

Abstract
The human antioxidant protein, HAH1, is an important participant in a Cu(I) transport chain, delivering one Cu(I) ion to the Wilson’s (WND) or Menkes Disease protein (MNK). Full geometry optimizations and second derivative calculations were performed on several binding site models using the B3LYP functional to derive parameters for the construction of a novel MM force field for Cu(I) and its ligating residues in HAH1. MM minimization and MD calculations were then performed using the AMBER suite to validate the newly-generated force field. The X-ray crystal structure of the protein and the geometry of the Cu(I) binding site within the protein were reproduced by the MD simulations on the protein based on rmsd and visual inspection, validating the new force field parameters. The results from the QM and MD simulations suggest that either a two- or three-coordinate exchange reaction is preferred and that it is unlikely that a four-coordinate Cu(I) species plays a role in copper exchange.
Because of its ability to readily cycle between oxidation states, Cu(I) is an important catalyst within the body. However, because of its high reactivity, Cu(I) is also prone to facilitate unhealthy side reactions in the body. One such reaction is the formation of toxic peroxide radicals. Hence, the concentration of Cu(I) is tightly regulated in biological systems. As a result, the [Cu(I)]free within the body has been estimated to be only 10−18 M, with [Cu(I)]total is on the micromolar range,(1, 2) indicating that there are a significant number of high-affinity Cu(I)-binding sites within a cell.
The Cu(I) transport pathway that involves the transmembrane multi-metal transport protein hCtr1, the human antioxidant protein (HAH1 or Atox1), and the Wilson’s or Menkes Disease protein (WND and MNK, respectively) is essential for the regulation of the availability of Cu ions within the body, in a process known as homeostasis.(2–11) Problems within the copper-transport structure of the cell result in Menkes Syndrome, Wilson’s Disease, familial amyotrophic lateral sclerosis (fALS) disorders, and Alzheimer’s disease.(3) The proposed mechanism for Cu(I) transfer from HAH1 to its target protein suggests that the metal ion is passed from the donor to the target domain via a series of multi-coordinate Cu(I) intermediates.(12, 13) Figure 1 illustrates the numerous possible copper coordination states including a two-coordinate donor state, multiple three-coordinate states in which both the Cu donor and acceptor bind the ion, and the terminal product. Direct protein-protein contact between the donor and target is necessary for Cu(I) transfer to occur, and it has been suggested that the chaperone donates its metal ion in an enzymatic fashion, lowering the energy barrier for inter-protein ion transport.(2, 5)
Figure 1.

The proposed mechanism for Cu(I) transfer between HAH1 and MNK4. A possible four-coordinate intermediate that would reside between the two three-coordinate states in the middle has been left out. In the HAH1 homodimer used as a mimic of the HAH1:MNK4 heterodimer in MD simulations, C14 and C17 of the target domain are replaced by Cys 12B and Cys 15B, respectively.
HAH1 is part of a family of copper-binding proteins that share a similar MT/CXXC Cu(I) binding motif. Cu(I) is bound to the metallochaperone via the S atoms of Cys residues in the binding loop. Wernimont et al. elucidated the structure of HAH1 using X-ray crystallography (PDB ID 1FEE), and determined the protein to be dimeric in the crystalline state, with one Cu(I) ion bound per dimer in a near-tetrahedral configuration.(13) Although it was clear that Cu was bound by residues from each monomer, the data from the crystal structure supported either a four-coordinate or three-coordinate Cu(I) state with a loosely-bound fourth residue.(12) A four-coordinate Cu(I) site would also produce an electrostatic environment with a net charge of -3 in the active site, which is unfavorable.(13) NMR studies performed by Anastassopoulou et al. concluded that Atx1, the yeast homolog of HAH1, exists as a monomer in solution, binding one Cu(I) ion per monomer in a nearly linear two-coordinate fashion (PDB ID 1TL4).(14) XAS data also suggested a two-coordinate Cu(I) site in the HAH1 monomer that would be amenable to further coordination to exogenous thiols and phosphines.(15) It is believed that a monomer of HAH1 interacts favorably with the fourth domain of MNK during Cu(I) transfer due in part to the highly complementary structures of the donor and target along the protein-protein interface.(4) The solution structure of the fourth domain of the Menkes ATPase, the target domain for Cu(I) transfer from HAH1, was determined by Gitchier and coworkers in 1998.(16) A sequence analysis of numerous metal-transporting ATPases by Arnesano and coworkers revealed sequence homology between Atx1 and Ccc2, the yeast homologs HAH1 and MNK, respectively. The primary structure similarity of the two proteins suggested that their complimentary tertiary structures could be important to inducing or facilitating Cu(I) transfer.(17) Furthermore, since no crystal structure of the donor-target complex exists, Larin et al. used the solution structure of MNK4 and the crystal structure of HAH1 to artificially create a complex whose structure was compared to the X-ray structure of the HAH1 homodimer by rmsd that showed a high level of structural homology to the HAH1:MNK4 heterodimer.(18) A similar docking study was undertaken by Arnesano in which the artificially docked Atx1:Ccc2 heterodimer was compared to available crystal structures of Atx1, Ccc2 and HAH1. (19) In this study, it was noted that the Atx1:Ccc2 complex, the yeast homolog to the human HAH1:MNK4 complex, is remarkably similar to the crystal structure of the HAH1 dimer as a whole and with respect to the geometry of the Cu(I) binding region of the heterodimer. In light of these studies, the use of the HAH1 homodimer structure as a model of the HAH1:MNK4 heterodimer is considered valid.
In 2006, Banci et al. released a collection of NMR structures of the Atx1:Ccc2 Cu(I) transfer complex.(20) In light of this new structure, the solvent-exposed Cys residues of the metal binding site were identified as more important than the Cys residues nearer the interface of the donor and target, and a “dominant” three-coordinate Cu(I) species was described in which Cu(I) is bound to the two solvent exposed Cys residues and the buried Cys of the target domain.(21) Furthermore, a nearby Lys residue was singled out as having a role in accessibility of the binding site to the target domain and for forming important interactions with the metal-binding residues near the protein interface. The plausibility of a four-coordinate intermediate was further reduced due to the nature of the metal binding site in the NMR structures. While our work supports some of the conclusions drawn in these articles, the NMR structure of the Atx1:Ccc2 complex does not exclude other tricoordinate Cu(I) species in the Cu(I) transfer mechanism between HAH1 and MNK4.
The first goal of this work was to generate geometrically optimized structures of the models of the Cu(I) binding site of the HAH1 dimer in several Cu(I) coordination states, and to calculate the bonding and charge parameters of the systems in order to create a molecular mechanics force field for the Cu(I) binding site in HAH1. The second phase of this work was to validate the new parameters by performing long timescale molecular dynamics simulations on the HAH1 homdimer as a model for the HAH1:MNK4 Cu(I) transfer heterodimer and to compare the results to the crystal structure of HAH1. MM calculations have been previously generated and reported for many transition metals, including Cu(II) (blue copper),(22) and there have been some reports of reliable MM Cu(I) force field parameters in the literature.(23–25) Very recently, Dalosto published Cu(I) parameters for the Atx1 system based on QM/MM calculations.(23) While the parameters published there are similar to those reported in this paper, our study incorporates dynamics, solvent interaction, and hydrogen bonding with the simulated protein, and energetics from QM calculations on model Cu(I) thiolate clusters to address the mechanism of Cu(I) transfer. The parameters presented here should be generally applicable to Cu(I) binding proteins that employ Cys residues to ligate the metal ion.
Computational Details
Ab initio calculations were performed on numerous multi-coordinate models of the Cu(I) binding site from HAH1. Ligating Cys residues were contracted to methylthiolate (CH3S−) groups and unionized Cys residues were reduced to methylthiols (CH3SH). Three model clusters were generated based on the X-ray crystal structure of the Cu(I) binding site from the HAH1 dimer. Those models include Cu(I)(CH3S−)43−, Cu(I)(CH3S−)3[CH3SH]2−, and Cu(I)(CH3S−)2[CH3SH]21−. Gaussian 03 rev C.01(26) was used to perform the calculations using the B3LYP(27–30) functional. Default grid spacing and convergence criteria were employed, and no symmetry constraints were applied. Cu(I) is a closed-shell system with electron configuration Ar[3d10]. Therefore singlet multiplicity was specified for each calculation. For C, H, and S atoms, the split-valence 6-311++G** basis was employed,(31–33) while the enhanced double-zeta basis set of Ryde (DZpdf) was used for Cu(I).(34) Figure 2 shows the X-ray crystal structure of the Cu(I) binding site of the HAH1 dimer including Cys 12 and Cys 15 of both monomers. Also shown are the initial structures of the model clusters used. The models were created using WebLab ViewerPro version 3.7(35) Force constant calculations were performed on the geometry-optimized models using internal coordinates and the IOp(7/33=1) keyword. Charge populations were also determined from the optimized structures using the Merz-Kollman-Singh method(36, 37), and the IOp(6/32=2) keyword. Default van der Waals radii were used for all atoms except Cu(I), for which a radius of 0.91 Å was specified based on the crystal radius of an Oh Cu(I) ion.(38)
Figure 2.
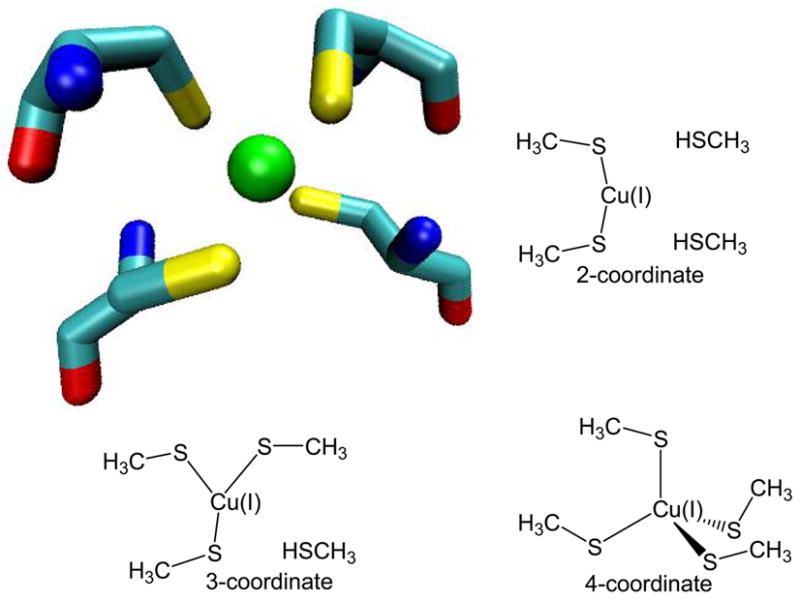
The Cu(I)-binding site from the HAH1 crystal structure including Cys side chains, and the three QM models used to represent two-, three-, and four-coordinate Cu(I) environments.
Simulations were performed in AMBER(39) using the 1994 parameter set of Cornell et al. for the protein atoms(40) The X-ray crystal structure of the HAH1 dimer was used as the starting structure for the force field based simulations. New atom types were specified for the Cu(I) ion, and any Cu-binding S atoms. Force field parameter files were created for each of the new atom types. Mass, bonding, angular, torsion, and non-bonding parameters for Cu(I) and for Cu-bound S are listed in Tables 1 and 2. Default charges were used for all atoms except Cu(I) and for atoms within the Cu-binding residues. Charges for those atoms were adapted from the MKS potentials generated in Gaussian 03. The espgen and respgen programs in the antechamber package within AMBER were used to convert the charges into AMBER format. These charges were modified to ensure integer charges on the systems, and the same charges for Cu(I) and Cu(I)-bound Cys residues were imposed in the MD simulations regardless of the coordination state of the metal ion. The total electrostatic potential derived charge of a Cu-bound Cys residue was approximately −1.1, and the charge imposed on Cu(I) was +1.36. AMBER ESP charge values are listed in Table 3. The apo-HAH1 dimer is neutral, so the total charges of the Cu(I)-bound protein systems are equal to that of the Cu(I) binding site alone.
Table 1.
Atom type, atomic mass, van der Waals radii, and van der Waals well-depths for Cu(I) and Cu(I)-bound S in HAH1.
| 2-coordinate structure | ||||
|---|---|---|---|---|
| Atom | Atom type | Mass (au) | van der Waals radius (Å) | Well-depth (ε,kcal/mol) |
| Cu | PP | 63.55 | 1.70 | 0.05 |
| S (Cys 12A) | SA | 32.06 | 2.00 | 0.25 |
| S (Cys 15A) | SB | 32.06 | 2.00 | 0.25 |
| 3-coordinate structure | ||||
|
| ||||
| Cu | PP | 63.55 | 1.70 | 0.05 |
| S (Cys 12A) | SA | 32.06 | 2.00 | 0.25 |
| S (Cys 15A) | SB | 32.06 | 2.00 | 0.25 |
| S (Cys 12B) | SC | 32.06 | 2.00 | 0.25 |
| 4-coordinate structure | ||||
|
| ||||
| Cu | PP | 63.55 | 1.70 | 0.05 |
| S (Cys 12A) | SA | 32.06 | 2.00 | 0.25 |
| S (Cys 15A) | SB | 32.06 | 2.00 | 0.25 |
| S (Cys 12B) | SC | 32.06 | 2.00 | 0.25 |
| S (Cys 15B) | SD | 32.06 | 2.00 | 0.25 |
Table 2.
Bond lengths, bond angles, and associated force constants for the HAH1 Cu(I) binding site.
| Bond | kbond (kcal/mol·Å2) | r0 (Å) |
|---|---|---|
| Cu-S | 60.00 | 2.19 |
| CT-S | 219.3 | 1.84 |
| Angle | kt (kcal/mol·rad2) | θ0 (deg) |
|
| ||
| S-Cu-S | 50.00 | 109.5 |
| C-S-Cu | 93.98 | 95.91 |
Table 3.
Cys and Cu(I) RESP charges used for the HAH1 Cu(I) binding site.
| Atom | CYS charge | Atom type | RESP charge in AMBER |
|---|---|---|---|
| N | −0.4157 | N | −0.3630 |
| H | 0.2719 | H | 0.2520 |
| Cα | 0.0213 | CT | 0.0350 |
| Hα | 0.1124 | H1 | 0.0480 |
| Cβ | −0.1231 | CT | −0.5720 |
| Hβ | 0.1112 | H1 | 0.2440 |
| Sγ | −0.3119 | SA, SB, SC, or SD | −1.0918 |
| C | 0.5973 | C | 0.6160 |
| O | −0.5679 | O | −0.5040 |
| Hγ | 0.1933 | HS | n/a |
| Cu | n/a | PP | 1.3673 |
Although the Cu-S bond lengths were shorter in the gas-phase QM models than in the crystal structure, the force constants calculated in Gaussian 03 were found to be too weak to reproduce the binding site geometry of the HAH1 crystal structure during MD simulations. Therefore, the initial QM force constants were increased to the values shown in the Table to ensure that the Cu(I)-S distances closely reproduced those seen in the HAH1 homodimer. Cβ-S bond lengths were taken from the QM models and bond force constants were based on CT-SH values in the 1994 force field (219.3 kcal/mol and 237 kcal/mol). Hβ-Cβ-S and Cβ-S-Cu angles and bending force constants were taken from the QM model data. S-Cu-S angle values were also taken from the QM model data, with the angle force constants resembling those of Hβ-Cβ-S in the 1994 force field (50.0 kcal/mol rad2 and 43.0 kcal/mol rad2, respectively). Torsion values were modified slightly from the 1994 parameter library for Cys. S was given a nonbonding radius of 2.00 Å and a well-depth of 0.25 kcal/mol. The copper(I) ion was given a nonbonding radius of 1.70 Å and a well depth of 0.05 kcal/mol. The van der Waals parameters for S are the same as those in the parm94 library, and the well-depth values for Cu(I) are comparable to those in the parm91 library. The hard-sphere radius in the Cu(I) nonbonding parameters are larger in comparison to both the AMBER parm91 value of 1.2 Å and that of 1.09 Å used by Dalosto.(23) The larger radius used in these simulations was used to ensure that any S or H atoms from nearby unbound Cys residues did not interfere with the metal center.
Initial gas-phase molecular mechanical (MM) minimizations were performed on the Cu(I)-bound dimer with sander using the default minimization algorithm for 2000 steps. The nonbonded cutoff was set to 20.0 Å. Minimization was followed by a series of temperature equilibration MD simulations during which the temperature of the system was raised from 100 K to 300 K. Upon temperature equilibration, long timescale MD simulations were performed at a constant temperature of 300 K using the Berendsen algorithm.(41) The gas-phase temperature-equilibrated structures were then surrounded by an 8.0Å TIP3P water box for solvent-phase MD simulations. The solvated system was minimized with a nonbonded cutoff of 8.0Å using the default minimization method for 8000 steps. Again, the temperature of the system was brought to 300 K over a series of MD simulations. SHAKE was implemented for bonds involving hydrogen. Long timescale constant pressure MD simulations were performed on the equilibrated systems at 300 K with periodic boundary conditions in place. The reported MD runs were at least 3,600 ps in length, using a time step of 2.0 fs.
Five different Cu(I)-bound HAH1 dimers were simulated in this study, with each system mimicking a potential intermediate in the Cu(I) transport mechanism from the HAH1 monomer to MNK4. The five systems differed in the Cu(I)-coordination state and the identity of the Cu(I)-binding residues. The four-coordinate system is nearly identical to the crystal structure of Cu(I)-bound HAH1, with Cu(I) being bound by Cys 12 and Cys 15 from monomer A and by Cys 12 and Cys 15 from monomer B. Two three-coordinate systems included Cu(I) bound to Cys 12 A, Cys 15 A and Cys 12 B (referred to as 3-coord A) and Cu(I) bound to Cys 12 A, Cys 15 A, and Cys 15 B (referred to as 3-coord B). Two two-coordinate systems were Cu(I) bound to Cys 12 A and Cys 15 A (called 2-coord) and Cu(I) bound to Cys 15 A and Cys 15 B (called 2-coord-bridged). The ptraj program in AMBER was used to evaluate the validity of the new Cu(I) and S force field parameters on the basis of the geometries of the five HAH1 dimer systems compared to the crystal structure of the dimer. Root-mean-square deviation (rmsd), rms fluctuation (rmsf), protein/solvent radial distribution functions, and radius of gyration were calculated for each of the five HAH1 systems.
Results and Discussion
Potential intermediates in the multi-coordinate transport mechanism for Cu(I) include some two- and three-coordinate metal sites, with the possibility of a four-coordinate Cu site. In order to assess the relative energies of these three different coordination states, single point energy calculations were performed on the gas-phase optimized model clusters in both gas-phase and with implicit aqueous solvent. The energies of two model clusters cannot be directly compared since the number of atoms of each model decreases as the coordination number of Cu increases. Therefore, the energies of two isodesmic reactions were calculated to gauge the energy difference between different coordination states.
The single point energy of each molecule or ion in the isodesmic reactions was calculated separately before the ΔErxn was determined. The relative energies for the reactions above are shown in Table 4 and plotted in Figure 3. Based on the relative QM energies of the different model clusters, the four-coordinate state is shown to be energetically unfavorable. Table 4 gives the electronic energies of the products and reactants in Hartrees and the ΔErxn for both gas- and solvent-phase reactions in Hartrees and kcal/mol. As expected, the is stabilized relative to the gas-phase value for both reactions. For the two-coordinate to three-coordinate reaction and the three-coordinate to four-coordinate reaction, is +244 kcal/mol and +330. kcal/mol, respectively. For the solvent-stabilized reactions, is +47.7 kcal/mol for the two- to three-coordinate reaction and +63.8 kcal/mol when creating the four-coordinate state from the three-coordinate state. Overall, the four-coordinate state is 111 kcal/mol higher in energy than the two-coordinate state in implicit aqueous solvent. Higher energy transition states, not shown here, may exist between the different cluster models. These ΔErxn calculations support the hypothesis that a potential four-coordinate intermediate in the Cu(I) transport mechanism between HAH1 and MNK4 is not likely to exist due to the comparatively larger energy difference between the three-coordinate and four-coordinate states over the two-coordinate to three-coordinate states.
Table 4.
E and ΔE values for gas-phase and solvated model clusters. Energies given in hartrees except where indicated.
| 2-coord | H2O | 3-coord | H3O+ | |
|---|---|---|---|---|
| EGas | −3394.453 | −76.444 | −3393.791 | −76.718 |
| ESolvent ε=80 | −3394.524 | −76.460 | −3394.050 | −76.858 |
|
| ||||
| 0.0759 | 47.7 kcal/mol | |||
| 0.389 | 244. kcal/mol | |||
|
| ||||
| 3coord | H2O | 4coord | H3O+ | |
|
| ||||
| EGas | −3393.791 | −76.444 | −3392.992 | −76.718 |
| ESolvent ε=80 | −3394.050 | −76.460 | −3393.551 | −76.858 |
|
| ||||
| 0.102 | 63.8 kcal/mol | |||
| 0.525 | 330. kcal/mol | |||
Figure 3.

Relative energies for the species involved in the isodesmic reactions of increasing Cu(I) coordination by addition of methyltiolate residues. Protons donated by methylthiols are released to solvent at an infinite distance as they are bound by the metal ion.
The QM calculations do not take into account the effect of the protein environment on Cu(I) transport. Moreover, the symmetrical nature of the QM clusters excludes the possibility of creating different two-coordinate or three-coordinate states. In the protein, there are many different possible two-coordinate and three-coordinate states that can be investigated with molecular mechanics methods. To perform such calculations required the creation of parameters for the Cu(I) ion and its ligating residues.
Table 5 lists the geometrical parameters about the Cu center for the three QM model clusters. Bond lengths and bond angles generated by geometry optimization of each cluster were collected for inclusion in the MM force field. Force constants for Cu-S bonds and the electrostatic potentials, based on the optimized geometries of the clusters, were also collected for inclusion into the new set of Cu(I) MM parameters. It can be seen that Cu-S distances for unbound methylthiols are more than 4.98 Å from the metal ion, and that Cu-S bond lengths are on the order of 2.2 Å – 2.3 Å. The bond angles also conform to experimental results. The 1800 S-Cu-S bond angle in the two-coordinate model agrees with the NMR structure of Atx1, and bond angles in the four-coordinate model reveal near-tetrahedral geometry, which agrees with the X-ray structure of HAH1. There is no NMR or X-ray evidence of a three-coordinate state, although such a state has been suggested by EXAFS data of Atx1(42) and that the Cu environment would be nearly trigonal planar, which the QM model supports as well.
Table 5.
Geometry parameters of the DFT-optimized multi-coordinate models.
| Model | Cu-S (Å) | Cu-S | Cu-S | Cu-S | S-Cu-S (deg) | C-S-Cu |
|---|---|---|---|---|---|---|
| 2-coord | 2.23 | 2.23 | 5.25 | 6.00 | 180.0 | 101.8 |
| 3-coord | 2.31 | 2.35 | 2.41 | 4.98 | 114.6 | 105.2 |
| 4-coord | 2.19 | 2.19 | 2.19 | 2.24 | 109.5 | 108.9 |
The four-coordinate HAH1 system was the first to be simulated using the newly-created parameters shown in Tables 1 through 3. Table 6 compares Cu-S bond lengths and S-Cu-S bond angles for the four-coordinate QM model cluster, the four-coordinate simulated system, and the active site geometry from the HAH1 crystal structure. The Table shows that the Cu-S bond lengths are shorter in the gas-phase QM model than in either the crystal structure or the solvent-phase MD-simulated system, and that the geometrical parameters for the simulated protein adequately match those of the crystal structure. Results from the analyses of the trajectories of all five MD-simulated structures are shown in Table 7.
Table 6.
Comparison of the HAH1 active site between the four-coordinate model, the solvated, equilibrated protein, and the X-ray crystal structure of the Cu(I)-bound protein.
| Parameter | QM Model | Protein | X-ray |
|---|---|---|---|
| Cu-S (Cys 12A) | 2.19 Å | 2.29 Å | 2.30 Å |
| Cu-S (Cys 15A) | 2.19 Å | 2.14 Å | 2.39 Å |
| Cu-S (Cys 12B) | 2.19 Å | 2.33 Å | 2.30 Å |
| Cu-S (Cys 15B) | 2.24 Å | 2.39 Å | 2.32 Å |
| Cys12A-Cu-Cys15A | 109.0 deg | 117.5 deg | 115.7 deg |
| Cys12A-Cu-Cys12B | 109.5 deg | 112.5 deg | 109.4 deg |
Table 7.
Summary of rms deviation, rms flexibility, and radius of gyration for the five solvated HAH1 protein models and key active site residues.
| C12 B bound | C15 B bound | ||||
|---|---|---|---|---|---|
| 2-coord | 2-coord-bridge | 3-coord B | 3-coord A | 4-coord | |
| RMSD (Å) | |||||
| Total | 2.69 | 2.67 | 2.27 | 2.10 | 2.04 |
| Backbone | 1.89 | 1.88 | 1.30 | 1.34 | 1.17 |
| Binding loop | 0.89 | 1.29 | 0.87 | 0.94 | 1.31 |
| Bind. loop | |||||
| backbone | 0.34 | 0.71 | 0.37 | 0.38 | 0.54 |
| Radgyr (Å) | |||||
| Protein avg. | 29.38 | 27.26 | 29.35 | 29.30 | 29.43 |
| RMSF (Å) | |||||
|
| |||||
| Total | 4.98 | 4.85 | 3.60 | 4.54 | 5.46 |
| Backbone | 4.80 | 4.70 | 3.46 | 4.37 | 5.23 |
| Cu | 5.11 | 2.66 | 2.19 | 4.19 | 3.94 |
| Cys 12A | 5.28 | 3.83 | 2.40 | 4.95 | 5.08 |
| Cys 15A | 4.10 | 2.66 | 1.78 | 4.38 | 3.45 |
| Cys 12B | 6.14 | 3.62 | 3.44 | 3.64 | 4.68 |
| Cys 15B | 4.32 | 2.15 | 1.88 | 2.60 | 3.21 |
Four rmsd values are listed in Table 7. There are two values that encompass the entire protein and two values that focus only on the Cu-binding loop of HAH1. It can be seen that all-atom rmsd values are below 2.70 Å for all five systems and that the geometries of the binding loops of each system are indistinguishable from that of the crystal structure as binding loop rmsd values are all close to 1.00 Å. These rmsd values showcase the ability of the new parameters to replicate the geometry of the crystal structure of the whole protein and, more importantly, that of the metal binding site.
The rmsf values listed in Table 7 reveal details about the flexibility of certain residues as well as the complete protein and the protein backbone for the three models. The small difference between rmsf values for the complete protein compared to the backbone suggests that the flexibility of the protein is not limited to the side chains and that the backbone also moves freely. From the rmsf data for the Cu(I)-binding residues Cys 12A, Cys 15A, Cys 12B, and Cys 15B, it appears that Cys 12A and Cys 12B have comparable magnitudes in each model. The values for Cys 15A and Cys 15B are also similar for each model. The similarity is derived from the location of these residues on the binding loop. Cys 12A and Cys 12B are more solvent-exposed and move more freely due to solvent interactions and being further away from the monomer interface. On the other hand, Cys 15A and Cys 15B show less flexibility as they are close to the interface region and not generally solvent-exposed. The flexibility of the Cys 12 residues may play a role in Cu(I) transfer between binding domains. The rmsf data for Cu(I) show that Cu(I) is least mobile when bound by only three residues. In the four-coordinate model, Cu(I) and its ligands are more flexible. We hypothesize that this is observed because the four-coordinate Cu(I) complex is more strained than the three-coordinated system. In other words, the structure is pre-organized to bind, or is designed to stabilize, the three-coordinate state over the four-coordinated one. Evidence for this is seen in the rmsf data for each of the four Cu-binding residues in the 4-coordinate model. Cys 12A and Cys 12B, which are close to the protein surface, are seen to be more flexible than Cys 15A and B which are further from solvent in the monomer-monomer interface. Moreover, the overall protein rmsf is greatest for the four-coordinated state.
Figures 3 and 4 display the rmsd plots of the five simulated systems. The Figures show that the active site regions of all five models apparently reached an equilibrium state after about 400 ps. And while the entire protein is generally in good agreement with the crystal structure throughout, the complete protein structure did take longer to reach an equilibrium state. For the whole-protein models, rmsd values of between 2.0 Å and 2.5 Å were achieved by 2,500 ps and were maintained beyond that point in the simulations.
Figure 4.

Whole-protein rmsd plots for the five solvated protein systems. Plots include all-atom rmsd values and backbone atom only values.
One interesting observation from the simulations of the solvated protein systems was the behavior of the Cu-binding residues in the 2-coord-bridged, 3-coord A, and 3-coord B systems. Cys side chains have the ability to form hydrogen bonding interactions with the solvent around the protein. The Cu-binding site of the HAH1 monomer exists on a solvent-exposed loop. In the HAH1 dimer, the protein-protein interface prohibits Cys 15 A and Cys 15 B from interacting with the solvent. However, Cys 12 A and Cys 12 B are still able to contact the solvent. Figure 5 shows the orientation of the Cys residues in 3-coord A and 3-coord B after 3,600 ps of MD simulation. It can be seen that when Cys 15 B is bound to Cu along with the two Cys from monomer A, then Cys 12 B is left to interact with the solvent. However, when Cys 12 B is bound to Cu along with the two Cys from the monomer, then Cys 15 B is not able to form any favorable solvent contacts. Rather, the unbound Cys 15 B is buried deeper in the monomer-monomer interface. Similarly, in the 2-coord-bridged system, where both Cys 15 A and Cys 15 B are Cu-bound, Cys 12 A and Cys 12 B freely interact with the solvent. We believe that favorable Cys-solvent interactions may have a role in determining the manner in which the target domain binds to the incident Cu ion during transfer.
Figure 5.

Considering only the binding motifs of the two monomers (Met10 – Cys15), all-atom and backbone-only rmsd plots are shown for the five solvated protein systems.
To determine the average proximity to solvent of the Cys side chains in the Cu(I) binding site, protein-solvent radial distribution functions were calculated. Figure 7 shows g(r) plots for the five multi-coordinate HAH1 MD simulations. Each plot represents a series of snapshots between 1,500 and 3,000 ps during the MD simulation. The plot for the four-coordinate structure shows that the Cys 15B side chain is farther from solvent than any of the other three Cu-bound residues, with Cys 12B maintaining the highest population of solvent molecules within 4 Å from the terminal S atom. The plots for the 3-coord A and 3-coord B models are much different in the region from 0–10 Å. In the 3-coord A structure, Cys 15B is unbound from Cu, yet as the g(r) plot shows, water is unable to penetrate into the protein to hydrogen bond with 15B even when compared to the three Cu-ligating residues including the buried residue15A. Conversely, the plot for the 3-coord B protein reveals that when the surface exposed Cys 12B is unbound from Cu, its side chain comes in (not unexpectedly) close contact with solvent. This is represented by a broad peak in the g(r) plot from 3-6Å from the S atom on Cys 12B and a peak between 1.5 Å and 3 Å for the terminal H of that residue. The location and magnitude of the S and H peaks on 3-coord B relative to those on 3-coord A suggest that Cys 12B readily forms favorable solvent contacts when it is not bound to the metal ion, and that the Cys 15B side chain is not capable of such interactions and might be less likely to become unligated with Cu(I). A similar comparison can be made in the two-coordinate radial distribution plot. It can be seen that the magnitude of the Cys 15A S peak is much larger than that of the Cys 15B S peak. This suggests that even when Cys 15B is not bound to Cu, it is still less accessible to solvent than Cys 15A which is complexed to copper. Another feature of the two-coordinate plot is that unbound S atoms produce different curves than the bound S atoms. In the two-coordinate model, only residues from monomer A are bound to Cu. It can be seen that the g(r) plots from the two S atoms on monomer A resemble each other. S atoms from monomer B produce a different set of similar peaks. The curves for the S atoms in the Cu-bound residues (Cys 15A and 15B) in the two-coordinate-bridged g(r) plot closely resemble each other. However, some unexpected dissimilarities exist when the peaks from Cys 12A and Cys 12B are compared. While the peaks from the Cu-bound S atoms maintain their similarity throughout the 0–15 Å range, the peaks for unbound S atoms and their accompanying H atoms are similar in shape, but not in magnitude for distances less than 6Å. S and H from Cys 12B have much higher solvent population with 5 Å than the same atoms from Cys 12A. In fact, H from Cys 12A shows no solvent population with 4 Å. The lower peaks for the H atoms in the two-coordinate-bridge structure could reflect interactions between the hydrogens and the Cu-binding S atoms on the spatially adjacent Cys 15 residues on each monomer, or it could suggest that when the two HAH1 monomers share a Cu bridge in the two-coordinate state, that the coordination unsaturated metal ion attempts to form bonds with the unbound Cys residues nearby.
Figure 7.
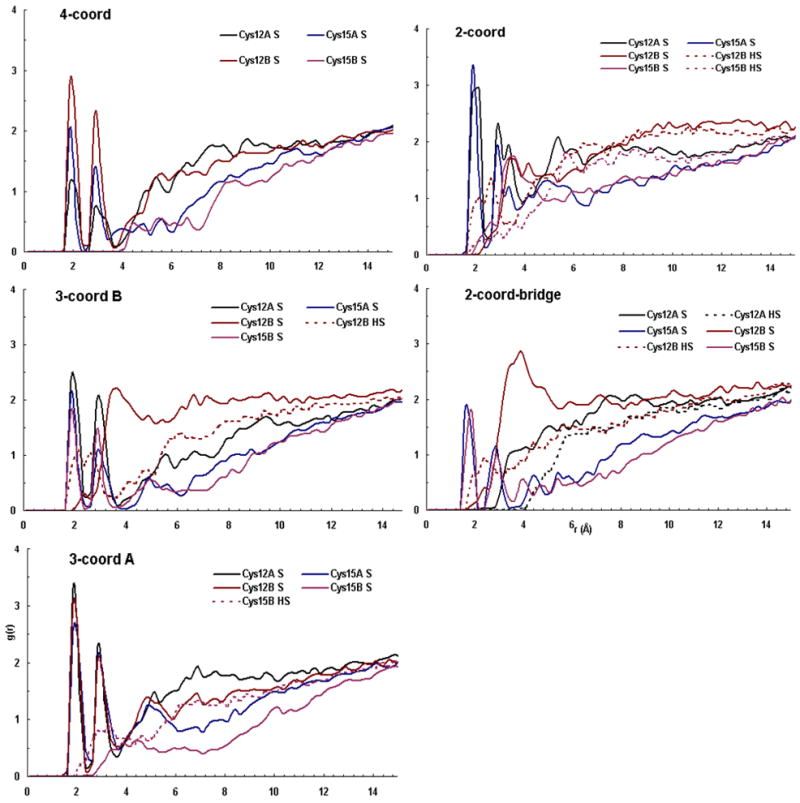
Radial distribution plots for protein-solvent interactions for the five different Cu(I) coordinations of the simulated HAH1 dimer active site. g(r) is plotted from 1500 – 3000 ps for each of the simulation windows above, representing protein-solvent interactions in the equilibrated systems.
Hydrogen bonding networks for Cys 15A and Cys 15B were studied from protein structures at the end of the MD simulations. The side chains Lys 60 of both monomers are arranged such that they interact closely with Cys 15A and Cys 15B at the bottom of the binding site. In the two-coordinate system, the distances between the S of Cys 15A and H from the end of Lys 60A and Lys 60B are 2.03 Å and 2.17 Å, respectively, suggesting strong H-bonding interactions between the two atoms. Likewise, the two-coordinate-bridged structure reveals that S from both Cys 15A and 15B are H-bonded to Lys 60A and Lys 60B. In this case, Lys 60A interacts with both Cys 15A and 15B with S ---H distances of less than 1.9 Å. Lys 60B interacts with Cys 15B at a distance of 1.95 Å. The three-coordinate systems also exhibit H-bonding between these lysines and the cysteines in the binding site. In the 3-coord A system, while Cys 12B is interacting with solvent at the protein surface, Cys 15B is bound to Cu and shares a H-bonding interaction with Lys 60B at 1.92 Å away. Cys 15A is also 1.92 Å from Lys 60B, but is also only 2.06 Å from Lys 60A. Both the hydrogen-bonding network of Cys 15B to lysine and that of Cys 12B to solvent in the 3-coord A model are favorable when compared to the arrangements observed in the 3-coord B model. The 3-coord B model, with Cys 12B bound to Cu and Cys 15B unbound, shows no H-bonding between 15B and any lysine. Furthermore, Cys 12B has no contact with either the solvent or a lysine residue. Cys 15A experiences H-bonding to both Lys 60A and 60B. Hydrogen bonding in the 4-coord structure is most similar to that of the 3-coord A system, with Cys 15A and 15B interacting with Lys 60A and 60B. However, in this case Cys 12B has no solvent interactions as in 3-coord A.
The combination of the solvent interactions shown by Cys 12B with the H-bonding interactions observed with Cys 15A and 15B in the HAH1 dimer model strengthen the suggestion that Cys 17 of the target MNK4 domain will bind Cu first in the transfer mechanism between HAH1 and MNK4.
Conclusion
The QM calculations done in the first part of this work created a foundation for the description of the Cu(I) binding site in HAH1. The main conclusion is the four-coordinate state is very energetically unfavorable and is probably an unlikely candidate for an intermediate along the reaction profile that involves exchanging ligating Cys residues around Cu(I). Moreover, the two to three coordinate transfer involves a large energetic penalty, but we suspect that the protein environment can play a role in decreases this energy difference quite significantly. Future QM/MM studies can better address this observation. The results from the MD simulations of the three Cu(I)-bound HAH1 dimer models show that the QM-derived parameters used to construct the MM force field adequately described the system based on comparisons to the HAH1 crystal structure. The rmsd data show that the computationally generated structures maintain the same fold and Cu(I) structure as the X-ray derived protein structure. After the MD simulations were completed, the question of deciphering the order of Cu(I) binding and release during Cu(I) transfer remains to be answered, but several hypotheses can be derived.
If one assumes that the protein environment cannot stabilize the exchange reaction sufficiently then one can propose a two coordinate only ligand exchange process. This proposal is given in Figure 8. Otherwise, the proposal of Figure 1 appears to be the best hypothesis based on the MD simulations in that the surface exposed Cys residues (12A and 12B) can be best stabilized by solvation effects while they interchange from the thiolate ↔ thiol forms along the reaction pathway and Cys residues (15A and 15B) maintain a stabilizing hydrogen bonding network with nearby Lys residues (60A and 60B). QM/MM studies aimed at refining the overall reaction profile in terms of the stability of two and three coordinates intermediates would aid in discriminating between these two pathways.
Figure 8.

The proposed mechanism for Cu(I) transfer between HAH1 and MNK4 involving only two-coordinated thiolate-Cu(I) species.
Figure 6.
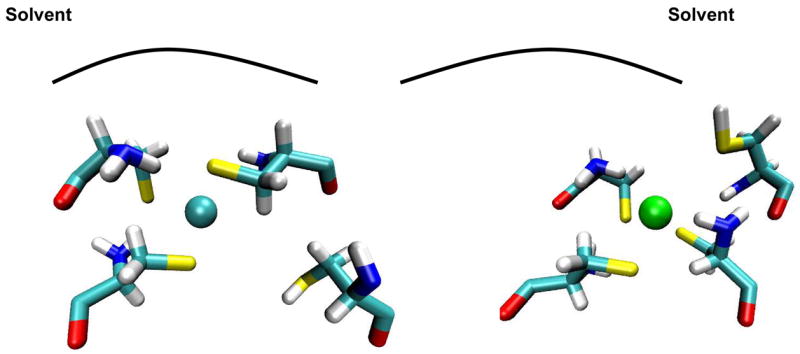
Snapshots of the 3-coord A (left) and 3-coord B solvated protein Cu(I) binding sites after 3,600 ps of simulation at 300 K. The unbound Cys residue in system 3-coord A (Cys 15 B) moves away from the metal ion and away from solvent, while the unbound residue in system 3-coord B (Cys 12 B) also moves away from the metal ion, but toward solvent.
Acknowledgments
We thank Amy Rosenzweig and Mike Weaver for helpful comments and suggestions.
Footnotes
This work was supported by a NIH grant (GM066859) to K.M.M., Jr.
References
- 1.Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O’Halloran TV. Undectectable Free Copper: The Requirement of a Copper Chaperone of Superoxide Dismutase. Science. 1999;284:805–808. doi: 10.1126/science.284.5415.805. [DOI] [PubMed] [Google Scholar]
- 2.O’Halloran TV, Culotta VC. Metallochaperones, an Intracellular Shuttle Service for Metal Ions. J Biol Chem. 2000;275:25057–25060. doi: 10.1074/jbc.R000006200. [DOI] [PubMed] [Google Scholar]
- 3.Gaggelli E, Kozlowski H, Valensin D, Valensin G. Copper Homeostasis and Neurogenerative Disorders (Alzheimer’s, Prion, and Parkinson’s Diseases and Amyotrophic Lateral Sclerosis) Chem Rev. 2006;106:1995–2044. doi: 10.1021/cr040410w. [DOI] [PubMed] [Google Scholar]
- 4.Harrison MD, Jones CE, Solioz M, Dameron CT. Intracellular Copper Routing: The Role of Copper Chaperones. TIBS. 2000;25:29–32. doi: 10.1016/s0968-0004(99)01492-9. [DOI] [PubMed] [Google Scholar]
- 5.Huffman DL, O’Halloran TV. Function, Structure, and Mechanism of Intracellular Copper Trafficking Proteins. Ann Rev Biochem. 2001;70:677–701. doi: 10.1146/annurev.biochem.70.1.677. [DOI] [PubMed] [Google Scholar]
- 6.Lieberman RL, Rosenzweig AC. Comprehensive Coordination Chemistry II: from biology to nanotechnology. 1. 2004. Metal Ion Homeostasis; pp. 195–211. [Google Scholar]
- 7.Puig S, Thiele DJ. Molecular Mechanisms of Copper Uptake and Distribution. Curr Opin Chem Biol. 2002;6:171–180. doi: 10.1016/s1367-5931(02)00298-3. [DOI] [PubMed] [Google Scholar]
- 8.Hamza I, Schaefer M, Klomp LWJ, Gitlin JD. Interaction of the copper chaperone HAH1 with the Wilson disease protein is essential for copper homeostasis. Proc Natl Acad Sci. 1999;96:13363–13368. doi: 10.1073/pnas.96.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mercer JFB, Barnes N, Stevenson J, Strausak D, Llanos RM. Copper-induced trafficking of the Cu-ATPases: A key mechanism for copper homeostasis. BioMetals. 2003;16:175–184. doi: 10.1023/a:1020719016675. [DOI] [PubMed] [Google Scholar]
- 10.Valentine JS, Gralla EB. Delivering Copper Inside Yeast and Human Cells. Science. 1997;278:817–818. doi: 10.1126/science.278.5339.817. [DOI] [PubMed] [Google Scholar]
- 11.Tanchou V, Gas F, Urvoas A, Cougouluègne F, Ruat S, Averseng O, Quéméneur E. Copper-mediated homo-dimerisation for the HAH1 metallochaperone. Biochem Biophys Res Comm. 2004;325:388–394. doi: 10.1016/j.bbrc.2004.10.048. [DOI] [PubMed] [Google Scholar]
- 12.Rosenzweig AC. Copper Delivery by Metallochaperone Proteins. Acc Chem Res. 2001;34:119–128. doi: 10.1021/ar000012p. [DOI] [PubMed] [Google Scholar]
- 13.Wernimont AK, Huffman DL, Lamb AL, O’Halloran TV, Rosenzweig AC. Structural Basis for Copper Transfer by the Metallochaperone for the Menkes/Wilson Disease Proteins. Nat Struct Biol. 2000;7:766–771. doi: 10.1038/78999. [DOI] [PubMed] [Google Scholar]
- 14.Anastassopoulou A, Banci L, Bertini I, Cantini F, Katsari E, Rosato A. Solution Structure of the Apo and Copper(I)-Loaded Human Metallochaperone HAH1. Biochem. 2004;43:13046–13053. doi: 10.1021/bi0487591. [DOI] [PubMed] [Google Scholar]
- 15.Ralle M, Lutsenko S, Blackburn N. X-ray absorption spectroscopy of the copper chaperone HAH1 reveals a linear 2-coordinate Cu(I) center capable of adduct formation with exogenous thiols and phosphines. J Biol Chem. 2003;278:23163–23170. doi: 10.1074/jbc.M303474200. [DOI] [PubMed] [Google Scholar]
- 16.Gitschier J, Moffat B, Reilly D, Wood WI, Fairbrother WJ. Solution structure of the fourth metal-binding domain from the Menkes copper-transporting ATPase. Nat Struct Biol. 1998;5:47–54. doi: 10.1038/nsb0198-47. [DOI] [PubMed] [Google Scholar]
- 17.Arnesano F, Banci L, Bertini I, Huffman DL, O’Halloran TV. Solution Structure of the Cu(I) and Apo Forms of the Yeast Metallochaperone, Atx1. Biochem. 2001;40:1528–1539. doi: 10.1021/bi0014711. [DOI] [PubMed] [Google Scholar]
- 18.Larin D, Mekios C, Das K, Ross B, Yang A, Gilliam TC. Characterization of the Interaction between the Wilson and Menkes Disease Proteins and the Cytoplasmic Copper Chaperone, HAH1. J Biol Chem. 1999;274:28497–28504. doi: 10.1074/jbc.274.40.28497. [DOI] [PubMed] [Google Scholar]
- 19.Arnesano F, Banci L, Bertini I, Bonvin AMJJ. A docking approach to the study of copper trafficking proteins: Interaction between metallochaperones and soluble domains of copper ATPases. Structure. 2004;12:669–676. doi: 10.1016/j.str.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 20.Banci L, Bertini I, Cantini F, Felli IC, Gonnelli L, Hadjiliadis N, Pierattelli R, Rosato A, Voulgaris P. The Atx1-Ccc2 complex is a metal-mediated protein-protein interaction. Nat Chem Biol. 2006;2:367–368. doi: 10.1038/nchembio797. [DOI] [PubMed] [Google Scholar]
- 21.Cobine PA, Winge DR. Visualizing tricoordinate copper transfer. Nat Chem Biol. 2006;2:352–353. doi: 10.1038/nchembio0706-352. [DOI] [PubMed] [Google Scholar]
- 22.Comba P, Remenyi R. A New Molecular Mechanics Force Field for the Oxidized form of Blue Copper Proteins. J Comp Chem. 2002;23:697–705. doi: 10.1002/jcc.10084. [DOI] [PubMed] [Google Scholar]
- 23.Dalosto SD. Computer simulation of the interaction of Cu(I) with Cys residues at the binding site of the yeast metallochaperone Cu(I)-Atx1. J Phys Chem B. 2007;111:2932–2940. doi: 10.1021/jp066689x. [DOI] [PubMed] [Google Scholar]
- 24.Gresh N, Policar C, Giessner-Prettre C. Modeling Cu(I) Complexes: SIBFA Molecular Mechanics versus ab Initio Energetics and Geometrical Arrangements. J Phys Chem A. 2002;106:5660–5670. [Google Scholar]
- 25.Ungar LW, Scherer NF, Voth GA. Classical molecular dynamics simulation of the photoinduced electron transfer dynamics of plastocyanin. Biophys J. 1997;72:5–17. doi: 10.1016/S0006-3495(97)78642-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery JA, Jr, Stratmann RE, Burant JC, Dapprich S, Millam JM, Daniel AD, Kudin KN, Strain MC, Farkas Ö, Tomasi J, Barone V, Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochterski J, Petersson GA, Ayala PY, Cui Q, Morokuma K, Salvador P, Dannenberg JJ, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Cioslowski J, Ortiz JV, Baboul AG, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komáromi I, Gomperts R, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Andres JL, Gonzalez C, Head-Gordon M, Replogle ES, Pople JA. Gaussian 03. C.01. Gaussian, Inc; Pittsburgh, PA: 2003. [Google Scholar]
- 27.Becke AD. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 28.Hertwig RH, Koch W. On the parameterization of the local correlation functional. What is Becke-3-LYP? Chem Phys Lett. 1997;268:345–351. [Google Scholar]
- 29.Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 30.Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields . J Phys Chem. 1994;98:11623–11627. [Google Scholar]
- 31.Gordon MS, Defrees DJ, Pople JA. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J Chem Phys. 1982;77:3654–3665. [Google Scholar]
- 32.Hariharan PC, Pople JA. The influence of polarization functions on molecular orbital hydrogenation energies. Theor Chem Acct. 1973;28:213–222. [Google Scholar]
- 33.Hehre WJ, Ditchfield R, Pople JA. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J Chem Phys. 1972;56:2257–2261. [Google Scholar]
- 34.Olsson MHM, Ryde U. Geometry, Reduction Potential, and Reorganization Energy of the Binuclear CuA Site, Studied by Density Functional Theory. J Am Chem Soc. 2001;123:7866–7876. doi: 10.1021/ja010315u. [DOI] [PubMed] [Google Scholar]
- 35.Molecular Simulations, I. WebLab ViewerPro. (3.7) 2000 [Google Scholar]
- 36.Besler BH, Merz KMJ, Kollman PA. Atomic charges derived from semiempirical methods. J Comp Chem. 1990;11:431–439. [Google Scholar]
- 37.Singh UC, Kollman PA. An approach to computing electrostatic charges for molecules. J Comp Chem. 1984;5:129–145. [Google Scholar]
- 38.Shannon RD. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystall. 1976;A32:751–767. [Google Scholar]
- 39.Case DA, Darden TA, Cheatham TEI, Simmerling CL, Wang J, Duke RE, Luo R, Merz KMJ, Pearlman DA, Crowley M, Walker RC, Zhang W, Wang B, Hayik S, Roitberg AE, Seabra G, Wong KF, Paesani F, Wu X, Brozell S, Tsui V, Gohlke H, Yang L, Tan C, Mongan J, Hornak V, Cui G, Beroza P, Mathews DH, Schafmeister C, Ross WS, Kollman PA. AMBER 9. 9. University of California; San Francisco, San Francisco: 2006. [Google Scholar]
- 40.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A second generation force field. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 41.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 42.Pufahl RA, Singer CP, Peariso KL, Lin S-J, Schmidt PJ, Fahrni CJ, Culotta VC, Penner-Hahn JE, O’Halloran TV. Metal ion chaperone function of the soluble Cu(I) receptor Atx1. Science. 1997;278:853–856. doi: 10.1126/science.278.5339.853. [DOI] [PubMed] [Google Scholar]