

Abstract
Two approaches to the aza-tricyclo dodecane skeleton of (−)-FR901483 are reported. Both routes utilized a Grignard addition to an N-acylpyridinium salt to establish the absolute stereochemistry at C-6 and a highly diastereoselective conjugate allylation reaction to form the quaternary center at C-1 of the natural product in excellent yield. Although the desired polysubstituted piperidine intermediates were prepared regio- and stereoselectively, the construction of the C-8/C-9 bond connectivity could not be achieved. All attempts at a pinacol cyclization or an intramolecular 6-exo-tet epoxide opening were unsuccessful due to unfavorable A1,3 strain inherent in the molecule.
Introduction
The immunosuppressive natural products cyclosporine A and FK 506 are leading therapeutic agents in the organ transplant field.1,2 The mechanism of action of both cyclosporin A and FK 506 is the inhibition of calcineurin, a calcium-dependent serine/threonine phosphatase. Calcineurin, in turn, leads to the inhibition of interleukin-2 (IL-2) production, which is a signal molecule that induces cytotoxic T-cells. In high doses these two compounds exhibit serious side effects such as neurotoxicity and adverse reactions in diabetic patients.3 Because of this, the search for less toxic immunosuppressants with a different mechanism of action has emerged. These efforts led to the isolation of (−)-FR901483 (1) from the fermentation broth of the fungal strain Cladobotrium sp. No. 11231 by the Fujisawa research group, and it was shown to significantly prolong the graft survival time in the rat skin allograft model.4 More importantly, it was suggested that the mechanism of action is quite different as compared to cyclosporin A and FK 506, namely (−)-FR901483 inhibits the adenylosuccinate synthetase and/or adenylosuccinate lyase enzymes in the purine biosynthesis. Because of the high degree of bioactivity and the unprecedented azatricyclic structure of (−)-FR901483, it has become a popular target of synthetic interest.5
In the previous syntheses of FR901483 (1), the pyrrolidine portion of the aza-tricyclo dodecane nucleus was installed utilizing a nitrone cycloaddition/hydrogenolysis,5a an oxidative spiroannulation,5b–c a Diels-Alder cycloaddition/aldol cyclization,5d a Michael addition/hydrogenolysis,5e or an aza-Cope rearrangement/Mannich cyclization tactic.5f Most of the approaches to the bicyclic [3.3.1] ring system of 1 utilized an intramolecular aldol cyclization reaction.5a–e Alternatively, the bicyclic [3.3.1]-ring system can be installed through an intermediate bridgehead imminium ion, followed by an intramolecular aza-Cope rearrangement5f or an intermolecular alkylation.5h The [3.3.1]-nonane nucleus was also successfully introduced utilizing an intramolecular carboradical cyclization.5g, 5m Our approach to the synthesis of 1 significantly differs from the previous synthetic endeavors in both strategy and execution. Retrosynthetically, we envisioned the target 1 could be derived via a reductive amination of ketone 2, followed by phosphorylation at C-9 (Scheme 1). Ketone 2 in turn could be synthesized from diol 3 by the selective formation and reduction of a tertiary carbocation at C-8. Unlike the other approaches to 1, we chose to exploit a Mitsunobu-type cyclization to construct the pyrrolidine portion of the aza-tricyclo dodecane nucleus. This would then be followed by an oxidative cleavage of the double bond to give compound 2. Diol 3, in turn, could be obtained by performing an intramolecular pinacol cyclization of keto-aldehyde 4, which could be accessed by a conjugate addition of an allylsilane or allyltin reagent such as 6 to dihydropyridone 5. Vinylogous amide 5 can be accessed via a tandem conjugate addition/oxidation in dihydropyridone 7. Finally, Grignard addition to the chiral 1-acylpyridinium salt 8 is expected to furnish enantiopure 7.
Scheme 1.
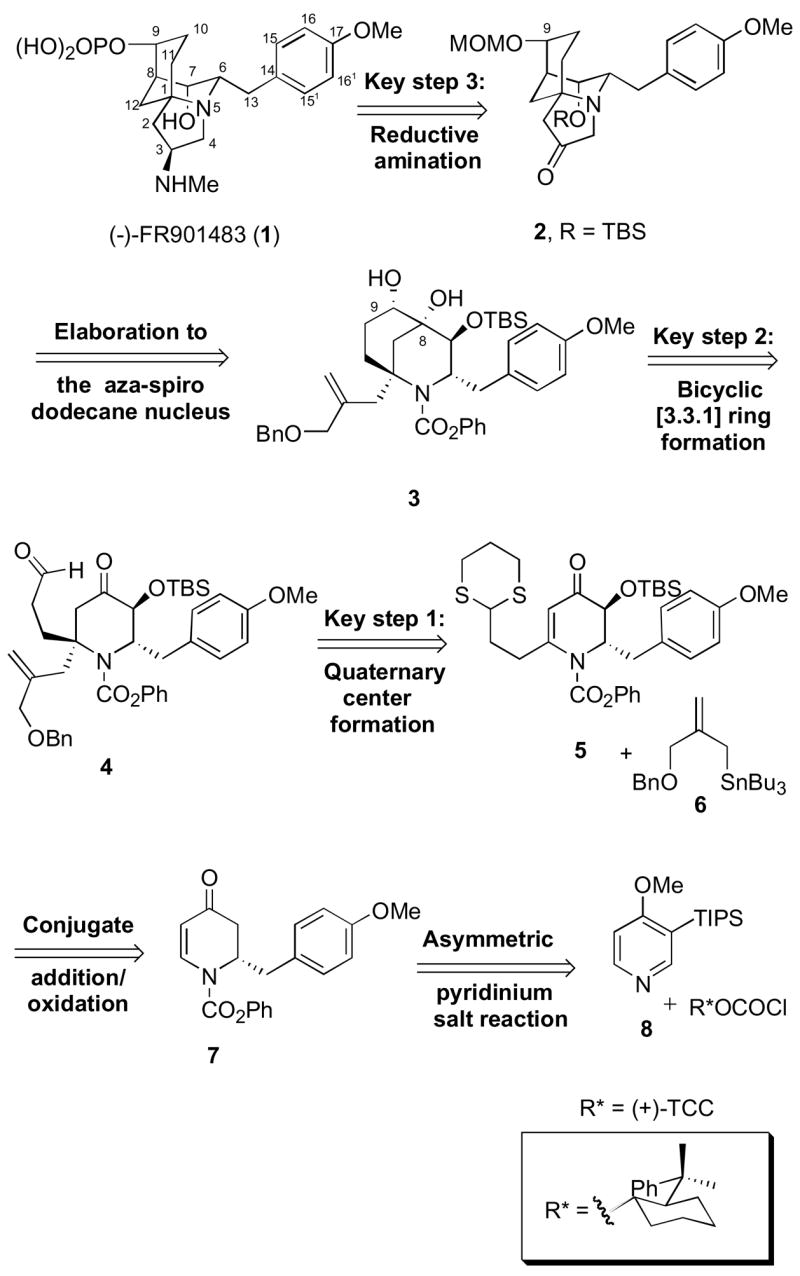
First Generation Retrosynthetic Analysis of (−)-FR901483
Results and Discussion
Our approach to the synthesis of (−)-FR901483 (1) commenced with Grignard addition to the chiral 1-acylpyridinium salt prepared from 3-triisopropylsilyl-4-methoxypyridine (8) and the chloroformate of (+)-trans-2-(α)-cumylcyclohexanol (TCC), thus establishing the absolute stereochemistry at the C-6 position of the resulting dihydropyridone 9 (90%, dr 95:5, Scheme 2).6,7 The resulting dihydropyridone 9 was obtained as a single diastereomer after recrystallization from ethyl acetate/hexanes. A one-pot reaction with sodium methoxide in methanol to remove the chiral auxiliary, followed by protodesilylation with 10% aqueous HCl, yielded an intermediate vinylogous amide,8 which in turn was reacylated by exposure to n-BuLi and phenyl chloroformate to give dihydropyridone 7 as a single enantiomer.
Scheme 2.
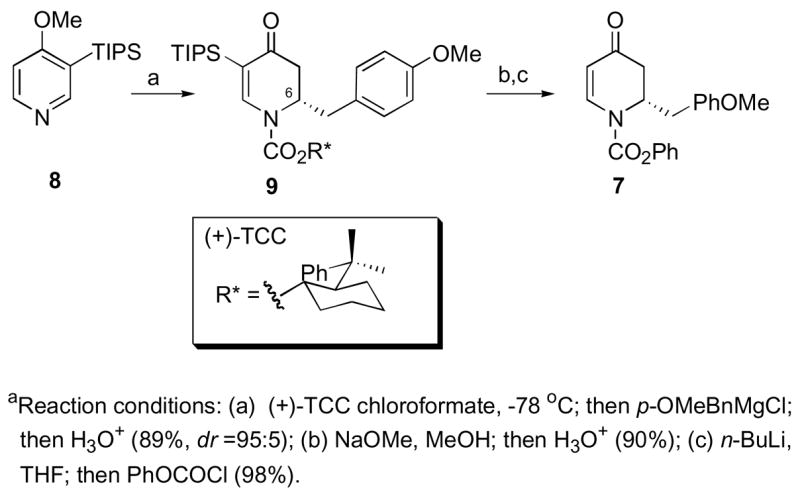
Synthesis of Dihydropyridone 6a
After establishing a practical route to the optically active dihydropyridone 7, we turned our attention to the synthesis of racemic intermediates for initial studies of the feasibility of the proposed pinacol coupling and reductive amination reactions (Scheme 1). Use of the enantiopure 7 would lead to the asymmetric synthesis of 1, since once the absolute stereochemistry at C-6 is established, the rest of the stereocenters will be introduced relative to the one at C-6 in the same manner as in the racemic route. The addition of p-methoxybenzylmagnesium chloride to the 1-acylpyridinium salt, prepared from 4-methoxypyridine (10) and phenyl chloroformate, provided dihydropyridone 11 in 77% yield (Scheme 3). 6 Copper-mediated addition of the Grignard reagent derived from 2-(2-bromoethyl)-1,3-dioxolane to compound 11 in the presence of trimethylsilylchloride afforded the desired silyl enol ether 12, which was then subjected to catalytic Pd(OAc)2 oxidative rearrangement conditions to give vinylogous amide 13 in 70% yield (2 steps).9 Reaction of dihydropyridone 13 with Pb(OAc)4 resulted in C-3 acetoxylation to give acetate 15a as the major diastereomer (75%, dr 15:1).10 The resulting trans stereochemistry can be explained by stereoelectronic control: in order to maintain a chair-like transition state, intramolecular delivery of the acetate group from the enol-lead triacetate intermediate 14 takes place from the axial direction (Scheme 3).10 The C-6 substituent is in an axial orientation due to A(1,3) strain with the N-acyl group leading to the observed major diastereomer 15a.11 The stereochemistry of the major diastereomer 15a was confirmed by subjecting vinylogous amide 13 to a mixture of Pb(OAc)4 and 5% AcOH for 3 days at reflux to give a 4:1 mixture of C-7 acetates (Scheme 4). A large NOE (9%) between H-6 and H-7 was observed in cis-acetate 15b. No NOE was observed in the trans-acetate 15a between H-6 and H-7. In addition, JH6-H7 = 4.9 Hz in 15b and JH6-H7 = 2.0 Hz in 15a were observed, confirming the assignment of 15a.
Scheme 3.
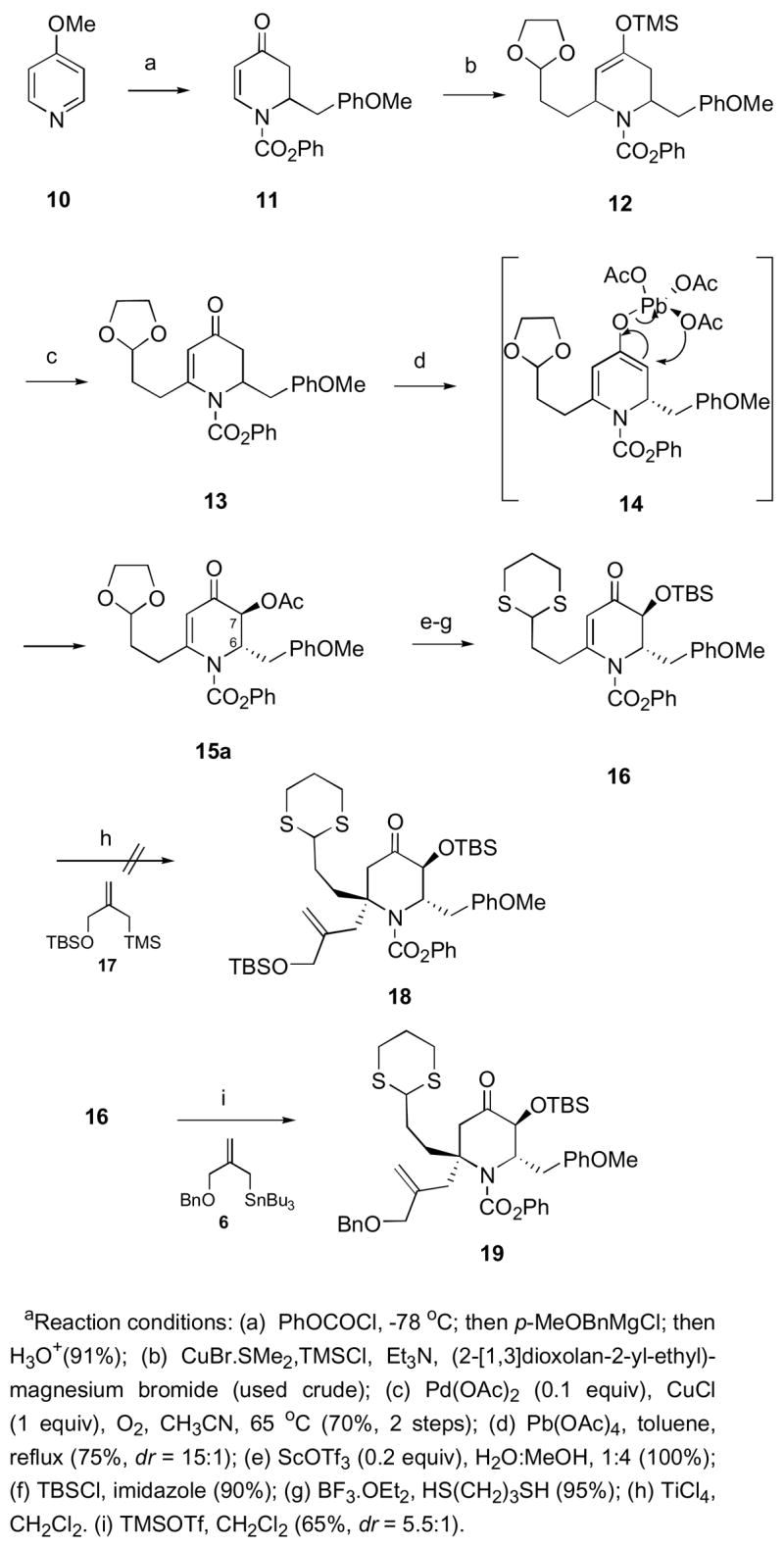
C-1 Quaternary Center Installation Via Conjugate Allylationa
Scheme 4.
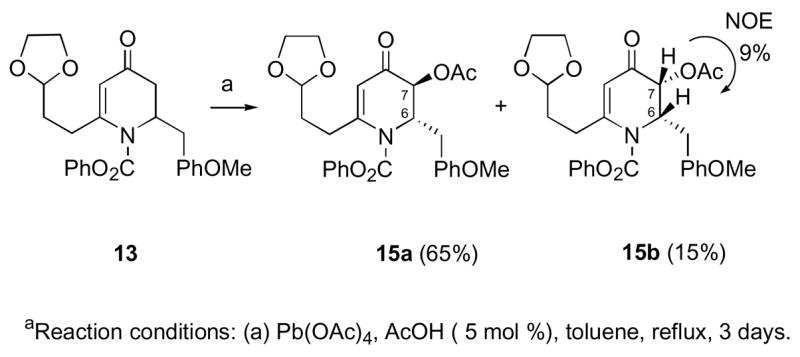
Confirmation of the C-7 Stereochemistrya
After considerable experimentation, hydrolysis of trans-acetate 15a was achieved by treatment with Sc(OTf)3 in a 4:1 MeOH/H2O mixture to afford the corresponding α-hydroxy ketone (Scheme 3).12,13 Protection of the resulting secondary alcohol as a TBS ether, followed by acetal exchange gave dihydropyridone 16 in 86% yield (3 steps).14 We then turned our attention to finding suitable reaction conditions to effect the conjugate allylation of compound 16 with the known allylsilane reagent 17.15 To our disappointment, the use of Sakurai’s conjugate allylation conditions resulted only in the recovery of starting materials, even at temperatures as high as −20 °C.16
In order to enhance the reactivity of the allylation reagent in the conjugate addition, we prepared the known allylstannane compound 6.17 The choice of compound 6 as the nucleophilic partner was deliberate as allylstannane reagents of this type are known to be more reactive than the corresponding allylsilanes. This is due to the presence of more anionic character on the carbon atom in the C-Sn bond, which makes it a weaker bond.18 To our delight, conjugate allylation of dihydropyridone 16 with stannane 6 gave the desired piperidone 19 as the major product in 65% yield (dr 5.5:1).19 The observed stereochemistry of the major diastereomer can be explained by stereoelectronic-controlled axial approach of the nucleophile, where the incoming allyl group adds syn to the axial substituent at C-6.20 The use of TMSOTf as the Lewis acid source in this type of transformation proved to be extremely important, as both the employment of BF3·OEt2 and TiCl4 resulted in isolation of starting material 16. This, to the best of our knowledge, represents the first example of Lewis-acid mediated conjugate allylation performed on a substituted dihydropyridone to give the requisite quaternary carbon α to the nitrogen atom (C-1 in this case).21 The conjugate allylation is unplagued by 1,2- addition and gives exclusively the 1,4-product. The reaction proceeds with good stereocontrol, making it an attractive way to establish quaternary centers stereoselectively in appropriately substituted dihydropyridone systems.
Next, we turned our attention to the epimerization of the C-7 silyl ether in piperidone 19, since there is a cis stereochemical relationship between the two substituents at C-6 and C-7 in the natural product (Scheme 5).
Scheme 5.
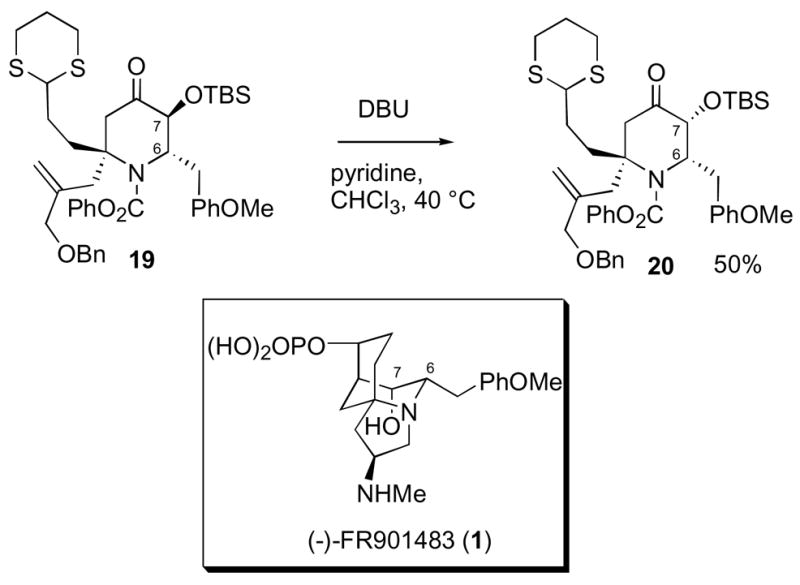
Epimerization of the Silyl Ether in Dihydropyridone 19
After considerable experimentation,22a we obtained the desired cis-epimer 20 on treatment with DBU, but only as a 1:1 mixture of stereoisomers at C-7. In an attempt to increase the yield of the desired compound, piperidone 19 was subjected to more forcing epimerization conditions (DBU, pyridine, CHCl3, 80 °C); however, a complex mixture of products was observed and the ratio of 20 to 19 did not improve. Apparently, our optimal conditions led to a 1:1 thermodynamic mixture of epimers at C- 7.23 At this point we decided to epimerize the C-7 center at a later stage in the synthesis.24
Next, hydrolysis of piperidone 19 in an aqueous iodomethane solution gave keto-aldehyde 21 in a quantitative yield, which was subsequently used without further purification (Scheme 6).25 We now were in position to investigate the proposed intramolecular pinacol coupling which would simultaneously set the stereocenters at C-8 and C-9 of diol 3.26 Unfortunately, after considerable effort using various conditions (VCl3/Zn; TiCl3/Zn-Cu; SmI2), we were unable to achieve the desired transformation. All attempts led to the recovery of starting materials or a complex mixture of products.
Scheme 6.
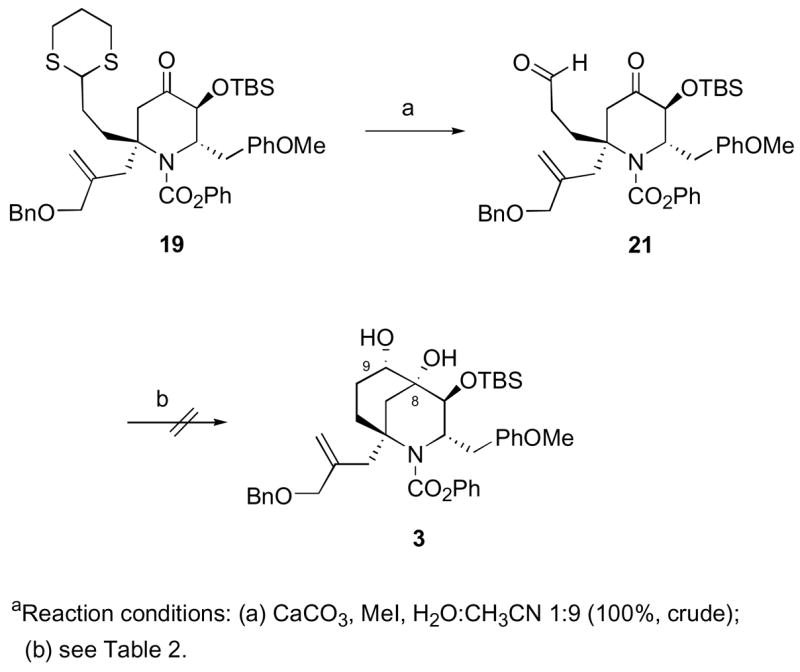
Attempted Pinacol Coupling of Keto-Aldehyde 21
Due to our inability to obtain the desired pinacol cyclization product 3, we turned our attention to a second generation retrosynthetic approach to (−)-FR901483 (1) (Scheme 7). Our analysis suggested that the targeted compound 1 could be derived via reductive amination in ketone 22, followed by phosphorylation at C-9. The C-8/C-9 bond connectivity would this time be established by taking advantage of an intramolecular 6-exo-tet epoxide opening of vinyl bromide 23. This will again be followed by a Mitsunobu-type cyclization to construct the pyrrolidine portion of the aza-tricyclo dodecane nucleus in ketone 22. Vinyl bromide 23 in turn would arise from α-hydroxy ketone 24 via a stereoselective carbonyl reduction, monomesylation of the less hindered alcohol at C-7, and epoxide formation. Piperidone 24 would be accessed by the conjugate addition of allyltin reagent 6 to dihydropyridone 25, once again providing the C-1 quaternary center stereoselectively. Vinylogous amide 25 would be synthesized from dihydropyridone 7 by our tandem conjugate addition/oxidative rearrangement protocol, followed by a regioselective installment of the vinyl bromide group and installation of the alcohol function at C-7.
Scheme 7.
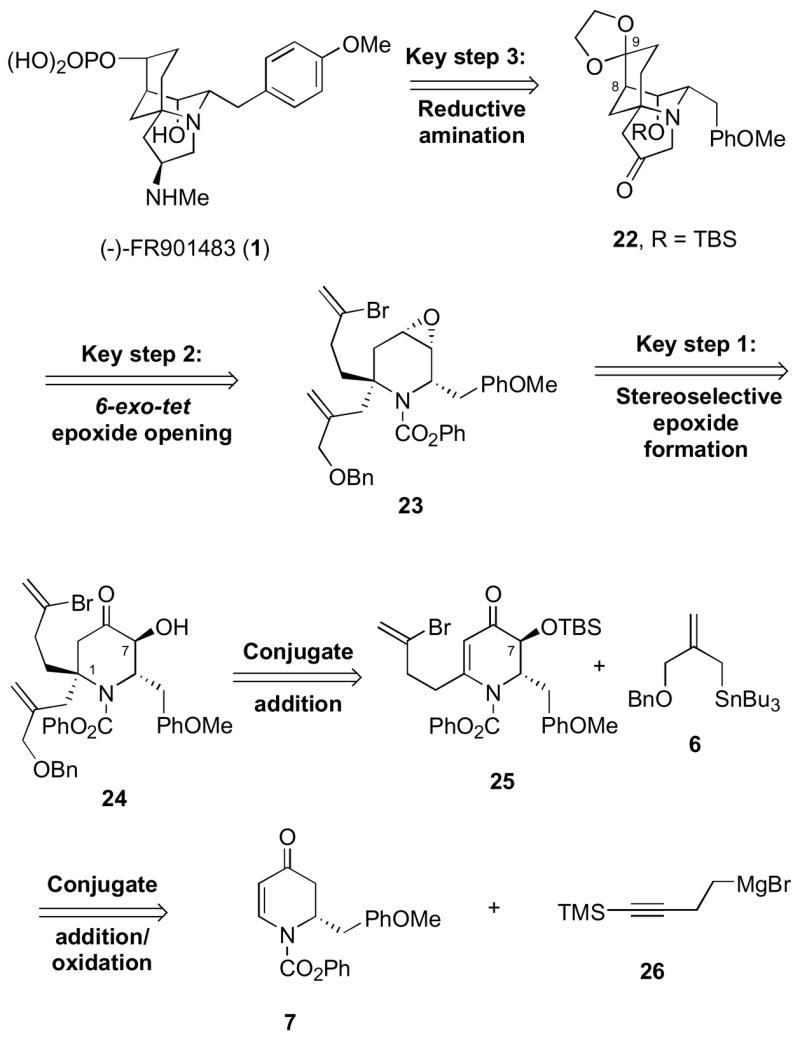
Second Generation Retrosynthetic Analysis
Our second generation approach to the target began with exposure of dihydropyridone 11 to copper-mediated addition of known Grignard reagent 2627 in the presence of trimethylsilyl chloride to give silyl enol ether 27 in a quantitative yield (Scheme 8). Subjection of 27 to premixed o-iodoxybenzoic acid (IBX) and 4-methoxypyridine N-oxide (MPO) afforded the desired dihydropyridone 28 in 55% yield.28,29 The versatility of this transformation is exemplified by the fact that the sensitive acetylene functionality is well tolerated under the reaction conditions.30 Removal of the terminal trimethylsilyl group in 28 yielded the corresponding acetylene, which was then subjected to B-bromo-9-BBN to yield vinyl bromide 29 as a single regioisomer.31 Reaction of 29 with Pb(OAc)4 resulted in the corresponding trans-acetate.32 Sc(OTf)3-mediated hydrolysis of the acetate group was followed by protection of the resulting secondary alcohol as the TBS ether to yield dihydropyridone 25 (57%, 3 steps). Conjugate allylation with allylstannane 6 gave the desired trimethylsilyl enol ether 30 as the a single diastereomer at C-1. Interesting, compound 30 proved to be stable to silica gel chromatography and had to be subjected to acidic hydrolysis to yield the desired piperidone 31 in 85% yield (2 steps). We proceeded to establish the stereochemical relationship between the allylic group at C-1 and the benzylic substituent at C-6 in compound 31. Irradiation of H6 showed a large NOE correlation to H11(5.8%), thus confirming the assigned stereochemistry. This result confirmed our working model that the conjugate addition of allylstannane 6 to C-6 substituted dihydropyridones indeed operated under stereoelectronic-approach control, where the incoming allylgroup adds syn to the axial substituent at C-6 in order to maintain a chair-like transition state.20
Scheme 8.
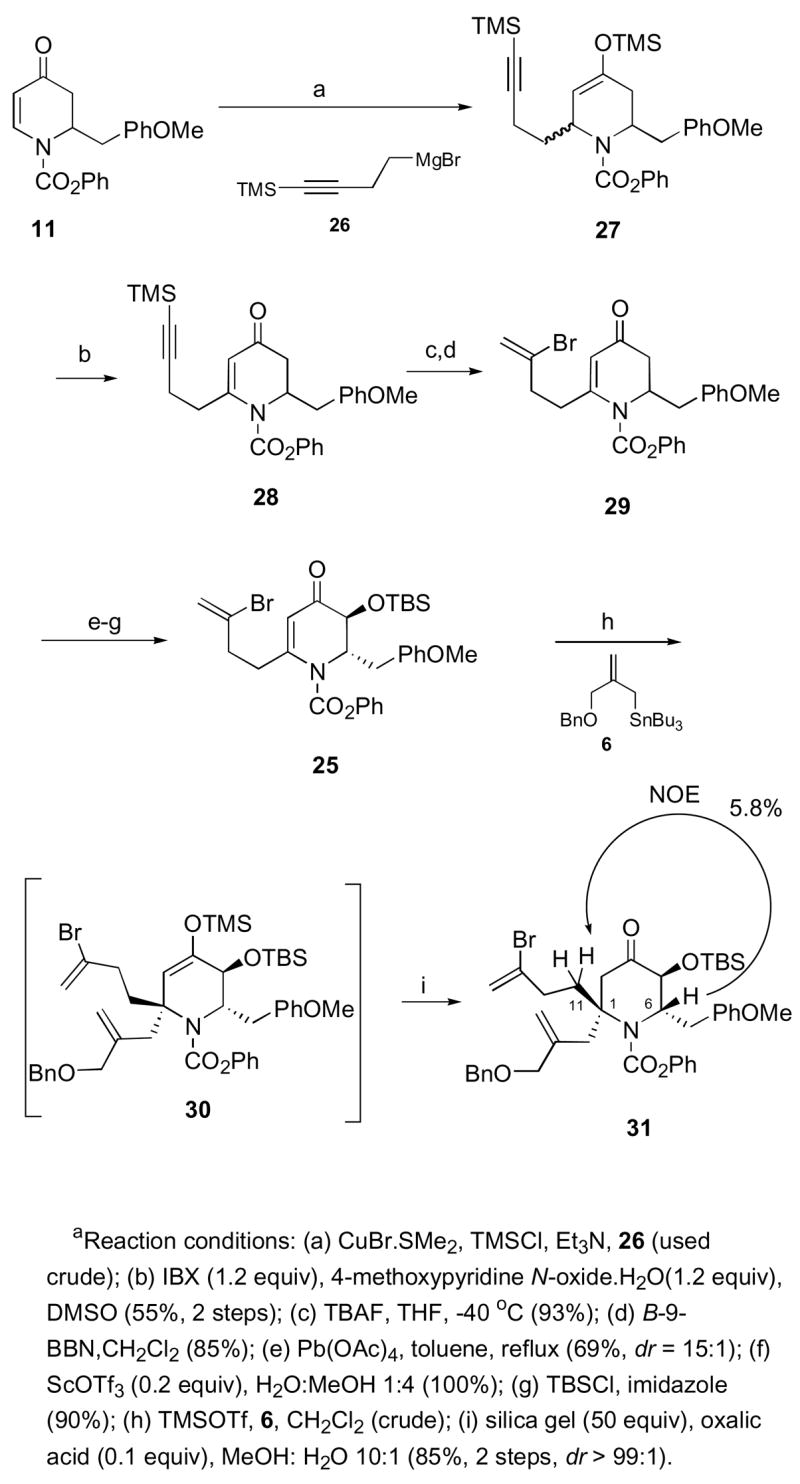
C-1 Quaternary Center Installation in Piperidone 31a
Having successfully installed the C-1 quaternary center with the desired stereochemical orientation, we then focused on the synthesis of key epoxide intermediate 23 (Scheme 9). Deprotection of the TBS ether in 31 was followed by intramolecular, stereocontrolled reduction at C-8 with tetramethylammonium triacetoxyborohydride to give the 1,2-diol 32 as a single stereoisomer. The selective derivatization of the C-8 position of trans-diol 32 by reaction with benzoic anhydride yielded the C-8 benzoate ester 33 as the major product of a 3:1 mixture of C-8/C-7 regioisomers.33 The direction of the acylation was determined by examination of the 1H NMR spectra of diol 32 and benzoate ester 33. The H8 hydrogen atom frequency in 33 shifted downfield as compared to the chemical shift of H7. This is to be expected as the electron withdrawing benzoate ester group exerts a larger deshielding effect on the H8 than on the H7 causing the observed downfield shift in its resonance. During the course of the acylation we also discovered that subjection of the corresponding undesired C-7 benzoate ester to methanolic NaOH resulted in the quantitative formation of diol 32, allowing the C-7 isomer to be recycled. Mesylation of 33 was followed by subjection of the benzoate ester moiety in 34 to basic hydrolysis to give the tetrahedral intermediate 35, which upon collapse yielded the desired epoxide 23 in a one-pot event.34
Scheme 9.

Synthesis of Key Epoxide Intermediate 23a
With epoxide 23 in hand, we turned our attention to finding suitable conditions to afford the C-8/C- 9 bond connectivity in FR901483 by effecting an intramolecular epoxide ring opening (Scheme 10). This type of intramolecular opening of an epoxide with an aryllithium to form a six-membered ring has been reported.35 Unfortunately, all our efforts to achieve the desired 6-exo-tet-epoxide opening to afford product 36 were unsuccessful.22b Exposure to n-butyllithium or dibutylcopper lithium led to the recovery of starting materials together with products resulting from attack at the carbamate. Similar results were observed when tert-butyllithium was used at −78 °C. Gratifyingly, lithium bromide exchange was achieved by reaction with t-BuLi at −120 °C for 1 hour; however, all attempts to obtain the desired epoxide opening product 36 via formation of a higher-order cuprate or a lower-order cuprate gave the corresponding debrominated product exclusively. A likely reason for the cyclization failure is an inherent A(1,3) strain in the molecule that produces a conformation unfavorable to the desired bond formation.
Scheme 10.

Attempted 6-exo-tet-Epoxide Opening in 23
Summary
Although the investigated routes to the bicyclic [3.3.1] ring system of FR901483 did not result in the formation of the C-8/C-9 bond connectivity, diastereoselective methodology for the installation of the quaternary center at C-1 utilizing a conjugate allylation reaction was developed, and several highly substituted piperidine derivatives were prepared regio- and stereoselectively. Most of the stereoselective transformations were designed based on a conformational bias caused by A(1,3) strain present in the molecules. Unfortunately, failure of the attempted C-8/C-9 formation is also attributed to allylic strain. If an N-acyl group of a late intermediate can be selectively removed to relieve A(1,3) strain, ring-closure may be feasible. Further studies will be needed to determine if the synthetic routes attempted can be modified to allow the key bond formation.
Experimental Section
6-(2-[1,3]Dioxolan-2-yl-ethyl)-2-(4-methoxybenzyl)-4-oxo-3,4-dihydro-2H-pyridine-1- carboxylic acid phenyl ester (13)
Magnesium turnings (7.57g, 311 mmol) were mechanically activated by stirring at rt overnight under argon, and then 50 mL of anhydrous THF was added. A portion of 2-(2-bromoethyl)-1,3-dioxolane (9.7 mL, 83 mmol) in 50 mL of anhydrous THF was slowly added to the mixture. Once the reaction was initiated, the mixture was cooled to 10 °C, and the remaining bromide was continuously added over a period of 2 h. After the addition was complete, the reaction mixture was stirred at 10 °C for 9 h to form the corresponding Grignard reagent. In a separate flask, copper bromide dimethyl sulfide complex (22.6 g, 110 mmol) was mixed with 50 mL of freshly distilled THF under argon and cooled to −78 °C. A solution of 2-(4- methoxybenzyl)-4-oxo-3,4-dihydro-2H-pyridine-1-carboxylic acid phenyl ester (11) (7.40 g, 20.7 mmol) in THF (30 mL) was added, followed by a premixed solution of TMSCl (30.6 mL, 241 mmol) and Et3N (35.2 mL, 252 mmol). A solution of the above prepared Grignard reagent in THF was then added dropwise over a period of 1 h. Once the addition was complete, the resulting slurry was stirred at −78 °C for 2 h and then warmed to −45 °C. After 24 h, the reaction mixture was quenched with a carefully buffered solution of 100 mL of NH4OH and NH4Cl (pH 8), and the phases were separated. The aqueous phase was extracted with Et2O (3 × 150 mL), and the combined organic layers were washed with 10% NH4OH until no further blue color was observed in the aqueous layer. The organic layers were combined, dried over K2CO3, filtered through Celite, and concentrated in vacuo to yield crude silyl enol ether 12.
To a solution of the above enol ether 12 in 100 mL of anhydrous CH3CN under argon was added Pd(OAc)2 (15 mol %, 0.74 g, 3.3 mmol), followed by CuCl (2.95 g, 29.8 mmol). The solution was purged with oxygen, warmed to 60 °C under an O2-balloon pressure atmosphere, and stirred vigorously overnight. Upon completion, the reaction mixture was cooled to rt, filtered through Celite with CH2Cl2, and the solvent was removed in vacuo to yield the crude product 13. Purification by silica gel chromatography (20 to 30 % EtOAc in hexanes) gave 6.75 g (70%, 2 steps) of the desired product 13 as a yellow foam: IR (neat) 2922, 2851, 1719, 1512, 1494, 1453, 1248, 1203, 834 cm−1; 1H NMR (CDCl3, 300 MHz) δ 7.38 (dt, J = 1.2 Hz, 5.7 Hz, 2 H), 7.26 (d, J = 3.9 Hz, 1 H), 7.10 (d, J = 6.0 Hz, 2 H) 7.00 (d, J = 6.0 Hz, 2 H), 6.85 (d, J = 6.9 Hz, 2 H), 5.63 (s, 1 H), 5.07 (q, J = 5.1 Hz, 1 H), 4.93 (t, J = 3.3 Hz, 1 H), 3.96 (m, 2 H), 3.86 (m, 2 H), 3.79 (s, 3 H), 3.08 (m, 2 H), 2.81 (m, 3 H), 2.43 (d, J = 12.6 Hz, 1 H), 1.97 (m, 2 H); 13C NMR (CDCl3, 75 MHz) δ 193.0, 158.5, (157.3 r), 151.5, 150.3, 130.5, 129.4, 128.8, 126.1, 121.3, 114.0, 113.2, 103.2, 64.9, 57.0, 57.2, 55.2, 39.7, 35.5, 32.3, 30.1; HRMS (M+H)+ calcd for C25H27NO6 + H: 438.1917, found 438.1904.
(2S*,3S*)-3-Acetoxy-6-(2-[1,3]dioxolan-2-yl-ethyl)-2-(4-methoxybenzyl)-4-oxo-3,4-dihydro- 2H-pyridine-1-carboxylic acid phenyl ester (15a)
To a stirred solution of 13 (121 mg, 0.277 mmol) in 8 mL of toluene at rt was added lead (IV) acetate (160 mg, 0.36 mmol). The resulting mixture was refluxed for 3 h, cooled to rt, and additional lead (IV) acetate (160 mg, 0.36 mmol) was added. The mixture was refluxed for 3 h. After cooling to rt, the solution was filtered through Celite with CH2Cl2. The solution was washed with saturated aqueous NaHCO3 (30 mL), and the aqueous phase extracted with CH2Cl2 (3 × 30 mL). The combined organic phase was dried over Na2SO4, filtered through Celite, and concentrated in vacuo to yield crude 15a. Purification by silica gel chromatography (20 to 30% EtOAc in hexanes) gave 103 mg (75%) of 15a as a colorless oil: IR (neat) 2934, 1741, 1678, 1592, 1513, 1401, 1248, 1181, 1032, 911, 750 cm−1; 1H NMR (CDCl3, 300 MHz) δ 7.36 (t, J = 7.4 Hz, 2 H), 7.22 (d, J = 7.3 Hz, 1 H), 7.12 (d, J = 8.3 Hz, 2 H), 6.86 (t, J = 8.7 Hz, 4 H), 5.70 (s, 1 H), 5.23 (dt, J = 1.5, 6.6 Hz,1 H), 4.93 (m, 2 H), 3.96 (m, 2 H), 3.86 (m, 2 H), 3.79 (s, 3 H), 3.13 (m, 1 H), 2.97 (m, 2 H), 2.82 (m, 1 H), 2.09 (s, 3 H), 2.01 (m, 2 H); 13C NMR (CDCl3, 75 MHz) δ 187.6, 169.9, 159.4, (159.0 r), 151.9, 150.3, 130.6, 129.7, 127.8, 126.5, 121.3, 114.4, 112.1, 103.4, 71.4, 65.2, 65.1, 61.9, 55.4, 33.5, 32.3, 30.5, 21.0; HRMS (M+H)+ calcd for C27H29NO8 + H: 496.1971, found 496.1959.
(2S*,3R*)-3-Acetoxy-6-(2-[1,3]dioxolan-2-yl-ethyl)-2-(4-methoxybenzyl)-4-oxo-3,4-dihydro- 2H-pyridine-1-carboxylic acid phenyl ester (15b)
IR (neat) 2934, 1738, 1682, 1593, 1513, 1397, 1289, 1249, 1177, 1033, 913 cm−1; 1H NMR (CDCl3, 300 MHz) δ 7.24 (m, 5 H), 6.86 (d, J = 6.3 Hz, 2 H), 6.72 (d, J = 6.3 Hz, 2 H), 5.83 (d, J = 4.5 Hz, 1 H), 5.63 (s, 1 H), 5.25 (m, 1 H), 4.91 (t, J = 3.3 Hz, 1 H), 3.96 (m, 2 H), 3.84 (m, 2 H), 3.79 (m, 3 H), 2.98 (m, 3 H), 2.81 (m, 1 H), 2.19 (s, 3 H), 2.05 (m, 1 H), 1.90 (m, 1 H); 13C NMR (CDCl3, 75 MHz) δ 189.1, 169.9, 158.7 (158.8 r), 151.3, 150.3, 130.7, 129.6, 129.1, 126.4, 121.5, 114.2, 111.6, 103.4, 73.0, 65.2, 65.1, 60.7, 55.5, 32.4, 30.7, 30.0, 20.9; HRMS (M+H)+ calcd for C27H29NO8 + H: 496.1971, found 496.1961.
(2S*,3S*)-3-(tert-Butyldimethylsilanyloxy)-6-(2-[1,3]dithian-2-yl-ethyl)-2-(4-methoxybenzyl)- 4-oxo-3,4-dihydro-2H-pyridine-1-carboxylic acid phenyl ester (16)
To a solution of 15a (23.0 mg, 0.046 mmol) in H2O:MeOH (1.0 mL, 1:4) was added ScOTf3 (20 mol %, 5.0 mg, 0.0093 mmol). The resulting mixture was stirred at rt for 40 h. Upon completion, the reaction mixture was poured into H2O (5 mL), and extracted with CH2Cl2 (3 × 10 mL). The combined extracts were dried over Na2SO4, filtered through Celite with CH2Cl2, and the solvent removed in vacuo. Purification by silica gel chromatography (20 to 30 % EtOAc in hexanes) gave 21.0 mg (100 %) of the desired α-hydroxy ketone as a white solid which was used directly in the next step. Mp 152–154 °C; IR (neat) 3394, 2919, 1737, 1664, 1513, 1410, 1320, 1248, 1179, 1033 cm−1; 1H NMR (CDCl3, 300 MHz) δ 7.40 (t, J = 5.7 Hz, 1 H), 7.27 (d, J = 3.9 Hz, 2 H), 7.10 (d, J = 6.3 Hz, 2 H), 7.01 (d, J = 6.0 Hz, 2 H), 6.83 (d, J = 6.9 Hz, 2 H), 5.60 (s, 1 H), 5.09 (dt, J = 2.1, 8.1 Hz, 1 H), 4.92 (t, J = 7.2 Hz, 1 H), 3.96 (m, 2 H), 3.87 (m, 2 H), 3.79 (s, 3 H), 3.04 (m, 2 H), 2.81 (m, 2 H), 2.70 (d, J = 4.2 Hz, 1 H), 1.97 (m, 2 H); 13C NMR (CDCl3, 75 MHz) δ 192.9, 158.4 (159.2 r), 152.2, 150.4, 130.6, 129.6, 128.4, 126.3, 121.5, 114.3, 110.3, 103.4, 70.7, 65.1, 65.0, 64.2, 55.4, 33.5, 32.3, 30.5; HRMS (M+H)+ calcd for C25H27NO7 + H: 454.1866, found 454.1873.
To a solution of the above α-hydroxy ketone (550 mg, 1.22 mmol) in DMF (20 mL) at 20 °C was added imidazole (165 mg, 2.43 mmol) and tert-butyldimethylsilyl chloride (275 mg, 1.82 mmol). The resulting mixture was stirred at rt for 12 h. The reaction mixture was quenched with saturated aqueous NaHCO3 (50 mL) and extracted with CH2Cl2 (3 × 40 mL). The combined organic layers were washed with brine (30 mL), dried over Na2SO4, filtered through Celite with CH2Cl2, and the solvent was removed in vacuo. Purification by silica gel chromatography (30 to 40% EtOAc in hexanes) gave 620 mg (90 %) of the desired TBS-protected alcohol as a colorless oil which was used directly in the next step. IR (neat) 2953, 1738, 1674, 1597, 1512, 1323, 1249, 1184, 1100, 1035, 841 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.37 (dt, J = 1.0, 7.4 Hz, 2 H), 7.23 (t, J = 8.4 Hz, 1 H), 7.10 (d, J = 8.4 Hz, 2 H), 6.95 (d, J = 7.7 Hz, 2 H), 6.85 (d, J = 6.6 Hz, 2 H), 5.59 (s, 1 H), 5.09 (dt, J = 2.0, 7.2 Hz, 1 H), 4.92 (t, J = 4.4 Hz, 1 H), 3.95 (m, 2 H), 3.84 (m, 2 H), 3.79 (s, 3 H), 3.69 (s, 1 H), 3.11 (m, 1 H), 3.01 (m, 1 H), 2.85 (m, 2 H), 1.99 (m, 2 H), 0.83 (s, 9 H), 0.07 (s, 3 H), 0.02 (s, 3 H); 13C NMR (CDCl3, 100 MHz) δ 191.9, 157.8 (158.8 r), 152.5, 150.5, 130.5, 129.7, 128.7, 126.3, 121.6, 114.3, 111.0, 103.6, 71.6, 65.7, 65.2, 65.1, 55.5, 33.2, 32.5, 30.5, 25.8, 18.2, −4.6, −5.0; HRMS (M+H)+ calcd for C31H41NO7Si + H: 568.2731, found 568.2741.
To a solution of the above TBS-protected alcohol (620 mg, 1.04 mmol) in CH2Cl2 (10 mL) at 20 °C was added neat BF3·OEt2 (60 μl, 0.44 mmol) and then 1,3-propanedithiol (170 μl, 1.64 mmol). The resulting mixture was stirred at rt for 8 h. The reaction mixture was quenched with saturated aqueous NaHCO3 (50 mL) and extracted with CH2Cl2 (3 × 40 mL). The combined organic layers were washed with brine, dried over Na2SO4, filtered through Celite with CH2Cl2, and the solvent was removed in vacuo to yield the crude product 16. Purification by silica gel chromatography (10 to 20% EtOAc in hexanes) gave 637 mg (95%) of 16 as a colorless oil: IR (neat) 2929, 1738, 1673, 1597, 1249, 1182, 1096, 837 cm−1; 1H NMR (CDCl3, 300 MHz) δ 7.37 (t, J = 7.6 Hz, 2 H), 7.22 (t, J = 6.8 Hz, 1 H), 7.10 (d, J = 8.4 Hz, 2 H), 6.92 (d, J = 6.4 Hz, 2 H), 6.84 (d, J = 6.8 Hz, 2 H), 5.56 (s, 1 H), 4.99 (dt, J = 3.2, 11.2 Hz, 1 H), 4.00 (t, J = 9.6 Hz, 1 H), 3.80 (s, 3 H), 3.71 (d, J = 2.4 Hz, 1 H), 3.19 (m, 1 H), 2.91 (m, 1 H), 2.81 (m, 6 H), 2.05 (m, 3 H), 1.97 (m, 1 H), 0.84 (s, 9 H), 0.04 (s, 3 H), 0.00 (s, 3 H); 13C NMR (CDCl3, 100 MHz) δ 191.8, 158.9, 157.0, 152.4, 150.6, 130.5, 129.7, 128.54, 126.3, 121.6, 114.4, 111.2, 71.7, 65.5, 55.5, 46.7, 34.1, 33.2, 33.1, 30.3, 26.0, 25.8, 18.3, −4.6, −5.0; HRMS (M+H)+ calcd for C32H43NO5S2Si + H: 614.2430, found 614.2439.
(2R*,5S*,6S*)-2(2-Benzyloxymethylallyl)-5-(tert-butyldimethylsilanyloxy)-2-(2-[1,3]dithian-2- yl-ethyl)-6-(4-methoxybenzyl)-4-oxo-piperidine-1-carboxylic acid phenyl ester (19)
A well-stirred solution of dihydropyridone 16 (0.560 g, 0.912 mmol) in 20 mL of dry CH2Cl2 was cooled to −78 °C. TMSOTf (490 μl, 2.73 mmol) was added in one portion, giving rise to a deep orange color. After 5 min, neat (2-benzyloxymethylallyl)tributylstannane reagent 6 (1.00 g, 1.82 mmol) was added dropwise over a 10 min period. The reaction mixture was stirred for 4 h at −78 °C, warmed up to −45 °C, and stirred for additional 6 h. The reaction mixture was quenched with saturated aqueous NaHCO3 (50 mL) and warmed to rt, during which time it became colorless. The mixture was extracted with CH2Cl2 (3 × 100 mL). The organic extracts were dried over Na2SO4 and filtered through Celite. The solvent was removed in vacuo to yield the crude product 19. Purification by silica chromatography (0 to 20% EtOAc in hexanes) gave 458 mg (65%) of 19 as a colorless oil: IR (neat) 2929, 2856, 1720, 1513, 1348, 1250, 1201, 1098, 838 cm−1; 1H NMR (CDCl3, 300 MHz) δ 7.39 (m, 12 H), 6.82 (d, J = 9.2 Hz, 2 H), 5.34 (d, J = 1.2 Hz, 1 H), 5.08 (s, 1 H), 4.67 (dd, J = 1.8, 10.8 Hz, 1 H), 4.50 (s, 2 H), 4.00 (t, J = 6.4 Hz, 1 H), 3.96 (s, 2 H), 3.78 (s, 3 H), 3.74 (s, 1 H), 3.21 (bs, 1 H), 3.19 (d, J = 14.0 Hz, 1 H), 2.98 (bs, 1 H), 2.80 (m, 6 H), 2.52 (bs, 1 H), 2.13 (m, 2 H), 1.90 (m, 2 H), 1.85 (m, 2 H), 0.78 (s, 9 H), −0.10 (s, 3 H), −0.24 (s, 3 H); 13C NMR (CDCl3, 75 MHz) δ 206.3, 158.8, 151.1, 141.8, 141.4, 138.5, 130.3, 129.7, 129.4, 128.6, 128.5, 127.6, 125.9, 122.2, 120.7, 114.4, 73.9, 72.5, 71.3, 70.6, 64.2, 55.5, 47.4, 30.8, 30.4, 30.3, 30.2, 26.1, 25.8, 25.6, 18.1, −4.9, −5.6; HRMS (M+H)+ calcd for C43H57NO6S2Si + H: 776.3475, found 776.3439.
(2R*,5R*,6S*)-2-(2-Benzyloxymethylallyl)-5-(tert-butyldimethylsilanyloxy)-2-(2-[1,3]dithian- 2-yl-ethyl)-6-(4-methoxybenzyl)-4-oxo-piperidine-1-carboxylic acid phenyl ester (20)
Compound 19 (120 mg, 0.155 mmol) in CH2Cl2 (5 mL) was treated with pyridine (1 μl, 0.02 mmol) and DBU (20 μl, 0.077 mmol), and the resulting mixture was refluxed for 60 h. The solution was cooled to rt and quenched with 1 N aqueous HCl (1 mL). The mixture was extracted with CH2Cl2 (3 × 15 mL), and combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. The crude product, a 1:1 mixture of piperidones 19 and 20, was purified by silica gel chromatography (10 in 20% EtOAc in hexanes) to give 60 mg (50 %) of the desired product 20 as a colorless oil: IR (neat) 2927, 2853, 1717, 1512, 1248, 1202, 1161, 1110, 838 cm−1; 1H NMR (CDCl3, 300 MHz) δ 7.32 (s, 8 H), 7.20 (d, J = 9.2 Hz, 2 H), 6.80 (d, J = 9.2 Hz, 2 H), 6.62 (bs, 1 H), 5.34 (d, J = 1.6 Hz, 1 H), 5.02 (s, 1 H), 4.93 (m, 1 H), 4.62 (d, J = 5.6 Hz, 1 H), 4.50 (s, 2 H), 3.99 (t, J = 7.6 Hz, 1 H), 3.92 (s, 2 H), 3.74 (s, 3 H), 3.06 (bs, 1H), 3.04 (d, J = 14.4 Hz, 1 H), 2.86 (m, 6 H), 2.67 (d, J = 19.2 Hz, 1 H), 2.59 (dt, J = 3.6, 12.4 Hz, 1 H), 2.48 (m, 1 H), 2.13 (m, 1 H), 1.87 (m, 5 H), 0.84 (s, 9 H), 0.08 (s, 3 H), 0.00 (s, 3 H); 13C NMR (CDCl3, 100 MHz) δ 206.1, 158.4, 150.8, 142.2, 138.1, 131.2, 130.7, 129.7, 129.4, 128.6, 127.9, 125.7, 122.2, 122.0, 118.2, 114.2, 75.8, 73.5, 72.8, 62.2, 60.4, 55.6, 47.5, 47.0, 37.8, 31.2, 30.6, 26.2, 25.9, 18.6, 0.2, −4.4, −5.4; HRMS (M+H)+ calcd for C43H57NO6S2Si + H: 776.3475, found 776.3439.
(2R*,5S*,6S*)-2-(2-Benzyloxymethylallyl)-5-(tert-butyldimethylsilanyloxy)-6-(4- methoxybenzyl)-4-oxo-2-(3-oxo-propyl)-piperidine-1-carboxylic acid phenyl ester (21)
To a mixture of 19 (32 mg, 0.41 mmol) and calcium carbonate (100 mg, 0.743 mmol) in aqueous H2O:CH3CN (2.0 mL, 1:9) was added CH3I (2.0 mL, 33 mmol). The mixture was stirred at room temperature overnight, diluted with EtOAc (10 mL), and filtered through Celite. The filtrate was washed with brine (2 × 10 mL), dried over Na2SO4, and the solvent was removed in vacuo to yield 28 mg (100 %) of the crude product 21 as a colorless oil. The product was used crude in the next step without further purification: IR (neat) 2927, 2854, 1720, 1611, 1512, 1251, 1201, 1097, 841 cm−1; 1H NMR (CDCl3, 400 MHz) δ 9.74 (q, J = 7.7 Hz, 1H), 7.32 (m, 8H), 7.12 (t, J = 7.7 Hz, 4H), 6.84 (d, J = 8.8 Hz, 2H), 5.36 (q, J = 1.4 Hz, 1 H), 5.10 (s, 1H), 4.69 (dd, J = 2.2, 9.16 Hz, 1H), 4.50 (s, 2H), 3.95 (s, 2H), 3.78 (s, 3H), 3.76 (s, 1 H), 3.23 (d, J = 12.1 Hz, 1 H), 3.18 (d, J = 14.0 Hz, 1 H), 2.86 (d, J = 13.2 Hz, 2 H), 2.74 (dd, J = 1.5, 15.8 Hz, 2H), 2.53 (m, 2H), 1.7 (bs, 1H), 1.55 (bs, 1H), 0.77 (s, 9H), −0.08 (s, 3H), −0.10 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 205.9, 159.0, 151.1, 141.3, 138.4, 130.5, 130.3, 129.8, 129.8, 128.57, 128.0, 127.9, 127.8, 126.1, 122.1, 114.6, 74.0, 72.6, 71.3, 70.6, 64.3, 55.6, 45.1, 41.2, 39.7, 29.9, 25.9, 18.1, −4.8, −5.5; HRMS (M+H)+ calcd for C40H51NO7Si + H: 685.3435, found 685.3411.
2-(4-Methoxybenzyl)-4-oxo-6-[4-(trimethylsilanyl)but-3-ynyl]-3,4-dihydro-2H-pyridine-1- carboxylic acid phenyl ester (28)
Magnesium turnings (2.16 g, 889 mmol) were mechanically activated by stirring at rt overnight under argon and then 40 mL of anhydrous ether was added. A portion of 4-bromo-1-trimethylsilyl-1-butyne (3.65 g, 178 mmol) in 20 mL of anhydrous ether was slowly added to the mixture. Once the reaction was initiated, the mixture was cooled to 10 °C, and the remaining bromide was continuously added over a period of 2 h. After the addition was complete, the reaction mixture was stirred at 10 °C for 3 h, warmed up to rt, and stirred for an additional 4 h to form the Grignard reagent 26. In a separate flask, copper bromide dimethyl sulfide complex (6.09 g, 296 mmol) was mixed with 20 mL of freshly distilled THF under argon and cooled to −78 °C. To a solution of 2-(4-methoxybenzyl)-4-oxo-3,4-dihydro-2H-pyridine-1- carboxylic acid phenyl ester (11) (2.00 g, 593 mmol) in THF (10 mL) was added a premixed solution of TMSCl (8.30 mL, 652 mmol) and Et3N (9.5 mL, 682 mmol). A solution of the above prepared Grignard reagent 26 in Et2O (diluted with 50 mL of THF) was added dropwise over a period of 1 h. Once the addition was complete, the resulting slurry was stirred at −78 °C for 2 h and then warmed to −45 °C. After 24 h, the reaction mixture was quenched with a carefully buffered solution (pH 8) of NH4OH and NH4Cl (100 mL) and the phases were separated. The aqueous phase was extracted with Et2O (3 × 50 mL), and the combined organic layers were washed with 10% NH4OH until no further blue color was observed in the aqueous layer. After drying over anhydrous K2CO3 and filtering through Celite, the solvent was removed under reduced pressure to give 3.4 g of crude 27, which was dissolved in 10 mL of anhydrous DMSO. In a separate flask, IBX (9.96 g, 356 mmol) and MPO (4.45 g, 356 mmol) were dissolved in 20 mL of DMSO at 10 °C. The IBX/MPO mixture was stirred until complete dissolution and was then added in one portion at 0 °C to the crude silyl enol ether 27 solution. The mixture was stirred vigorously at rt overnight. The reaction mixture was carefully diluted with 5% NaHCO3 (50 mL) and extracted with Et2O (3 × 100 mL). The combined organic phase was dried over anhydrous MgSO4, filtered through Celite, and the solvent was removed in vacuo to yield the crude product 28. Purification by silica gel chromatography (10 to 25% EtOAc in hexanes) gave 2.3 g (55%, 2 steps) of 28 as a yellow foam: IR (neat) 2955, 2171, 1725, 1666, 1513, 1248, 1201, 1162, 1037, 889 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.39 (d, J = 7.7 Hz, 2 H), 7.27 (d, J = 7.0 Hz, 1 H), 7.15 (dd, J = 1.8, 6.6 Hz, 2 H), 6.93 (d, J = 7.3 Hz, 2 H), 6.82 (dd, J = 1.8, 6.2 Hz, 2 H), 5.66 (s, 1 H), 5.10 (dq, J = 1.5, 6.2 Hz, 1 H), 3.79 (s, 3H), 3.29 (m, 1 H), 3.10 (dd, J = 7.3, 7.3 Hz, 1 H), 2.89 (dd, J = 8.1, 6.2 Hz, 1 H), 2.84 (d, J = 5.9 Hz, 1 H), 2.71 (m, 1 H), 2.55 (t, J = 6.6 Hz, 2 H), 2.46 (d, J = 17.2 Hz, 1 H), 0.11 (s, 9 H); 13C NMR (CDCl3, 100 MHz) δ 193.2, 158.8, 155.4, 151.8, 150.4, 130.8, 129.6, 129.2, 126.5, 121.6 (122.0 r), 114.7, 114.3, 105.0, 87.3, 58.5, 55.5, 40.3, 36.1, 35.0, 19.1, 0.2; HRMS (M+H)+ calcd for C27H31NO4Si + H: 462.2101, found 462.2085.
6-(3-Bromobut-3-enyl)-2-(4-methoxybenzyl)-4-oxo-3,4-dihydro-2H-pyridine-1-carboxylic acid phenyl ester (29)
A solution of 28 (1.43 g, 3.10 mmol) in THF (35 mL) was cooled to −78 °C. Tetrabutylammonium fluoride (1.0 M in THF, 3.72 mL, 3.72 mmol) was added dropwise over a period of 10 min. After the addition was complete, the reaction mixture was stirred at −78 °C for 30 min, warmed to rt, and stirred for an additional 1 h at rt. The reaction mixture was poured into saturated aqueous NH4Cl (100 mL) and then extracted with Et2O (3 × 250 mL). The combined organic extracts were dried over MgSO4, filtered through Celite, and concentrated in vacuo to yield the crude acetylene product. Purification by silica gel chromatography (10 to 25% EtOAc in hexanes) gave 1.11 g (93%) of the desired acetylene as a colorless oil which was used directly in the next step. IR (neat) 3286, 2932, 1738, 1665, 1598, 1512, 1402, 1333, 1301, 1247, 1162, 1040, 829, 749, 689 cm−1; 1H NMR (CDCl3, 100 MHz) δ 7.39 (t, J = 8.0 Hz, 2 H), 7.26 (m, 1 H), 7.13 (d, J = 8.4 Hz, 2 H), 6.94 (d, J = 7.6 Hz, 2 H), 6.84 (d, J = 8.4 Hz, 2 H), 5.68 (s, 1 H), 5.11 (q, J = 7.6 Hz, 1 H), 3.79 (s, 3 H), 3.27 (m, 1 H), 3.10 (dd, J = 7.2, 6.4 Hz, 1 H), 2.90 (dd, J = 8.0, 5.6 Hz, 1 H), 2.87 (dd, J = 6.0, 11.2 Hz, 1 H), 2.76 (m, 1 H), 2.52 (m, 2 H), 2.45 (s, 1 H), 2.07 (t, J = 2.0 Hz, 1 H); 13C NMR (CDCl3, 100 MHz) δ 193.2, 158.8, 155.3, 151.7, 150.4, 130.7, 129.7, 129.1, 128.5, 126.5, 121.5 (121.9 r), 114.3 (114.6 r), 82.4, 7.6, 58.3, 55.5, 40.2, 36.0, 34.7, 17.7; HRMS (M+H)+ calcd for C24H23NO4 + H: 390.1705, found 390.1711.
A solution of the above acetylene (44.0 mg, 0.113 mmol) in CH2Cl2 (1.0 mL) at 0 °C under argon was treated with 9-bromo-9-borobicyclo[3.3.1]nonane (1.0 M in CH2Cl2, 0.17 mL, 0.17 mmol). The resulting mixture was allowed to warm to rt and stirred for 24 h. After this time, the mixture was cooled to 0 °C and ethanolamine (1.1 mL) was added, followed by MeOH (2.5 mL). The mixture was then diluted with Et2O (15 mL) and shaken with saturated aqueous potassium sodium tartrate (15 mL). The aqueous phase was extracted with Et2O (2 × 15 mL), and the combined organic extracts were dried over MgSO4, filtered through Celite, and concentrated in vacuo to yield the crude product 29. Purification by silica gel chromatography (10–15% EtOAc in hexanes) gave 45.0 mg (85%) of 29 as a colorless oil: IR (neat) 2927, 1736, 1666, 1597, 1513, 1406, 1301, 1248, 1202, 1164, 1035, 890 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.38 (dt, J = 2.0, 8.8 Hz, 2 H), 7.27 (t, J = 1.2 Hz, 1 H), 7.08 (dd, J = 2.8, 8.8 Hz, 2 H), 6.97 (dd, J = 2.0, 2.8 Hz, 2 H), 6.84 (dd, J = 2.8, 8.8 Hz, 2 H), 5.60 (dd, J = 0.8, 1.6 Hz, 2 H), 5.44 (d, J = 2.4 Hz, 1 H), 5.07 (dq, J = 2.0, 8.0 Hz, 1 H), 3.78 (s, 3 H), 3.20 (m, 1 H), 3.04 (dd, J = 8.8, 9.2 Hz, 1 H), 2.81 (m, 4 H), 2.45 (dt, J = 1.6, 17.2 Hz, 2 H); 13C NMR (CDCl3, 100 MHz) δ 192.9, 158.7, 155.8, 151.6, 150.3, 132.2, 130.6, 129.7, 128.8, 126.5, 121.4, 118.3, 114.3, 114.0, 58.3, 55.6, 40.34, 40.0, 36.1, 34.4; HRMS (M+H)+ calcd for C24H24BrNO4 + H: 470.0967, found 470.0959.
(2S*,3S*)-6-(3-Bromobut-3-enyl)-3-(tert-butyldimethylsilanyloxy)-2-(4-methoxybenzyl)-4- oxo-3,4-dihydro-2H-pyridine-1-carboxylic acid phenyl ester (25)
To a stirred solution of 29 (224 mg, 0.477 mmol) in 5 mL of toluene at rt was added lead (IV) acetate (275 mg, 0.620 mmol). The resulting mixture was refluxed for 3 h, cooled to rt, and additional lead (IV) acetate (275 mg, 0.620 mmol) was added. The mixture refluxed for 3 h. After cooling to rt, the solution was filtered through Celite with CH2Cl2. The filtrate was washed with saturated aqueous NaHCO3 (40 mL), and the aqueous phase was extracted with CH2Cl2 (3 × 40 mL). The combined organic phase was dried over Na2SO4, filtered through Celite, and concentrated in vacuo to yield the crude acetate. Purification by silica gel chromatography (20 to 30% EtOAc in hexanes) gave 173 mg (69%) of the desired product as a colorless oil which was used directly in the next step. IR (neat) 2926, 1743, 1678, 1591, 1514, 1248, 1219, 1182, 1032, 839 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.38 (dt, J = 1.6, 7.2 Hz, 2 H), 7.12 (dd, J = 2.4, 6.8 Hz, 2 H), 6.84 (m, 5 H), 5.70 (s, 1 H), 5.62 (m, 1 H), 5.47 (d, J = 2.0 Hz, 1 H), 5.25 (dt, J = 2.8, 7.2 Hz, 1 H), 4.94 (t, J = 2.0 Hz, 1 H), 3.20 (m, 1 H), 3.00 (dd, J = 8.8, 8.8 Hz, 1 H), 2.93 (m, 4 H), 2.76 (m, 1 H), 2.65 (m, 2 H), 2.11 (s, 3 H); 13C NMR (CDCl3, 100 MHz) δ 187.6, 169.6, 159.1, 157.6, 150.2 (152.0 r), 132.1, 130.7, 130.5, 129.9, 127.6, 126.7, 121.3, 118.4, 114.5, 112.6, 71.3, 61.9, 55.5, 40.1, 34.5, 33.5, 21.0; HRMS (M+H)+ calcd for C26H26BrNO6 + H: 528.1022, found 528.1022.
To a solution of the above acetate (390 mg, 0.734 mmol) in H2O:MeOH (20 mL, 1:4) was added ScOTf3 (20 mol %, 72.0 mg, 0.147 mmol). The resulting mixture was stirred at 60 °C for 24 h. The reaction mixture was cooled to rt, poured into H2O (20 mL), and extracted with CH2Cl2 (3 × 20 mL). The combined organic extracts were dried over Na2SO4, filtered through Celite with CH2Cl2, and the solvent was removed in vacuo to yield the crude product. Purification by silica gel chromatography (20 to 30 % EtOAc in hexanes) gave 355 mg (100 %) of the desired alcohol as a white foam which was used directly in the next step. IR (neat) 3376, 2919, 1738, 1666, 1589, 1513, 1248, 1179, 838 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.39 (t, J = 8.0 Hz, 2 H), 7.26 (t, J = 7.2 Hz, 1 H), 7.10 (d, J = 8.8 Hz, 2 H), 7.00 (d, J = 7.6 Hz, 2 H), 6.85 (d, J = 8.4 Hz, 2 H), 5.62 (d, J = 0.8 Hz, 1 H), 5.60 (s, 1 H), 5.45 (d, J = 1.2 Hz, 1 H), 5.11 (dt, J = 1.6, 8.0 Hz, 1 H), 3.79 (s, 4 H), 3.20 (m, 1 H), 2.97 (m, 6 H); 13C NMR (CDCl3, 100 MHz) δ 192.3, 159.0, 157.3, 152.3, 150.5, 132.2, 130.6, 129.8, 128.2, 126.6, 121.6, 118.4, 114.4, 111.1, 70.9, 64.1, 55.5, 40.2, 34.5, 38.8; HRMS (M+H)+ calcd for C24H24BrNO5 + H: 486.0916, found 486.0911.
To a solution of the above alcohol (355 mg, 0.733 mmol) in DMF (10 mL) at 20 °C was added imidazole (100 mg, 1.47 mmol) and tert-butyldimethylsilyl chloride (166 mg, 1.10 mmol). The resulting mixture was stirred at rt for 24 h. The reaction mixture was quenched with saturated aqueous NaHCO3 (50 mL) and extracted with CH2Cl2 (3 × 50 mL). The combined organic layers were washed with brine (30 mL), dried over Na2SO4, filtered through Celite with CH2Cl2, and the solvent was removed in vacuo to yield the crude TBS ether 25. Purification by silica gel chromatography (30 to 40% EtOAc in hexanes) gave 397 mg (90 %) of 25 as a colorless oil: IR (neat) 2929, 2856, 1737, 1676, 1598, 1513, 1249, 1182, 841 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.38 (dt, J = 1.6, 7.2 Hz, 2 H), 7.26 (m, 1 H), 7.10 (dd, J = 2.0, 6.8 Hz, 2 H), 6.94 (dd, J = 1.2, 7.5 Hz, 2 H), 6.86 (dd, J = 2.0, 6.4 Hz, 2 H), 5.60 (m, 1 H), 5.55 (s, 1 H), 5.44 (d, J = 1.6 Hz, 1 H), 4.98 (dt, J = 1.6, 8.0 Hz, 1 H), 3.80 (s, 3 H), 3.70 (d, J = 1.2 Hz, 1 H), 3.21 (m, 1 H), 2.79 (m, 5 H), 0.84 (s, 9 H), 0.04 (s, 3 H), 0.02 (s, 3 H); 13C NMR (CDCl3, 100 MHz) δ 191.7, 158.9, 155.9, 152.5, 150.5, 132.4, 130.5, 129.8, 128.5, 126.4, 121.5, 118.3, 114.3, 111.6, 71.5, 65.6, 55.5, 40.3, 34.3, 33.2, 25.8, 18.2, −4.6, −5.0; HRMS (M+H)+ calcd for C30H38BrNO5Si + H: 600.1781, found 600.1800.
(2R*,5S*,6S*)-2-(2-Benzyloxymethylallyl)-2-(3-bromobut-3-enyl)-5-(tert-butyldimethylsilanyloxy)- 6-(4-methoxybenzyl)-4-oxo-piperidine-1-carboxylic acid phenyl ester (31)
A well-stirred solution of dihydropyridone 25 (57 mg, 0.097 mmol) in 3 mL of dry CH2Cl2 was cooled to −78 °C. TMSOTf (40 μl, 0.22 mmol) was added in one portion, giving rise to a deep orange color. After 5 min, neat (2-benzyloxymethylallyl)tributylstannane reagent 6 (76 mg, 0.15 mmol) was added dropwise over a 10-min period. The reaction mixture was stirred for 4 h at −78 °C, warmed to −45 °C, and stirred for additional 6 h. The reaction mixture was quenched with saturated aqueous NaHCO3 (20 mL) and allowed to warm up to rt, during which time it became colorless. The mixture was extracted with CH2Cl2 (3 × 20 mL). The combined organic extracts were dried over Na2SO4 and filtered through Celite. The solvent was removed in vacuo to yield 110 mg of the crude silyl enol ether product 30, which was dissolved in wet MeOH (5 mL). Silica gel (100 mg) was added, followed by H2O (0.5 mL) and oxalic acid (20 mg, 0.22 mmol). The reaction mixture was stirred at rt for 16 h. The reaction mixture was filtered and then extracted with CH2Cl2 (3 × 20 mL). The combined organic extracts were dried over Na2SO4, filtered through Celite, and the solvent was removed in vacuo to yield the crude piperidone product 31. Purification by silica gel chromatography (0 to 20% EtOAc in hexanes) gave 62 mg (85%) of 31 as a colorless oil: IR (neat) 2926, 2853, 1722, 1512, 1379, 1250, 1199, 1072, 844 cm−1; 1H NMR (C6D6, 400 MHz) δ 7.20 (m, 10 H), 6.89 (t, J = 7.6 Hz, 2H), 6.67 (d, J = 7.6 Hz, 2H), 5.38 (s, 1H), 5.26 (m, 3H), 4.84 (m, 1H), 4.31 (s, 2H), 3.92 (s, 1H), 3.90 (s, 2H), 3.50 (d, J = 17.2 Hz, 1H), 3.27 (s, 1H), 3.25 (s, 3H), 3.07 (dd, J = 3.2, 13.6 Hz, 1H), 2.84 (d, J = 17.2 Hz, 1H), 2.54 (m, 2H), 2.24 (m, 2H), 1.72 (m, 1H), 1.54 (m, 1H), 0.90 (s, 9 H), 0.00 (s, 3H), −0.20 (s, 3H); 13C NMR (C6D6, 100 MHz) δ 205.6, 159.1, 151.7, 142.0, 138.8, 133.7, 130.5, 129.7, 129.5, 128.4, 128.2, 127.9, 127.7, 127.5, 127.1, 125.5, 122.1, 117.2, 74.1, 72.2, 70.9, 64.5, 62.5, 54.7, 45.2, 41.3, 37.5, 37.4, 25.7, 25.5, 18.2, −4.9, −5.7; HRMS (M+H)+ calcd for C41H52NO6BrSi + H: 761.2747, found 761.2709.
(2R*,4S*,5S*,6S*)-2-(2-Benzyloxymethylallyl)-2-(3-bromobut-3-enyl)-4,5-dihydroxy-6-(4- methoxybenzyl)piperidine-1-carboxylic acid phenyl ester (32)
A solution of 31 (57.0 mg, 0.0758 mmol) in THF (4 mL) was cooled to −40 °C. Tetrabutylammonium fluoride (1.0 M in THF, 91 μl, 0.091 mmol) was added dropwise over a period of 5 min. After the addition was complete, the reaction mixture was stirred at −40 °C for 1 h, warmed to −20 °C, and stirred for an additional 2 hours. The reaction mixture was poured into saturated aqueous NH4Cl (10 mL) and then extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were dried over Na2SO4, filtered through Celite, and concentrated in vacuo to yield the crude alcohol. To a solution of tetramethylammonium triacetoxyborohydride (80.0 mg, 0.303 mmol), in 4.0 mL of freshly distilled acetone at rt, was added acetic acid (35.0 μl, 0.607 mmol). After stirring for 15 min, the above crude alcohol compound in 4.0 mL of freshly distilled acetone was added dropwise over a period of 5 min. The reaction mixture was stirred at rt for 16 h. The reaction mixture was quenched with saturated aqueous ammonium chloride (25 mL), and one half of the acetone was removed in vacuo. The solution was extracted with CH2Cl2 (3 × 25 mL). The combined organic extracts were washed with brine (25 mL), dried over Na2SO4, filtered through Celite, and the solvent was removed in vacuo to yield the crude product. Purification by silica gel chromatography (30 to 50% EtOAc in hexanes) gave 45 mg (92% over 2 steps) of 32 as a colorless oil: IR (neat) 3456, 2924, 1693, 1512, 1454, 1248, 1203, 1034 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.40 (t, J = 7.6 Hz, 1H), 7.28 (m, 11H), 7.14 (dd, J = 1.6, 7.2 Hz, 2H), 5.58 (d, J = 1.6 Hz, 1H), 5.40 (d, J = 2.0 Hz, 1H), 5.33 (d, J = 1.2 Hz, 1H), 5.16 (s, 1H), 4.50 (dd, J = 11.6, 6.0 Hz, 2H), 4.42 (bd, J = 12.0 Hz, 1H), 4.14 (bs, 1H), 4.04 (dd, J = 12.8, 8.0 Hz, 2H), 3.79 (s, 4H), 3.12 (dd, J =2.8, 12.8 Hz, 1H), 3.02 (dd, J = 14.0, 6.8 Hz, 2H), 2.83 (t, J = 12.4 Hz, 1H), 2.73 (m, 1H), 2.48 (m, 2H), 2.14 (dd, J = 6.0, 14.8 Hz, 1H), 1.95 (m, 2H), 1.83 (m, 1H), 1.63 (d, J = 3.2 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 158.6, 154.6, 151.2, 142.4, 138.2, 133.9, 130.9, 130.2, 129.7, 128.65, 128.1, 128.0, 125.8, 122.1, 118.0, 117.2, 114.3, 74.4, 74.3, 72.4, 69.7, 64.2, 61.2, 55.5, 41.5, 40.0, 38.0, 37.4, 36.8; HRMS (M+H)+ calcd for C35H40BrNO6 + H: 650.2117, found 650.2101.
(2R*,4S*,5S*,6S*)-4-Benzoyloxy-2-(2-benzyloxymethylallyl)-2-(3-bromobut-3-enyl)-5- hydroxy-6-(4-methoxybenzyl)piperidine-1-carboxylic acid phenyl ester (33)
A solution of 32 (23.9 mg, 36.9 μmol) in CH2Cl2 (1.5 mL) was cooled to −20 °C. Et3N (6.0 μL, 41 μmol) was added dropwise, followed by a catalytic amount of DMAP. The reaction mixture was stirred at −20 °C for 5 min and then cooled to 0 °C. A freshly prepared solution of benzoic anhydride (0.044 M in CH2Cl2, 0.84 mL, 37 μmol) was added over a period of 10 min. The reaction mixture was stirred at 0 °C for 2 h, warmed up to rt, and stirred for an additional 2 h. The reaction mixture was quenched with saturated aqueous NaHCO3 (10 mL) and extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were dried over Na2SO4, filtered through Celite, and the solvent was removed in vacuo to yield crude 33. Purification by silica gel chromatography (10 to 25% EtOAc in hexanes) gave 15.5 mg (56 %) of 33 as a colorless oil: IR (neat) 3434, 2910, 2848, 1719, 1510, 1451, 1272, 1243, 1200, 1104, 1066, 1024, 860 cm−1; 1H NMR (CDCl3, 400 MHz) δ 8.10 (d, J = 8.4 Hz, 2H), 7.65 (t, J = 7.6 Hz, 1H), 7.53 (t, J = 7.6 Hz, 2H), 7.41 (t, J = 7.6 Hz, 2H), 7.26 (m, 9H), 6.70 (d, J = 8.8 Hz, 2H), 5.59 (s, 1H), 5.41 (d, J = 1.6 Hz, 1H), 5.37 (m, 2H), 5.26 (s, 1H), 4.58 (bd, J = 10.4 Hz, 1H), 4.51 (s, 2H), 4.08 (dq, J = 4.4, 8.8 Hz, 2H), 3.96 (bs, 1H), 3.72 (s, 4H), 3.14 (m, 3H), 2.84 (m, 2H), 2.53 (t, J = 8.4 Hz, 3H), 2.40 (d, J = 7.2, 6.8 Hz, 1H), 2.36 (t, J = 9.6 Hz, 1H), 1.80 (m, 1H); 13C NMR (CDCl3, 100 MHz) δ 166.8, 158.6, 142.1, 138.3, 133.8, 133.7, 130.5, 130.4, 129.9, 129.8, 129.7, 128.8, 128.2, 128.6, 128.0, 127.8, 125.8, 122.1, 118.0, 117.3, 114.3, 75.1, 74.3, 72.4, 71.0, 62.4, 60.4, 55.4, 41.2, 39.0, 37.3, 33.0, 29.9; HRMS (M+H)+ calcd for C42H44BrNO7 + H: 754.2379, found 754.2385.
(2R*,4S*,5S*,6S*)-4-Benzoyloxy-2-(2-benzyloxymethylallyl)-2-(3-bromobut-3-enyl)-5- methanesulfonyloxy-6-(4-methoxybenzyl)piperidine-1-carboxylic acid phenyl ester (34)
A solution of 33 (20.0 mg, 26.0 μmol) in CH2Cl2 (1.0 mL) was cooled to 0 °C. Et3N (4.0 μl, 29 μmol) was added dropwise, followed by a catalytic amount of DMAP. The reaction mixture was stirred at 0 °C for 10 min. A freshly prepared solution of p-methanesulfonyl chloride (0.253 M in CH2Cl2, 0.15 mL, 40 μmol) was added over a period of 10 min. The reaction mixture was stirred at 0 °C for 2 h, warmed to 20 °C, and stirred for an additional 4 h. The reaction mixture was quenched with saturated aqueous NaHCO3 (10 mL) and extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were dried over Na2SO4, filtered through Celite, and the solvent was removed in vacuo to yield crude 34. Purification by silica gel chromatography (10 to 25% EtOAc in hexanes) gave 21.0 mg (97 %) of 34 as a colorless oil: IR (neat) 2923, 1722, 1513, 1450, 1344, 1268, 1177, 1032, 949 cm−1; 1H NMR (CDCl3, 400 MHz) δ 8.10 (dd, J = 0.8, 8.0 Hz, 2H), 7.66 (dt, J = 1.2, 7.2 Hz, 1H), 7.54 (t, J = 8.4 Hz, 2H), 7.41 (t, J = 7.2 Hz, 2H), 7.26 (m, 9H), 6.79 (d, J = 8.8 Hz, 2H), 5.65 (t, J = 8.4 Hz, 1H), 5.58 (d, J = 1.2 Hz, 1H), 5.41 (d, J = 2.0 Hz, 2H), 5.33 (bs, 1H), 4.91 (m, 1H), 4.88 (s, 1H), 4.52 (s, 2H), 4.14 (dd, J = 12.8, 40.4 Hz, 2H), 3.77 (s, 3H), 3.24 (m, 2H), 2.92 (m, 3H), 2.77 (s, 3H), 2.53 (m, 3H), 2.37 (t, J = 11.6 Hz, 1H), 1.80 (m, 1H); 13C NMR (CDCl3, 100 MHz) δ 165.5, 159.0, 151.0, 141.6, 138.3, 133.9, 133.3, 130.48, 130.0, 129.7, 129.4, 128.9, 128.6, 128.5, 128.0, 127.8, 126.0, 122.1, 118.8, 118.7, 117.4, 114.6, 78.9, 74.5, 72.41, 71.2, 61.1, 60.4, 55.4, 40.7, 38.6, 37.3, 33.2, 32.1, 29.9; HRMS (M+H)+ calcd for C43H46BrNO9S + H: 832.2155, found 832.2145.
(2S*,4R*,6S*,7R*)-4-(2-Benzyloxymethylallyl)-4-(3-bromobut-3-enyl)-2-(4-methoxybenzyl)-7-oxa-3-aza-bicyclo[4.1.0]heptane-3-carboxylic acid phenyl ester (23)
A solution of 34 (14.0 mg, 16.8 μmol) in MeOH (1.0 mL) was cooled to 0 °C. Solid NaOH (4.7 mg, 0.12 mmol) was added in one portion. The reaction mixture was stirred at 0 °C for 30 min, warmed up to rt, and stirred for an additional 10 min. Solid K2CO3 (100 mg) was added followed by CH2Cl2 (15 mL). The resulting heterogeneous mixture was filtered through Celite, and the solvent was removed in vacuo to yield crude 23. Purification by silica gel chromatography (5 to 15% EtOAc in hexanes) gave 10 mg (94%) of 23 as a colorless oil: IR (neat) 2923, 2855, 1713, 1515, 1456, 1379, 1250, 1203 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.39 (t, J = 7.6 Hz, 1H), 7.30 (m, 9H), 7.12 (d, J = 7.2 Hz, 2H), 6.83 (d, J = 8.8 Hz, 2H), 5.58 (s, 1H), 5.39 (d, J = 2.0 Hz, 1H), 5.32 (d, J = 1.2 Hz, 1H), 5.10 (s, 1H), 4.78 (m, 1H), 4.49 (dd, J = 18.4, 5.2 Hz, 2H), 3.98 (q, J = 12.8 Hz, 2H), 3.80 (s, 3H) 3.38 (m, 1H), 3.09 (dq, J = 4.4, 2.8 Hz, 1H), 2.93 (d, J = 13.6 Hz, 1H), 2.87 (dd, J = 3.2, 12.0 Hz, 1H), 2.75 (m, 2H), 2.58 (d, J = 13.6 Hz, 1H), 2.37 (m, 3H), 1.80 (dd, J = 4.0, 4.4 Hz, 1H), 1.60 (m, 1H); 13C NMR (CDCl3, 100 MHz) δ 158.5, 151.1, 142.8, 138.1, 133.8, 130.7, 130.1, 129.7, 129.7, 128.7, 128.1, 128.0, 125.9, 122.1, 117.4, 117.2, 114.1, 73.6, 72.6, 59.9, 55.9, 55.4, 49.3, 48.8, 42.3, 39.0, 38.3, 37.1, 34.7 (29.9 r); HRMS (M+H)+ calcd for C35H38BrNO5 + H: 632.2012, found 632.2033.
Supplementary Material
General experimental methods, Tables 1 and 2, and 1H and 13C NMR spectra for compounds 13, 15a-b, 16, 19-21, 23, 25, 28-29 and 31-34. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We express appreciation to the National Institutes of Health (Grant GM 34442) for financial support of this research. D.G. also thanks the Burroughs-Wellcome Fellowship Fund for a second year Graduate Assistantship. The authors thank Dr. Sabapathy Sankar for NMR interpretation. NMR and mass spectra data were obtained at NCSU instrumentation laboratories, which were established by grants from the North Carolina Biotechnology Center and the National Science Foundation (Grants CHE-9509532 and CHE- 0078253).
References
- 1.Borel JF, editor. Cyclosporin A. Elsevier Biochemical Press; Amsterdam: 1982. [Google Scholar]
- 2.Schreiber SL. Cell. 1992;70:365. doi: 10.1016/0092-8674(92)90158-9. [DOI] [PubMed] [Google Scholar]
- 3.(a) Schreiber SL. Science. 1991;251:283. doi: 10.1126/science.1702904. [DOI] [PubMed] [Google Scholar]; (b) Kannedy MS, Deeg HJ, Storb N, Thomas ED. Transplant Proc. 1983;15:471. [Google Scholar]
- 4.Sakamoto K, Tsujii E, Abe F, Nakanishi T, Yamashita M, Shigematsu N, Izumi S, Okuhara M. J Antibiot. 1996;46:37. doi: 10.7164/antibiotics.49.37. [DOI] [PubMed] [Google Scholar]
- 5.For total syntheses of (−)-FR901483, see: Snider BB, Lin H. J Am Chem Soc. 1999;121:7778.Scheffler G, Seike H, Sorensen EJ. Angew Chem, Int Ed. 2000;39:4593. doi: 10.1002/1521-3773(20001215)39:24<4593::aid-anie4593>3.0.co;2-x.Ousmer M, Braun NA, Ciufolini MA. Org Lett. 2001;3:765. doi: 10.1021/ol015526i.. For the total syntheses of (+)-FR901483, see: Maeng JH, Funk RL. Org Lett. 2001;3:1125. doi: 10.1021/ol015506g.Kan T, Fujimoto T, Ieda S, Asoh Y, Kitaoka H, Fukujama T. Org Lett. 2004;6:2729. doi: 10.1021/ol049074w.Brummond KM, Hong SP. J Org Chem. 2005;70:907. doi: 10.1021/jo0483567.. For approaches to FR901483 and its analogues, see: Quirante J, Escolano C, Massot M, Bonjoch J. Tetrahedron. 1997;53:1391.Yamazaki N, Suzuki H, Kibajashi C. J Org Chem. 1997;62:8280. doi: 10.1021/jo9715579.Braun NA, Ciufolini MA, Peters K, Peters EM. Tetrahedron Lett. 1998;39:4667.Snider BB, Lin H, Foxman BM. J Org Chem. 1998;63:6442.Bonjoch J, Diaba F, Puigbo E, Sole D, Segarra V, Santamaria L, Beleta J, Ryder H, Palacios JM. Biorg Med Chem. 1999;7:2891. doi: 10.1016/s0968-0896(99)00250-3.Brummond KM, Lu J. Org Lett. 2001;3:1347. doi: 10.1021/ol010029n.Wardrop DJ, Zhang W. Org Lett. 2001;3:2353. doi: 10.1021/ol0161514.Bonjoch J, Diaba F, Puigbo E, Sole D. Tetrahedron Lett. 2003;44:8387.Panchaud P, Ollivier C, Renaud P, Zigmantas S. J Org Chem. 2004;69:2755. doi: 10.1021/jo035843y.
- 6.(a) Comins DL, Zhang YM, Joseph SP. Org Lett. 1999;1:657. doi: 10.1021/ol990738p. [DOI] [PubMed] [Google Scholar]; (b) Comins DL, Joseph SP, Goehring RR. J Am Chem Soc. 1994;116:4719. [Google Scholar]
- 7.The numbering scheme adopted is based on the accepted numbering for the natural product, (−)-FR901483 ( compound 1, Scheme 1).
- 8.Comins DL, Brown JD. Tetrahedron Lett. 1986;27:4549. [Google Scholar]
- 9.Larock RC, Hightower TR, Kraus GA, Hahn P, Zheng D. Tetrahedron Lett. 1995;36:2423. [Google Scholar]
- 10.Comins DL, Stoltze DA, Thakker P, McArdle CL. Tetrahedron Lett. 1998;39:5693. [Google Scholar]
- 11.For reviews on A(1,3) strain, see: Hoffmann RW. Chem Rev. 1989;89:1841.Johnson F. Chem Rev. 1968;68:375.
- 12.Kajiro H, Mitamura S, Mori A, Hiyama T. Tetrahedron Lett. 1999;40:1689. [Google Scholar]
- 13.A variety of basic hydrolysis conditions (K2CO3, MeOH; KCN, MeOH) resulted in acetate hydrolysis together with carbamate removal or significant decomposition (DBU, MeOH; NH3, MeOH). Other acidic type hydrolysis conditions (10% aqueous HCl, EtOH; 10% aqueous H2SO4, EtOH; HBF4·OEt2, MeOH) resulted in significant polymerization upon scale up.
- 14.Acetal exchange was necessary as subjection of the (1,3)-dioxolane acetal protected version of compound 16 to the Sakurai conjugate allylation conditions (allyl silane 17, TiCl4, CH2Cl2) resulted in the Lewis acid coordinating to the acetal moiety leading to attack at the side chain.
- 15.(a) Trost BM, Chan DMT, Nanninga TN. Org Synth. 1984;62:58. [Google Scholar]; (b) Marko IE, Plancher JM. Tetrahedron Lett. 1999;40:5259. [Google Scholar]; (c) Dumeunier R, Markó IE. Tetrahedron Lett. 2000;41:10219. [Google Scholar]
- 16.Sakurai H, Hosomi A, Hayashi J. Org Synth. 1984;62:86. [Google Scholar]
- 17.(a) Evans DA, Rajapakse HA, Stenkamp D. Angew Chem Int Ed. 2002;41:4569. doi: 10.1002/1521-3773(20021202)41:23<4569::AID-ANIE4569>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]; (b) Kelly DR, Mahdi JG. Tetrahedron Lett. 2002;43:511. [Google Scholar]
- 18.Hagen G, Mayr H. J Am Chem Soc. 1991;113:4954. [Google Scholar]
- 19.The stereochemistry of the major diastereomer 19 is believed to be as depicted based on analogy with the allylation reaction in 25 yielding the structurally similar compound 31 with unambiguously established stereochemical correlation of the substituents on the piperidone ring (See Scheme 8).
- 20.Comins DL, LaMunyon DH, Chen X. J Org Chem. 1997;62:8182. doi: 10.1021/jo971448u.Kuethe JT, Comins DL. Org Lett. 1999;1:1031. doi: 10.1021/ol9908124.. For discussions on stereoelectronic control in reactions of this type, see: Deslongchamps P. Stereoelectronic Effects in Organic Chemistry. Pergamon; New York: 1983. Chapter 6Stevens RV. Acc Chem Res. 1984;17:289.
- 21.For a few examples of conjugate allylations in dihydropyridones unsubstituted α to the nitrogen at the vinylogous amide portion of the molecule, see: Sato M, Aoyagi S, Yago S, Kibayashi C. Tetrahedron Lett. 1996;37:9063.Comins DL, Killpack MO, Despagnet E, Zeller E. Heterocycles. 2002;58:505.Kranke B, Hebrault D, Schultz-Kukula M, Kunz H. Syn Lett. 2004;4:671.
- 22.(a) See Table 1 in Supporting Information. (b) See Table 2 in Supporting Information.
- 23.MacSpartan Pro minimization employing MMFF conformer distribution of ketone 19 revealed that the compound existed in a twist-boat conformation with no apparent thermodynamic bias for the desired epimerization to take place.
- 24.We could epimerize the C-7 acetate functionality 15a, by prolonged exposure to acetic acid, and prepare the C-7 epimer of 16. However, all our attempts at the conjugate allylation reaction to install the C-1 quaternary center failed, presumably due to the fact that the bulky TBS ether (which is now epimeric at C-7 in dihydropyridone 16) blocks the α-face during the addition step. Since this is the face the nucleophile has to approach in order to maintain a low energy, chair-like transition state, attack does not occur.
- 25.White JD, Hanselmann R, Wardrop DJ. J Am Chem Soc. 1999;121:1106. [Google Scholar]
- 26.(a) Dobler MR, Bruce I, Cederbaum F, Cooke NG, Diozario LJ, Hall RG, Irving E. Tetrahedron Lett. 2001;42:8281. [Google Scholar]; (b) Takahara PM, Freudenberger JH, Konradi AW, Pedersen SF. Tetrahedron Lett. 1989;30:7177. [Google Scholar]; (c) Matsamuto T, Yamaguchi H, Tanabe M, Yasui Y, Suzuki K. Tetrahedron Lett. 2000;41:8393. [Google Scholar]; (d) Kawatsura M, Kishi E, Kito M, Sakai H, Shirahama H, Matsuda F. Synlett. 1997;55:479. [Google Scholar]; (e) Molander GA, Kenny C. J Org Chem. 1988;53:2134. [Google Scholar]; (f) Fleming M, McMurry JJ. Org Synth. 1982;60:113. [Google Scholar]; (g) McMurry J. Chem Rev. 1989;89:1513. [Google Scholar]; (h) Molander GA, Harris CR. Tetrahedron. 1998;54:3321. [Google Scholar]
- 27.(a) Crimmins MT, Gould LD. J Am Chem Soc. 1987;109:6199. [Google Scholar]; (b) Rossi R, Carpita A, Ciofalo M, Lippolis V. Tetrahedron. 1991;47:8443. [Google Scholar]
- 28.Nicolaou KC, Gray DLF, Montagnon T, Harrison ST. Angew Chem Int Ed. 2002;41:996. doi: 10.1002/1521-3773(20020315)41:6<996::aid-anie996>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 29.During the course of the oxidation of 27 to 28, we observed a small amount (15%) of the piperidone product resulting from hydrolysis of the silyl enol ether functionality.
- 30.During the course of the oxidation of 27 to 28 we were surprised to see that exposure to our earlier catalytic oxidation conditions (Pd(OAc)2 (0.1 equiv), CuCl (1 equiv), O2, CH3CN, 65 °C) gave only the undesired piperidone byproduct resulting from the hydrolysis of the silyl enol ether moiety.
- 31.(a) White JD, Blakemore PR, Browder CC, Hong J, Lincoln CM, Nagornyy PA, Robarge LA, Wardrop DL. J Am Chem Soc. 2001;123:8593. doi: 10.1021/ja011256n. [DOI] [PubMed] [Google Scholar]; (b) Hara S, Dojo H, Takinami S, Suzuki A. Tetrahedron Lett. 1983;24:731. [Google Scholar]
- 32.The product showed a small coupling constant (JH6-H7 = 2.0 Hz) in its 1H-NMR, consistent with its trans-stereochemistry.
- 33.In a previous experiment we had discovered that exposure of diol 32 to MsCl (1.0 equiv) in the presence of Et3N and DMAP resulted in the formation of the C-7 mesylate as the major compound of a 3:1 mixture of C-8/C-7 regioisomers. Thus, in order to obtain the desired epoxide 23 we needed to selectively derivatize the C-8 hydroxyl first. This led to the C-8 benzoate ester formation/C-7 mesylation route.
- 34.Mashimo K, Sato Y. Tetrahedron. 1970;26:803. [Google Scholar]
- 35.(a) Gauthier DR, Jr, Bender SL. Tetrahedron Lett. 1996;37:13. [Google Scholar]; (b) Hudlickly T, Rinner U, Gonzalez D, Akgun H, Schilling S, Siengalewicz P, Martinot TA, Pettit GR. J Org Chem. 2002;67:8726. doi: 10.1021/jo020129m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
General experimental methods, Tables 1 and 2, and 1H and 13C NMR spectra for compounds 13, 15a-b, 16, 19-21, 23, 25, 28-29 and 31-34. This material is available free of charge via the Internet at http://pubs.acs.org.