

Abstract
Whereas the important role of free radicals in diabetes-associated complications is well established, the contributions of the highly reactive oxidant peroxynitrite have not been properly explored. The present study used a pharmacological approach to evaluate the role of peroxynitrite in peripheral diabetic neuropathy. Control and STZ-diabetic mice were maintained with or without treatment with the potent peroxynitrite decomposition catalyst Fe(III) tetra-mesitylporphyrin octasulfonate (FeTMPS), at doses of 5 or 10 mg/kg/day in the drinking water for 3 weeks after an initial 3 weeks without treatment. Mice with a 6-week duration of diabetes developed clearly manifest motor (MNCV) and sensory nerve conduction velocity (SNCV) deficits, thermal hypoalgesia (paw withdrawal, tail-flick, and hot plate tests), mechanical hypoalgesia (tail pressure Randall-Sellito test), tactile allodynia (flexible von Frey filament test), and ~44% loss of intraepidermal nerve fibers. They also had increased nitrotyrosine and poly(ADP-ribose) immunofluorescence in sciatic nerve, grey matter of the spinal cord, and dorsal root ganglion neurons. FeTMPS treatment alleviated or essentially corrected (at a dose of 10 mg/kg/day) MNCV and SNCV deficits, and was associated with less severe small sensory nerve fiber dysfunction and degeneration. Nitrotyrosine and poly(ADP-ribose) immunofluorescence in sciatic nerve, spinal cord, and dorsal root ganglion neurons in peroxynitrite decomposition catalyst-treated diabetic mice was markedly reduced. In conclusion, peroxynitrite contributes to large motor, large sensory, and small sensory fiber neuropathy in streptozotocin-diabetic mice. The findings provide rationale for development of potent peroxynitrite decomposition catalysts for the treatment of diabetic neuropathy.
Keywords: nitrosative stress, nerve conduction, peripheral diabetic neuropathy, peroxynitrite decomposition catalysts, poly(ADP-ribose) polymerase
Introduction
Diabetic distal symmetric sensorimotor polyneuropathy affects at least 60% of patients with type 1 (insulin-dependent) diabetes in the US, and is the leading cause of foot amputation (1). Although the important role of oxidative stress in type 1 peripheral diabetic neuropathy (PDN) is well established (2–4), the contribution of the nitrosative component of free radical and oxidant-induced injury, and, in particular, of the highly reactive oxidant peroxynitrite (5), to diabetes-associated neuropathic changes has not been properly explored. Recent clinical studies revealed increased plasma nitrotyrosine levels and their correlation with endothelial dysfunction and redistribution of sudomotor responses, an early sign of sympathetic nerve dysfunction, in type 1 diabetic subjects (6,7). Furthermore, plasma peroxynitrite generation assessed by the pholasin chemiluminescence test correlated with the diabetic neuropathy impairment score of the lower limbs (8). Two experimental studies with the peroxynitrite decomposition catalysts, Fe(III)tetrakis-2-(N-triethylene glycol monomethyl ether)pyridyl porphyrin (FP15) and Fe(III)tetrakis-(1-methyl-4-pyridyl)porphyrin penta-chloride (FeTMPyP) (9,10), demonstrated the involvement of peroxynitrite in motor (MNCV) and sensory nerve conduction velocity (SNCV) deficits, and impaired nitrergic innervation, known to contribute to gastroparesis and impotence associated with autonomic neuropathy, in STZ-diabetic mice, and thermal hypoalgesia in non-obese diabetic (NOD) mice. Here, we report the effects of the highly effective peroxynitrite decomposition catalyst Fe(III) tetra-mesitylporphyrin octasulfonate (FeTMPS) (11) on nitrosative stress and poly(ADP-ribose) polymerase activation in peripheral nerve, grey matter (dorsal horn) of the spinal cord, and dorsal root ganglion neurons, and on indices of peripheral nerve function and degeneration in STZ-diabetic mice, a model of type 1 PDN.
Materials and methods
Reagents
Unless otherwise stated, all chemicals were of reagent-grade quality and were purchased from Sigma Chemical Co., St. Louis, MO. Fe(III) tetra-mesitylporphyrin octasulfonate (FeTMPS) was prepared by a slight modification of published procedures (11). The rabbit polyclonal anti-nitrotyrosine (NT) antibody was purchased from Upstate, Lake Placid, NY, and mouse monoclonal anti-poly(ADP-ribose) from Trevigen, Inc., Gaithersburg, MD. Secondary Alexa Fluor 488 goat anti-rabbit and Alexa Fluor 488 goat anti-mouse antibodies as well as Prolong Gold Antifade reagent were purchased from Invitrogen, Eugene, OR. The Avidin/Biotin Blocking kit, the M.O.M. Basic kit, the Standard Vectastain Elite ABC kit, the DAB Substrate kit, and 3,3′-diaminobenzidine were obtained from Vector Laboratories, Burlingame, CA. Rabbit polyclonal anti-protein gene product 9.5 (ubiquitin c-terminal hydrolase) antibody was purchased from Chemicon International, Inc, Temecula, CA. Other reagents for immunohistochemistry were purchased from Dako Laboratories, Inc., Santa Barbara, CA.
Animals
The experiments were performed in accordance with regulations specified by the National Institutes of Health ‘Principles of Laboratory Animal Care, 1985 Revised Version’ and Pennington Biomedical Research Center Protocol for Animal Studies. Mature male C57Bl6/J mice were purchased from Jackson Laboratories (Bar Harbor, ME). They were fed with standard mouse chow (PMI Nutrition International, Brentwood, MO) and had access to water ad libitum. Diabetes was induced by a single injection of STZ, 100 mg/kg/day; i.p., to non-fasted animals. Blood samples for glucose measurements were taken from the tail vein three days after STZ injection and the day before the animals were sacrificed. Mice with blood glucose ≥13.8 mM were considered diabetic. The injected mice that had blood glucose concentrations in the non-diabetic range were given low-dose STZ injections (40 mg/kg/day; i.p.) until they developed hyperglycemia (typically, one to three additional injections). The experimental groups comprised the control and diabetic mice treated with or without the peroxynitrite decomposition catalyst FeTMPS. The agent was administered at two doses of 5 and 10 mg/kg/day in the drinking water for 3 weeks after an initial 3 weeks without treatment. This dose was selected from our previous experiments with approximately equally potent peroxynitrite decomposition catalyst Fe(III)tetrakis-2-(N-triethylene glycol monomethyl ether)pyridyl porphyrin (FP15) in the same animal model (10). At the end of the study, the physiological and behavioral tests were performed in the following order: tactile responses to flexible von Frey filaments (first day), tail pressure Randall-Sellito test (second day), thermal algesia by tail-flick test (third day), thermal algesia by paw withdrawal test (fourth day), thermal algesia by hot plate test (fifth day), SNCV and MNCV (sixth day). Measurements of MNCV and SNCV were performed in mice anaesthetized with a mixture of ketamine and xylazine (45 mg/kg body weight and 15 mg/kg body weight, respectively; i.p.). The C57Bl6/J mice with the 3-week duration of STZ-diabetes displayed MNCV and SNCV deficits (MNCV: 43.1±0.5 vs 51.4±0.6 m/sec in the controls, p<0.01; SNCV: 34.2±0.3 vs 39.3±0.2 m/sec, p<0.01), thermal hypoalgesia (paw withdrawal latencies: 15.56±0.61 vs 10.36±0.48 sec in the controls, p<0.01; tail flick response latencies: 3.27±0.12 vs 2.8±0.08 sec in the controls, p<0.01), mechanical hypoalgesia (withdrawal threshold in the tail pressure Randall-Selitto test: 200±2.5 vs 166±2.5 g in the controls, p<0.01), tactile allodynia (tactile response threshold: 1.35±0.10 vs 2.23±0.26 g in the controls, p<0.05), but no intraepidermal nerve fiber loss (23.2±2.3 nerve fiber profiles per mm vs 24.3±2.8 in the controls).
Anesthesia, euthanasia and tissue sampling
The animals were sedated by CO2 and immediately sacrificed by cervical dislocation. Sciatic nerves, spinal cord, dorsal root ganglia (DRG), and foot pads were fixed in normal buffered 4% formalin for assessment of nitrotyrosine and poly(ADP-ribose) by immunofluorescent histochemistry and intra-epidermal nerve fiber density by conventional immunohistochemistry. Poly(ADP-ribose) abundance is a measure of PARP activity (12–14).
Physiological tests
Sciatic MNCV and hind-limb digital SNCV were measured as described elsewhere (10,15). In all measurements, body temperature was monitored by a rectal probe and maintained at 37°C with a warming pad. Hind-limb skin temperature was also monitored by a thermistor, and maintained between 36 and 38°C by radiant heat.
Behavioral tests
Tactile responses and mechanical algesia
Tactile responses were evaluated by quantifying the withdrawal threshold of the hindpaw in response to stimulation with flexible von Frey filaments as previously described (16). Tail pressure thresholds were registered with the Paw/Tail Pressure Analgesia meter for the Randall-Selitto test (37215-Analgesy-Meter, UGO-Basile, Comerio VA, Italy). Pressure increasing at a linear rate of 10 g with the cut-off of 250 g to avoid tissue injury, was applied to the base of the tail. The applied tail pressure that evoked biting or licking behavior was registered by an analgesia meter and expressed in g. Three tests separated by at least 15 min were performed for each animal, and the mean value of these tests was calculated.
Thermal algesia
The paw withdrawal latency in response to radiant heat (15% intensity which produced a heating rate of ~1.3°C per sec, cut-off time 30 sec) was determined as we described (16,17) using the IITC model 336 TG combination tail-flick and paw algesia meter (IITC Life Science) with a floor temperature ~32–33°C (manufacturer’s set-up). For assessment of tail flick response latencies, the device was set at 40% heating intensity (heating rate ~ 2.5°C per sec) with a cut-off at 10 sec. In the hot plate test (IITC Model 39 Hot Plate Analgesia Meter, IITC Life Science) the unit had a plate preset temperature of 55°C. In all three tests, at least three readings per animal were taken at 15-min intervals, and the average was calculated.
Immunohistochemical studies
All sections were processed by a single investigator and evaluated blindly. Low-power observations of skin sections stained for PGP 9.5 were conducted using a Zeiss Axioskop microscope. Color images were captured with a Zeiss AxioCam HRc CCD camera at 1300×1030 resolution. Low-power images were generated with a ×40 acroplan objective using the automatic capturing feature of the Zeiss Axiovision software (ver. 3.1.2.1). Low-power observations of sciatic nerve, grey matter of the spinal cord (dorsal horn) and DRG sections stained for NT and poly(ADP-ribose) were performed using a Zeiss Axioplan 2 imaging microscope. Color images were captured with a Photometric CoolSnap™ HQ CCD camera at 1392×1040 resolution. Low-power images were generated with a ×40 acroplan objective using the RS Image™ 1.9.2 software.
NT immunoreactivity in sciatic nerve, spinal cord, and DRG neurons
NT immunoreactivities in sciatic nerve, grey matter (dorsal horn) of the spinal cord, and dorsal root ganglia were assessed by immunofluorescent histochemistry. In brief, sections were deparaffinized in xylene, hydrated in decreasing concentrations of ethanol and washed in water. For immunofluorescent histochemistry, rabbit polyclonal anti-NT antibody was used in a working dilution 1:400. Primary antibody was omitted in the negative controls. Secondary Alexa Fluor 488 goat anti-rabbit antibody was applied in a working dilution 1:200. Sections were mounted in Prolong Gold Antifade reagent. The intensity of fluorescence was quantified using the Image J 1.32 software (National Institutes of Health, Bethesda, MD) and expressed as the mean ± SEM for each experimental group.
Poly(ADP-ribose) immunoreactivity
Poly(ADP-ribose) immunoreactivity was assessed as described (12,15) with minor modifications. In brief, sections were deparaffinized in xylene, hydrated in decreasing concentrations of ethanol and washed in water. Non-specific binding was blocked with the mouse IG blocking reagent supplied with the Vector M.O.M. Basic Immunodetection kit. Then mouse monoclonal anti-poly(ADP-ribose) antibody was diluted 1:100 in 1% BSA in TBS, and applied overnight at 4°C in the humidity chamber. Primary antibody was omitted in the negative controls. Secondary Alexa Fluor 488 goat anti-mouse antibody was diluted 1:200 in TBS and applied for 2 h at room temperature. Sections were mounted in Prolong Gold Antifade reagent. At least ten fields of each section were examined to select one representative image. Representative images were microphotographed, and the number of poly(ADP-ribose)-positive nuclei was calculated for each microphotograph.
Intraepidermal nerve fiber density (INFD)
INFD was assessed as described (17). Three randomly chosen 5-μm sections from each mouse were deparaffinized in xylene, hydrated in decreasing concentrations of ethanol, and washed in water. Non-specific binding was blocked by 10% goat serum containing 1% BSA in TBS (Dako, Carpinteria, CA) for 2 h, and the Avidin/Biotin Blocking kit, according to the manufacturer’s instructions. Then, rabbit polyclonal anti-protein gene product 9.5 (ubiquitin c-terminal hydrolase) antibody was applied in 1:2000 dilution. Secondary bio-tinylated goat anti-rabbit IgG (H+L) antibody was applied in 1:400 dilution, and the staining was performed with the Standard Vectastain Elite ABC kit. For visualization of specific binding sites, the DAB Substrate kit containing 3,3-diaminobenzidine was used. Sections were counterstained with Gill’s hematoxylin, dehydrated and mounted in Micromount mounting medium (Surgipath Medical Ind., Richmond, IL). Intraepidermal nerve fiber profiles were counted in a blinded manner by three independent investigators, under an Olympus BX-41 microscope, and the average values were used. Microphotographs of stained sections were taken on Axioscop 2 microscope (Zeiss) at ×4 magnification, and the length of epidermis was assessed with the ImagePro 3.0 program (Media Cybernetics). An average of 2.8±0.3 mm of the sample length was investigated to calculate a number of nerve fiber profiles per mm of epidermis.
Statistical analysis
The results were expressed as the mean ± SEM. Data were subjected to the equality of variance F test, and then to log transformation, if necessary, before one-way analysis of variance. Where overall significance (p<0.05) was attained, individual between group comparisons were made using the Student-Newman-Keuls multiple range test. Significance was defined at p≤0.05. When between-group variance differences could not be normalized by log transformation (datasets for body weights and plasma glucose), the data were analyzed by the nonparametric Kruskal-Wallis one-way analysis of variance, followed by the Bonferroni/Dunn test for multiple comparisons.
Results
Whereas initial body weights were similar in the control and diabetic mice, final body weights were 14% lower in the diabetic group (p<0.01, Table I). Initial (after STZ injection) blood glucose concentrations were 82% higher in diabetic mice compared with the controls. Hyperglycemia progressed with the prolongation of diabetes, and the difference between final blood glucose concentrations in the two groups exceeded 4-fold. FeTMPS, at either 5 or 10 mg/kg/day, did not affect weight gain or blood glucose concentrations in the control or diabetic mice.
Table I.
Initial and final body weights and blood glucose concentrations in control mice and diabetic mice with and without the peroxynitrite decomposition catalyst FeTMPS treatment.
| Body weight (g)
|
Blood glucose (mmol/l)
|
|||
|---|---|---|---|---|
| Initial | Final | Initial | Final | |
| Control | 20.6±0.40 | 29.0±0.95 | 7.90±0.17 | 8.00±0.20 |
| Control + FeTMPS (5 mg/kg/day) | 20.3±0.50 | 27.0±0.70 | 7.70±0.33 | 7.90±0.12 |
| Control + FeTMPS (10 mg/kg/day) | 20.7±0.47 | 28.0±0.90 | 8.35±0.27 | 8.05±0.24 |
| Diabetic | 20.2±0.33 | 24.9±0.53a | 14.40±0.70a | 33.00±0.20a |
| Diabetic + FeTMPS (5 mg/kg/day) | 20.9±0.33 | 25.0±0.40a | 15.30±0.70a | 32.40±0.50a |
| Diabetic + FeTMPS (10 mg/kg/day) | 20.6±0.60 | 24.6±0.80a | 14.20±0.90a | 32.60±0.40a |
Data are the means ± SEM, n=8–14 per group.
Significantly different from the controls (p<0.01)
Sciatic MNCV was ~20% lower in diabetic mice compared with the controls (Fig. 1A). FeTMPS dose-dependently reversed diabetes-induced MNCV deficit, with normal MNCV achieved after treatment with the 10 mg/kg/day dose. In a similar fashion, hind-limb digital SNCV was reduced by 16% in diabetic mice compared with the controls (Fig. 1B). Both doses of FeTMPS alleviated (p<0.01 vs untreated diabetic group), but did not normalize (p<0.01 and p<0.05 vs the control group for diabetic mice treated with 5 and 10 mg/kg/day FeTMPS, respectively) sensory nerve conduction slowing in diabetic mice. A peroxynitrite decomposition catalyst treatment did not affect MNCV or SNCV in the control mice.
Figure 1.
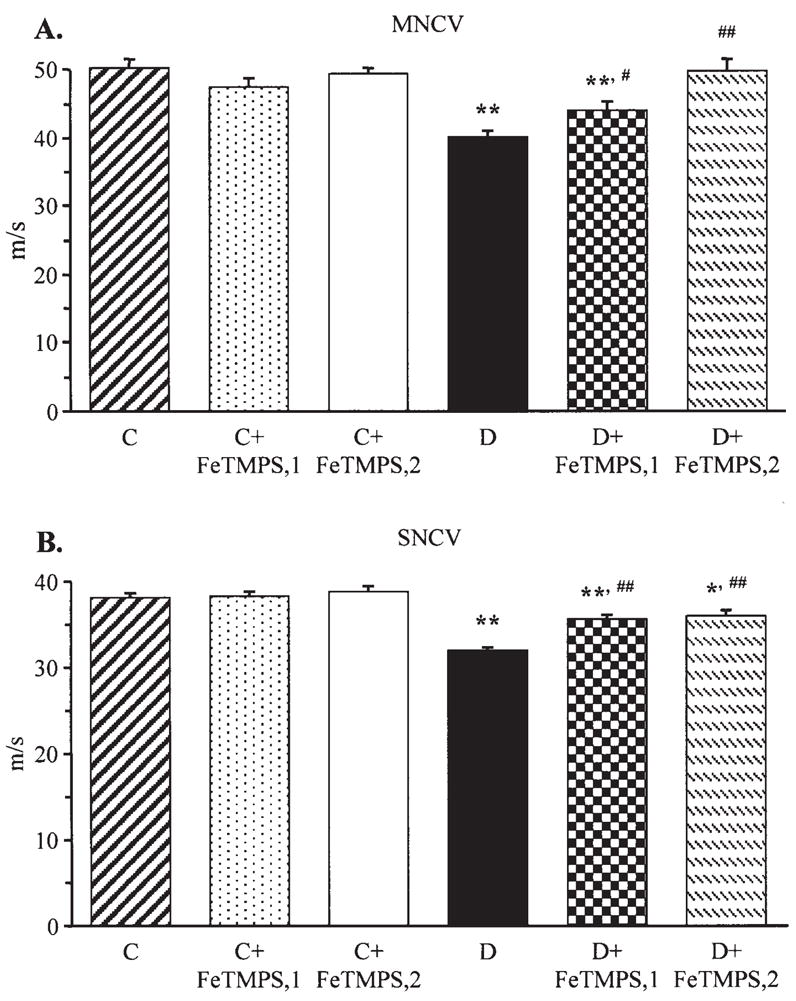
Final sciatic motor nerve conduction velocities (MNCV) (A) and hind-limb digital sensory nerve conduction velocities (SNCV) (B) in the control and diabetic mice with and without the peroxynitrite decomposition catalyst FeTMPS treatment. Mean ± SEM, n=8–14 per group. C, control mice; D, diabetic mice. FeTMPS,1 and FeTMPS,2 administered at 5 and 10 mg/kg/day, respectively. *p<0.05 and **p<0.01 vs the non-diabetic control mice; #p<0.05 and ##p<0.01 vs untreated diabetic mice.
The latencies of hind-paw withdrawal in response to radiant heat were increased by 103% in diabetic mice compared with the control group (p<0.01), consistent with clearly manifest thermal hypoalgesia (Fig. 2A). This is in agreement with the results of the tail-flick and hot plate tests which also revealed increased thermal response latencies in the diabetic group (Fig. 2B and C). All three tests detected virtually identical responses to thermal noxious stimuli in untreated and peroxynitrite decomposition catalyst-treated non-diabetic mice. FeTMPS at either 5 or 10 mg/kg/day alleviated, but did not completely prevent, diabetes- associated thermal hypoalgesia registered by the paw withdrawal and tail-flick tests. In the hot plate test, thermal response latencies in diabetic mice treated with FeTMPS, 5 mg/kg/day, did not differ significantly from those in the untreated diabetic group. In diabetic mice treated with FeTMPS, 10 mg/kg/day, the hot plate test response latencies were significantly reduced compared with those in the untreated diabetic group (p<0.05).
Figure 2.
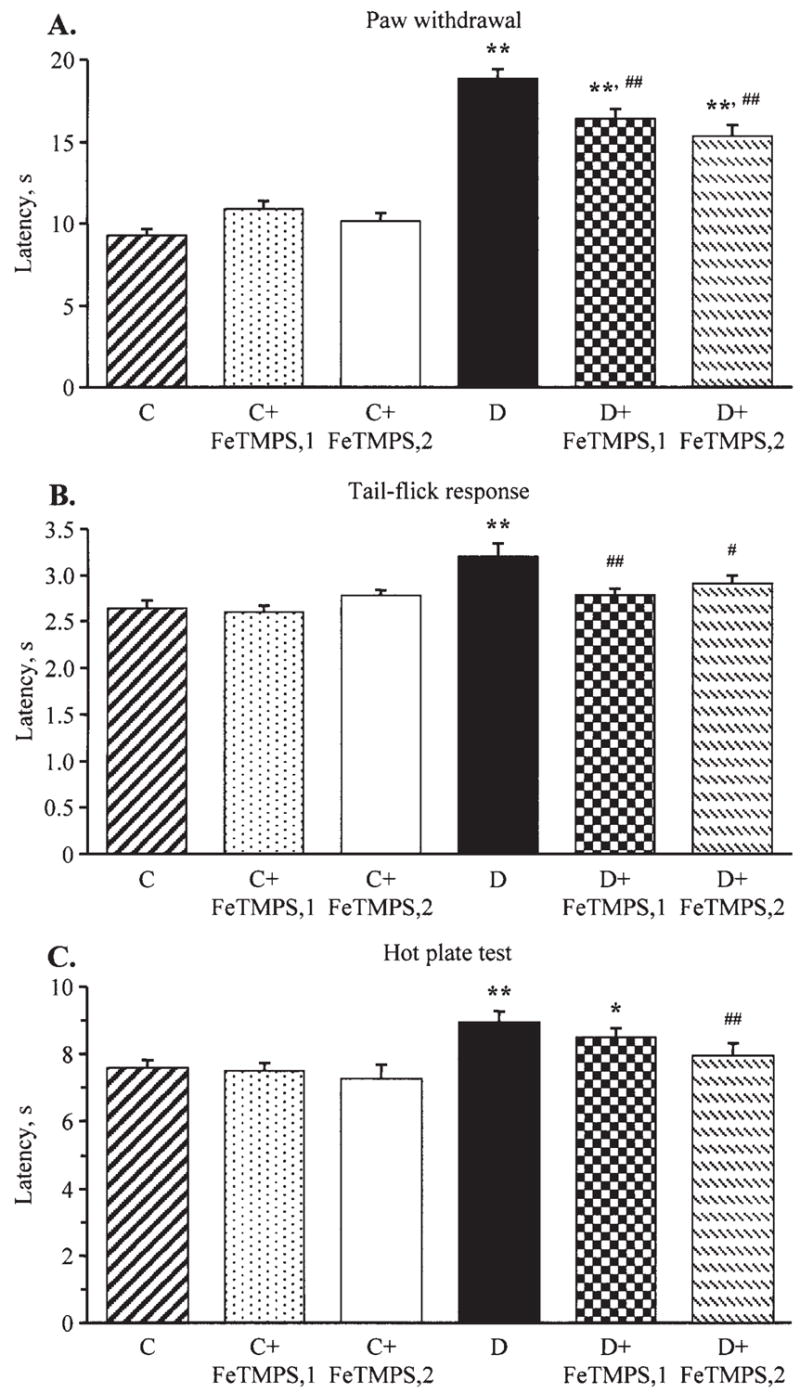
Paw withdrawal latencies in response to radiant heat (A), tail-flick test response latencies (B), and hot plate test response latencies (C) in the control and diabetic mice with or without the peroxynitrite decomposition catalyst FeTMPS treatment. Mean ± SEM, n=8–14 per group. C, control mice; D, diabetic mice. FeTMPS,1 and FeTMPS,2 administered at 5 and 10 mg/kg/day, respectively. *p<0.05 and **p<0.01 vs the non-diabetic control mice; #p<0.05 and ##p<0.01 vs untreated diabetic mice.
Diabetic mice with the 6-week duration of STZ-diabetes also had mechanical hypoalgesia detected with the tail pressure Randall-Sellito test (Fig. 3A). The tail pressure threshold was increased by 19% in diabetic mice compared with the control group. Neither of the doses of FeTMPS affected the tail pressure thresholds in the control mice. The tail pressure thresholds in diabetic mice treated with FeTMPS, 5 mg/kg/day, did not differ significantly from those in the untreated diabetic group. In diabetic mice treated with FeTMPS, 10 mg/kg/day, the tail pressure thresholds were significantly reduced compared with those in the untreated diabetic group (p<0.05). Another sensory abnormality developing in diabetic mice was tactile allodynia. The tactile withdrawal threshold in response to light touch with flexible von Frey filaments was reduced by 69% in diabetic mice compared with the controls (p<0.01). FeTMPS, 5 mg/kg/day, tended to increase the tactile withdrawal threshold in diabetic mice, but the difference with the corresponding untreated group did not achieve statistical significance (p<0.01 vs the controls and p=0.17 vs the untreated diabetic group). FeTMPS, 10 mg/kg/day, increased the tactile response thresholds by 35% in diabetic mice compared with the untreated diabetic group (p<0.05); however, they still remained 59% lower than in the non-diabetic control group (p<0.01). A peroxynitrite decomposition catalyst treatment did not affect this variable in the control mice.
Figure 3.
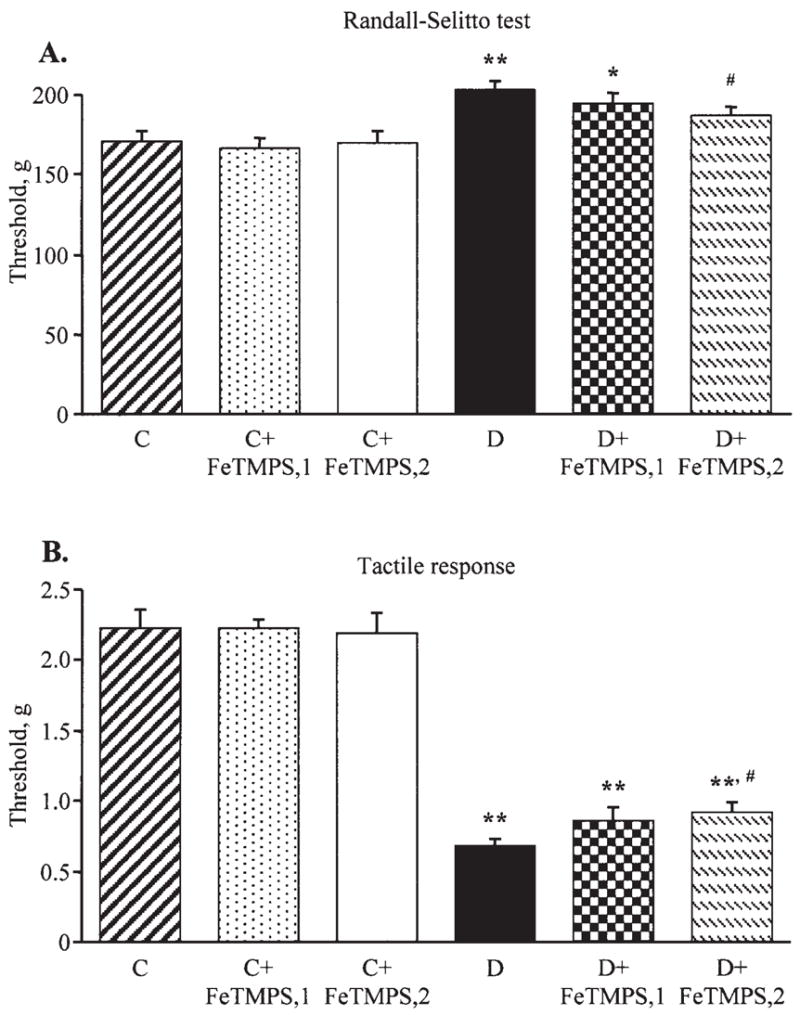
Mechanical withdrawal thresholds in the tail-pressure Randall-Sellito test (A) and tactile response thresholds in response to stimulation with flexible von Frey filaments (B) in the control and diabetic mice with or without the peroxynitrite decomposition catalyst FeTMPS treatment. Mean ± SEM, n=8–14 per group. C, control mice; D, diabetic mice. FeTMPS,1 and FeTMPS,2 administered at 5 and 10 mg/kg/day, respectively. *p<0.05 and **p<0.01 vs the control mice; #p<0.05 vs untreated diabetic mice.
INFD was reduced by 44% in diabetic mice compared with the controls (p<0.01, Fig. 4A and B). INFDs in the diabetic mice treated with 5 and 10 mg/kg/day FeTMPS were similar to those in the non-diabetic control group. Neither of the two doses of FeTMPS affected INFD in the control mice.
Figure 4.
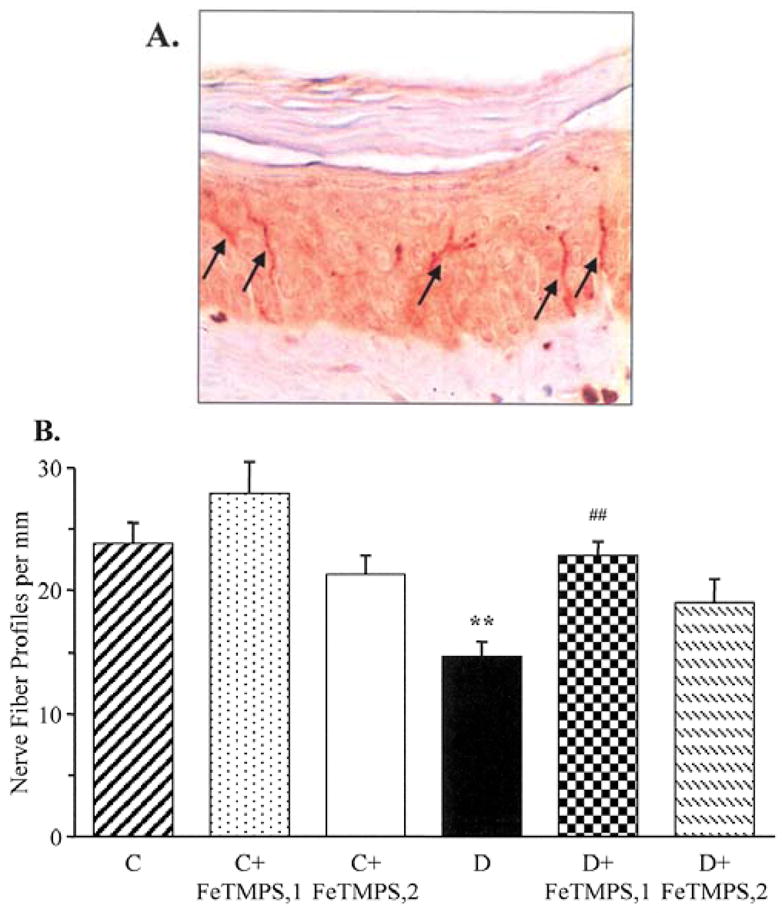
Intraepidermal nerve fiber profiles in the control and diabetic mice with and without the peroxynitrite decomposition catalyst FeTMPS treatment. (A) Representative image of intraepidermal nerve fiber profiles, magnification ×200; and (B) skin fiber density. Mean ± SEM, n=8–14 per group. C, control mice; D, diabetic mice. FeTMPS,1 and FeTMPS,2 administered at 5 and 10 mg kg−1 day−1, respectively. **p<0.01 vs the control mice; ##p<0.01 vs untreated diabetic mice.
NT immunofluorescence was increased by 49% in the sciatic nerve of diabetic mice compared with the control group (Fig. 5A and B). Neither 5 nor 10 mg/kg/day doses of FeTMPS affected NT immunofluorescence in the control mice. Both doses of FeTMPS reduced diabetes-associated increase in NT immunofluorescence (p<0.01 and p<0.05 vs the untreated diabetic group, for the diabetic mice treated with 5 and 10 mg/kg/day FeTMPS, respectively; p<0.05 vs the non-diabetic controls for the diabetic mice treated with 10 mg/kg/day FeTMPS). The number of sciatic nerve poly(ADP-ribose)-positive nuclei was increased by 105% in diabetic mice compared with the controls (p<0.01, Fig. 5C and D). Neither of the two doses of FeTMPS affected poly(ADP-ribose) immunofluorescence in the control mice. Both doses reduced (p<0.05 vs the untreated diabetic group for both), but did not normalize (p<0.05 vs the non-diabetic controls for both), the numbers of sciatic nerve poly(ADP-ribose)-positive nuclei in the diabetic mice.
Figure 5.
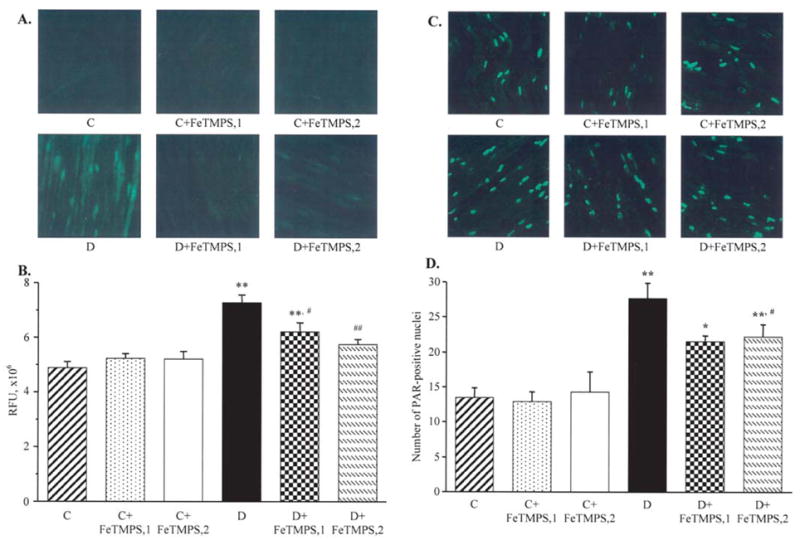
Representative microphotographs of immunofluorescent staining of nitrotyrosine (A) and poly(ADP-ribose) (C) in sciatic nerves of the control and diabetic mice with and without the peroxynitrite decomposition catalyst FeTMPS treatment. Magnification, ×40. Scores of nitrotyrosine immunofluorescence (B) and the numbers of poly(ADP-ribose)-positive nuclei (D) in sciatic nerves of the control and diabetic mice with and without the peroxynitrite decomposition catalyst FeTMPS treatment. Mean ± SEM, n=8–14 per group. C, control mice; D, diabetic mice. FeTMPS,1 and FeTMPS,2 administered at 5 and 10 mg/kg/day, respectively. *p<0.05 and **p<0.01 vs the non-diabetic control mice; #p<0.05 and ##p<0.01 vs the untreated diabetic mice.
Similar patterns were observed in the dorsal horn of the spinal cord (Fig. 6) and DRG (Fig. 7). In the spinal cord, NT immunofluorescence and the number of poly(ADP-ribose)-positive nuclei were increased by 191 and 157%, respectively, in diabetic mice compared with the control group (p<0.01 for both comparisons). Peroxynitrite decomposition catalyst treatment did not affect spinal cord NT and poly(ADP-ribose) immunofluorescence in the control mice. FeTMPS dose-dependently reduced NT and poly(ADP-ribose) immunofluorescence in the diabetic mice (p<0.01 vs the untreated diabetic group for all comparisons).
Figure 6.
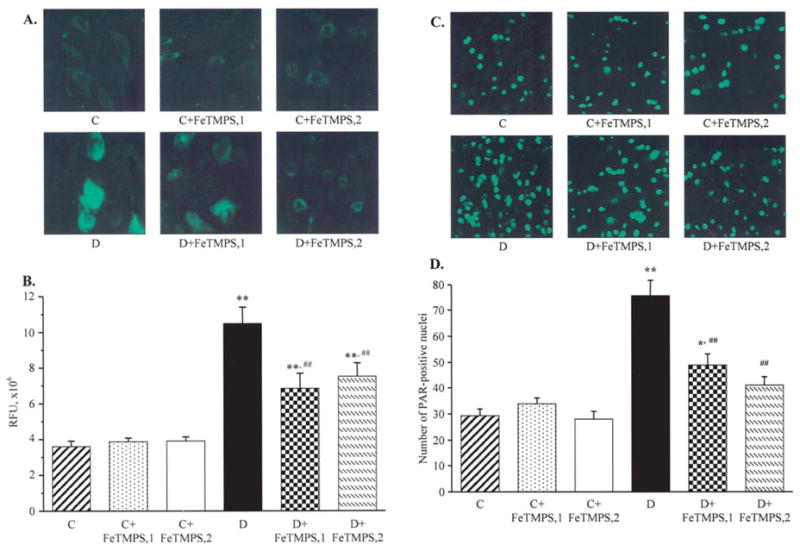
Representative microphotographs of immunofluorescent staining of nitrotyrosine (A) and poly(ADP-ribose) (C) in the spinal cord of the control and diabetic mice with and without the peroxynitrite decomposition catalyst FeTMPS treatment. Magnification, ×40. Scores of nitrotyrosine immunofluorescence (B) and the numbers of poly(ADP-ribose)-positive nuclei (D) in the spinal cord of the control and diabetic mice with and without peroxynitrite decomposition catalyst FeTMPS treatment.. Mean ± SEM, n=8–14 per group. C, control mice; D, diabetic mice. FeTMPS,1 and FeTMPS,2 administered at 5 and 10 mg/kg/day, respectively. *p<0.05 and **p<0.01 vs the non-diabetic control mice; ##p<0.01 vs untreated diabetic mice.
Figure 7.
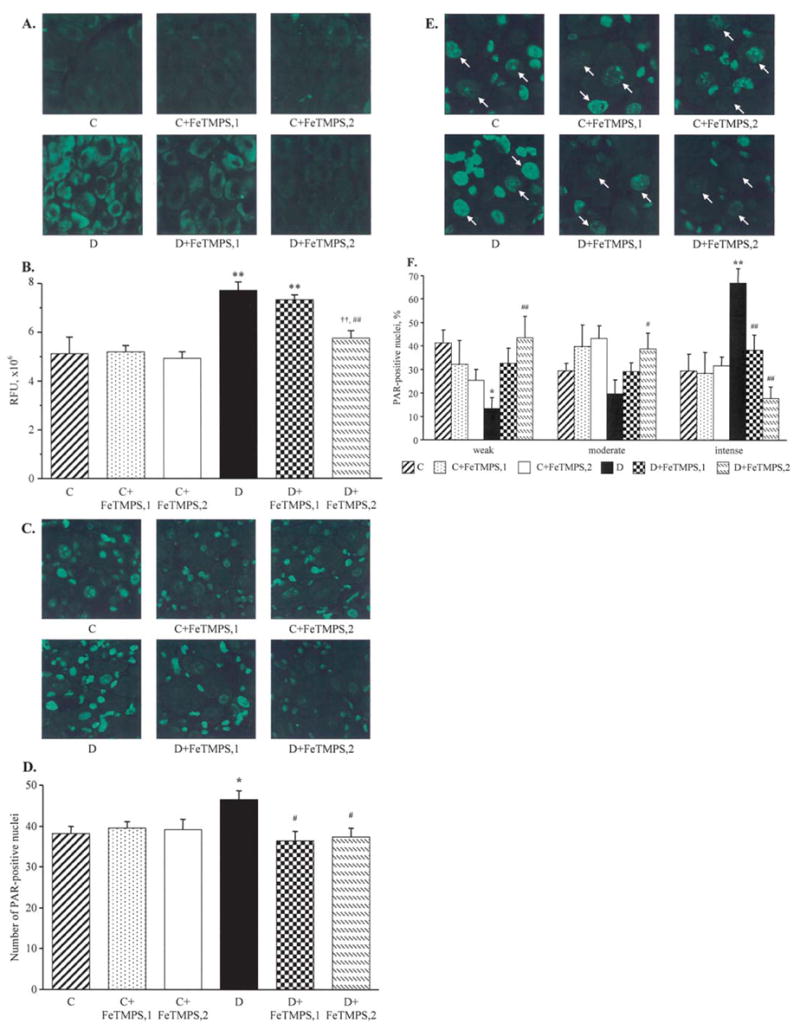
Representative microphotographs of immunofluorescent staining of nitrotyrosine (A) and poly(ADP-ribose) (C) in the dorsal root ganglia, and poly(ADP-ribose) in the dorsal root ganglion neurons (E) of the control and diabetic mice with and without the peroxynitrite decomposition catalyst FeTMPS treatment. Magnification, ×40. Scores of nitrotyrosine immunofluorescence in the dorsal root ganglion neurons (B), counts of dorsal root ganglion poly(ADP-ribose)-positive nuclei (D), and percentage of dorsal root ganglion neurons with weak, moderate, and intense poly(ADP-ribose) immunofluorescence (F), in the experimental groups. The number of dorsal root ganglion neurons with weak, moderate and intense poly(ADP-ribose) immunofluorescence was expressed as the percentage of neurons with identifiable poly(ADP-ribose) immunofluorescence in the dorsal root ganglia of the control and diabetic mice with and without the peroxynitrite decomposition catalyst FeTMPS treatment. Mean ± SEM, n=8–12 per group. C, control mice; D, diabetic mice. FeTMPS,1 and FeTMPS,2 administered at 5 and 10/mg/kg/day, respectively. *p<0.05 and **p<0.01 vs the non-diabetic control mice; #p<0.05 and ##p<0.01 vs untreated diabetic mice.
NT immunofluorescence in DRG neurons was increased by 51% in diabetic mice compared with the control group (p<0.01, Fig. 7A and B). The total number of DRG poly (ADP-ribose)-positive nuclei was 22% higher in the diabetic than in the control mice (p<0.05), with poly(ADP-ribose) immunofluorescence localized primarily in satellite cells (Fig. 7C and D). The percentage of DRG neurons with weak and moderate poly(ADP-ribose) immunofluorescence was lower, and the percentage of DRG neurons with intense immunofluorescence was higher in diabetic mice compared with the control group (Fig. 7F). A peroxynitrite decomposition catalyst treatment did not result in significant changes in neuronal NT immunofluorescence and the total number of DRG poly(ADP-ribose)-positive nuclei in the control mice, but dose-dependently reduced these two variables in the diabetic mice. The percentage of DRG neurons with intense poly(ADP-ribose) immunofluorescence was reduced and the percentage of those with weak and moderate immunofluorescence was increased in FeTMPS-treated diabetic mice compared with the corresponding untreated group. Again, the effect was dose-dependent. Both doses of a peroxynitrite decomposition catalyst did not cause statistically significant changes in the percent distribution of DRG neurons with weak, moderate and intense poly(ADP-ribose) immunofluorescence in the control mice.
Discussion
Our findings indicate that nitrosative stress is an early and characteristic feature of PDN in experimental type 1 diabetes. Accumulation of NT, a footprint of peroxynitrite and other RNS-induced injury (5,18), has been observed in peripheral nerve, dorsal horn of the spinal cord, and DRG neurons in STZ-diabetic mice. Note that in contrast with short-lived free radicals, peroxynitrite is relatively stable and can migrate from one cell type to another (5). Therefore, it is reasonable to suggest that diabetes-induced oxidative-nitrosative stress is present in all cell types of the peripheral nervous system. Indeed, high glucose- and diabetes-induced oxidative-nitrosative stress has been documented in Schwann cells of peripheral nerve (19), endothelial cells of vasa nervorum (4,19), and DRG neurons (20,21). We previously reported PARP activation manifest by poly(ADP-ribose) accumulation in endothelial and Schwann cells of peripheral nerve in STZ-diabetic rats (15). The present study (Fig. 7) revealed that PARP activation was also present in DRG neurons and satellite cells of STZ-diabetic mice, and was corrected by a peroxynitrite decomposition catalyst treatment.
Our findings are in agreement with other studies (22–24) demonstrating suitability of the STZ-diabetic C57Bl6/J mouse for studying type 1 PDN. C57Bl6/J mice with a 6-week duration of STZ-diabetes displayed clearly manifest MNCV and SNCV deficits, consistent with other reports (22,24). Nerve conduction slowing has been documented in rodents with both short-term (2–4,10,15,22,24) and long-term (25) STZ-diabetes and other models of type 1 PDN (26). MNCV and SNCV deficits in diabetic rats and mice have been reported to be prevented or reversed by numerous pharmacological agents including, but not limited by, inhibitors of aldose reductase, non-enzymatic glycation, protein kinase C, angiotensin-converting enzyme, p38 MAPK, PARP, NAD(P)H oxidase, cyclooxygenase-2, α1-adrenoceptor antagonists, β2-adrenoceptor agonists, antioxidants and metal chelators (27,28). At this point, it is unclear whether such amenability to correction by various pharmaceutical agents points to equal importance of multiple pathogenetic mechanisms or a single, yet unidentified, common mechanism where the effects of apparently unrelated agents converge. Our findings with FP15 (10) and now FeTMPS suggest that peroxynitrite decomposition catalysts should be added to the list of agents counteracting MNCV and SNCV deficits in experimental type 1 PDN.
Assessment of indices of sensory neuropathy in diabetic animal models have resulted in contradictory findings. Thermal hyperalgesia (3,16), hypoalgesia (29), and the unchanged response to thermal noxious stimuli (23) have been reported in STZ-diabetic rats and mice. Note that thermal hyperalgesia is sometimes observed in human subjects with mild PDN (30), whereas the advanced stage of the disease is characterized by increased thermal perception thresholds (hypoalgesia) that progress to sensory loss, occurring in conjunction with degeneration of all types of peripheral nerve fibers. Thus, studies of the mechanisms underlying both thermal hyperalgesia and hypoalgesia are clinically relevant. In the present study, mice with a 6-week duration of STZ-diabetes had thermal hypoalgesia, consistent with our previous findings in the non-obese diabetic (NOD) mouse model (10). A peroxynitrite decomposition catalyst treatment reversed diabetes-induced increase in the paw withdrawal and tail-flick test response latencies which implicates nitrosative stress in thermal hypoalgesia of type 1 PDN.
In contrast to STZ-diabetic rats that display mechanical hyperalgesia (3,16,31), STZ-diabetic mice have increased mechanical withdrawal thresholds i.e., the condition consistent with sensory loss in human subjects with advanced PDN. Our findings suggest that nitrosative stress contributes to the development of mechanical hypoalgesia associated with experimental type 1 PDN.
Painful diabetic neuropathy in human subjects is sometimes complicated by tactile allodynia, a condition where light touch is perceived as painful (32). Tactile allodynia was observed in STZ-diabetic rats that displayed reduced tactile withdrawal thresholds in the flexible von Frey filament test (16). The data in STZ-diabetic mice are contradictory, and, in contrast to our findings, increased rather than reduced tactile response thresholds have been identified by others (23,33). In the present study, tactile response thresholds in the peroxynitrite decomposition catalyst-treated STZ-diabetic mice were slightly higher than in the untreated group, but still much lower than in the non-diabetic controls. Note that in contrast to STZ-diabetic wild-type mice (129S1/SvImJ background), diabetic PARP−/− mice did not develop tactile allodynia (29). Furthermore, a more effective (than with FeTMPS) prevention of diabetes-induced tactile allodynia was achieved with the PARP inhibitor 10-(4-methyl-piperazin-1-ylmethyl)-2H-7-oxa-1,2-diazabenzo[de] anthracen-3-one (GPI-15427) treatment that essentially normalized PARP activity in the peripheral nerve and spinal cord (29). Note that only partial correction of poly(ADP-ribose) immunofluorescence in both tissues was achieved with either 5 or 10 mg/kg/day of FeTMPS. Higher doses of the peroxynitrite decomposition catalyst are likely needed for a complete normalization of PARP activity and prevention of tactile allodynia in STZ-diabetic mice.
The recent development of a technique for the assessment of small sensory nerve fiber degeneration i.e., quantitation of epidermal nerve fiber density (34), stimulated studies of this phenomenon in animal models of PDN and human diabetic subjects (17,23,33,35,36). Note that the skin of human diabetic subjects produces significant amounts of nitric oxide, a peroxynitrite precursor (37). Furthermore, our unpublished data showed that nitrotyrosine content (ELISA) was ~2-fold higher in the skin of rats with a 12-week duration of STZ-diabetes than in the non-diabetic group, consistent with findings of increased cutaneous nitrotyrosine immuno-reactivity in diabetic patients (38). In the present study, mice with a 6-week duration of STZ-diabetes displayed ~44% epidermal nerve fiber loss [consistent with other reports (23,33)] which was alleviated by both doses of FeTMPS. These findings are in agreement with another study from our group (29), in which STZ-diabetic PARP+/+ mice with a 10-week duration of STZ-diabetes displayed ~53% intraepidermal nerve fiber loss, whereas diabetic PARP−/− mice preserved normal nerve fiber density. Furthermore, diabetes-induced intraepidermal nerve fiber loss in rats with a 12-week duration of STZ-diabetes was prevented by the PARP inhibitor GPI-15427. All these findings suggest that peroxynitrite contributes to small sensory nerve fiber degeneration in type 1 PDN via PARP activation. PARP activation results in profound metabolic abnormalities, changes in transcriptional regulation and gene expression, altered signal transduction, inflammatory response, translocation of apoptosis-inducing factor from the mitochondria to the nucleus, and cell death signaling (12–14,39,40). Further studies are needed to elucidate which of these, or newly discovered, mechanisms are responsible for diabetes-associated small sensory nerve fiber degeneration. The role of tyrosine nitration in PDN remains unclear. Studies with the tyrosine nitration inhibitor epicatechin are in progress in our laboratory.
Evaluation of FeTMPS on PDN in leptin-deficient (ob/ob) mice, a model of type 2 diabetic neuropathy, resulted in divergent findings (41). Whereas a peroxynitrite decomposition catalyst treatment alleviated diabetes-induced sensory nerve dysfunction, it had no effect on intraepidermal nerve fiber loss. At this point, it is unclear whether these findings suggest that higher doses of a peroxynitrite decomposition catalyst are needed to prevent small sensory nerve fiber degeneration in ob/ob mice, or are indicative of different roles for nitrosative stress in type 1 and type 2 PDN. Note that substantial differences in the pathogenetic mechanisms underlying type 1 and type 2 PDN have previously been reported by others (42).
In conclusion, peroxynitrite plays an important role in MNCV and SNCV deficits, sensory disorders, and small sensory nerve fiber degeneration in streptozotocin-diabetic mice. A peroxynitrite decomposition catalyst, at the doses of 5–10 mg/kg/day which are ~100- to 500-fold lower than the effective doses of conventional antioxidants, corrected nerve conduction slowing, and partially prevented small sensory nerve dysfunction and degeneration. It remains to be established whether this class of pharmacological agents can reverse advanced sensory neuropathy, and, in particular, restore normal epidermal nerve fiber density by promoting small sensory nerve fiber regeneration. The results provide the rationale for development of peroxynitrite decomposition catalysts for treatment of PDN. Furthermore, assessment of correlations between nitrotyrosine levels in easily accessible biological materials i.e., serum (plasma), peripheral blood monocytes and skin, and indices of nerve function and morphology in human diabetic subjects can lead to identification of new biomarkers of PDN with diagnostic and prognostic values.
Acknowledgments
This study was supported by the American Diabetes Association Research Grant 7-05-RA-102, the Juvenile Diabetes Research Foundation International Grant 1-2005-223, the National Institutes of Health Grant DK 071566-01 (all to I.G.O.), and the Intramural Research Program of the National Institutes of Health/National Institute of Alcohol Abuse and Alcoholism (to P.P.). FeTMPS synthesis was supported by the National Institutes of Health Grant GM 36298 and the New Jersey Commission on Science and Technology (both to JTG). The authors thank Nazar Mashtalir and Jeho Shin for their expert technical assistance.
References
- 1.Boulton AJ, Vinik AI, Arezzo JC, Bril V, Feldman EL, Freeman R, Malik RA, Maser RE, Sosenko JM, Ziegler D American Diabetes Association. Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes Care. 2005;28:956–962. doi: 10.2337/diacare.28.4.956. [DOI] [PubMed] [Google Scholar]
- 2.Nagamatsu M, Nickander KK, Schmelzer JD, Raya A, Wittrock DA, Tritschler H, Low PA. Lipoic acid improves nerve blood flow, reduces oxidative stress, and improves distal nerve conduction in experimental diabetic neuropathy. Diabetes Care. 1995;18:1160–1167. doi: 10.2337/diacare.18.8.1160. [DOI] [PubMed] [Google Scholar]
- 3.Cameron NE, Tuck Z, McCabe L, Cotter MA. Effect of the hydroxyl radical scavenger, dimethylthiourea, on peripheral nerve tissue perfusion, conduction velocity and nociception in experimental diabetes. Diabetologia. 2001;44:1161–1169. doi: 10.1007/s001250100626. [DOI] [PubMed] [Google Scholar]
- 4.Coppey LJ, Gellett JS, Davidson EP, Dunlap JA, Lund DD, Yorek MA. Effect of antioxidant treatment of streptozotocin-induced diabetic rats on endoneurial blood flow, motor nerve conduction velocity, and vascular reactivity of epineurial arterioles of the sciatic nerve. Diabetes. 2001;50:1927–1937. doi: 10.2337/diabetes.50.8.1927. [DOI] [PubMed] [Google Scholar]
- 5.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxy-nitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoeldtke RD. Nitrosative stress in early Type 1 diabetes. David H.P Streeten Memorial Lecture. Clin Auton Res. 2003;13:406–421. doi: 10.1007/s10286-003-0139-x. [DOI] [PubMed] [Google Scholar]
- 7.Ceriello A, Piconi L, Esposito K, Giugliano D. Telmisartan shows an equivalent effect of vitamin C in further improving endothelial dysfunction after glycemia normalization in type 1 diabetes. Diabetes Care. 2007;30:1694–1698. doi: 10.2337/dc07-0318. [DOI] [PubMed] [Google Scholar]
- 8.Ziegler D, Sohr CG, Nourooz-Zadeh J. Oxidative stress and antioxidant defense in relation to the severity of diabetic polyneuropathy and cardiovascular autonomic neuropathy. Diabetes Care. 2004;27:2178–2183. doi: 10.2337/diacare.27.9.2178. [DOI] [PubMed] [Google Scholar]
- 9.Nangle MR, Cotter MA, Cameron NE. Effects of the peroxynitrite decomposition catalyst, FeTMPyP, on function of corpus cavernosum from diabetic mice. Eur J Pharmacol. 2004;502:143–148. doi: 10.1016/j.ejphar.2004.08.033. [DOI] [PubMed] [Google Scholar]
- 10.Obrosova IG, Mabley JG, Zsengeller Z, Charniauskaya T, Abatan OI, Groves JT, Szabo C. Role for nitrosative stress in diabetic neuropathy: evidence from studies with a peroxy-nitrite decomposition catalyst. FASEB J. 2005;19:401–403. doi: 10.1096/fj.04-1913fje. [DOI] [PubMed] [Google Scholar]
- 11.Shimanovich R, Groves JT. Mechanisms of peroxynitrite decomposition catalyzed by FeTMPS, a bioactive sulfonated iron porphyrin. Arch Biochem Biophys. 2001;387:307–317. doi: 10.1006/abbi.2000.2247. [DOI] [PubMed] [Google Scholar]
- 12.Garcia Soriano F, Virag L, Jagtap P, Szabo E, Mabley JG, Liaudet L, Marton A, Hoyt DG, Murthy KG, Salzman AL, Southan GJ, Szabo C. Diabetic endothelial dysfunction: the role of poly(ADP-ribose) polymerase activation. Nat Med. 2001;7:108–113. doi: 10.1038/83241. [DOI] [PubMed] [Google Scholar]
- 13.Jagtap P, Szabo C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov. 2005;4:421–440. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]
- 14.Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M, Klaus JA, Otsuka T, Zhang Z, Koehler RC, Hurn PD, Poirier GG, Dawson VL, Dawson TM. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci USA. 2006;103:18308–18313. doi: 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Obrosova IG, Li F, Abatan OI, Forsell MA, Komjati K, Pacher P, Szabo C, Stevens MJ. Role of poly(ADP-ribose) polymerase activation in diabetic neuropathy. Diabetes. 2004;53:711–720. doi: 10.2337/diabetes.53.3.711. [DOI] [PubMed] [Google Scholar]
- 16.Ilnytska O, Lyzogubov VV, Stevens MJ, Drel VR, Mashtalir N, Pacher P, Yorek MA, Obrosova IG. Poly(ADP-ribose) polymerase inhibition alleviates experimental diabetic sensory neuropathy. Diabetes. 2006;55:1686–1694. doi: 10.2337/db06-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drel VR, Mashtalir N, Ilnytska O, Shin J, Li F, Lyzogubov VV, Obrosova IG. The leptin-deficient (ob/ob) mouse: a new animal model of peripheral neuropathy of type 2 diabetes and obesity. Diabetes. 2006;55:3335–3343. doi: 10.2337/db06-0885. [DOI] [PubMed] [Google Scholar]
- 18.Levrand S, Vannay-Bouchiche C, Pesse B, Pacher P, Feihl F, Waeber B, Liaudet L. Peroxynitrite is a major trigger of cardiomyocyte apoptosis in vitro and in vivo. Free Radic Biol Med. 2006;41:886–895. doi: 10.1016/j.freeradbiomed.2006.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Obrosova IG, Pacher P, Szabo C, Zsengeller Z, Hirooka H, Stevens MJ, Yorek MA. Aldose reductase inhibition counteracts oxidative-nitrosative stress and poly(ADP-ribose) polymerase activation in tissue sites for diabetes complications. Diabetes. 2005;54:234–242. doi: 10.2337/diabetes.54.1.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng C, Zochodne DW. Sensory neurons with activated caspase-3 survive long-term experimental diabetes. Diabetes. 2003;52:2363–2371. doi: 10.2337/diabetes.52.9.2363. [DOI] [PubMed] [Google Scholar]
- 21.Schmeichel AM, Schmelzer JD, Low PA. Oxidative injury and apoptosis of dorsal root ganglion neurons in chronic experimental diabetic neuropathy. Diabetes. 2003;52:165–171. doi: 10.2337/diabetes.52.1.165. [DOI] [PubMed] [Google Scholar]
- 22.Yagihashi S, Yamagishi SI, Wada Ri R, Baba M, Hohman TC, Yabe-Nishimura C, Kokai Y. Neuropathy in diabetic mice overexpressing human aldose reductase and effects of aldose reductase inhibitor. Brain. 2001;124:2448–2458. doi: 10.1093/brain/124.12.2448. [DOI] [PubMed] [Google Scholar]
- 23.Christianson JA, Riekhof JT, Wright DE. Restorative effects of neurotrophin treatment on diabetes-induced cutaneous axon loss in mice. Exp Neurol. 2003;179:188–199. doi: 10.1016/s0014-4886(02)00017-1. [DOI] [PubMed] [Google Scholar]
- 24.Ho EC, Lam KS, Chen YS, Yip JC, Arvindakshan M, Yamagishi S, Yagihashi S, Oates PJ, Ellery CA, Chung SS, Chung SK. Aldose reductase-deficient mice are protected from delayed motor nerve conduction velocity, increased c-Jun NH2-terminal kinase activation, depletion of reduced glutathione, increased superoxide accumulation, and DNA damage. Diabetes. 2006;55:1946–1953. doi: 10.2337/db05-1497. [DOI] [PubMed] [Google Scholar]
- 25.Kato N, Mizuno K, Makino M, Suzuki T, Yagihashi S. Effects of 15-month aldose reductase inhibition with fidarestat on the experimental diabetic neuropathy in rats. Diabetes Res Clin Pract. 2000;50:77–85. doi: 10.1016/s0168-8227(00)00164-9. [DOI] [PubMed] [Google Scholar]
- 26.Stevens MJ, Zhang W, Li F, Sima AA. C-peptide corrects endoneurial blood flow but not oxidative stress in type 1 BB/Wor rats. Am J Physiol Endocrinol Metab. 2004;287:E497–E505. doi: 10.1152/ajpendo.00048.2004. [DOI] [PubMed] [Google Scholar]
- 27.Cameron NE, Eaton SE, Cotter MA, Tesfaye S. Vascular factors and metabolic interactions in the pathogenesis of diabetic neuropathy. Diabetologia. 2001;44:1973–1988. doi: 10.1007/s001250100001. [DOI] [PubMed] [Google Scholar]
- 28.Obrosova IG. Update on the pathogenesis of diabetic neuropathy. Curr Diabetes Rep. 2003;3:439–445. doi: 10.1007/s11892-003-0005-1. [DOI] [PubMed] [Google Scholar]
- 29.Obrosova IG, Lyzogubov V, Ilnytska O, Mashtalir N, Vareniuk I, Pavlov I, Xu W, Zhang J, Drel VR. PARP inhibition or gene deficiency counteract intraepidermal nerve fiber loss and neuropathic pain associated with Type 1 diabetes. Diabetes. 2007;56(suppl 1):A1. doi: 10.1016/j.freeradbiomed.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dyck PJ, Dyck PJ, Larson TS, O’Brien PC, Velosa JA. Patterns of quantitative sensation testing of hypoesthesia and hyperalgesia are predictive of diabetic polyneuropathy: a study of three cohorts. Nerve growth factor study group. Diabetes Care. 2000;23:510–517. doi: 10.2337/diacare.23.4.510. [DOI] [PubMed] [Google Scholar]
- 31.Stevens MJ, Li F, Drel VR, Abatan OI, Kim H, Burnett D, Larkin D, Obrosova IG. Nicotinamide reverses neurological and neurovascular deficits in streptozotocin diabetic rats. J Pharmacol Exp Ther. 2007;320:458–464. doi: 10.1124/jpet.106.109702. [DOI] [PubMed] [Google Scholar]
- 32.Vinik AI, Suwanwalaikorn S, Stansberry KB, Holland MT, McNitt PM, Colen LE. Quantitative measurement of cutaneous perception in diabetic neuropathy. Muscle Nerve. 1995;18:574–584. doi: 10.1002/mus.880180603. [DOI] [PubMed] [Google Scholar]
- 33.Christianson JA, Ryals JM, Johnson MS, Dobrowsky RT, Wright DE. Neurotrophic modulation of myelinated cutaneous innervation and mechanical sensory loss in diabetic mice. Neuroscience. 2007;145:303–313. doi: 10.1016/j.neuroscience.2006.11.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sumner CJ, Sheth S, Griffin JW, Cornblath DR, Polydefkis M. The spectrum of neuropathy in diabetes and impaired glucose tolerance. Neurology. 2003;60:108–111. doi: 10.1212/wnl.60.1.108. [DOI] [PubMed] [Google Scholar]
- 35.Pittenger G, Burkus N, McNulty P, Basta B, Vinik A. Intra-epidermal nerve fibers are indicators of small fiber neuropathy in both diabetic and non-diabetic patients. Diabetes Care. 2004;27:1974–1979. doi: 10.2337/diacare.27.8.1974. [DOI] [PubMed] [Google Scholar]
- 36.Toth C, Brussee V, Zochodne DW. Remote neurotrophic support of epidermal nerve fibres in experimental diabetes. Diabetologia. 2006;49:1081–1088. doi: 10.1007/s00125-006-0169-8. [DOI] [PubMed] [Google Scholar]
- 37.Vinik AI, Ullal J, Parson HK, Barlow PM, Casellini CM. Pioglitazone treatment improves nitrosative stress in type 2 diabetes. Diabetes Care. 2006;29:869–876. doi: 10.2337/diacare.29.04.06.dc05-0517. [DOI] [PubMed] [Google Scholar]
- 38.Szabo C, Zanchi A, Komjati K, Pacher P, Krolewski AS, Quist WC, LoGerfo FW, Horton ES, Veves A. Poly(ADP-ribose) polymerase is activated in subjects at risk of developing type 2 diabetes and is associated with impaired vascular reactivity. Circulation. 2002;106:2680–2686. doi: 10.1161/01.cir.0000038365.78031.9c. [DOI] [PubMed] [Google Scholar]
- 39.Ha HC, Hester LD, Snyder SH. Poly(ADP-ribose) polymerase-1 dependence of stress-induced transcription factors and associated gene expression in glia. Proc Natl Acad Sci USA. 2002;99:3270–3275. doi: 10.1073/pnas.052712399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- 41.Vareniuk I, Pavlov IA, Drel VR, Lyzogubov VV, Ilnytska O, Bell SR, Tibrewala J, Groves JT, Obrosova IG. Nitrosative stress and peripheral diabetic neuropathy in leptin-deficient (ob/ob) mice. Exp Neurol. 2007;205:425–436. doi: 10.1016/j.expneurol.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 42.Sima AA, Kamiya H. Diabetic neuropathy differs in type 1 and type 2 diabetes. Ann NY Acad Sci. 2006;1084:235–249. doi: 10.1196/annals.1372.004. [DOI] [PubMed] [Google Scholar]