

Abstract
In an ongoing effort to develop new and potent antituberculosis agents, a second generation series of nitrofuranyl amides was synthesized based on the lead compound 5-nitro-furan-2-carboxylic acid 3,4-dimethoxy-benzylamide. The primary design consideration was to improve the solubility and consequently bioavailability of the series by the addition hydrophilic rings to the benzyl and phenyl B ring core. The synthesis of 27 cyclic, secondary amine substituted phenyl and benzyl nitrofuranyl amides is described and their activity against M. tuberculosis reported. The series showed a strong structure-activity relationship as the benzyl nitrofuranyl amides were significantly more active than similarly substituted phenyl nitrofuranyl amides. Para-substituted benzyl piperazines showed the most antituberculosis activity. Compounds in the series were subsequently selected for bioavailability and in vivo testing. This study lead to the successful discovery of novel compounds with increased antituberculosis activity in vitro and a better understanding of the requisite pharmacological properties to advance this class.
Introduction
Someone in the world is newly infected with TB bacilli every second. Overall one third of the world’s population is currently infected with tuberculosis and it has been estimated that 5 – 10% of those people are expected to become sick or infectious at some point their lifetime.1 According to World Health Organization, in 2003 8.8 million new TB cases arose and an estimated 1.7 million deaths resulted from TB.2 The major challenges for tuberculosis control are the development of multidrug-resistant tuberculosis (MDRTB) strains and the increasing numbers of immunocompromised individuals with HIV infections who are highly susceptible to the disease.3 Consequently, there is an urgent need to develop new, potent, fast-acting antituberculosis drugs with low toxicity profiles that can be used in conjunction with drugs used to treat HIV infections.
Recently, we described a novel set of nitrofuranyl amides with potent antituberculosis activity.4 Compounds in this series were easily synthesized, and exhibited good therapeutic indices. They are members of an emerging new class of nitroaromatic antibiotics that are currently being intensively investigated as new antituberculosis drugs.5 Most importantly, one of the compounds 5-nitro-furan-2-carboxylic acid 3,4-dimethoxy-benzylamide (1) (fig. 1), has demonstrated significant oral activity in a mouse model of tuberculosis infection. However, formulation of these compounds for oral administration and in vivo experimentation was problematic due to their poor solubility. This suggested that poor bioavailability may have hindered the activity of several compounds tested in vivo. Such problems have previously been encountered in the development of other synthetic antimicrobial agents and most notably in the development of the fluoroquinolone class of antibiotics. The addition of a piperazine ring to the quinolone core of the first generation quinolones lead to a significant improvement in oral activity and tissue penetration found in second generation fluoroquinolone Norfloxacin.6 This strategy also proved fruitful in the development of the orally bioavailable Ansamycin anitibiotics such as the highly potent antituberculosis drug Rifampin, which was developed from RifamycinSV by formylation and addition of a piperazinyl hydrazine side chain.7
Figure 1.
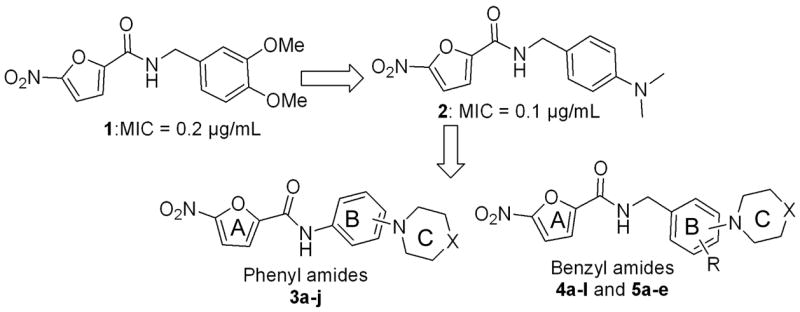
Target lead compounds and target phenyl and benzyl nitrofuranyl amides.
Accordingly, we chose to explore this strategy in the development of the nitrofuranyl amide series. During exploratory experiments, the methoxy group in the lead compound 1 (MIC = 0.2 μg/mL) was substituted with a dimethyl amino group 2 (Fig. 1). Compound 2 retained good MIC activity (0.1 μg/mL) suggesting that substitution of cyclic secondary amines such as piperazine derivatives would be tolerated within the structure activity relationship of this series and that these compounds might have increased the biological activity and pharmacological properties.
In this paper we describe the synthesis and evaluation of several classes of cyclic secondary amine substituted phenyl and benzyl nitrofuranyl amides as novel anti-tuberculosis agents.
Chemistry and in vitro SAR
Synthesis of compound 2 was carried out by a simple acid chloride amide forming reaction. The 5-nitro-furan-2-carbonyl chloride (11) was treated with (4-aminomethyl-phenyl)-dimethyl-amine in presence of Et3N to give the desired amide 2 in 89% yield.
Synthesis and evaluation of substituted phenyl nitrofuranyl amides
Synthesis of the substituted phenyl amides (3a–j) involved a three reaction sequence of nucleophilic aromatic substitution, nitro reduction and acylation with the nitrofuranoic acid chloride (Scheme 1). The fluorine of 3 or 4-fluoro nitrobenzene (6 and 7) was first substituted by conventional aromatic nucleophilic displacement with secondary amides morpholine, 1-methyl-piperazine, 1-benzyl piperazine, 4-benzyl piperadine and 1-(2-pyridyl) piperazine to give corresponding substituted nitrobenzenes 12a–j with yields ranging 78%–95%.8 It was noted that the substitution of p-fluoro nitrobenzene was faster than m-fluoro nitrobenzene, 8 hrs compared with 24 hrs, respectively. The nitro functional group of compounds 12a–j, except compounds 12c and 12h, was reduced by catalytic hydrogenation to give the corresponding anilines 13a–j in quantitative yields. Due to sensitivity of the benzyl substituted piperazines to hydrogenation, the amines 12c and 12h were reduced to their corresponding amides 13c and 13h (both in 82% yields) using SnCl2.2H2O. Finally, all the amines, 13a–j, were treated with 5-nitro-furan-2-carbonyl chloride (11) to give the desired phenyl amides 3a-j in 82–90% yields.
Scheme 1.
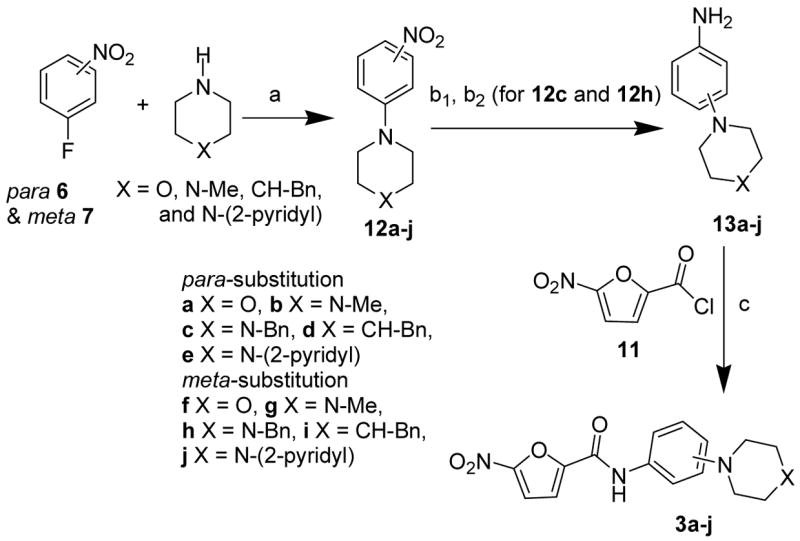
Reagents: Reagents: a) K2CO3, DMSO, 90 °C, 8 h for 6 and 24 h for 7; b1) H2, pd/c, EtOAc-MeOH, room temp., 8 h; b2) For 12c and 12h; SnCl2.2H2O, EtOAc, refluxm 2 h.; c) 11, CH2Cl2, Et3N, room temp.
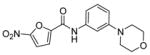
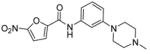
The antituberculosis activities of 3a–j (Table 1) showed overall lower activity than initial leads 1 and 2. Pyridinyl and benzyl subsitituted piperazines 3e and 3c showed the greatest activity when placed in the para-position. The same substitution to the meta-position, 3j and 3h, led to a significant decrease in activity. It was interesting to note that for the smaller ring systems in 3a and 3b vs 3f and 3g no difference in activity with the placement of rings meta or para was observed.
Table 1.
Cyclic secondary amine substituted phenyl nitrofuranyl amides and their anti-tuberculosis activity
| No. | Compound | M. tb H37Rv MIC90 (μg/mL) |
|---|---|---|
| 3a |
|
3.13 |
| 3b |
|
12.5 |
| 3c |
|
0.8 |
| 3d |
|
3.13 |
| 3e |
|
0.4 |
| 3f |
|
3.13 |
| 3g |
|
12.5 |
| 3h |
|
12.5 |
| 3i |
|
12.5 |
| 3j |
|
3.13 |
Synthesis and evaluation of substituted benzyl nitrofuranyl amides
The benzyl amide series was prepared using a similar synthetic pattern of nucleophilic aromatic substitution: reduction followed by acylation (Scheme 2). In this case, a cyano group was used as an electron-withdrawing group to facilitate the nucleophilic aromatic substitution. Accordingly, the fluorine of the 3-or 4-fluoro benzonitrile (8 or 9 respectively) was substituted with corresponding cyclic secondary amines in DMSO at 90° C in the presence of potassium carbonate to give compounds 14a–h in 83% to 96% yields.9 The substituted benzonitriles were subjected to reduction using Red-Al reagent to afford corresponding crude amines,10 which were further reacted immediately with 5-nitro-furan-2-carbonyl chloride to give benzyl amides 4a–h in yields ranging from 69% to 96%.
Scheme 2.

Reagents: a) K2CO3, DMSO, 90 °C, 8 h; b) Red-Al, 0 °C, dry THF, 3 h.; c) 11, CH2Cl2, Et3N, room temp.
Compounds 4a–h were made from commercially available piperazine and morpholine starting materials. In order to expand the diversity of substitution, a method to synthesize novel piperazine derivatives was developed from the starting material 4-piperazin-1-yl-benzonitrile (15), which upon alkylation with different alkyl halides provided alternative piperazine substitutions. Accordingly, alkylation of 15 with bromomethyl-cyclopropane gave the nitrile 16 (93% yield),11 which on reduction with Red-Al followed by treatment with acid chloride 11 in presence of base gave the desired amide 4k in 82% overall yield (Scheme 3).
Scheme 3.
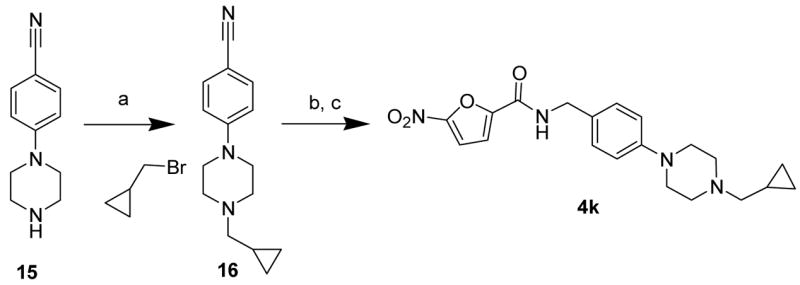
Reagents: a) K2CO3, DMF, room temp., 7 h; b) Red-Al, 0 °C-room temp., dry THF, 3 h.; c) 11, CH2Cl2, Et3N, room temp.
Further fuctionalization of the sulfur group in the thiomorpholine ring of amide 4h was performed to create compounds with lower potential serum binding.12 This was achieved by stepwise oxidation of 4h with m-chloroperbenzoic acid to give the sulfoxide 4i and the sulfone 4j derivatives in 15% and 20% yields respectively (Scheme 4).1
Scheme 4.
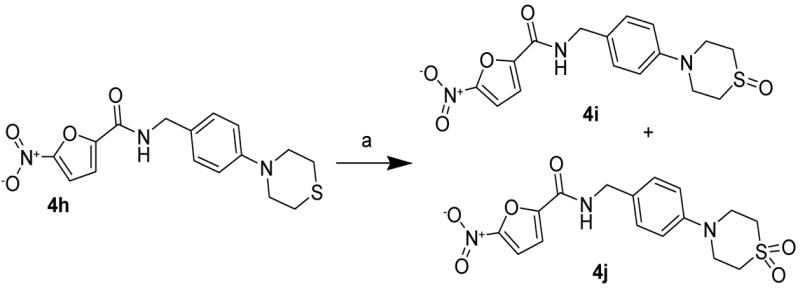
Reagents: a) m-CPBA, NaHCO3, CH2Cl2, room temp., 30 min.
To further explore the SAR of these compounds, the piperazine group was separated from the phenyl ring with a hydrazone link creating a similar substitution to the modification found in the derivatization of rifampin from rifamycin. Amine 17 was reacted with acid chloride 11 to give amide 18 in 65% yield. The benzyl alcohol group of 18 was oxidized to give the corresponding aldehyde,14 which was further treated with 4-methyl-piperazin-1-yl-amine in ethanol to give the hydrazone derivative 4l in 74% yield (Scheme 5).15
Scheme 5.
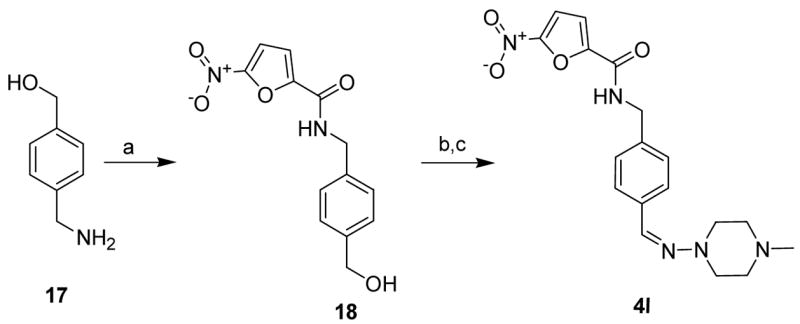
Reagents: a) 11, NEt3, CH2Cl2, room temp., 14 h; b) Dess-Martin periodinane, pyridine, CH2Cl2, room temp., 2 h; c) 4-methyl-piperazin-1-yl-amine, ethanol, 1 h reflux, 14 h room temp.
The antituberculosis activity of 4a–l (Table 2) showed interesting SAR. Compounds 4c and 4k had significantly increased activity over the initial leads 1 and 2. In this series para-substitution was again favored over meta (4c vs 4f, 4b vs 4e, 4d vs 4g). The thiomorpholine analogue 4h (0.1 μg/mL) had slightly better activity than the morpholine analogue 4a (0.2 μg/mL). However, further derivatization of 4a to its oxides 4i and 4j, reduced the MIC activity. Replacing the benzyl group on the piperazine in 4c with a simple cyclopropyl methyl substitution (4k) maintained good activity at 0.05 μg/mL. The rifampin side chain hydrazone derivative 4l showed moderate activity with increased hydrophilicity.
Table 2.
Cyclic secondary amine substituted benzyl nitrofuranyl amides and their anti-tuberculosis activity
| No. | Compound | M. tb H37Rv MIC90 (μg/mL) |
|---|---|---|
| 4a |
|
0.2 |
| 4b |
|
0.2 |
| 4c |
|
0.0125 |
| 4d |

|
0.8 |
| 4e |
|
3.125 |
| 4f |
|
0.1 |
| 4g |
|
1.56 |
| 4h |
|
0.1 |
| 4i |
|
3.13 |
| 4j |
|
12.25 |
| 4k |
|
0.05 |
| 4l |
|
0.4 |
Synthesis and evaluation of fluorine substituted benzyl amides
After synthesis of amides 4a–l it was proposed to explore the addition of more functionality on the phenyl ring of the benzyl amine moiety. This was achieved by starting from a di-substituted benzonitrile such as 3,4-difluorobenzonitrile (10).16 The reaction of nitrile 10 with cyclic secondary amines, in presence of a base, substituted the fluorine group in the para-position, which generated the corresponding tertiary amines 19a–e in 75% to 88% yields. Subsequently, the nitrile group in 19a–e was reduced with Red-Al to give corresponding benzylamines, which were then treated with 5-nitro-furan-2-carbonyl chloride to give the final targeted amides 5a–e in 77% to 89% yields (Scheme 6).
Scheme 6.

Reagents: a) K2CO3, DMSO, 90 °C, 8 h; b) Red-Al, 0 °C-room temp., dry THF, 3 h.; c) 11, CH2Cl2, Et3N, room temp.
The antituberculosis activity of 5a–e, with a fluorine substitution at the meta-position (Table 3), demonstrated a similar SAR to the benzyl series with only slightly lower MIC values in general. The benzyl piperazine analog 5a exhibited the best activity.
Table 3.
Fluorine substituted benzyl nitrofuanyl amides and their anti-tuberculosis activity
| No. | Compound | M. tb H37Rv MIC90 (μg/mL) |
|---|---|---|
| 5a |
|
0.025 |
| 5b |
|
0.8 |
| 5c |
|
0.8 |
| 5d |
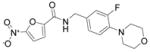
|
0.4 |
| 5e |
|
0.2 |
Oral bioavailability and in vivo efficacy studies
The MIC values of compounds in these series are good and compare favorably with other known antituberculosis agents. Preliminary in vitro MIC testing in presence of protein was performed to evaluate whether protein binding might affect the antituberculosis activity of the compounds. In vitro MIC50 values were established with and without the presence of 10% mouse serum for M. tuberculosis H37Rv, according to NCCLS guidelines (Table 4).17 Serial drug dilutions were performed 1:3, and therefore a 3-fold decrease in MIC in this assay was not considered significant. The results showed that only compound 4g had potential protein binding issues. Since in vivo testing of novel compounds occurs primarily via the oral route, the ability of the compounds to cross the intestinal tract needed to be assessed. Therefore, compounds were selected for advancement into animal studies based on bioavailability assays that determined which compounds achieved sufficient serum levels able to eradicate the disease. Where possible the selected compounds were formulated as hydrochloride salts to maximize dissolution and bioavailability. The compounds were orally dosed to mice and subsequently the mice were bled at set intervals. The serum collected from these mice was then serially diluted and tested for anti-tuberculosis activity in a bioassay using M. tuberculosis.18 The results from the bioassay reflect approximate concentrations of unbound bioactive product in the serum rather than providing total drug levels. It was clear that compounds 2 and 4g had the best absorption (Cmax) and compounds 4c and 4g had the best combination of low MIC and Cmax values. A further subset of compounds was advanced for in vivo testing in a mouse model of tuberculosis infection (Table 5).19–21
Table 4.
Results of the in vivo biological activity and basic bioavailability assay
| Comp | MIC50 H37Rv (μg/ml) | Results from serum Assay | Tmax (hours) | Half-life (hours)3 | Bioavailability4 | ||
|---|---|---|---|---|---|---|---|
| No serum | +10% serum | Dilution factor1 | Cmax (μg/mL) Approximate drug concentration in mouse serum2 | ||||
| INH | 0.041 | 0.12 | >1:320 | >38.4 | ND | ND | High |
| 2.HCl | 0.041 | 0.12 | 1:40–1:80 | 7.2 | 0.5 | 4.9 | Medium |
| 4a | 0.041 | 0.2 | 1:10–1:20 | 3 | ND | ND | Medium |
| 4b.HCl | 0.041 | 0.12 | 1:10–1:40 | 2.4 | 0.5 | 3.5 | Low |
| 4c.HCl | 0.014 | 0.014 | 1:80–1:160 | 2.24 | 0.5 | 1.1 | High |
| 4f.HCl | 0.014 | 0.041 | 1:40 | 1.64 | 0.5 | 1.3 | Medium |
| 4g | 0.041 | 0.2 | 1:20–1:40 | 6 | ND | ND | High |
| 4h | 0.014 | 0.041 | 1:20–1:40 | 0.82 | ND | ND | Low |
| 4k.HCl | 0.041 | 0.041 | 1:40–1:80 | 3.28 | 0.5 | 1.3 | Medium |
| 5a | 0.014 | 0.041 | 1:40 | 1.64 | ND | ND | Medium |
| 5b.HCl | 0.37 | 0.37 | 1:10–1:80 | 7.4 | 0.5 | 3.5 | Low |
The dilution factor represents the last dilution step of the serum samples in which drug activity was still observed in the bioassay
Drug levels in the mouse serum are estimated by multiplying the dilution factor by the MIC value of the drug in the presence of 10% serum;
Estimated based on graph of concentration vs. time curve;
Bioavailability factor (BF) = Cmax/MIC in presence of serum; Bioavailability: Low = BF 1–20; Medium = BF 21–80; High = BF >80 30
Table 5.
Viable number of M. tuberculosis bacilli in the lungs of infected mice after 9 days of drug treatment (SEM= standard error).
| Compound | Lungs (Log10CFU +/−SEM) | Log10 reduction untreated | CPU versus controls |
|---|---|---|---|
| 2 | 7.46 +/− 0.25 | 1.04 | |
| 4c | 7.92 +/− 0.27 | 0.58 | |
| 4h | 7.53 +/− 0.14 | 0.97 | |
| 5a | 7.65 +/− 0.11 | 0.85 | |
| INH | 3.88 +/− 0.36 | 4.62 |
In this rapid mouse model, the compounds were administered via oral gavage for nine consecutive days at 300 mg/kg. Four compounds gave a significant reduction of the bacterial load in the lungs (Table 5) and in spleen (results not shown) (P > 0.05). The bacterial numbers in the lung were reduced between 0.5 and 1.04 Log10 CFU (up to 90% killing of the bacterial load). The control drug isoniazid (INH) at 25 mg/kg reduced the bacterial load in this experiment with 4.62 Log10CFU in the lungs.
Discussion
In this study we detail the synthesis and evaluation of an advanced series of nitrofuranyl amides. The compounds were produced in good yields and the synthesis offered no barrier to scale-up synthesis for larger quantities required for in vivo testing. The compounds were tested for MIC activity against M. tuberculosis and clear structure-activity relationships were observed for this series. The substituted benzyl series had much greater antituberculosis activity than the substituted phenyl series. In both the phenyl and the benzyl amides, para-substitution with the cyclic secondary amine produced better antituberculosis activity. Compounds from the benzyl series are extremely potent and are the most active compounds developed in this class to date. Among them, 4c with benzyl-piperazine at para-position is the most active compound with an MIC value of 0.0125 μg/mL against M. tuberculosis H37Rv. The addition of a fluorine to the meta position of the benzyl ring of 4c produced 5a which also had excellent activity (0.025 μg/mL)
The in vivo testing shed new light on the relative bioavailability of compounds in this series. It was noted that while some compounds in this series were well absorbed, the compounds appear to be rapidly eliminated with short half lives, as was seen for the compound with best in vitro activity 4c (T1/2 1.1hrs). 4c contains both an amide and benzyl piperazine bonds, both likely candidates for rapid metabolism. It is our current hypothesis that rapid degradation is limiting the in vivo efficacy. Current studies are ongoing to understand the metabolism and distribution of this series and to design a new generation of compounds with a longer serum half life to retain potent antituberculosis activity.
Experimental
All the anhydrous solvents and starting materials were purchased from Aldrich Chemical Company (Wilwaukee, WI). All reagent grade solvents used for chromatography were purchased from Fisher Scientific (Suwanee, GA) and Flash column chromatography silica cartridges were obtained from Biotage Inc. (Lake Forest, VA). The reactions were monitored by thin layer chromatography (TLC) on pre-coated Merck 60 F254 silica gel plates and visualized using UV light (254 nm). A Biotage FLASH 25+ column chromatography system was used to purify mixtures. All 1H and 13C NMR spectra were recorded on a Bruker ARX-300 (300 and 75 MHz for 1H and 13C NMR, repectively) or Varian INOVA-500 (500 and 125 MHz for 1H and 13C NMR, respectively) spectrometers. Chemical shifts (δ) are reported in ppm relative to the residual solvent peak or internal standard (tetramethylsilane), and coupling constants (J) are reported in hertz (Hz). Mass spectra were recorded on a Bruker Esquire LCMS using ESI. Purity of the final products was confirmed before testing by analytical HPLC using an Alltech platinum C-18 reverse phase column (4.5×150mm) and an H2O (0.1% TFA) to acetonitrile 0–100% linear gradient at a flow rate of 1.0 mL min−1 and UV detection at 254nm.
General Procedure I: for preparation of 12a–j and 14a–h: Secondary amine (2 eq) was added to a mixture of substituted fluoro benzene (1 eq.) and K2CO3 (1.5 eq.) in dimethyl sulfoxide (7 mL/g). The reaction mixture was stirred at 90° C and followed by TLC. After completion of the reaction, the mixture was diluted with ethyl acetate (60 mL/g), and washed with water (2 × 50 mL/g), followed by brine (50 mL/g). The ethyl acetate fraction was dried over Na2SO4 and concentrated. The crude products were purified by flash column chromatography to afford pure products.
1-Benzyl-4-(4-nitro-phenyl)-piperazine (12c)
1-Benzyl piperazine (1.04 mL, 6.02 mmol) was added to a mixture of 4-fluoro nitro benzene 6 (425 mg, 3.01 mmol) and K2CO3 (623 mg, 4.52 mmol) in dimethyl sulfoxide (3 mL) and the reaction continued as described in general procedure I to afford 805 mg of amine 12c in 90% yield. 1H-NMR (500 MHz, CDCl3): δ 2.59 (4Hs, t, J = 4.88 Hz), 3.42 (4Hs, t, J = 5.12 Hz), 3.57 (2Hs, s), 6.81 (2Hs, d, J = 7.32 Hz), 7.29 (1H, sextet, J = 1.22 Hz), 7.34 (4Hs, d, J = 7.39 Hz), 8.12 (2Hs, d, J = 7.32 Hz); ESI-MS: 298.2 (M+1).
4-Benzyl-1-(4-nitro-phenyl)-piperidine (12d)
4-Benzyl piperidine (1.06 mL, 6.02 mmol) was added to a mixture of 4-fluoro nitro benzene 6 (425 mg, 3.01 mmol) and K2CO3 (623 mg, 4.52 mmol) in dimethyl sulfoxide (3 mL) and the reaction continued as described in general procedure I to afford 847 mg of amine 12d in 95% yield. 1H-NMR (500 MHz, CDCl3): δ 1.33 (2Hs, dq, J = 3.9, 12.45 Hz), 1.74–1.88 (3Hs, m), 2.57 (2Hs, d, J = 6.83 Hz), 2.91 (2Hs, t, J = 15.13 Hz), 3.93 (2Hs, d, J = 13.18 Hz), 6.78 (2Hs, d, J = 9.52 Hz), 7.15 (2Hs, d, J = 7.08 Hz), 7.22 (1H, t, J = 7.32 Hz), 7.30 (2Hs, t, J = 7.56 Hz), 8.1 (2Hs, d, J = 9.52 Hz); ESI-MS: 319.1 (M+23).
4-(3-Nitro-phenyl)-morpholine (12f)
Morpholine (0.52 mL, 6.02 mmol) was added to a mixture of 3-fluoro nitro benzene 7 (425 mg, 3.01 mmol) and K2CO3 (623 mg, 4.52 mmol) in dimethyl sulfoxide (3 mL) and the reaction continued as described in general procedure I to afford 589 mg of amine 12f in 94% yield. 1H-NMR (300 MHz, CDCl3): δ 3.27 (4Hs, t, J = 4.83 Hz), 3.9 (4Hs, t, J = 4.95 Hz), 7.21 (1H, ddd, J = 1.03, 2.25, 9.06 Hz), 7.42 (1H, t, J = 8.19 Hz), 7.68–7.76 (2Hs, m); ESI-MS: 231.0 (M+23).
1-Methyl-4-(3-nitro-phenyl)-piperazine (12g)
1-Methyl piperazine (0.66 mL, 6.02 mmol) was added to a mixture of 3-fluoro nitro benzene 7 (425 mg, 3.01 mmol) and K2CO3 (623 mg, 4.52 mmol) in dimethyl sulfoxide (3 mL) and the reaction continued as described in general procedure I to afford 619 mg of amine 12g in 93% yield. 1H-NMR (300 MHz, CDCl3): δ 2.38 (3Hs, s), 2.6 (4Hs, t, J = 5.0 Hz), 3.32 (4Hs, t, J = 5.14 Hz), 7.2 (1H, dd, J = 2.16, 8.26 Hz), 7.39 (1H, t, J = 8.14 Hz), 7.67 (1H, dd, J = 1.43, 8.01 Hz); ESI-MS: 222.4 (M+1).
General Procedure II: for preparation of 13a–j, except 13c and 13h
The substituted nitro compound (1 eq. in a mixture of methanol-ethyl acetate, 1:2, 20 mL) was treated with 10% Pd-carbon (5% w/w). The reaction was subjected to hydrogenation under 50 Psi hydrogen gas pressure at room temperature and the reaction was monitored by TLC. After completion of the reaction, the mixture was filtered thorough a celite bed and concentrated in a vacuum to afford pure product in quantitative yields.
4-(4-Benzyl-piperidin-1-yl)-phenylamine (13d)
4-Benzyl-1-(4-nitro-phenyl)-piperidine 12d, (600 mg, 2.02 mmol) was treated with 10% Pd-carbon (5% w/w) under the conditions described in general procedure II to afford amine 13d in quantitative yield. 1H-NMR (500 MHz, CD3OD): δ 1.44 (2Hs, q, J = 9.27, 21.23 Hz), 1.6–1.7 (1H, m), 1.75 (1H, d, J = 12.2 Hz), 2.55 (2Hs, t, J = 11.47 Hz), 2.6 (2Hs, d, J = 7.08 Hz), 3.39 (2Hs, d, J = 11.23 Hz), 6.71 (2Hs, d, J = 7.81 Hz), 6.87 (2Hs, d, J = 8.05 Hz), 7.16–7.21 (3Hs, m), 7.28 (2Hs, t, J = 7.07 Hz); ESI-MS: 267.1 (M+1).
3-Morpholin-4-yl-phenylamine (13f)
4-(3-Nitro-phenyl)-morpholine 12f, (600 mg, 2.88 mmol) was treated with 10% Pd-carbon (5% w/w) under the conditions described in general procedure II to afford amine 13f in quantitative yield. 1H-NMR (300 MHz, CDCl3): δ 3.145 (4Hs, t, J = 4.79 Hz), 3.86 (4Hs, t, J = 4.89 Hz), 6.24–6.29 (2Hs, m), 6.37 (1H, ddd, J = 0.73, 2.21, 8.20 Hz), 7.08 (1H, t, J = 8.28); ESI-MS: 201.3 (M+23).
3-(4-Methyl-piperazin-1-yl)-phenylamine (13g)
1-Methyl-4-(3-nitro-phenyl)-piperazine 12g, (600 mg, 2.71 mmol) was treated with 10% Pd-carbon (5% w/w) under the conditions described in general procedure II to afford amine 13g in quantitative yield. 1H-NMR (300 MHz, CDCl3): δ 2.36 (3Hs, s), 2.57 (4Hs, t, J = 4.94 Hz), 3.2 (4Hs, t, J = 5.11 Hz), 3.59–3.67 (2Hs, bs), 6.22 (1H, dd, J = 2.07, 8.41 Hz), 6.28 (1H, t, J = 2.21 Hz), 6.39 (1H, dd, J = 1.89, 7.82 Hz), 7.06 (1H, t, J = 8.02 Hz); ESI-MS: 192.1 (M+1).
General Procedure III: For preparation of 13c and 13h
SnCl2.H2O (1.125 g/mmol) was added to a solution of the substituted nitro benzene in ethyl acetate (10 mL/mmol). The solution was refluxed for 2 hours. The cooled solution was diluted with water and the pH was adjusted to 8 by addition of a saturated NaHCO3 solution. The aqueous phase was extracted with EtOAc (3 × 75 mL) and the combined organic extracts were thoroughly washed with brine and dried over Na2SO4. The products obtained after the removal of the solvent were used without further purification.
4-(4-Benzyl-piperazin-1-yl)-phenylamine (13c)
1-Benzyl-4-(4-nitro-phenyl)-piperazine 12c (750 mg, 2.52 mmol) was treated with SnCl2.H2O (14.4 g, 63.9 mmol) under the conditions described in general procedure III to afford 552 mg of amine 13c in 82% yield. 1H-NMR (300 MHz, CDCl3): δ 2.6–2.8 (4Hs, bs), 3.11 (4Hs, t, J = 4.38 Hz), 3.64 (2Hs, s), 6.66 (2Hs, d, J = 8.76 Hz), 6.82 (2Hs, d, J = 8.76 Hz), 7.25–7.45 (5Hs, m); ESI-MS: 268.2 (M+1).
General Proceure IV: For preparation of 2 and 3a–j
5-Nitro-furan-2-carbonyl chloride (438 mg, 2.5 mmol) in CH2Cl2 (10 mL) was added to a mixture of amine (2.0 mmol) in Et3N (1.04 mL, 7.5 mmol). The mixture was stirred for 12 hrs at room temperature and was followed by TLC. After completion of reaction, 100 mL of ethyl acetate was added to the reaction mixture, and it was sequentially washed with saturated aq. NaHCO3 (2 × 50 mL), water (2 × 50 mL) and brine (2 × 50 mL). The organic phase was dried over Na2SO4, filtered, concentrated and purified by flash column chromatography to provide the corresponding pure amides.
5-Nitro-furan-2-carboxylic acid [4-(4-benzyl-piperazin-1-yl)-phenyl]-amide (3c)
5-Nitro-furan-2-carbonyl chloride (438 mg, 2.5 mmol) in CH2Cl2 (10 mL) was added to amine 13c (534 mg, 2.0 mmol) in Et3N (1.04 mL, 7.5 mmol) and the reaction continued as described in general procedure IV to afford 755 mg of amide 3c in 93% yield. 1H-NMR (500 MHz, CDCl3): δ 2.66–2.76 (4Hs, bs), 3.26–3.33 (4Hs, bs), 3.64–3.7 (2Hs, bs), 7.99 (2Hs, d, J = 9.03 Hz), 7.32–7.37 (2Hs, m), 7.38–7.46 (4Hs, m), 7.47 (1H, d, J = 3.90), 7.61 (2Hs, d, J = 9.03 Hz), 8.14 (1H, bs); 13C-NMR (300 MHz, CDCl3-DMSO-D6, 5:1): 48.13, 51.98, 61.85, 111.94, 114.87, 114.91, 121.36, 126.12, 127.29, 128.12, 128.67, 137.17, 147.65, 148.11, 150.69, 153.39; ESI-MS: 407.5 (M+1). Anal. (C22H22N4O4) C, H, N.
5-Nitro-furan-2-carboxylic acid [4-(4-benzyl-piperidin-1-yl)-phenyl]-amide (3d)
5-Nitro-furan-2-carbonyl chloride (438 mg, 2.5 mmol) in CH2Cl2 (10 mL) was added to a mixture of amine 13d (532 mg, 2.0 mmol) in Et3N (1.04 mL, 7.5 mmol) and the reaction continued as described in general procedure IV to afford 728 mg of amide 3d in 90% yield. 1H-NMR (500 MHz, CDCl3): δ 1.50–1.65 (3Hs, m), 1.65–1.85 (2Hs, m), 2.6–2.8 (4Hs, m), 3.68 (2Hs, d, J = 10.98 Hz), 6.9–7.02 (2Hs, bs), 7.2–7.3 (3Hs, m), 7.3–7.39 (2Hs, m), 7.41 (1H, d, J = 3.66), 7.47 (1H, t, J = 2.1, 3.6), 7.515–7.65 (2Hs, bs), 8.12–8.22 (1H, bs); 13C-NMR 300 MHz, (CDCl3): 31.37, 37.26, 42.58, 49.36, 112.13, 115.74, 116.16, 121.15, 125.38, 127.34, 127.70, 128.59, 139.84, 147.74, 149.11, 153.06; ESI-MS: 406.4 (M+1); Anal. (C23H23N3O4) C, H, N.
5-Nitro-furan-2-carboxylic acid (3-morpholin-4-yl-phenyl)-amide (3f)
5-Nitro-furan-2-carbonyl chloride (438 mg, 2.5 mmol) in CH2Cl2 (10 mL) was added to a mixture of amine 13f (356 mg, 2.0 mmol) in Et3N (1.04 mL, 7.5 mmol) and the reaction continued as described in general procedure IV to afford 494 mg of amide 3f in 78% yield. 1H-NMR (500 MHz, CDCl3): δ 3.23 (4Hs, t, J = 4.63 Hz), 3.89 (4Hs, t, J = 4.88 Hz), 6.77 (1H, dd, J = 2.19, 8.3 Hz), 7.1 (1H, dd, J = 1.46, 7.81 Hz), 7.30 (1H, t, J = 8.05 Hz), 7.38 (1H, d, J = 3.66 Hz), 7.46–7.5 (2Hs, m), 8.20 (1H, bs); 13C-NMR (300 MHz, CDCl3): 51.75, 69.37, 110.78, 114.99, 115.26, 115.31, 119.17, 132.16, 140.47, 150.80, 154.44, 157.39; ESI-MS: 318.3 (M+1); Anal. (C15H15N3O5) C, H, N.
5-Nitro-furan-2-carboxylic acid [3-(4-methyl-piperazin-1-yl)-phenyl]-amide (3g)
5-Nitro-furan-2-carbonyl chloride (438 mg, 2.5 mmol) in CH2Cl2 (10 mL) was added to a mixture of amine 13g (382 mg, 2.0 mmol) in Et3N (1.04 mL, 7.5 mmol) and the reaction continued as described in general procedure IV to afford 593 mg of amide 3g in 90% yield. 1H-NMR (500 MHz, CDCl3): δ 2.38 (3Hs, s), 2.60 (4Hs, t, J = 4.64 Hz), 3.28 (4Hs, t, J = 4.51 Hz), 6.8 (1H, d, J = 8.3 Hz), 7.08 (1H, d, J = 8.05 Hz), 7.25–7.32 (2Hs, m), 7.37–7.45 (2Hs, m), 7.48 (1H, dd, J = 1.22, 3.66 Hz), 8.17 (1H, bs); 13C-NMR (300 MHz, CD3OD): 44.16, 47.92, 53.98, 107.99, 111.48, 111.70, 112.26, 115.49, 128.50, 137.57, 147.55, 151.16, 154.73; ESI-MS: 331.3 (M+1). Anal. (C16H18N4O4) C, H, N.
4-(4-Methyl-piperazin-1-yl)-benzonitrile (14b)
1-Methyl piperazine (1.36 mL, 12.38 mmol) was added to a mixture of 4-fluoro-benzonitrile 8 (1.0 g, 8.25 mmol) and K2CO3 (2.27 mg, 16.51 mmol) in dimethyl sulfoxide (7 mL) and the reaction continued as described in general procedure I to afford amine 1.54 g of 14b in 93% yield. 1H-NMR (500 MHz, CDCl3): δ 2.36 (3Hs, s), 2.55 (4Hs, t, J = 4.88 Hz), 3.35 (4Hs, t, J = 4.88 Hz), 6.87 (2Hs, d, J = 8.78 Hz), 7.49 (2Hs, d, J = 8.78 Hz); ESI-MS: 202.1 (M+1).
3-(4-Benzyl-piperidin-1-yl)-benzonitrile (14g)
1-Methyl piperazine (2.17 mL, 12.38 mmol) was added to a mixture of 3-fluoro-benzonitrile 9 (1.0 g, 8.25 mmol) and K2CO3 (2.27 mg, 16.51 mmol) in dimethyl sulfoxide (7 mL) and the reaction continued as described in general procedure I to afford amine 2.05 g of 14g in 90% yield. 1H-NMR (500 MHz, CDCl3): δ 1.43 (2Hs, dq, J = 3.9, 12.45 Hz), 1.73–1.86 (3Hs, m), 2.65 (2Hs, d, J = 6.83 Hz), 2.88 (2Hs, dt, J = 2.68, 12.45 Hz), 3.73 (2Hs, d, J = 12.45 Hz), 7.1 (1H, td, J = 0.97, 7.56 Hz), 7.14–7.17 (2Hs, m), 7.22 (2Hs, d, J = 6.83 Hz), 7.27 (1H, t, J = 7.32 Hz), 7.34–7.38 (3Hs, m); ESI-MS: 299.5 (M+23).
4-Thiomorpholin-4-yl-benzonitrile (14h)
Thio morpholine (2.35 mL, 24.79 mmol) was added to a mixture of 4-fluoro-benzonitrile 8 (2.0 g, 16.52 mmol) and K2CO3 (4.56 mg, 33.05 mmol) in dimethyl sulfoxide (10 mL) and the reaction continued as described in general procedure I to afford amine 2.93 g of 14h in 87% yield. 1H-NMR (300 MHz, CDCl3): δ 2.71 (4Hs, t, J = 5.07 Hz), 3.78 (4Hs, t, J = 5.11 Hz), 6.82 (2Hs, d, J = 9.0 Hz), 7.51 (2Hs, d, J = 9.05 Hz); ESI-MS: 227.4 (M+23).
General Procedure V: Preparation of (4a–l) and (5a–e)
(a) 65% Red-Al in toluene was added drop wise to a mixture of nitrile in THF (5 mL) at 0° C while stirring under argon atmosphere. After, the reaction was stirred for 3 hrs at room temperature; it was quenched by adding 5 mL water, drop wise, at 0° C. The reaction mixture was then filtered through a celite bed, washed with THF, and the combined fractions were concentrated under vacuum. The resulting crude mixture was used in further reactions without further purification and characterization. (b) 5-Nitro-furan-2-carbonyl chloride was added to the crude amine in THF and Et3N. The mixture was stirred for 12 hrs at room temperature and was followed by TLC After completion of reaction, 100 mL of ethyl acetate was added to the reaction mixture, and it was sequentially washed with saturated aq. NaHCO3 (2 × 50 mL), water (2 × 50 mL) and brine (2 × 50 mL). The organic phase was dried over Na2SO4, filtered, concentrated and purified by flash column chromatography to provide the corresponding pure amides.
5-Nitro-furan-2-carboxylic acid 4-(4-methyl-piperazin-1-yl)-benzylamide (4b)
65% Red-Al in toluene (5.79 mL, 18.6 mmol) was reacted with nitrile 14b (1.25 g, 6.21 mmol) as described in general procedure V(a) to yield a crude amine mixture. 5-Nitro-furan-2-carbonyl chloride (1.63 mg, 9.29 mmol) was added to the crude amine in THF (10 mL) and Et3N (2.58 mL, 18.58 mmol). The reaction was then carried out as described in general procedure V(b) to afford 1.49 g of amide 4b in 70% yield. 1H-NMR (500 MHz, CDCl3): δ 2.41 (3Hs, s), 2.63 (4Hs, t, J = 4.88 Hz), 3.27 (4Hs, t, J = 4.88 Hz), 4.6 (2Hs, d, J = 5.61 Hz), 6.78–6.83 (1H, bs), 6.97 (2Hs, d, J = 8.78 Hz), 7.31 (2Hs, d, J = 8.78 Hz), 7.33 (1H, d, J = 3.90 Hz), 7.41 (1H, d, J = 3.90 Hz); 13C-NMR (300 MHz, CDCl3): 41.71, 44.02, 48.06, 53.94, 111.31, 114.88, 115.52, 127.89, 128.91, 147.54, 149.95, 156.61; ESI-MS: 345.3 (M+1); Anal. (C17H20N4O4) C, H, N.
5-Nitro-furan-2-carboxylic acid 3-(4-benzyl-piperidin-1-yl)-benzylamide (4g)
65% Red-Al in toluene (3.81 mL, 12.28 mmol) was reacted with nitrile 14g (1.13 g, 4.09 mmol) as described in general procedure V(a) to yield a crude amine mixture. 5-Nitro-furan-2-carbonyl chloride (1.06 g, 6.1 mmol) was added to the crude amine in THF (10 mL) and Et3N (1.7 mL, 12.28 mmol), and the reaction was carried out as described in general procedure V(b) to afford 1.42 g of amide 4g in 83% yield. 1H-NMR (500 MHz, CDCl3): δ 1.44 (2Hs, dq, J = 3.66, 11.71, 23.92 Hz), 1.68–1.77 (1H, m), 1.78 (2Hs, d, J = 13.18 Hz), 2.62 (2Hs, d, J = 2.83 Hz), 2.7 (2Hs, dt, J = 2.19, 12.20 Hz), 3.72 (2Hs, d, J = 12.45 Hz), 4.61 (2Hs, d, J = 5.61 Hz), 6.81–6.88 (2Hs, m), 6.9–6.94 (2Hs, m), 7.21 (2Hs, d, J = 7.07 Hz), 7.22–7.36 (5Hs, m), 7.40 (1H, d, J = 3.66 Hz); 13C-NMR (300 MHz, CDCl3): 31.41, 37.30, 42.57, 43.54, 49.17, 111.84, 115.27, 115.48, 118.09, 125.28, 127.70, 128.59, 129.10, 137.20, 139.85, 147.52, 151.74, 155.52; ESI-MS: 420.4 (M+1); Anal. (C24H25N3O4) C, H, N.
5-Nitro-furan-2-carboxylic acid 4-thiomorpholin-4-yl-benzylamide (4h)
65% Red-Al in toluene (6.84 mL, 22.02 mmol) was reacted with nitrile 14h (1.5 g, 7.32 mmol) as described in general procedure V(a) to yield a crude amine mixture. 5-Nitro-furan-2-carbonyl chloride (1.92 mg, 11.02 mmol) was added to the crude amine in THF (10 mL) and Et3N (3.0 mL, 21.92 mmol), and reaction was carried out as described in general procedure V(b) to afford 1.96 g of amide 4h in 79% yield. 1H-NMR (500 MHz, CDCl3): δ 2.75–2.82 (4Hs, m), 3.61–3.64 (4Hs, m), 4.6 (2Hs, d, J = 5.85 Hz), 6.8–6.85 (1H, bs), 6.93 (2Hs, d, J = 8.78 Hz), 7.31 (2Hs, d, J = 8.78 Hz), 7.33 (1H, d, J = 3.66 Hz), 7.41 (1H, d, J = 3.66 Hz); 13C-NMR (300 MHz, CDCl3): 26.04, 42.6, 51.37, 111.86, 115.42, 116.55, 127.26, 128.82, 147.57, 150.33, 155.48; ESI-MS: 346.0 (M+1).
4-(4-Cyclopropylmethyl-piperazin-1-yl)-benzonitrile (16)
Bromomethyl-cyclopropane (0.64 mL, 6.63 mmol) was added drop wise to a mixture of 4-piperazin-1-yl-benzonitrile (1 g, 4.42 mmol) and K2CO3 (915 mg, 6.63 mmol) in dry N,N-dimethylformamide (5 mL), under argon atmosphere at 0° C. The reaction mixture was stirred at room temperature for 12 h, diluted with water (50 mL) and extracted with ethyl acetate (3 × 40 mL). The combined organic fractions were washed with saturated brine (50 mL) and dried over Na2SO4 followed by solvent evaporation under vacuum which gave the crude product. The crude product was subsequently purified by column chromatography (4:1 petroleum ether: ethyl acetate) to afford 991 mg of product 16 (93% yield). 1H-NMR (500 MHz, CDCl3): δ0.01 (2Hs, q, J = 4.63 Hz), 0.43 (2Hs, q, J = 5.85 Hz), 0.73 (1H, m), 2.19 (2Hs, d, J = 6.59 Hz), 2.54 (4Hs, t, J = 5.51 Hz), 3.23 (4Hs, t, J = 5.12 Hz), 6.74 (2Hs, d, J = 9.03 Hz), 7.37 (2Hs, d, J = 9.03 Hz), 7.89 (1H, bs); ESI-MS: 242.5 (M+1).
5-Nitro-furan-2-carboxylic acid 4-(4-cyclopropylmethyl-piperazin-1-yl)-benzylamide (4k)
65% Red-Al in toluene (3.09 mL, 9.9 mmol) was reacted with nitrile 16 (800 mg, 3.3 mmol) as described in general procedure V(a) to yield a crude amine mixture. 5-Nitro-furan-2-carbonyl chloride (873 mg, 4.9 mmol) was added to the crude amine and Et3N (1.3 mL, 9.9 mmol) in CH2Cl2 (10 mL), and the reaction was carried out as described in general procedure V(b) to afford mg of amide 4k in 82% yield. 1H-NMR (500 MHz, CDCl3): δ0.0 – 0.04 (2Hs, m), 0.38 – 0.45 (2Hs, m), 0.74 – 0.82 (1H, m), 2.19 (2Hs, d, J = 6.34 Hz), 2.56 (4Hs, t, J = 5.12 Hz), 3.07 (1H, t, J = 5.12 Hz), 3.11 (3Hs, t, J = 5.12 Hz), 4.42 (2Hs, d, J = 5.85 Hz), 6.62 – 6.68 (1H, bs), 6.8 (2Hs, d, J = 8.54 Hz), 7.12 (2Hs, d, J = 8.54 Hz), 7.13 (1H, d, J = 3.66 Hz), 7.23 (1H, d, J = 3.66 Hz); 13C-NMR (300 MHz, CDCl3): 2.94, 4.42, 41.63, 46.0, 50.83, 60.72, 111.35, 114.89, 116.05, 128.05, 130.25, 147.49, 148.56, 156.68; ESI-MS: 385.6 (M+1).
5-Nitro-furan-2-carboxylic acid 4-(1-oxo-1λ4-thiomorpholin-4-yl)-benzylamide (4i) and 5-Nitrofuran-2-carboxylic acid 4-(1,1-dioxo-1 λ6-thiomorpholin-4-yl)-benzylamide (4j)
A mixture of compound 4k (1 g, 2.87 mmol) and NaHCO3 (1.2 g, 14.39 mmol) in CH2Cl2 (10 mL) at 0° C was treated with m-chloroperbenzoicacid (1.29 g, 5.75 mmol) and stirred at room temperature for 30 min. The reaction mixture was quenched with 10% aqueous NH4OH solution (10 mL) and diluted with CH2Cl2 (30 mL). The organic layer was washed with 10% aqueous NH4OH solution (30 mL), water (30 mL), brine (30 mL), and dried over Na2SO4. The organic solution was concentrated in a vacuum followed by flash column purification with petroleum ether and ethyl acetate in 5:1 ratio, which afforded 261 mg of 4i and 173 mg of 4j in 25% and 15% yields respectively. 4i: 1H-NMR (500 MHz, CDCl3): δ 2.87 – 2.99 (4Hs, m), 3.63 (2Hs, td, J = 3.66, 14.89 Hz), 4.04 (2Hs, dt, J = 2.68, 13.82 Hz), 4.6 (2Hs, d, J = 5.85 Hz), 6.98 (3Hs, d, J = 8.54 Hz), 7.32 – 7.36 (3Hs, m), 7.41 (2Hs, d, J = 3.66 Hz); 13C-NMR (300 MHz, CDCl3): 40.31, 42.49, 43.92, 111.89, 115.49, 116.09, 128.1, 129.04, 147.56, 148.48, 155.53; ESI-MS: 386.1 (M+23); 4j: 1H-NMR (300 MHz, CDCl3): δ3.12 (4Hs, t, J = 5.43 Hz), 3.88 (4Hs, t, J = 5.28 Hz), ), 4.57 (2Hs, d, J = 5.9 Hz), 6.81 – 6.90 (1H, bs), 6.92 (2Hs, d, J = 8.71 Hz), 7.27 – 7.36 (3Hs, m), 7.38 (2Hs, d, J = 3.79 Hz); 13C-NMR (300 MHz, CDCl3): 42.41, 47.08, 49.98, 111.86, 115.56, 116.04, 128.71, 129.25, 146.89, 147.41, 155.52; ESI-MS: 402.2 (M+1).
5-Nitro-furan-2-carboxylic acid 4-hydroxymethyl-benzylamide (18)
Acid chloride 11 in CH2Cl2 (10 mL) was added to (4-aminomethyl-phenyl)-methanol 17 (200 mg, 1.45 mmol) in Et3N (205 μL). The reaction mixture was stirred for 14 hrs at room temperature. After completion of the reaction, 100 mL of ethyl acetate was added and washed sequentially with 10% aqueous NaHCO3 (2 × 50 mL), water (2 × 50 mL) and brine (2 × 50 mL). The organic phase was dried over Na2SO4, filtered and concentrated followed by flash column purification to yield 260 mg (65%) of compound 18; TLC: Rf 0.5 (ethyl acetate); 1HNMR (500 MHz, CDCl3): δ 4.6 (2H, d, J = 5.85 Hz), 4.68 (2H, s), 7.15–7.25 (1H, bs), 7.27–7.29 (1H, m), 7.3–7.36 (4H, m), 7.37 (1H, d, J = 2.68 Hz); ESI-MS, m/z: 275 (M−1).
5-Nitro-furan-2-carboxylic acid 4-[(4-methyl-piperazin-1-ylimino)-methyl]-benzylamide (4l)
Pyridine (175 μL) was added to alcohol 18 (120 mg, 0.434 mmol) in CH2Cl2 (10 mL) followed by Dess-Martin periodinane reagent (184 mg, 0.434 mmol). The resulting reaction mixture was stirred at room temperature for two hours and was quenched with aqueous NaHCO3. The reaction mixture was extracted with CH2Cl2, dried over Na2SO4, and concentrated under vacuum to give 80 mg of the corresponding aldehyde (5-nitro-furan-2-carboxylic acid 4-formyl-benzylamide). 4-Methyl-piperazin-1-ylamine (36 μL, 0.291 mmol) was added to the crude aldehyde (80 mg, 0.291 mmol) in 10 mL ethanol. The reaction mixture was refluxed for an hour, then stirred at room temperature for 14 hrs. The reaction mixture was concentrated in vacuum, column purified and recrystallized to yield 80 mg (74%) of compound 4l; TLC: Rf 0.4 (9:1 chloroform: methanol); 1HNMR (500 MHz, CDCl3): δ 2.38 (3H, s), 2.58–2.72 (4H, bs), 3.32–3.19 (4H, bs), 4.64 (2H, d. J = 5.85 Hz), 6.82–6.88 (1H, bs), 7.31 (1H, d, J = 3.66 Hz), 7.34 (2H, d, J = 8.3 Hz), 7.38 (1H, d, J = 3.66 Hz), 7.54 (1H, s), 7.61 (2H, d, J = 8.05 Hz); 13CNMR (300 MHz, CDCl3): 42.84, 45.39, 50.39, 53.92, 111.82, 115.65, 125.98, 127.66, 134.31, 135.65, 136.11, 147.4, 155.59; ESI-MS, m/z: 372.1 (M+1).
General procedure VI: For preparation of 19a–e
Secondary amine (1 eq.) was added to a mixture of 3,4-difluoro-benzonitrile (1 eq.) and K2CO3 (1.5 eq.) in dimethyl sulfoxide (7 mL/g). The reaction mixture was stirred at 90° C and followed by TLC. After completion of the reaction, the mixture was diluted with ethyl acetate (60 mL/g), and washed with water (2 × 50 mL/g), followed by brine (50 mL/g). The ethyl acetate fraction was dried over Na2SO4 and concentrated. The crude products were purified by flash column chromatography to afford pure products.
4-(4-Benzyl-piperazin-1-yl)-3-fluoro-benzonitrile (19a)
1-Benzyl piperazine (1.25 mL, 7.19 mmol) was added to a mixture of 3,4-difluoro-benzonitrile 10 (1.0 g, 7.19 mmol) and K2CO3 (1.48 g, 10.78 mmol) in dimethyl sulfoxide (7 mL) and the reaction continued as described in general procedure VI to afford amine 1.86 g of 19a in 88% yield. 1H-NMR (300 MHz, CDCl3): δ 2.64 (4Hs, t, J = 4.84 Hz), 3.25 (4Hs, t, J = 4.8 Hz), 3.59 (2Hs, s), 6.92 (1H, t, J = 8.54 Hz), 7.25–7.42 (7Hs, m); ESI-MS: 296.4 (M+1).
3-Fluoro-4-thiomorpholin-4-yl-benzonitrile(19c)
Thiomarpholine (0.68 mL, 7.19 mmol) was added to a mixture of 3,4-difluoro-benzonitrile 10 (1.0 g, 7.19 mmol) and K2CO3 (1.48 g, 10.78 mmol) in dimethyl sulfoxide (7 mL) and the reaction continued as described in general procedure VI to afford amine 1.2 g of 19c in 81% yield. 1H-NMR (300 MHz, CDCl3): δ2.8 (4Hs, t, J = 5.06 Hz), 3.5 (4Hs, t, J = 5.12 Hz), 6.94 (1H, t, J = 8.51 Hz), 7.28 (1H, dd, J = 1.92, 12.35 Hz), 7.37 (1H, ddd, J = 0.86, 1.86, 8.38 Hz); ESI-MS: 245.5 (M+23).
5-Nitro-furan-2-carboxylic acid 4-(4-benzyl-piperazin-1-yl)-3-fluoro-benzylamide (5a)
65% Red-Al in toluene (3.15 mL, 10.15 mmol) was reacted with nitrile 19a (1 g, 3.38 mmol) as described in general procedure V(a). 5-Nitro-furan-2-carbonyl chloride (1.12 g, 6.75 mmol) was added to the crude amine and Et3N (1.4 mL, 10.15 mmol) in THF (10 mL), and the reaction was carried out as described in general procedure V(b) to afford 1.21 g of amide 5a in 82% yield. 1H-NMR (500 MHz, CDCl3): δ2.74 – 2.82 (4Hs, m), 3.09 – 3.18 (4Hs, m), 3.62 (2Hs, s), 4.59 (2Hs, d, J = 5.85 Hz), 6.85 – 6.9 (1H, bs), 6.96 (1H, t, J = 8.3 Hz), 7.08 (2H, dt, J = 1.95, 12.93 Hz), 7.3 – 7.42 (7Hs, m); 13C-NMR (300 MHz, CDCl3): 42.22, (49.91, 49.95), 52.54, 62.53, 111.83, 115.23, (115.51, 115.62), (118.56, 118.61), (123.5, 123.54), 126.6, 127.72, 128.69, (130.53, 130.62), 137.41, 147.34, 153.4, 155.55, 156.68; ESI-MS: 437.1 (M−1).
5-Nitro-furan-2-carboxylic acid 3-fluoro-4-thiomorpholin-4-yl-benzylamide (5c)
65% Red-Al in toluene (4.14 mL, 13.49 mmol) was reacted with nitrile 19c (1 g, 4.49 mmol) as described in general procedure V(a) to yield a crude amine mixture. 5-Nitro-furan-2-carbonyl chloride (1.5 g, 8.99 mmol) was added to the crude amine and Et3N (1.8 mL, 13.49 mmol) in THF (10 mL), and the reaction was carried out as described in general procedure V(b) to afford 1.3 g of amide 5c in 79% yield. 1H-NMR (500 MHz, CDCl3): δ 2.82 – 2.88 (4Hs, m), 3.32 – 3.4 (4Hs, m), 4.6 (2Hs, d, J = 5.85 Hz), 6.86 – 6.91 (1H, bs), 6.98 (1H, t, J = 8.3 Hz), 7.09 (2H, dt, J = 1.95, 12.93 Hz), 7.34 (1H, d, J = 3.9 Hz), 7.41 (1H, d, J = 3.9 Hz); 13C-NMR (300 MHz, CDCl3): 27.41, (42.18, 42.19), (52.63, 52.67), 111.85, 115.32, (115.60, 115.65), 118.39, (119.71, 119.75), (123.49, 123.54), (131.26, 131.35), (140.12, 140.23), 147.3, 153.55, 155.59, 156.83; ESI-MS: 363.9 (M−1); Anal. Calcd. for C16H16FN3O4S: C, 52.59, H, 4.41; N, 11.50. Found: C, 51.90; H, 4.38; N, 11.08.
MIC determinations against M. tuberculosis H37Ra and H37Rv
MIC values of the nitrofuranyl amides against M. tuberculosis H37Rv were determined by the micro broth dilution method according to NCCLS guidelines. A broth culture of M. tuberculosis was grown in Middlebrook 7H9 medium with 10% ADC supplement to an OD600 of 0.4 – 0.6. The culture was diluted with 7H9 medium to an OD600 of 0.01 and 100 μL of these cells were then added to a microtiter plate containing serial dilutions of the nitrofuranyl amides for a final volume of 200 μL. The plates were incubated at 37° C for 8 days. The MIC90 was determined by visual inspection for wells with greater than 90% inhibition of growth.
Maximum Tolerated Dose Assay (MTD)
Three healthy mice were given one single dose of the compound by oral gavage and were observed at regular times for any adverse effects. Three different concentrations were tested, generally at 100, 300 and 500 mg/kg. The latter dose was about twice to five times the dose used for efficacy testing of the compound in mice. After 7 days of observation the mice are sacrificed and the organs are studied by gross necropsy.
Determination of the in vivo biological activity and basic bioavailability after oral administration
8–10 Week old female C57BL/6 mice were dosed (at a dose lower than the maximum tolerated dose of the compound used, generally 300 mg/kg), via oral gavage. At 20 min., 2 hr, 4 hr and 8 hr after dosing, 3 mice were bled from the tail vein. Blood samples were collected aseptically, stored on ice, and centrifuged to collect serum. The drug concentration in the collected serum samples was determined by a microdilution MIC method using M. tuberculosis H37Rv. Serum samples were prepared as two-fold dilutions using serum from naïve mice as diluent (ranging from 10% to 0.312% from collected mouse serum samples). The serum dilutions were subsequently added in 10 μL to the 96-well assay plate, starting with a maximum of 10% serum collected from drug treated mice in the top well. Standards of the tested compounds were tested on the same 96-well microtiter plate using 3-fold dilutions of the compound ranging from 30 μg/mL up to 0.51 ng/mL final concentrations, in presence and without 10% mouse serum. Compounds in standard lanes were diluted in 100% DMSO to avoid any solubility problems (2% final DMSO concentration). The M. tuberculosis H37Rv suspension was grown as a mid-Log culture and frozen in aliquots until further use. A frozen stock was added at 104 CFU per well in a volume of 50 μL 7H9 medium to the 96-well plates (total volume per well is adjusted to 100 μL with 7H9 culture medium). The plates were incubated at 35 °C for 2 weeks. Optical density was measured after 3, 6, 9 and 12 days at 600 nm, and results were confirmed by visual inspection at 10 days. Inhibition of bacterial growth in the bioassay indicated sufficient high concentrations of bioactive product in the bloodstream. Wells were scored as positive (drug containing) wells when the OD600 values were less than 50% of the OD600 value of the untreated control wells. An estimation of serum drug levels (in μg per mL serum) was obtained by using the MIC data from the standard drug lanes.
GKO Mouse Model
Mice were infected via low dose aerosol with M. tuberculosis to reproducibly deliver approximately 50 bacilli in the lungs as described previously.19 Treatment is initiated 18 days post infection for 9 daily treatments for one single dose (at 300 mg/kg). Bacterial load is determined 27 days post infection in lungs and spleens of the mice by serial dilution of the tissue homogenates on nutrient Middlebrook 7H11 agar plates (GIBCO BRL, Gaithersburg, Md.). The plates were incubated at 37° C in ambient air for 4 weeks prior to the counting of viable M. tuberculosis colonies (CFU). The viable bacterial numbers were converted to logarithms, which were then evaluated by multiplecomparison analysis of variance by a one-way Dunnett test (SigmaStat software program). Differences were considered significant at the 95% level of confidence.
Supplementary Material
Acknowledgments
We acknowledge Veronica Gruppo, Karen Marietta and Christine Johnson for their excellent technical assistance. This work is supported by grant AI062415 from the National Institutes of Health and with help from Tuberculosis Antimicrobial Acquisition and Coordinating Facility (TAACF) program.
References
- 1.Dye C, Scheele S, Dolin P, et al. Global Burden of Tuberculosis-Estimated Incidence, Prevalence, and Mortality by Country. J Am Med Assoc. 1999;2827:677–686. doi: 10.1001/jama.282.7.677. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization Global Tuberculosis Programme 2005, WHO Report on the Tuberculosis Epidemic 2005, W.H.O. Geneva
- 3.de Jong BC, Israelski DM, Corbett EL, Small PMTB. AIDS. Clinical management of tuberculosis in the context of HIV infection. Annu Rev Med. 2004;55:283–301. doi: 10.1146/annurev.med.55.091902.103753. [DOI] [PubMed] [Google Scholar]
- 4.Tangallapally RP, Lee RE, Yendapally R, Hevener K, Jones VC, Lenaerts AJM, McNeil MR, Wang Y, Franzblau S, Lee RE. Synthesis amd evaluation of Nitrofuranyl amides as novel inhibitors of UDP-Gal mutase and new antituberculosis agents. J Med Chem. 2004;47:5276–5283. doi: 10.1021/jm049972y. [DOI] [PubMed] [Google Scholar]
- 5.Barry CE, Boshoff HIM, Dowd CS. Prospects for Clinical Introduction of Nitroimidazole Antibiotics for the Treatment of Tuberculosis. Curr Pharm Des. 2004;10:3239–3262. doi: 10.2174/1381612043383214. [DOI] [PubMed] [Google Scholar]
- 6.Brighty KE, Gootz TD. Chemistry and mechanism of action of the quinolone antibacterials. In: Andriole VT, editor. The Quinolones. 3. Academic Press Inc; San Diego: 1998. pp. 34–98. [Google Scholar]
- 7.Sensi P, Maggi N, Furesz S, Maffii G. Chemical modifications and biological properties of rifamycins. Antimicrobial Agents Chemother. 1966;6:699–714. [PubMed] [Google Scholar]
- 8.Lauteslager XY, Van Stokkum IHM, Van Ramesdonk, et al. Conformational Dynamics of Charge-Transfer States in Donor-Bridge-Acceptor Systems. Euro J Org Chem. 2001;16:3105–3118. [Google Scholar]
- 9.Brown GR, Foubister AJ, Ratcliffe PD. High Yields of meta-Substituted Amination Products in the SNAr Substitution of Benzenes. Tet lett. 1999;40(6):1219–1222. [Google Scholar]
- 10.Valenta V, Protiva M. Potential neuroleptics of the orthopramide series; synthesis of N-substituted 5-(aminosulfonyl)-2-methoxybenzamides. Collection of Czechoslovak Chemical Communications. 1987;52(8):2095–2106. [Google Scholar]
- 11.Takahashi H, Chida Y, Yoshii T, Suzuki T, Yanaura S. Asymmetric α-substituted phenethylamines. VI. Synthesis and analgesic activity of optically pure (R)- and (S)-N-alkyl-1-cyclohexyl-2-phenylethylamines. Chem & Pharm Bull. 1986;34(5):2071–2077. doi: 10.1248/cpb.34.2071. [DOI] [PubMed] [Google Scholar]
- 12.Gadwood RC, Shinabarger DA. Progressin the Oxazolidinone Antibacterials. Ann Rep in Med Chem. 2000;35:135–144. [Google Scholar]
- 13.Hart DJ, Li J, Wu W, Kozikowski AP. Applications of Organosulfur Chemistry to Organic Synthesis: Total Synthesis of (+)-Himbeline and (+)-Himbacine. J Org Chem. 1997;62(15):5023–5033. [Google Scholar]
- 14.Dess DB, Martin JC. Readily accessible 12-I-5 oxidant for the conversion of primary and secondary alcohols to aldehydes and ketones. J Org Chem. 1983;48(22):4155–4156.Dess DB, Martin JC. A useful 12-I-5 triacetoxyperiodinane (the Dess-Martin periodinane) for the selective oxidation of primary or secondary alcohols and a variety of related 12-I-5 species. J Am Chem Soc. 1991;113(19):7277–7287. For to improved procedures for the preparation of DMP, see: Ireland REL. An improved procedure for the preparation of the Dess-Martin periodinane. J Org Chem. 1993;58(10):2899.Meyer SD, Schreiber L. Acceleration of the Dess-Martin Oxidation by water. J Org Chem. 1994;59(24):7549–7552.
- 15.John MD, Lori DH, Carl LH, Marland PH, Thomas FM, Joseph PS, Marjorie S. New structure-activity relationships of the quinolone antibacterials using the target enzyme. The development and application of a DNA gyrase assay. J Med Chem. 1986;29(3):394–404. doi: 10.1021/jm00153a015. [DOI] [PubMed] [Google Scholar]
- 16.Tokuyama R, Takahashi Y, Tomita Y, Tsubouchi M, Yoshida T, Iwasaki N, Kado N, Okezaki E, Nagata O. Structure-activity relationship (SAR) studies on oxazolidinone antibacterial agents 2. Relationship between lipophilicity and antibacterial activity in 5-thiocarbonyl oxazolidinones. Chem & Pharm Bull. 2001;49(4):353–360. doi: 10.1248/cpb.49.353. [DOI] [PubMed] [Google Scholar]
- 17.Lee RE, Protopopova M, Crooks E, Slayden RA, Terrot M, Barry CE., III Combinatorial Lead Optimization of [1,2]-Diamines Based on Ethambutol as Potential Antituberculosis Preclinical Candidates. J Comb Chem. 2003;5:172–187. doi: 10.1021/cc020071p. [DOI] [PubMed] [Google Scholar]
- 18.Gruppo V, Johnson CM, Marietta KS, Scherman H, Zink EE, Crick DC, Adams LB, Orme IM, Lenaerts AJ. New strategies for the rapid screening of experimental compounds against Mycobacterium tuberculosis in the preclinical stage submitted 05/05 to Antimicrob. Agents Chemother. doi: 10.1128/AAC.50.4.1245-1250.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lenaerts AJ, Gruppo VJ, Brooks V, Orme IM. Rapid in vivo Screening of Experimental Drugs for Tuberculosis Using Gamma Interferon Gene-Disrupted Mice. Antimicrob Agents Chemother. 2003;47(2):783–785. doi: 10.1128/AAC.47.2.783-785.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelly BP, Furney SK, Jessen MT, Orme IM. Low-dose Aerosol Infection Model for Testing Drugs for Efficacy Against Mycobacterium tuberculosis. Antimicrob Agents Chemother. 1996;40(12):2809–2812. doi: 10.1128/aac.40.12.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orme I. Search for New Drugs for Treatment of Tuberculosis. Tuberculosis Drug Screening Program. Antimicrob Agents Chemother. 2001;45:1943–1946. doi: 10.1128/AAC.45.7.1943-1946.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.