboji T, Long C, Chilkoti A, Chen G, Harris JM, Hoffman AS. Nature. 1995;378:472–474. doi: 10.1038/378472a0. [DOI] [PubMed] [Google Scholar]
- 12.Twaites BR, Alarcon CD, Cunliffe D, Lavigne M, Pennadam S, Smith JR, Gorecki DC, Alexander C. J. Control. Release. 2004;97:551–566. doi: 10.1016/j.jconrel.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 13.Kurisawa M, Yokoyama M, Okano T. J. Control. Release. 2000;68:1–8. doi: 10.1016/s0168-3659(00)00246-7. [DOI] [PubMed] [Google Scholar]
- 14.Chen YJ, Huang LW, Chiu HC, Lin SC. Enzyme Microb. Technol. 2003;32:120–130. [Google Scholar]
- 15.Luo QZ, Mutlu S, Gianchandani YB, Svec F, Frechet JMJ. Electrophoresis. 2003;24:3694–3702. doi: 10.1002/elps.200305577. [DOI] [PubMed] [Google Scholar]
- 16.Harmon ME, Tang M, Frank CW. Polymer. 2003;44:4547–4556. [Google Scholar]
- 17.Huber DL, Manginell RP, Samara MA, Kim BI, Bunker BC. Science. 2003;301:352–354. doi: 10.1126/science.1080759. [DOI] [PubMed] [Google Scholar]
- 18.Grabstain V, Bianco-Peled H. Biotechnol. Prog. 2003;19:1728–1733. doi: 10.1021/bp034161t. [DOI] [PubMed] [Google Scholar]
- 19.Ista LK, Mendez S, Perez-Luna VH, Lopez GP. Langmuir. 2001;17:2552–2555. [Google Scholar]
- 20.Ratner BD, Cheng XH, Wang YB, Hanein Y, Bohringer K. Abstr. Pap. Am. Chem. Soc. 2003;225:U582–U582. [Google Scholar]
- 21.Liu HC, Ito Y. J. Biomed. Mater. Res. Part A. 2003;67A:1424–1429. doi: 10.1002/jbm.a.20004. [DOI] [PubMed] [Google Scholar]
- 22.Canavan HE, Cheng X, Graham DJ, Ratner BD, Castner DG. Langmuir. 2004 [Google Scholar]
- 23.Tsuda Y, Kikuchi A, Yamato M, Sakurai Y, Umezu M, Okano T. J. Biomed. Mater. Res. Part A. 2004;69A:70–78. doi: 10.1002/jbm.a.20114. [DOI] [PubMed] [Google Scholar]
- 24.Shimizu T, Yamato M, Isoi Y, Akutsu T, Setomaru T, Abe K, Kikuchi A, Umezu M, Okano T. Circ.Res. 2002;90:E40–E48. doi: 10.1161/hh0302.105722. [DOI] [PubMed] [Google Scholar]
- 25.Maeda Y, Higuchi T, Ikeda I. Langmuir. 2001;17:7535–7539. [Google Scholar]
- 26.Katsumoto Y, Tanaka T, Sato H, Ozaki Y. J. Phys. Chem. A. 2002;106:3429–3435. doi: 10.1021/jp0124903. [DOI] [PubMed] [Google Scholar]
- 27.Boutris C, Chatzi EG, Kiparissides C. Polymer. 1997;38:2567–2570. [Google Scholar]
- 28.Cai WS, Gan LH, Tam KC. Colloid Polym. Sci. 2001;279:793–799. [Google Scholar]
- 29.Reichelt R, Schmidt T, Kuckling D, Arndt KF. Macromol. Symp. 2004;210:501–511. [Google Scholar]
- 30.Chern JM, Lee WF, Hsieh MY. J. Appl. Polym. Sci. 2004;92:3651–3658. [Google Scholar]
- 31.Tam KC, Wu XY, Pelton RH. Polymer. 1992;33:436–438. [Google Scholar]
- 32.Hirotsu S. J. Chem. Phys. 1991;94:3949–3957. [Google Scholar]
- 33.Shibayama M, Morimoto M, Nomura S. Macromolecules. 1994;27:5060–5066. [Google Scholar]
- 34.Suzuki Y, Nozaki K, Yamamoto T, Itoh K, Nishio I. J. Chem. Phys. 1992;97:3808–3812. [Google Scholar]
- 35.Takeoka Y, Watanabe M. Adv. Mater. 2003;15:199–201. [Google Scholar]
- 36.Reese CE, Mikhonin AV, Kamenjicki M, Tikhonov A, Asher SA. J. Am. Chem. Soc. 2004;126:1493–1496. doi: 10.1021/ja037118a. [DOI] [PubMed] [Google Scholar]
- 37.Bergbreiter DE, Tao GL, Franchina JG, Sussman L. Macromolecules. 2001;34:3018–3023. [Google Scholar]
- 38.Bohanon T, Elender G, Knoll W, Koberle P, Lee JS, Offenhausser A, Ringsdorf H, Sackmann E, Simon J, Tovar G, Winnik FM. J. Biomater. Sci.-Polym. Ed. 1996;8:19–39. doi: 10.1163/156856297x00551. [DOI] [PubMed] [Google Scholar]
- 39.Akiyama Y, Kikuchi A, Yamato M, Okano T. Langmuir. 2004;20:5506–5511. doi: 10.1021/la036139f. [DOI] [PubMed] [Google Scholar]
- 40.Takei YG, Aoki T, Sanui K, Ogata N, Sakurai Y, Okano T. Macromolecules. 1994;27:6163–6166. [Google Scholar]
- 41.Pan YV, Wesley RA, Luginbuhl R, Denton DD, Ratner BD. Biomacromolecules. 2001;2:32–36. doi: 10.1021/bm0000642. [DOI] [PubMed] [Google Scholar]
- 42.Wang YB, Cheng XC, Hanein Y, Shastry A, Denton DD, Ratner BD, Bohringer KF. The 12th International Conference on Solid-State Sensors and Actuators (Transducers'03); Boston, MA. 2003. [Google Scholar]
- 43.Cheng XH, Wang YB, Hanein Y, Bohringer KF, Ratner BD. J. Biomed. Mater. Res. Part A. 2004;70A:159–168. doi: 10.1002/jbm.a.30053. [DOI] [PubMed] [Google Scholar]
- 44.Sheiko SS. Advances in Polymer Science. 2000;151:61–174. [Google Scholar]
- 45.Reich Z, Kapon R, Nevo R, Pilpel Y, Zmora S, Scolnik Y. Biotechnol. Adv. 2001;19:451–485. doi: 10.1016/s0734-9750(01)00077-5. [DOI] [PubMed] [Google Scholar]
- 46.Hartley PG, Thissen H, Vaithianathan T, Griesser HJ. Plasmas Polym. 2000;5:47–60. [Google Scholar]
- 47.Schild HG, Tirrell DA. Langmuir. 1991;7:665–671. [Google Scholar]
- 48.Jackson JE. J. Qual. Technol. 1980;12:201–213. [Google Scholar]
- 49.Wold S, Esbensen K, Geladi P. Chemometrics Intell. Lab. Syst. 1987;2:37–52. [Google Scholar]
- 50.Tsukruk VV, Sidorenko A, Gorbunov VV, Chizhik SA. Langmuir. 2001;17:6715–6719. [Google Scholar]
- 51.Chizhik SA, Huang Z, Gorbunov VV, Myshkin NK, Tsukruk VV. Langmuir. 1998;14:2606–2609. [Google Scholar]
- 52.Tsukruk VV, Huang Z, Chizhik SA, Gorbunov VV. J. Mater. Sci. 1998;33:4905–4909. [Google Scholar]
- 53.Harmon ME, Kuckling D, Pareek P, Frank CW. Langmuir. 2003;19:10947–10956. [Google Scholar]
- 54.Domke J, Radmacher M. Langmuir. 1998;14:3320–3325. [Google Scholar]
- 55.Chen Z, Shen YR, Somorjai GA. Annu. Rev. Phys. Chem. 2002;53:437–465. doi: 10.1146/annurev.physchem.53.091801.115126. [DOI] [PubMed] [Google Scholar]
- 56.Miranda PB, Shen YR. J. Phys. Chem. B. 1999;103:3292–3307. [Google Scholar]
- 57.Opdahl A, Hoffer S, Mailhot B, Somorjai G. Chem. Rec. 2001;1:101–122. doi: 10.1002/tcr.2. [DOI] [PubMed] [Google Scholar]
- 58.Kim J, Koffas TS, Lawrence CC, Somorjai GA. Langmuir. 2004;20:4640–4646. doi: 10.1021/la0498511. [DOI] [PubMed] [Google Scholar]
- 59.d'Agostino Riccardo., editor. Plasma deposition, treatment, and etching of polymers. Academic Press; Boston: 1990. [Google Scholar]
- 60.Lopez GP, Ratner BD. Langmuir. 1991;7:766–773. [Google Scholar]
- 61.Lopez GP, Chilkoti A, Briggs D, Ratner BD. J. Polym. Sci. Pol. Chem. 1992;30:2427–2441. [Google Scholar]
- 62.Lopez GP, Ratner BD. J. Polym. Sci. Pol. Chem. 1992;30:2415–2425. [Google Scholar]
- 63.Matzelle TR, Ivanov DA, Landwehr D, Heinrich LA, Herkt-Bruns C, Reichelt R, Kruse N. J. Phys. Chem. B. 2002;106:2861–2866. [Google Scholar]
- 64.Munz M, Cappella B, Sturm H, Geuss M, Schulz E. Filler-Reinforced Elastomers Scanning Force Microscopy. 2003;164:87–210. [Google Scholar]
- 65.Janicijevic A, Ristic D, Wyman C. J. Microsc.-Oxf. 2003;212:264–272. doi: 10.1111/j.1365-2818.2003.01254.x. [DOI] [PubMed] [Google Scholar]
- 66.Jandt KD. Mater. Sci. Eng. R-Rep. 1998;21:221–295. [Google Scholar]
- 67.Baker SP. Thin Solid Films. 1997;308:289–296. [Google Scholar]
- 68.Senff H, Richtering W. Colloid Polym. Sci. 2000;278:830–840. [Google Scholar]
- 69.Takahashi K, Takigawa T, Masuda T. J. Chem. Phys. 2004;120:2972–2979. doi: 10.1063/1.1636695. [DOI] [PubMed] [Google Scholar]
- 70.Kim SH, Opdahl A, Marmo C, Somorjai GA. Biomaterials. 2002;23:1657–1666. doi: 10.1016/s0142-9612(01)00292-7. [DOI] [PubMed] [Google Scholar]
- 71.Chen Z, Ward R, Tian Y, Eppler AS, Shen YR, Somorjai GA. J. Phys. Chem. B. 1999;103:2935–2942. [Google Scholar]
- 72.Lin SY, Chen KS, Liang RC. Polymer. 1999;40:2619–2624. [Google Scholar]
- 73.Chen Q, Zhang D, Somorjai G, Bertozzi CR. J. Am. Chem. Soc. 1999;121:446–447. [Google Scholar]
- 74.Bruscolini P, Casetti L. Phys. Rev. E. 2001;64(Part 1) doi: 10.1103/PhysRevE.64.051805. Art. No. 051805. [DOI] [PubMed] [Google Scholar]