

Abstract
The atomic-resolution crystal structures of human carbonic anhydrases I and II complexed with “two-prong” inhibitors are reported. Each inhibitor contains a benzenesulfonamide prong and a cupric iminodiacetate (IDA-Cu2+) prong separated by linkers of different lengths and compositions. The ionized NH− group of each benzenesulfonamide coordinates to the active-site Zn2+ ion; the IDA-Cu2+ prong of the tightest-binding inhibitor, BR30, binds to H64 of CAII and H200 of CAI. This work provides the first evidence verifying the structural basis of nanomolar affinity measured for two-prong inhibitors targeting the carbonic anhydrases.
Introduction
The α-carbonic anhydrases (EC 4.2.1.1) catalyze the reversible hydration of CO2 to form HCO3−, and these zinc enzymes are involved in a number of biosynthetic reactions such as gluconeogenesis, ureagenesis, lipogenesis, the biosynthesis of certain amino acids, and pyrimidine nucleotide biosynthesis.1 These enzymes are also implicated in cell proliferation, spermatozoan motility, pH homeostasis in bodily fluids, and aqueous humor production.2 Human carbonic anhydrase II (CAII) is the best studied of the twelve catalytically-active mammalian carbonic anhydrases; with kcat/KM = 1.5 × 108 M−1 s−1, CAII is also among the most catalytically efficient enzymes, operating near the limit of diffusion control.3 CAII is also a drug target for the treatment of glaucoma, since CAII inhibitors lower the high intraocular pressure that characterizes this disease.1,2 The crystal structure of CAII was first reported by Liljas and colleagues4 and has since been refined at 1.54 Å resolution.5 The structure of H64A CAII has been refined at 1.05 Å resolution.6 These structures show that the catalytic zinc ion resides at the bottom of a ∼15 Å-deep conical active site cleft, where it is tetrahedrally coordinated by H94, H96, H119, and a hydroxide ion; zinc-bound hydroxide donates a hydrogen bond to T199. About half-way out of the active site cleft is H64, which serves as a proton shuttle during catalysis.6
Inhibition of carbonic anhydrase by arylsulfonamides was first described in the 1940s7 and has been an active focus of research ever since. Arylsulfonamide inhibitors have potential uses as diverse as glaucoma therapy or zinc biosensing.7-9 A wide array of structural data show that the sulfonamide moiety ionizes upon binding to the enzyme to yield an NH− group that displaces the zinc-bound hydroxide ion and donates a hydrogen bond to T199.9,10
The diverse biological roles of mammalian carbonic anhydrase isozymes inspire continuing interest in the design of isozyme-specific inhibitors.9d, 11-12, 14-16 The structure-based design of carbonic anhydrase inhibitors has led to the study of “two-prong” benzenesulfonamide inhibitors that achieve enhanced binding affinity by targeting the active site Zn2+ ion as well as a second structural feature of the active site cavity, such as the hydrophobic wall.12 This approach has been limited, however, by the lack of strong specific interactions, such as hydrogen bonds, that can be targeted on a hydrophobic surface. Even so, coupling two molecules that independently bind to two separate, specific sites, e.g., the active site zinc ion and a polar residue 10-15 Å away, can significantly enhance enzyme-inhibitor affinity. For example, coupling cupric iminodiacetate (IDA-Cu2+; designed to bind to solvent-exposed histidine residues of CAII13) to benzenesulfonamide results in up to 100-fold increases in binding affinity.14-16 Moreover, altering the length of the linker between the benzenesulfonamide prong and the IDA-Cu2+ prong of these inhibitors modulates affinity and isozyme selectivity. Enhanced binding affinity results from the reduced entropic cost of binding two covalently linked prongs in comparison with the binding of two unlinked prongs (Figure 1). There is precedent for such binding enhancement in nature: polyvalency has long been recognized as an important feature of antibody-antigen affinity.17
Figure 1.

Favorable interactions of both the primary and secondary prongs of the inhibitor lead to tighter binding of the two-prong inhibitor than of either prong alone. For the inhibitors discussed in this paper, one prong is the benzenesulfonamide moiety that coordinates to the active site zinc ion, and the second prong is the IDA-Cu2+ moiety that targets a histidine residue on the protein surface. Covalent linkage of the two prongs decreases the entropic cost of binding such that KD < KP•KS.
In order to confirm the structural rationale motivating the design of two-prong inhibitors of the carbonic anhydrases, we now report the atomic resolution crystal structures of CAI and CAII complexed with two-prong inhibitors (Table 1). One of the tightest-binding inhibitors, BR30, clearly binds with both prongs making the predicted intermolecular interactions, and structural comparison of the CAII-BR30 and CAI-BR30 complexes suggests an explanation for the differing affinities of BR30 for CAI and CAII. Inhibitors BR15, BR17, and BR22, which contain longer linkers between the benzenesulfonamide and IDA-Cu2+ prongs, are partially disordered in the crystal structures. Surprisingly, in all CAII structures a second inhibitor binding site is observed at the rim of the active site near the N-terminus of the enzyme, and a third binding site is observed in the CAI-BR30 complex. Notably, in all structures in which the IDA-Cu2+ moiety is resolved, a histidine residue coordinates to the Cu2+ ion. Thus, this work provides the first X-ray crystallographic demonstration of IDA-Cu2+-histidine coordination interactions in a protein.
Table 1.
Carbonic Anhydrase Inhibitors*
| Inhibitor | KD (nM) | |
|---|---|---|
| CAI | CAII | |
| 4030 | 660 | |
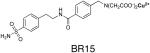 |
200 | 235 |
| 76 | 80 | |
| 230 | 27 | |
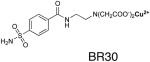 |
120 | 28 |
Results and Discussion
Inhibitor binding does not trigger any major structural changes in any enzyme-inhibitor complex, and the root-mean-square deviations of backbone Cα atoms between the native enzyme and each enzyme-inhibitor complex range from 0.228 Å to 0.387 Å. The benzenesulfonamide moiety of each inhibitor binds identically in the active sites of CAI and CAII: the ionized sulfonamide NH− group coordinates to Zn2+ and donates a hydrogen bond to Oγ of T199, and one sulfonamide oxygen accepts a hydrogen bond from the backbone NH group of T199. In the structure of the CAII-BR15 complex, the portion of the inhibitor observed in the electron density map includes only the benzenesulfonamide moiety and part of the linker up to the carbonyl oxygen of the amide group (Figure 2a), indicative of substantial molecular disorder. The binding of BR17 is slightly more ordered, with electron density extending through the ethylene glycol linker (Figure 2b). The electron density map of the CAII-BR22 complex indicates that the inhibitor tail is disordered between two alternative conformations (Figure 2c). However, both conformations extend along the hydrophobic side of the active-site cleft, away from the N-terminal histidines and the bound Cu2+ ions. Finally, the electron density map of the CAII-BR30 complex reveals a fully ordered inhibitor bound in the enzyme active site (Figure 2d). The benzenesulfonamide prong coordinates to Zn2+ and the IDA-Cu2+ prong binds to H64.
Figure 2.
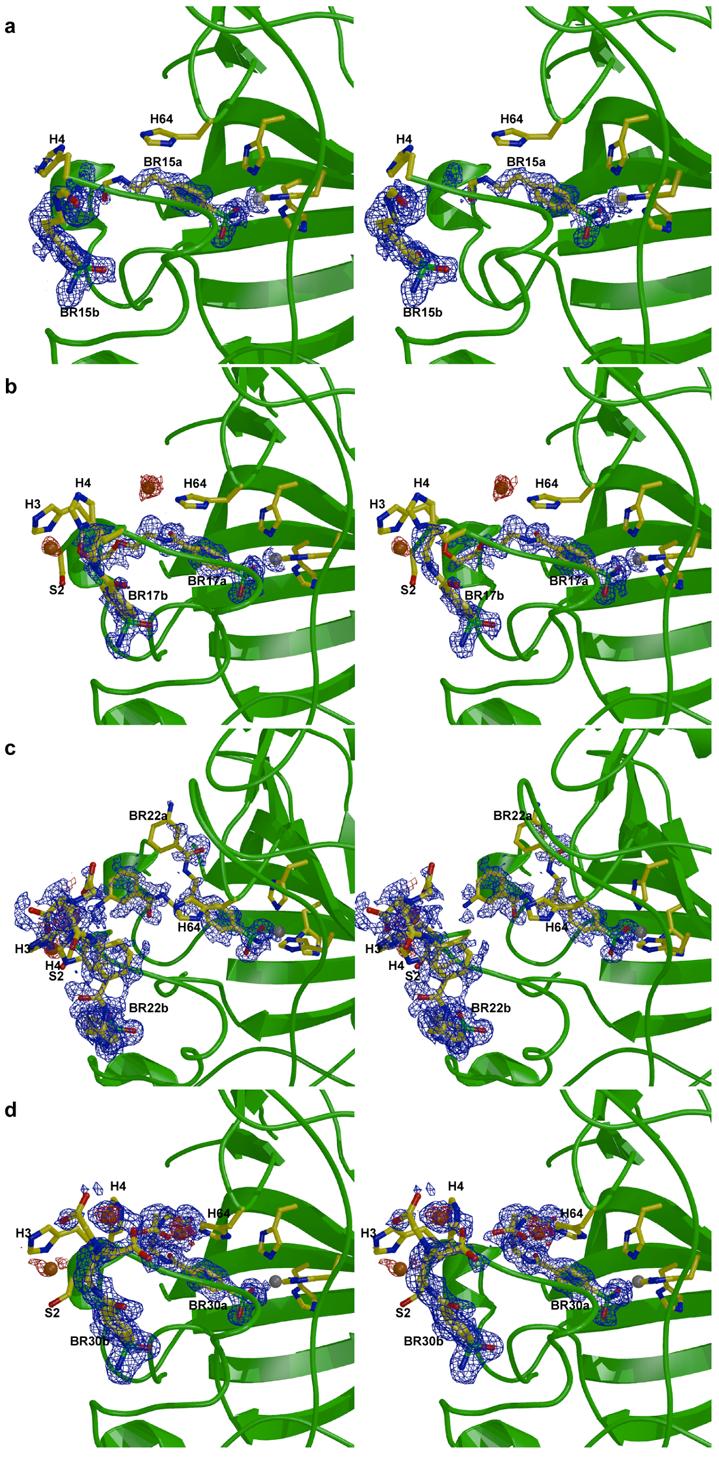
Stereo figures illustrating CAII-ligand interactions, including anomalous difference maps used for identifying copper ions (red, contoured at 2 σ) and simulated-annealing omit maps of ligands (blue, contoured at 1.5 σ (c), 2 σ (a) or 2.5 σ (b, d)). Zn2+ ions are shown in gray, Cu2+ ions in orange. (a) CAII-BR15 complex, (b) CAII-BR17 complex, (c) CAII-BR22 complex, (d) CAII-BR30 complex. Inhibitors bound in the active site with ionized sulfonamide coordinated to Zn2+ are labeled “a” and inhibitors in the second binding site at the rim of the active site are labeled “b”. Zn2+ ions are shown in gray, Cu2+ ions in orange. Figure prepared with Bobscript and Raster3D.18
Unexpectedly, a second inhibitor molecule binds near the N-terminus of the enzyme and the rim of the active site cavity in each of the complexes with CAII. This second binding site, indicated as the “b” site in Figure 2, is identical for the four inhibitors described herein but differs from the alternative binding site previously observed by Kim and colleagues.19 In the second binding site, one sulfonamide oxygen atom hydrogen bonds to a solvent molecule, which in turn hydrogen bonds to Oγ of S2, while the other oxygen atom has no interactions. The nitrogen atom of the sulfonamide hydrogen bonds to the backbone O of H15 and to Oδ of D19. Unlike inhibitor binding in the “a” site, inhibitor binding in the “b” site does not appear to require ionization of the benzenesulfonamide moiety (Figure 2). In the structure of the CAII-BR15 complex, only the benzenesulfonamide prong is discernible, while in the BR17 complex part of the tail is also observed. In the BR22 and BR30 complexes, the complete inhibitor can be modeled in the second binding site, although the only interactions with the protein are those described above. Although no major structural changes occur as a result of inhibitor binding to the “a” site, the three N-terminal residues, M1-H3, undergo some rearrangement upon binding to the “b” site in the BR22 and BR30 complexes; furthermore, S2, which is disordered in wild-type CAII, is visible in the electron density of these two structures and coordinates to a Cu2+ ion.
Due to the exothermic nature of the binding of benzenesulfonamide and its derivatives to CAI and CAII, enzyme-inhibitor interactions are easily probed by isothermal titration calorimetry (ITC).14,20,21 Recently, we observed that the binding isotherm for the interaction of a two-prong inhibitor to CAI was consistent with two different binding sites with enzyme:inhibitor stoichiometry ≈ 1:2.14 This observation is in marked contrast to the binding of benzenesulfonamide or IDA-Cu2+ to either CAI or CAII; each of these ligands binds to a single site with enzyme:inhibitor stoichiometry ≈ 1:1 (see below and ref 14). We initially conjectured that the multiple binding modes of two-prong inhibitors accounted for two different binding sites. The X-ray crystal structures provide clear evidence for such multiple binding modes, i.e., the inhibitor binding sites “a” and “b” identified in CAII (Figure 4). However, to ensure that the biphasic ITC binding profile is not limited to our previously employed two-prong inhibitor, we performed a comparative ITC titration for the binding of benzenesulfonamide and BR30 to CAII. As shown in Figure 3, whereas the ITC titration profile for the binding of benzenesulfonamide to CAII is best fit by a “one site” binding model, that of BR30 to CAII requires at least a “two site” binding model to obtain a reasonable fit of the experimental data. Hence, unlike benzenesulfonamide, which exclusively binds in the active site pocket, BR30 clearly binds to at least two different sites on CAII in solution and in the crystal.
Figure 4.

Stereo figure illustrating CAI-BR30 interactions, including anomalous difference maps used for identifying copper ions (red, contoured at 3 σ) and simulated-annealing omit maps of ligands (blue, contoured at 2.5 σ). The inhibitor bound in the active site with ionized sulfonamide coordinated to Zn2+ is labeled “a” and the inhibitor bound in the second binding site is labeled “b”. Very weak electron density is also observed for a third inhibitor, “c”. Figure prepared with Bobscript and Raster3D.18
Figure 3.

Isothermal titration calorimetric studies for the binding of benzenesulfonamide (left panel) and BR30 (right panel) to CAII. The top panels show the raw data for the titration of 10 μM enzyme with increasing aliquots (4 μl each) of stock solution of the ligands. The bottom panels show the integrated heat signals as a function of molar ratios of ligands to CAII. The solid smooth lines are the best fit of the data for “one site” (left panel) and “two site” (right panel) classes of binding sites of benzenesulfonamide and BR30 to CAII, respectively. Following are the derived thermodynamic parameters for the best fit of the data. CAII-benzenesulfonamide complex (one binding site model): N = 0.76, Ka = 1.7 × 106 M−1, and ΔH° = −9.1 kcal/mol. CAII-BR30 complex (two binding site model): N1 = 1.2, Ka1 = 2.9 × 108 M−1, ΔH°1 = −42.2 kcal/mol; N2 = 0.28, Ka2 = 1.6 × 106 M−1, ΔH°2 = −9.8 kcal/mol.
In three of the CAII-inhibitor complexes (BR17, BR22, and BR30), ordered Cu2+, Zn2+, and Hg2+ positions are confirmed using Bijvoet differences in |F+| - |F−| maps (Figures 2a, c, d). In the CAII-BR17 complex, a single Cu2+ ion is coordinated by N∊ of H64 (which occupies the “out” conformation) and has a single ordered water ligand; there is no visible density for the rest of the inhibitor molecule in the vicinity of the metal ion. Two Cu2+ ions are observed in the CAII-BR22 complex. The first ion is coordinated with square planar geometry by Nδ and N of H3, Oγ of S2, and Oδ of D52 from a symmetry-related molecule. There is no visible electron density for the rest of the inhibitor molecule. The second Cu2+ ion is coordinated by N∊ of H4, and by the imino nitrogen and two acetate oxygens of the inhibitor in the second binding site. Three Cu2+ ions are observed in the CAII-BR30 complex. The first is coordinated with square pyramidal geometry by N∊ of H64 (which occupies the “in” conformation), by the imino nitrogen and two acetate oxygens of the active-site inhibitor, and by a solvent molecule in the apical position. The second Cu2+ ion is coordinated by the imino nitrogen and two acetate oxygens of a second inhibitor molecule and is positioned 2.91 Å from N∊ of H4. The third Cu2+ ion is coordinated with square planar geometry by Nδ and N of H3, Oγ of S2, and Oδ of D52 from a symmetry-related molecule.
In the structure of the CAI-BR30 complex, the inhibitor molecule binds in a mode similar to that observed in the structure of the CAII-BR30 complex except that the IDA-Cu2+ prong binds to N∊ of H200 rather than H64 (Figure 4). Least squares superposition of CAI and CAII reveals that the presence of H200 in CAI shifts the Cα of H64 1.5 Å from its position in CAII, which causes a ∼1.0 Å shift in the position of the IDA-Cu2+ prong (Figure 5). These structural differences, along with the difference in KD for binding of the benzenesulfonamide moiety to CAI and CAII (Table 1), contribute to the ∼4-fold higher affinity of BR30 for CAII compared with CAI.
Figure 5.

Least-square superposition of the CAI-BR30 structure (blue and green) with the CAII-BR30 structure (red and orange). The root-mean-square deviation of 252 Cα atoms between the two structures is 0.904 Å. Figure prepared with Bobscript and Raster3D.18
Two additional BR30 molecules bind to each CAI molecule in the asymmetric unit (Figure 4). The sulfonamide nitrogen atom of the second inhibitor hydrogen bonds to the backbone O of H243 and to a solvent molecule. A sulfonamide oxygen also hydrogen bonds to a solvent molecule, as does O7 of the inhibitor molecule. The Cu2+ ion is coordinated with distorted square planar geometry by N∊2 of H243 and by two oxygen and one nitrogen atoms of the iminodiacetate moiety. The third inhibitor is characterized by very weak electron density (Figure 4). One sulfonamide oxygen accepts hydrogen bonds from Oγ and the backbone nitrogen atoms of S99. The Cu2+ ion is coordinated with square planar geometry by N∊2 of H103 and by two oxygen and one nitrogen atoms of the iminodiacetate moiety.
In light of the results of earlier studies of similar inhibitors15,16, as well as the high affinities measured for BR17 and BR22 (Table 1), the disordered secondary prongs of these inhibitors in all complexes except those with BR30 are surprising. Potential protein ligands to IDA-Cu2+ within 8 Å of BR22 include Sδ of M1 (present in the recombinant protein but not in the protein purified from human cells) and N∊ of Q136, which are 5.86 Å and 5.95 Å from N17 of the A conformation of the inhibitor, and Oδ of D72 and Oδ of E69, which are 5.21 Å and 6.96 Å from N17 of conformation B. However, there are no anomalous difference peaks in the vicinity of these residues, indicating that the Cu2+ ion is completely disordered or, less likely, dissociated from these inhibitors. Inhibitors BR17 and BR22 bind more tightly to CAII than benzenesulfonamide22 by factors of 18-fold and 53-fold, respectively (Table 1), indicating that the second prong does indeed contribute to enhanced affinity. Possibly, the discrepancy between the solution and crystallographic results may arise from binding of free Cu2+ to the N-terminal histidine residues, which then might prevent optimal binding of the inhibitor. However, the origin of free Cu2+ is unclear in light of the stability of the IDA-Cu2+ complex (KD = 10−11 M) compared to the imidazole-Cu2+ complex (KD = 10−5 M).23 Additionally, the inhibitor concentration in the crystallographic experiments is much higher than in the solution experiments: at higher concentrations, inhibitor binding at the second, N-terminal site may affect the binding of the IDA-Cu2+ prong of the first inhibitor molecule in the enzyme active site.
It is important to note that the enzyme-inhibitor dissociation constants reported in Table 1 have been measured by the dansylamide displacement method.8a Since dansylamide binds only to Zn2+ in the active site, as confirmed by X-ray crystallography8c, it could be argued that the dissociation constants of the inhibitors shown in Table 1 only reflect the contribution of benzenesulfonamide-Zn2+ interactions in the primary binding site (i.e., the equilibrium described by KP in Figure 1) and not the contribution of IDA-Cu2+-histidine interactions in the secondary site (i.e., the equilibrium described by KS in Figure 1). However, in an earlier study of two-prong CAI inhibitors, Banerjee and colleagues conclusively demonstrate that affinity measurements made using the dansylamide displacement method are sensitive to enzyme-inhibitor interactions in the secondary binding site: the chelation of Cu2+ ions by ethylenediaminetetraacetate and resultant inactivation of the IDA-Cu2+ prong of an inhibitor causes a 76-fold increase in the dissociation constant as measured by the dansylamide displacement method.14 Hence, the dansylamide dissociation assay is indeed sensitive to binding interactions in both the primary and secondary sites and therefore measures the overall Kd for the inhibitors shown in Table 1.
Finally, the origin of free Cu2+ ions in three of the reported structures is unclear. Since dissociation from the IDA moiety of the inhibitor is very unfavorable, Cu2+ may have been present as a trace contaminant in either the synthetic or crystallographic preparations. Regarding the binding of free Cu2+, it is interesting to note that the structure of Cu2+-substituted CAII has been reported, in which a second Cu2+ ion binds to H4 and to H64 in the “out” conformation.24 Thus, the conformation of H64 can be used to distinguish between the binding of free Cu2+ and the binding of the IDA-Cu2+ prong of inhibitors such as BR30, which interacts with the “in” conformer of H64 (Figure 2d). Accordingly, since the “out” conformer of H64 coordinates to Cu2+ in the complex with BR17, it is possible that this reflects the binding of free Cu2+.
Concluding Remarks
We have shown that the design of two-prong inhibitors of CAII enhances enzyme-inhibitor affinity and that the two prongs of the tightest binding inhibitor, BR30, make the targeted interactions initially hypothesized in the design rationale.16 The IDA-Cu2+ moiety is an effective “warhead” for histidine imidazoles, and the structures of the CAII-BR30 and CAI-BR30 complexes reveal IDA-Cu2+ coordination to H64 of CAII (Figure 2d) and to H200 of CAI (Figure 4). Although inhibition of proton transfer by free metal ions has been suggested for certain metal-containing CA inhibitors24,25, inhibitors such as BR30 completely deactivate the enzyme by displacing the catalytic nucleophile (zinc-bound hydroxide), blocking substrate binding in the enzyme active site, and inhibiting the catalytic proton shuttle, H64. Further work will allow us to explore more detailed structure-affinity relationships for two-prong inhibitors targeting the greater family of carbonic anhydrase isozymes. Although the inhibitors presented in this study are slightly less potent than classic carbonic anhydrase inhibitors (e.g., ethoxzolamide, with Kd = 1.9 nM)26, the demonstrated selectivity for CAII is a key feature that we hope to enhance in future modifications of the IDA-Cu2+ prongs.
Experimental Section
Synthesis
BR15
The synthesis of this compound has been reported.16
BR17
The synthesis of this compound has been reported.15
BR22

Compound 6
To a solution of IDA acid 527 (1.0 g, 4.04 mmol) in ethylacetate (EtOAc) (30 ml) were added N-hydroxysuccinimide (NHS) (0.5 g, 0.43 mmol) and dicyclohexylcarbodiimide (DCC) (0.9 g, 4.36 mmol). The resulting solution was stirred at room temperature for 45 min. The white solid was filtered and washed with EtOAc. The solvent was removed under reduced pressure and the viscous liquid was again dissolved in CHCl3 (30 ml). The m-aminobenzoic acid (788 mg, 4.75 mmol) was dissolved in CHCl3 (10 ml) in presence of triethylamine (Et3N) (0.8 ml, 5.75 mmol) and added to the above solution. The reaction was continued another 8 h at 40°C. The organic layer was washed with water and dried over anhydrous Na2SO4. After solvent evaporation the solid obtained was recrystallized with CHCl3/hexane. Yield: 0.42 g (28%). 1H NMR (300 MHz, CDCl3): δ 1.28 (t, 6H, J = 7.1 Hz), 3.51 (s, 2H), 3.61 (s, 4H), 4.22 (q, 4H, J = 7.1 Hz), 7.21 (bs, 1H), 7.39 (m, 1H), 7.80 (d, 1H, J = 7.5 Hz), 7.99 (m, 1H), 8.24 (s, 1H).
BR22
The reaction between the acid 6 (0.2 g, 0.55 mmol) and p-aminobenzenesulfonamide HCl salt (0.112 g, 0.55 mmol) was carried out in the presence of benzotriazol-1-yl-oxy-tris-(dimethylamino)phosphonium hexafluorophosphate (BOP) (0.25 g, 0.56 mmol) and Et3N (0.2 ml, 1.4 mmol) in CHCl3/dimethylformamide (DMF) at room temperature. The reaction mixture was stirred at room temperature for 8 h. The reaction was quenched with brine and the solvent was removed under reduced pressure. Water was added to the residue and the product was extracted with ethylacetate, then washed successively with 4% citric acid, 4% NaHCO3 solution, and brine. The crude product was purified by silica gel column chromatography with 8% MeOH in CHCl3 (Rf = 0.4). Yield: 0.285 (95%). 1H NMR (300, CDCl3-CD3OD-D2O): δ 1.25-1.32 (m, 6H), 3.00 (t, 2H, J = 6.5 Hz), 3.52 (s, 2H), 3.59 (s, 4H), 3.69 (t, 2H, J = 6.5 Hz), 4.16-4.24 (m, 4H), 7.33 (d, 2H, J = 8.0 Hz), 7.39 (t, 1H, J = 8.0 Hz), 7.47 (d, 1H, J = 8.0 Hz), 7.58 (d, 1H, J = 8.0 Hz), 7.85 (d, 2H, J = 8.0 Hz), 8.03 (s, 1H). 13C NMR (125 MHz, CDCl3-CD3OD): δ 14.44, 35.45, 40.91, 56.69, 60.43, 61.65, 116.86, 122.51, 123.75, 127.17, 129.66, 129.92, 135.63, 138.33, 140.82, 144.51, 167.58, 170.29, 171.73.
The above ester (0.184 g, 0.337 mmol) was dissolved in methanol (MeOH)/CH2Cl2 (6/4 ml) and solid LiOH (50 mg, 1.20 mmol) was added and stirred for 6 h at room temperature. The pH of the solution was lowered to 3 by adding 2 N HCl when a precipitate appeared. The precipitate was filtered and washed with methanol/dichloromethane. Drying under vacuum afforded the acid as a white solid. Yield: 120 mg (73%). 1H NMR (300 MHz, D2O): δ 2.95 (t, 2H, J = 6.6 Hz), 3.30 (s, 4H), 3.47 (s, 2H), 3.62 (t, 2H, J = 6.6 Hz), 7.35-7.40 (m, 3H), 7.43 (t, 1H, J = 8.0 Hz), 7.56 (d, 1H, J = 8.0 Hz), 7.70-7.75 (m, 3H).
The above acid (110 mg, 0.223 mmol) was dissolved in MeOH (8 ml) and solid CuCl2.2H2O (38 mg, 0.223 mmol) was added. The mixture was stirred at room temperature for 6 h. Solvent was removed and residue dried under vacuum. It was redissolved in ethanol and precipitated with CH2Cl2. The precipitate was filtered and washed with ethanol/CH2Cl2 and dried under vacuum affording a blue solid. Yield: 110 mg (86%). Anal. Calcd. for C21H22CuN4O8S.H2O: C, 44.09; H, 4.23; N, 9.79. Found: C, 44.19; H, 4.56; N, 10.03.
BR30

To a solution of 4-carboxybenzenesulfonamide (0.46 g, 2.28 mmol) and Et3N (1.7 ml, 12.21 mmol) in CHCl3/DMF (1: 1, 20 ml) was added a solution of amine 728 (0.7 g, 2.28 mmol) in DMF (10 ml) followed by the addition of BOP (1.01 g, 2.29 mmol). The reaction mixture was stirred for 12 h at room temperature. The reaction was quenched with brine. Solvent was removed under reduced pressure. Water was added to the residue and the product was extracted with ethylacetate, washed with 4% citric acid and 4% NaHCO3 solution and finally with brine. The pure product was obtained by silica gel column chromatography with 3-5% MeOH in CHCl3 (Rf = 0.2 in 8% MeOH in CHCl3). Yield: 0.45 g (48%). 1H NMR (400 MHz, CDCl3): δ 1.24 (t, 6H, J = 14 Hz), 2.93-2.96 (m, 2H), 3.42-3.46 (m, 2H), 3.54 (s, 4H), 4.13-4.17 (m, 4H), 5.27 (br s, 2H), 7.90 (distorted doublet, 2H, J = 8 Hz), 8.01 (distorted doublet, 2H, J = 8 Hz), 8.25 (br s, 1H).
The above ester (0.21 g, 0.51 mmol) was dissolved in MeOH/CH2Cl2 (1:1, 12 ml) and solid LiOH (65 mg, 1.54 mmol) was added. The reaction mixture was stirred at room temperature for 12 h. The pH of the solution was lowered to 3 by adding 2 N HCl when a precipitate appeared. The precipitate was filtered and washed with methanol/dichloromethane. Drying under vacuum afforded the acid as a white solid. Yield: 0.14 g (77%). 1H NMR (400 MHz, D2O): δ 2.92-2.96 (m, 2H), 3.42-3.46 (m, 2H), 3.49 (s, 4H), 7.86 (distorted doublet, 2H, J = 8 Hz), 7.97 (distorted doublet, 2H, J = 8 Hz).
The acid (66 mg, 0.18 mmol) was dissolved in MeOH (10 ml) and CuCl2•2H2O (32 mg, 0.18 mmol) was added. Immediate color change was observed. No precipitation was obtained upon continuation of stirring for 6 more hours at room temperature. The solvent was removed and the residue was dried under vacuum. It was redissolved in ethanol and precipitated with CH2Cl2. The precipitate was filtered and washed with ethanol/CH2Cl2 and dried under vacuum affording a blue solid (66 mg). Yield: 81%. Anal. Calcd. for C13H15CuN3O7S.H2O: C, 35.58; H, 3.90; N, 9.57. Found: C, 35.23; H, 3.78; N, 9.88.
Isothermal titration calorimetry
Isothermal titration calorimetric (ITC) experiments were performed on a Microcal MCS calorimeter, and the data were analyzed as described by Wiseman and colleagues.21 The calorimeter was calibrated by known heat pulses as described in the MCS-ITC manual. During titration, the reference cell was filled with a 0.03% azide solution in water. Prior to titration, both carbonic anhydrase and inhibitor solutions were thoroughly degassed. The sample cell was filled either with 1.8 ml (effective volume = 1.36 ml) of buffer (for control) or with an appropriately diluted enzyme. The contents of the sample cell were titrated with increasing aliquots (first aliquot 1 μl, and subsequent aliquots 4 μl each) of inhibitors. During the titration, the reaction mixture was constantly stirred at 400 rpm. The enzyme concentration was adjusted by 2% (as recommended by the manufacturer) to include a dilution effect of the enzyme solution, which occurs following a buffer rinse.
All calorimetric titration data were presented after subtracting the background signal, deduced from the amplitude of heat pulses at the end of the titration. The raw experimental data were presented as the amount of heat produced per second following each injection of inhibitor into the enzyme solution (minus the blank) as a function of time. The amount of heat produced per injection was calculated by integration of the area under individual peaks by the Origin software. Final data are presented as the amount of heat produced per injection versus the molar ratio of inhibitor to enzyme. Whereas the binding isotherm for the CAII-benzenesulfonamide complex was analyzed according to the “one-site” binding model, that for the CAII-BR30 complex was analyzed by the “two-site” binding model, as described by Wiseman and colleagues.21
Crystallography
CAI and CAII were purified as described.14,29 Crystals of recombinant CAII were grown by the hanging drop method: 5 μl of protein solution (10 mg/ml protein, 1 mM methyl mercuric acetate, 50 mM Tris-sulfate, pH 8.0) and 5 μl of precipitant solution (2.5 M (NH4)2SO4, 50 mM Tris-sulfate, pH 7.7) were mixed and suspended over a reservoir containing 1 ml of precipitant solution at 4° C. Crystals formed within 3 days. Single crystals were transferred to fresh sitting drops containing inhibitor (1.4-2.0 mM inhibitor from 10 mM stock in dimethylsulfoxide:acetonitrile), 50 mM Tris-sulfate, pH 7.7, and 2.05 M (NH4)2SO4 over a well of 1 ml precipitant solution. After 2 days, crystals were transferred to a drop containing 30% glycerol, 70% precipitant solution and flash cooled in liquid nitrogen. Crystals of recombinant CAI were grown by the hanging drop method: 4.6 μl of protein solution (11 mg/ml protein, 50 mM Tris-sulfate, pH 8.7), 0.4 μl of inhibitor solution (13 mM BR30 in isopropanol:acetonitrile), and 5 μl of precipitant solution (23% polyethylene glycol (PEG) 3350, 200 mM NaCl, and 100 mM 2-(N-morpholino) ethanesulfonic acid (MES), pH 6.4) were mixed and suspended over a reservoir containing 1 ml of precipitant solution at 4° C. Crystals formed within 2 weeks. Single crystals were transferred to a drop containing 5% glycerol, 23% PEG 3350, 200 mM NaCl, and 100 mM MES, pH 6.4, then flash cooled in liquid nitrogen.
X-ray diffraction data were collected at 100 K (λ = 0.9124 Å) at the Cornell High Energy Synchrotron Source (Ithaca, NY), beamlines F1 (λ = 0.9124 Å) and A1 (λ = 0.9777 Å), using an ADSC Quantum-4 CCD detector.30 Data were integrated and reduced using HKL2000.31 Diffraction data for each CAII crystal were indexed in space group P21 with one molecule in the asymmetric unit. Diffraction data for CAI were indexed in space group P212121 with two molecules in the asymmetric unit. This is a new crystal form of this enzyme. Unit cell parameters and data reduction statistics are recorded in Table 2.
Table 2.
Crystallographic Data Collection and Refinement Statistics
| Complex | |||||
|---|---|---|---|---|---|
| CAI-BR30 | CAII-BR15 | CAII-BR17 | CAII-BR22 | CAII-BR30 | |
| Space Group | P212121 | P21 | P21 | P21 | P21 |
| Unit Cell constants (Å) |
a = 63.219(4) b = 72.608(5) c = 121.949(8) |
a = 42.346(8) b = 41.359(9) c = 72.17(2) β = 104.34(2)° |
a = 42.444(4) b = 41.640(4) c = 72.278(5) β = 104.240(5)° |
a = 42.557(3) b = 41.704(3) c = 72.710(4) β = 104.380(3)° |
a = 42.362(5) b = 41.482(5) c = 72.23(1) β = 104.340(6)° |
| No. measured reflections |
406,999 | 157,092 | 398,544 | 464,376 | 197,281 |
| No. unique reflections |
75,235 | 63,831 | 99,069 | 127,404 | 77,219 |
| Max resolution (Å) |
1.55 | 1.25 | 1.10 | 0.99 | 1.15 |
| Rmerge (outer shell) |
0.071 (0.540) |
0.086 (0.828) | 0.088 (0.413) | 0.093 (0.479) | 0.062 (0.581) |
| Completeness of data |
98.8% (92.5%) |
94.9% (87.9%) | 99.3% (99.2%) |
93.0% (53.9%) |
88.8% (87.1%) |
| No. reflections used in refinement |
73,375 | 60,541 | 98,072 | 126,056 | 75,642 |
| No. reflections in test set |
1860 | 1320 | 977 | 1328 | 1567 |
| Rwork | 0.219 | 0.1641 | 0.1272 | 0.1233 | 0.1433 |
| Rfree | 0.241 | 0.2120 | 0.1504 | 0.1349 | 0.1879 |
| No. nonhydrogen atoms |
4748 | 2547 | 2525 | 2484 | 2440 |
| No. solvent molecules |
574 | 239 | 326 | 287 | 269 |
| r.m.s.d. from ideality | |||||
| bond lengths (Å) |
0.006 | 0.013 | 0.015 | 0.016 | 0.012 |
| bond angles (°) |
1.3 | 2.4 | 2.3 | 2.2 | 2.3 |
| Dihedral angles (°) |
23.9 | 26.5 | 25.9 | 26.2 | 26.5 |
| Improper dihedral angles (°) |
0.84 | 1.40 | 1.65 | 1.77 | 1.56 |
| PDB accession codes |
2FOY | 2FOQ | 2FOS | 2FOU | 2FOV |
The atomic coordinates of CAII refined at 1.54 Å resolution5 were obtained from the Research Collaboratory for Structural Bioinformatic Protein Data Bank (RCSB PDB)32 (PDB code 2CBA) and used as a starting model for crystallographic refinement after deletion of solvent atoms. An initial round of rigid body refinement followed by simulated annealing and individual B factor refinement was performed using the program CNS 1.1.33 Model visualization and rebuilding was performed using the graphics program O 9.34 Locations of mercury and copper ions were identified from peaks in anomalous difference and |Fo| - |Fc| maps, and residue 206 was modeled as S-(methylmercury)-cysteine. Refinement was then continued using the program SHELX-97.35 Anisotropic refinement of all atoms led to a drop in Rfree of 0.0217 for the CAII-BR15 complex, 0.0464 for the CAII-BR17 complex, 0.0387 for the CAII-BR22 complex, and 0.0224 for the CAII-BR30 complex. The addition of riding hydrogen atoms resulted in an additional decrease in Rfree of 0.0142 for the CAII-BR17 complex, 0.0204 for the CAII-BR22 complex, and 0.0120 for the CAII-BR30 complex. Inhibitor molecules were identified from peaks in |Fo| - |Fc| maps and were gradually built into the models over several rounds of refinement; restraints on inhibitor bond angles and distances were taken from similar structures in the Cambridge Structural Database36 and standard restraints were used on protein bond angles and distances throughout refinement. Alternate conformations of disordered side chains and inhibitor molecules were modeled by fitting to positive and negative peaks in |Fo| - |Fc| maps. Water molecules were built into >3σ peaks in |Fo| - |Fc| maps that demonstrated appropriate hydrogen-bonding geometry.
The structure of the CAI-BR30 complex was solved by molecular replacement using the program Phaser37 and the coordinates of CAI refined at 1.60 Å resolution38 obtained from the Protein Data Bank (PDB code 1HCB). The two molecules in the asymmetric unit form an isologous dimer with cross-rotation function peaks at (α = 2.387°, β = 154.972°, γ = 187.725°) and (α = 176.555°, β = 24.434°, γ = 8.210°) and translation function peaks at (a = −0.63065, b = 0.27231, c = −0.40012) and (a = −0.17297, b = 0.26764, c = 0.21103). Crystallographic refinement was performed using the program CNS 1.1.
Final refinement statistics for all structures of carbonic anhydrase-inhibitor complexes are presented in Table 2. The atomic coordinates of each structure have been deposited in the PDB with the accession codes listed in Table 2.
Acknowledgements
This work is based upon research conducted at the Cornell High Energy Synchrotron Source (CHESS), which is supported by the National Science Foundation under award DMR 0225180, using the Macromolecular Diffraction at CHESS (MacCHESS) facility, which is supported by award RR-01646 from the National Institutes of Health, through its National Center for Research Resources. We thank the NIH for grant GM49758 (D.W.C.) in support of this work.
References
- 1.(a) Silverman DN, Lindskog S. Acc. Chem. Res. 1988;21:30–36. [Google Scholar]; (b) Sly WS, Hu PY. Annu. Rev. Biochem. 1995;64:375–401. doi: 10.1146/annurev.bi.64.070195.002111. [DOI] [PubMed] [Google Scholar]; (c) Christianson DW, Fierke CA. Acc. Chem. Res. 1996;29:331–339. [Google Scholar]; (d) Supuran CT, Scozzafava A, Casini A. Medical Research Reviews. 2003;23:146–189. doi: 10.1002/med.10025. [DOI] [PubMed] [Google Scholar]
- 2.(a) Chegwidden WR, Carter ND. In: The Carbonic Anhydrases: New Horizons. Chegwidden WR, Carter ND, Edwards YH, editors. Birkhäuser Verlag; Basel, Switzerland: 2000. pp. 13–28. [Google Scholar]; (b) Parkkila S. The Carbonic Anhydrases: New Horizons. Birkhäuser Verlag; Basel, Switzerland: 2000. pp. 79–93. [Google Scholar]
- 3.Khalifah RG. J. Biol. Chem. 1971;246:2561–2573. [PubMed] [Google Scholar]
- 4.Liljas A, Kannan KK, Bergstén P-C, Waara I, Fridborg K, Strandberg B, Carlbom U, Järup L, Lövgren S, Petef M. Nature New Biology. 1972;235:131–137. doi: 10.1038/newbio235131a0. [DOI] [PubMed] [Google Scholar]
- 5.Håkansson K, Carlsson M, Svensson LA, Liljas A. J. Mol. Biol. 1992;227:1192–1204. doi: 10.1016/0022-2836(92)90531-n. [DOI] [PubMed] [Google Scholar]
- 6.(a) Tu C, Silverman DN, Forsman C, Jonsson BH, Lindskog S. Biochemistry. 1989;28:7913–7918. doi: 10.1021/bi00445a054. [DOI] [PubMed] [Google Scholar]; (b) Duda D, Govindasamy L, Agbandje-McKenna M, Tu C, Silverman DN, McKenna R. Acta Cryst. 2003;D59:93–104. doi: 10.1107/s0907444902019455. [DOI] [PubMed] [Google Scholar]
- 7.(a) Mann T, Keilin D. Nature (London, United Kingdom) 1940;146:164–5. [Google Scholar]; (b) Krebs HA. Biochem. J. 1948;43:525–528. doi: 10.1042/bj0430525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Chen RF, Kernohan JC. J. Biol. Chem. 1967;242:5813–5823. [PubMed] [Google Scholar]; (b) Thompson RB, Patchan MW. Journal of Fluorescence. 1995;5:123–130. doi: 10.1007/BF00727528. [DOI] [PubMed] [Google Scholar]; (c) Elbaum D, Nair SK, Patchan MW, Thompson RB, Christianson DW. J. Am. Chem. Soc. 1996;118:8381–8387. [Google Scholar]; (d) Nair SK, Elbaum D, Christianson DW. J. Biol. Chem. 1996;271:1003–1007. doi: 10.1074/jbc.271.2.1003. [DOI] [PubMed] [Google Scholar]
- 9.(a) Baldwin JJ, Ponticello GS, Anderson PS, Christy ME, Murcko MA, Randall WC, Schwam H, Sugrue MF, Springer JP, Gautheron P, Grove J, Mallorga P, Viader M-P, McKeever BM, Navia MA. J. Med. Chem. 1989;32:2510–2513. doi: 10.1021/jm00132a003. [DOI] [PubMed] [Google Scholar]; (b) Smith GM, Alexander RS, Christianson DW, McKeever BM, Ponticello GS, Springer JP, Randall WC, Baldwin JJ, Habecker CN. Protein Science. 1994;3:118–125. doi: 10.1002/pro.5560030115. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Stams T, Chen Y, Boriack-Sjodin PA, Hurt JD, Liao J, May JA, Dean T, Laipis P, Silverman DN, Christianson DW. Protein Science. 1998;7:556–563. doi: 10.1002/pro.5560070303. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Vullo D, Innocenti A, Nishimori I, Pastorek J, Scozzafava A, Pastoreková S, Supuran CT. Bioorg. Med. Chem. Lett. 2005;15:963–969. doi: 10.1016/j.bmcl.2004.12.053. [DOI] [PubMed] [Google Scholar]
- 10.(a) Håkansson K, Liljas A. FEBS Lett. 1994;350:319–322. doi: 10.1016/0014-5793(94)00798-5. [DOI] [PubMed] [Google Scholar]; (b) Boriack-Sjodin PA, Zeitlin S, Chen H-H, Crenshaw L, Gross S, Dantanarayana A, Delgado P, May JA, Dean T, Christianson DW. Protein Science. 1998;7:2483–2489. doi: 10.1002/pro.5560071201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Scozzafava A, Supuran CT. Journal of Enzyme Inhibition. 1999;14:343–363. doi: 10.3109/14756369909030328. [DOI] [PubMed] [Google Scholar]; (b) Popescu A, Simion A, Scozzafava A, Briganti F, Supuran CT. Journal of Enzyme Inhibition. 1999;14:407–423. doi: 10.3109/14756369909030332. [DOI] [PubMed] [Google Scholar]; (c) Kim C-Y, Whittington DA, Chang JS, Liao J, May JA, Christianson DW. J. Med. Chem. 2002;45:888–893. doi: 10.1021/jm010163d. [DOI] [PubMed] [Google Scholar]; (d) Winum J-Y, Vullo D, Casini A, Scozzafava A, Supuran CT. J. Med. Chem. 2003;46:5471–5477. doi: 10.1021/jm030911u. [DOI] [PubMed] [Google Scholar]; (e) Ilies MA, Vullo D, Pastorek J, Scozzafava A, Ilies M, Caproiu MT, Pastorekova S, Supuran CT. J. Med. Chem. 2003;46:2187–2196. doi: 10.1021/jm021123s. [DOI] [PubMed] [Google Scholar]; (f) Garaj V, Puccetti L, Fasolis G, Winum J-Y, Montero J-L, Scozzafava A, Vullo D, Innocenti A, Supuran CT. Biorganic & Medicinal Chemistry Letters. 2004;14:5427–5433. doi: 10.1016/j.bmcl.2004.07.087. [DOI] [PubMed] [Google Scholar]; (g) Gupta SP, Kumaran S. Journal of Enzyme Inhibition & Medicinal Chemistry. 2005;20:251–9. doi: 10.1080/14756360500067439. [DOI] [PubMed] [Google Scholar]; (h) Innocenti A, Villar R, Martinez-Merino V, Gil MJ, Scozzafava A, Vullo D, Supuran CT. Bioorg. Med. Chem. Lett. 2005;15:4872–4876. doi: 10.1016/j.bmcl.2005.04.078. [DOI] [PubMed] [Google Scholar]; (i) Mincione F, Starnotti M, Masini E, Bacciottini L, Scrivanti C, Casini A, Vullo D, Scozzafava A, Supuran CT. Bioorg. Med. Chem. Lett. 2005;15:3821–3827. doi: 10.1016/j.bmcl.2005.06.054. [DOI] [PubMed] [Google Scholar]; (j) Özensoy Ö, Puccetti L, Fasolis G, Arslan O, Scozzafava A, Supuran CT. Bioorg. Med. Chem. Lett. 2005;15:4862–4866. doi: 10.1016/j.bmcl.2005.08.048. [DOI] [PubMed] [Google Scholar]
- 12.(a) Cappalonga Bunn AM, Alexander RS, Christianson DW. J. Am. Chem. Soc. 1994;116:5063–5068. [Google Scholar]; (b) Jain A, Huang SG, Whitesides GM. J. Am. Chem. Soc. 1994;116:5057–5062. [Google Scholar]; (c) Boriack PA, Christianson DW, Kingery-Wood J, Whitesides GM. J. Med. Chem. 1995;38:2286–2291. doi: 10.1021/jm00013a004. [DOI] [PubMed] [Google Scholar]
- 13.(a) Pack DW, Chen G, Maloney KM, Chen C-T, Arnold FH. J. Am. Chem. Soc. 1997;119:2479–2487. [Google Scholar]; (b) Fazal MA, Roy BC, Sun S, Mallik S, Rodgers KR. J. Am. Chem. Soc. 2001;123:6283–6290. doi: 10.1021/ja003193z. [DOI] [PubMed] [Google Scholar]
- 14.Banerjee AL, Eiler D, Roy BC, Jia X, Haldar MK, Mallik S, Srivastava DK. Biochemistry. 2005;44:3211–3224. doi: 10.1021/bi047737b. [DOI] [PubMed] [Google Scholar]
- 15.Banerjee AL, Swanson M, Roy BC, Jia X, Haldar MK, Mallik S, Srivastava DK. J. Am. Chem. Soc. 2004;126:10875–10883. doi: 10.1021/ja047557p. [DOI] [PubMed] [Google Scholar]
- 16.Roy BC, Banerjee AL, Swanson M, Jia XG, Haldar MK, Mallik S, Srivastava DK. J. Am. Chem. Soc. 2004;126:13206–13207. doi: 10.1021/ja047271k. [DOI] [PubMed] [Google Scholar]
- 17.Crothers DM, Metzger H. Immunochemistry. 1972;9:341–357. doi: 10.1016/0019-2791(72)90097-3. [DOI] [PubMed] [Google Scholar]; Holmes NJ, Parham P. J. Biol. Chem. 1983;258:1580–1586. [PubMed] [Google Scholar]
- 18.(a) Kraulis PJ. J. Appl. Crystallogr. 1991;24:946–950. [Google Scholar]; (b) Merritt EA, Murphy MEP. Acta Cryst. 1994;D50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]; (c) Esnouf RM. Journal Of Molecular Graphics & Modelling. 1997;15:132–134. doi: 10.1016/S1093-3263(97)00021-1. [DOI] [PubMed] [Google Scholar]
- 19.Kim C-Y, Chandra PP, Jain A, Christianson DW. J. Am. Chem. Soc. 2001;123:9620–9627. doi: 10.1021/ja011034p. [DOI] [PubMed] [Google Scholar]
- 20.(a) Fisher HF, Singh N. Methods Enzymol. 1995;259:194–221. doi: 10.1016/0076-6879(95)59045-5. [DOI] [PubMed] [Google Scholar]; (b) Day YSN, Baird CL, Rich RL, Myszka DJ. Protein Science. 2002;11:1017–1025. doi: 10.1110/ps.4330102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Myszka DG, Abdiche YN, Arisaka F, Byron O, Eisenstein E, Hensley P, Thomson JA, Lombardo CR, Schwarz F, Stafford W, Doyle ML. J. Biomol. Tech. 2003;14:247–269. [PMC free article] [PubMed] [Google Scholar]
- 21.Wiseman T, Williston S, Brandts JF, Lin L-N. Anal. Biochem. 1989;179:131–137. doi: 10.1016/0003-2697(89)90213-3. [DOI] [PubMed] [Google Scholar]
- 22.Taylor PW, King RW, Burgen AS. Biochemistry. 1970;9:2638–2645. doi: 10.1021/bi00815a012. [DOI] [PubMed] [Google Scholar]
- 23.(a) Martell AE, Smith RM. Critical Stability Constants. Vol. 1. Plenum Press; New York: 1974. [Google Scholar]; (b) Martell AE, Smith RM. Critical Stability Constants. Vol. 2. Plenum Press; New York: 1975. [Google Scholar]
- 24.Håkansson K, Wehnert A, Liljas A. Acta Cryst. 1994;D50:93–100. doi: 10.1107/S0907444993008790. [DOI] [PubMed] [Google Scholar]
- 25.(a) Eriksson EA, Jones TA, Liljas A, Bertini I. In: The Zinc Enzymes. Luchinat C, Maret W, Zeppezauer M, editors. Birkhäuser Boston, Inc.; Boston, MA: 1986. pp. 317–328. [Google Scholar]; (b) Sumalan SL, Casanova J, Alzuet G, Borrás J, Castiñeiras A, Supuran CT. J. Inorg. Biochem. 1996;62:31–39. doi: 10.1016/0162-0134(95)00086-0. [DOI] [PubMed] [Google Scholar]
- 26.Conroy CW, Maren TH. Mol. Pharmacol. 1995;48:486–491. [PubMed] [Google Scholar]
- 27.Burks E, Koshti N, Jacobs H, Gopalan A. Synlett. 1998:1285–1287. [Google Scholar]
- 28.del Olmo E, Macho A, Alves M, López JL, el Banoua F, Muñoz E, San Feliciano A. Bioorg. Med. Chem. Lett. 2002;12:2621–2626. doi: 10.1016/s0960-894x(02)00476-6. [DOI] [PubMed] [Google Scholar]
- 29.Banerjee AL, Swanson M, Mallik S, Srivastava DK. Protein Expression Purif. 2004;37:450–454. doi: 10.1016/j.pep.2004.06.031. [DOI] [PubMed] [Google Scholar]
- 30.Szebenyi DME, Arvai A, Ealick S, LaIuppa JM, Nielson C. Journal of Synchrotron Radiation. 1997;4:128–135. doi: 10.1107/S0909049597000204. [DOI] [PubMed] [Google Scholar]
- 31.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 32.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 34.Jones TA, Zou J-Y, Cowan SW, Kjeldgaard M. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 35.Sheldrick GM, Schneider TR. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 36.Allen FH. Acta Cryst. 2002;B58:380–388. [Google Scholar]
- 37.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Acta Cryst. 2005;D61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 38.Kumar V, Kannan KK. J. Mol. Biol. 1994;241:226–232. doi: 10.1006/jmbi.1994.1491. [DOI] [PubMed] [Google Scholar]