

Abstract
The genetic basis of bipolar disorder has long been thought to be complex, with the potential involvement of multiple genes, but methods to analyze populations with respect to this complexity have only recently become available. We have carried out a genome-wide association study of bipolar disorder by genotyping over 550,000 SNPs in two independent case-control samples of European origin. The initial association screen was performed using pooled DNA; selected SNPs were confirmed by individual genotyping. While DNA pooling reduces power to detect genetic associations, there is a substantial cost savings and gain in efficiency. A total of 88 SNPs representing 80 different genes met the prior criteria for replication in both samples. Effect sizes were modest: no single SNP of large effect was detected. Of 37 SNPs selected for individual genotyping, the strongest association signal was detected at a marker within the first intron of DGKH (p = 1.5 × 10−8, experiment-wide p<0.01, OR= 1.59). This gene encodes diacylglycerol kinase eta, a key protein in the lithium-sensitive phosphatidyl inositol pathway. This first genome-wide association study of bipolar disorder shows that several genes, each of modest effect, reproducibly influence disease risk. Bipolar disorder may be a polygenic disease.
Keywords: mania, DAG, polygenic, lithium, DFNB31, whirlin, Wnt, pooling, HapMap
Introduction
Genome-wide association studies are a relatively new approach to the genetics of complex disease. By use of dense maps of polymorphic markers (SNPs) and high-throughput genotyping methods, it is now possible to test essentially every gene and most inter-genic regions for evidence of association with disease. This approach offers a powerful alternative to genetic linkage studies, which are often underpowered to detect genes contributing to complex phenotypes (1) and to candidate gene association studies, which are biased by the choice of genes included and may not reveal novel pathways to disease (2). In view of the advantages of genome-wide association methods, major efforts are now underway to detect genes contributing to common disease. The largest such studies are the Genetic Association Information Network (GAIN) and the Wellcome Trust Case Control Consortium, both of which are designed to examine a variety of disorders.
Disadvantages of genome-wide association studies include the large number of hypotheses tested and the cost, which can be substantial. Because of the multiple-testing problem, genes that confer modest risk of disease may be overshadowed by false positives (3, 4). Large-scale replication studies are one solution, but cost considerations typically allow testing of only a subset of markers in the replication sample. A complete genome-wide association study in each of two or more independent samples would be expected to reveal the most reproducible signals, even if the effect sizes were not large, but the cost might be prohibitive.
DNA pooling offers a solution to this dilemma. By combining many individual DNA samples in pools, it is possible to perform genome-wide association studies at a fraction of the cost of individual genotyping. Modern laboratory methods of precise sample aliquotting and DNA quantification, along with robust genotyping platforms and novel, validated statistical methods, allow for remarkably accurate estimates of allele frequency differences in cases and controls (5-7). Confirmation by individual genotyping assures that positive results do not reflect error introduced by the pooling procedures. DNA pooling studies allow for the efficient screening of multiple independent samples within the same experiment, increasing the likelihood that the results obtained reflect important risk alleles, rather than idiosyncratic signals or artifacts of multiple testing.
We have performed a genome-wide association study of bipolar disorder using pooled DNA from two independent case-control samples of European ancestry. All pools were genotyped with over 550,000 SNP markers. Replicated associations were identified, and selected findings were confirmed by individual genotyping. Our results show that several genes, each of modest effect, reproducibly contribute to the risk for bipolar disorder and suggest that bipolar disorder is a polygenic disease. This first genome-wide association study of bipolar disorder has important implications for future genetic and biological studies of this common and disabling brain disease.
Methods
Test Sample
We used a test/replication design to address the problem of multiple testing and highlight alleles of general relevance. Our test sample (“NIMH”) consisted of 461 unrelated bipolar I probands selected from families with at least one affected sibling pair. Probands who reported exclusively European origins (excluding Ashkenazim) were drawn from the NIMH Genetics Initiative (http://nimhgenetics.org). All probands underwent a semi-structured diagnostic interview and were assigned a “confident” diagnosis of DSM-IV bipolar I disorder by each of 2 trained clinicians (8).
The matched control sample consisted of 562 unrelated persons who were ascertained by the NIMH Genetics Initiative through a marketing firm and screened by questionnaire for major depression, bipolar disorder, and psychosis (Table 1). Those who did not meet DSM-IV criteria for major depression, denied a history of bipolar disorder or psychosis, and reported exclusively European origins (excluding Ashkenazim) were included.
Table 1.
Study Samples
| NIMH | German | |
|---|---|---|
| Cases (n, % female) | 413 (64) | 772 (53) |
| Controls (n,% female) | 563 (44) | 876 (47) |
| Family history of bipolar disorder | 100% | 13% |
| German ancestry | 9% (cases), 13% (controls)a | 96% cases, 98% controls |
In this sample ancestry was described only as “Western European (e.g. French, German).”
All subjects in the NIMH sample were collected under protocols approved by the local Institutional Review Boards.
A subset of the cases and controls were checked for evidence of population stratification or cryptic relatedness prior to pooling. Unlinked (r2<0.3) SNP markers (n=2296 in cases, 344 in controls) were genotyped at Illumina, Inc. using their GoldenGate assay (unpublished data). Cryptic relatedness was assessed by use of GRR (9). No cryptically-related individuals were detected. Population stratification was assessed using STRUCTURE 2.0 (10), under an admixture model with 20,000 burn-ins and 20,000 replicates. All cases were assigned to one population with high probability, but 6 controls were excluded due to a >70% probability of membership in a second population.
Replication Sample
The replication (“German”) sample consisted of 772 bipolar I patients recruited from consecutive hospital admissions. Index patients from families previously collected for linkage studies were also included (11). All patients had a DSM-IV diagnosis of bipolar I disorder necessitating inpatient treatment. DSM-IV lifetime diagnoses were made by a consensus best-estimate procedure (12), based on all available information, including a structured interview, medical records, and family history. The details of the recruitment and phenotype characterization procedures are outlined elsewhere (13,14). Over 96% reported that their parents and all four grandparents were born in Germany.
The German case sample was matched with a population-based control sample collected by the same investigators. A total of 876 control individuals were randomly recruited from the list of registered inhabitants with the support of the local Census Bureau of the city of Bonn (North Rhine-Westphalia, Germany). Individuals with a personal history of affective disorder or schizophrenia were excluded. Over 98% were of exclusively German ancestry.
The study protocol for the German samples was approved by the Ethics Committees of the Faculties of Medicine of the Universities of Bonn and Heidelberg.
Power
Power was estimated using Genetic Power Calculator (15) on the basis of the total number of samples pooled, scaled down by 32% to account for the pooling procedure, as recommended by Barratt et. al.(16). The NIMH sample had 80% power to detect at p<0.05 a marker in linkage disequilibrium (D'=0.8) with a disease allele of 20% frequency that confers a 1.4-fold increased risk of BPAD (trait frequency 2%) under an additive model. Under the same assumptions, the larger German sample had close to 90% power to detect such an allele.
DNA Pooling Procedures
We created a total of 39 non-overlapping pools (NIMH sample: 7 case pools and 9 control pools, 50-80 subjects per pool; German sample: 13 case pools and 10 control pools, 42-60 subjects per pool) (17). Equimolar amounts of DNA, diluted to a concentration of approximately 10 ng/ul (Quant-iT PicoGreen dsDNA Assay Kit, Invitrogen, Oregon, USA), were pooled by manual pipetting. Samples with DNA concentrations outside our specified range of 5% around the plate mean were excluded from the pools (49 cases from the NIMH sample, 93 cases and 333 controls from the German sample). Excluded samples differed from the others only in concentration and were included at the individual genotyping stage.
Pooled DNA was concentrated using Micron YM-100 Filter columns (Millipore Corporation, Bedford, MA) to greater than 50ng/ul (NanoDrop Technologies, Wilmington, DE) before genotyping. Pools were loaded onto the HumanHap550 chip and processed on the BeadStation using the procedures recommended by the manufacturer (Illumina, Inc., La Jolla, CA). The HumanHap 550 chip samples about 90% of common variation in persons of European descent at an r2>0.8 (18). To assess variance in allele frequency attributable to the pooling procedure, each pool was created at least twice and the technical replicate pools were compared. Correction and normalization of the pooling data and tests of the robustness of the method are described in the Supplemental Methods. Since these correction procedures have not been validated with mitochondrial SNPs, these were excluded from the present study.
Whole-genome analysis
Analyses of data from the DNA pools were performed by locally-authored software using the Apophenia library (19) and available by request. We calculated normalized allele frequencies from raw intensity data and averaged data across replicate pools to obtain a relative allele frequency estimate (RAF) for each SNP in each pool. SNP-pool combinations with a variance between replicate pools of >2% were excluded. We tested (t-test) the null hypothesis that the transformed RAF for cases was equal to the transformed RAF for the controls. Details about the normalization and pooling procedures are presented in the Supplemental Methods.
Replication Criteria
In order to minimize false positives due to multiple testing, we employed a replication design. This design did not treat the two samples equally, due to differences in ascertainment and ancestry. The NIMH sample consisted only of cases with a sibling pair affected with bipolar disorder, unlike the German sample where less than 15% reported a family history of bipolar disorder. The NIMH sample, while selected for European ancestry, is drawn from the U.S. population and is more diverse than the German sample, the vast majority of whom report exclusively German ancestry. Thus we reasoned that the NIMH sample would be most likely to contain important risk alleles of relevance in broad European populations.
To be eligible for replication testing in the German sample, SNPs were required to fulfill three criteria in the NIMH sample. First, we required a SNP to have RAF over 0.05 in both cases and controls. Rare alleles were excluded because the accuracy of allele frequency estimates in pooled DNA is lower for rare alleles (see Supplemental Methods) and there would be low power to detect less common SNPs in the German sample. Second, we required that a SNP be associated with bipolar disorder at a nominal p-value of 0.05 or below and an odds ratio (OR) of at least 1.4. This assured reasonable power to replicate in the German sample even if the OR was over-estimated in the NIMH sample. Third, we required that the SNP map in or near a known gene, as determined by SNPper (20). This criterion increases the biological interpretability of any finding (3), albeit at the risk of overlooking associations with intergenic SNPs that may ultimately prove to lie in un-annotated genes or regulatory sequences. SNPs passing these criteria were then tested in the German sample. A SNP was declared replicated if and only if the same allele was associated in the German sample at the nominal p<0.05 level. The replication design and criteria were settled in advance of any analysis of the results.
Individual Genotyping
Replicated SNPs were prioritized for individual genotyping by several criteria. Priority was given to SNPs with a minor allele frequency > 0.05 and OR > 1.4 in both samples, SNPs that clustered with replicated SNPs within the same gene, non-synonymous SNPs, SNPs with the lowest RAF variances, and SNPs that mapped to chromosomal regions previously linked to bipolar disorder in multiple studies (Supplemental References). Functional relevance to known pathways was explicitly ignored in order to preserve the relatively unbiased nature of the whole-genome approach. We also individually genotyped 8 SNPs chosen to represent 1) rare alleles; 2) highly-significant results in the NIMH sample that were not replicated in the German sample; 3) replicated SNPs that, while significant, yielded an OR<1.4 in the German sample. We also genotyped a SNP in DGKH (rs9513885) that was in a conserved region near multiple significant signals in the pooled genotyping.
SNPs were individually genotyped using a modification of the 5′ nuclease (Taqman) assay, scored on a fluorescent plate reader by use of a clustering algorithm (kindly provided by Sam Chen, Virginia Commonwealth University). Individual genotyping was performed on the full sample, including those individuals excluded from the pools. Genotyping accuracy was ensured by genotyping duplicate samples on each plate (one discrepant genotype was detected and excluded from analysis), genotyping of trio samples to detect Mendelian errors (none found), and verification of Hardy-Weinberg equilibrium (HWE) using Pedstats (21). One SNP (rs4813030) had HWE probabilities <0.01 in the German sample and combined samples. Individually genotyped SNPs were analyzed with UNPHASED 2.404(22,23). Haplotypes with estimated frequency <1% were excluded from the multi-locus analyses.
Results
Figure 1 displays probability plots of the ranked p-values obtained from pooled DNA for the NIMH and German samples. The NIMH sample contains a number of SNPs that deviate slightly above the identity line, consistent with several weak association signals. The German sample shows some deviation below the identity line, suggesting that the p-values obtained from the pools were conservative in that sample. The mean t-statistic was close to zero in both samples (NIMH=−0.4602, German=−0.5091). This demonstrates that there is no overall deviation in the distribution of results from the expected values as would occur in the presence of population stratification, cryptic relatedness, or other systematic error (24,25).
Figure 1.
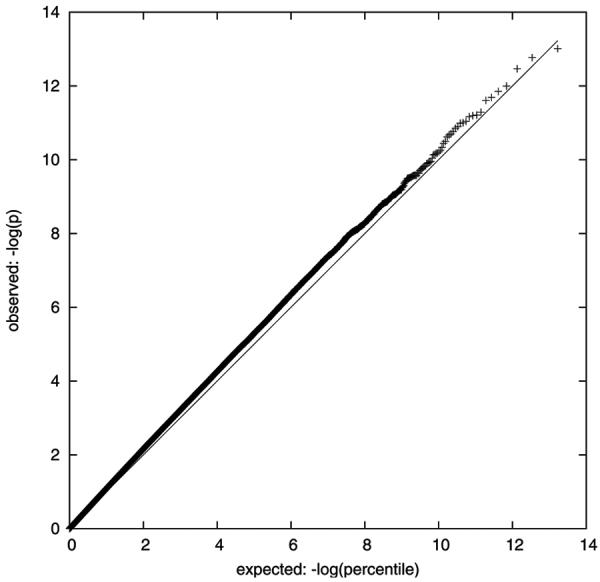
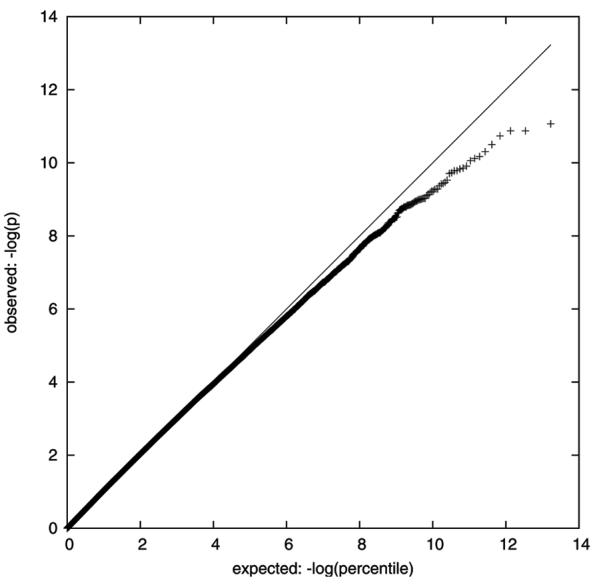
Probability plots of expected (line) vs observed (+) p-values derived from pooled DNA in the a) NIMH and b) German samples. X-axis: −ln(expected p-value); Y-axis: −ln(observed p-value).
Table 2 summarizes the results of the initial screen of pooled DNA from the NIMH sample. In total, 33,949 SNPs (6.1%) differed in frequency between NIMH cases and controls at p<0.05 and 5781 SNPs (1%) had an odds ratio >1.4. Of these, 4241 (73%) passed the RAF threshold of 0.05, and 1877 (32.5%) mapped in or near known genes.
Table 2.
Summary Results in Pooled DNA Samples
| NIMH | German | |
|---|---|---|
| #SNPs tested | 555235 | |
| #SNPs with two-tailed p < 0.05 | 33949 | 27979 |
| …OR >1.4 or <0.71 | 5781 | |
| …q>0.05 in cases and controls | 4241 | |
| …located in or near genes | 1877 | |
| Replicated SNPs | 88, in 80 genes | |
These 1877 SNPs were tested for replication in the German sample. A total of 88 SNPs representing 80 different genes met the prior criteria for replication (Supplemental Table 1). Among these, the largest OR observed was 2.34 in the NIMH sample and 1.86 in the German sample. Several replicated SNPs map to genes within previously-identified bipolar disorder linkage regions, some of which contain more than one replicated SNP.
Individual genotyping
Results of the individual genotyping are shown in Table 3. As expected, the differences in allele frequencies between cases and controls (delta) was similar in the pooled and individual data (mean absolute difference in delta = 0.031, 95% CI 0.022-0.039), while the individual allele frequency estimates were slightly more variable (mean absolute difference in allele frequency= 0.034, 95% CI 0.029-0.039). Overall, 28 of 37 (76%) SNPs remained significantly associated with bipolar disorder at the p<0.05 level when genotyped individually in the NIMH sample. Of these, 10 (36%) were significant when individually genotyped in the German sample, and 15 additional SNPs were significant in the combined dataset.
Table 3.
Individual genotyping results.
| SNP name |
Location | Gene (Allele) | NIMH | German | Combined | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Case freq |
Control freq |
p-value | Case freq |
Control freq |
p-value | p-value | OR (CI) | |||
| rs12410676a | 1p13.3 | VAV3 (G) | 0.895 | 0.867 | 0.061 | 0.884 | 0.857 | 0.024 | 0.004 | 1.28 (1.09-1.51) |
| rs13414801 | 2p23.2 | BRE (C) | 0.696 | 0.628 | 0.001 | 0.658 | 0.660 | 0.913 | 0.056 | ns |
| rs4148776a | 2q31.3 | ABCB11 (T) | 0.062 | 0.042 | 0.049 | 0.055 | 0.048 | 0.418 | 0.062 | ns |
| rs1131171 | 2q37.1 | NCL (G) | 0.842 | 0.809 | 0.048 | 0.828 | 0.805 | 0.104 | 0.013 | 1.20 (1.04-1.38) |
| rs7610043 | 3p24.1 | RBMS3 (G) | 0.567 | 0.492 | 0.0008 | 0.523 | 0.499 | 0.124 | 0.001 | 1.21 (1.08-1.35) |
| rs9863764a | 3p26.1 | GRM7 (G) | 0.131 | 0.109 | 0.137 | not genotyped | ||||
| rs3762685a | 3q24 | PLSCR4 (C) | 0.391 | 0.307 | 7.5×10−5 | 0.373 | 0.354 | 0.293 | 0.0009 | 1.22 (1.08-1.36) |
| rs4411993 | 4p16.1 | SORCS2 (C) | 0.907 | 0.864 | 0.003 | 0.888 | 0.862 | 0.030 | 0.0005 | 1.35 (1.14-1.61) |
| rs7683874b | 4p16.1 | SORCS2 (G) | 0.950 | 0.928 | 0.041 | 0.954 | 0.931 | 0.006 | 0.0006 | 1.51 (1.19-1.92) |
| rs10937823b | 4p16.1 | SORCS2 (C) | 0.949 | 0.912 | 0.001 | 0.954 | 0.930 | 0.004 | 1.4×10−5 | 1.67 (1.32-2.13) |
| rs2291182 | 4q13.3 | C4orf35 (G) | 0.103 | 0.070 | 0.011 | 0.089 | 0.080 | 0.436 | 0.027 | 1.25 (1.02-1.52) |
| rs7660807 | 4q13.3 | UNQ689 (G) | 0.111 | 0.070 | 0.001 | 0.096 | 0.084 | 0.266 | 0.004 | 1.33 (1.10-1.61) |
| rs2162126a | 4q28.2 | PHF17 (C) | 0.884 | 0.840 | 0.004 | 0.156 | 0.148 | 0.5516 | 0.210 | ns |
| rs3736456 | 4q35.1 | CYP4V2 (C) | 0.058 | 0.046 | 0.231 | not genotyped | ||||
| rs437179 | 6p21.32 | SKIV2L (C) | 0.747 | 0.700 | 0.018 | 0.745 | 0.714 | 0.053 | 0.003 | 1.21 (1.07-1.36) |
| rs13218303 | 6q24.3 | GRM1 (A) | 0.148 | 0.122 | 0.082 | not genotyped | ||||
| rs17350383a | 7p21.1 | HDAC9 (T) | 0.181 | 0.161 | 0.218 | not genotyped | ||||
| rs7812884 | 8p23.2 | CSMD1 (A) | 0.078 | 0.062 | 0.174 | not genotyped | ||||
| rs2255317a | 8q22.2 | MATN2 (C) | 0.898 | 0.874 | 0.095 | not genotyped | ||||
| rs942518 | 9q32 | DFNB31 (G) | 0.117 | 0.088 | 0.035 | 0.106 | 0.073 | 0.001 | 0.0001 | 1.44 (1.20-1.74) |
| rs17477151 | 11p12 | NGL1 (G) | 0.927 | 0.899 | 0.027 | 0.926 | 0.916 | 0.285 | 0.030 | 1.26 (1.03-1.55) |
| rs11021955 | 11p15.3 | GALNTL4 (C) | 0.898 | 0.855 | 0.003 | 0.883 | 0.860 | 0.059 | 0.001 | 1.33 (1.12-1.56) |
| rs7933829 | 11q24.3 | C11orf44 (C) | 0.819 | 0.762 | 0.002 | 0.779 | 0.769 | 0.538 | 0.023 | 1.18 (1.03-1.33) |
| rs9315885a | 13q14.11 | DGKH (T) | 0.737 | 0.680 | 0.005 | 0.701 | 0.654 | 0.006 | 0.0001 | 1.26 (1.12-1.42) |
| rs1012053a | 13q14.11 | DGKH (A) | 0.899 | 0.843 | 0.0002 | 0.882 | 0.826 | 1.5 × 10−5 | 1.5 × 10−8 | 1.59 (1.35-1.87) |
| rs1170191 | 13q14.11 | DGKH (G) | 0.868 | 0.820 | 0.003 | 0.849 | 0.800 | 0.0003 | 3.7 × 10−6 | 1.42 (1.23-1.65) |
| rs9513877 | 13q33.1 | VGCNL1 (G) | 0.676 | 0.601 | 0.0005 | 0.643 | 0.602 | 0.021 | 8.1 × 10−5 | 1.26 (1.12-1.41) |
| rs17125698a | 14q32.11 | CHES1 (C) | 0.858 | 0.821 | 0.028 | 0.868 | 0.843 | 0.049 | 0.003 | 1.26 (1.08-1.47) |
| rs1818290a | 16p13.2 | A2BP1 (G) | 0.014 | 0.010 | 0.657 | not genotyped | ||||
| rs7204975a | 16p13.2 | A2BP1 (C) | 0.886 | 0.836 | 0.001 | 0.857 | 0.851 | 0.542 | 0.533 | ns |
| rs10500336 | 16p13.3 | A2BP1 (G) | 0.146 | 0.110 | 0.018 | 0.142 | 0.130 | 0.380 | 0.022 | 1.20 (1.02-1.41) |
| rs4398100 | 16q23.2 | PLCG2 (C) | 0.834 | 0.802 | 0.069 | 0.851 | 0.829 | 0.106 | 0.014 | 1.20 (1.04-1.39) |
| rs2360111 | 17p13.1 | NXN (C) | 0.629 | 0.554 | 0.0007 | 0.592 | 0.556 | 0.048 | 0.0003 | 1.23 (1.10-1.37) |
| rs7212506 | 17q24.3 | ABCA6 (C) | 0.061 | 0.049 | 0.236 | not genotyped | ||||
| rs6501822a | 1725.1 | LLGL2 (G) | 0.918 | 0.901 | 0.0192 | not genotyped | ||||
| rs12608087 | 18q11.2 | LAMA3 (C) | 0.935 | 0.905 | 0.011 | 0.920 | 0.912 | 0.439 | 0.030 | 1.25 (1.02-1.52) |
| rs335929 | 18q11.2 | AQP4 (C) | 0.225 | 0.179 | 0.010 | 0.204 | 0.097 | 0.097 | 0.004 | 1.23 (1.07-1.41) |
| rs4813030c | 20p13 | PSMF1 (C) | 0.829 | 0.777 | 0.004 | 0.807 | 0.798 | 0.545 | 0.023 | 1.18 (1.03-1.35) |
Did not meet all allele frequency, p-value, or OR thresholds in both pooled samples
SNPs are in strong linkage disequilibrium: D' = 0.99, r2 = 0.96
SNP is not in HWE
The results confirmed a variety of genes. Effect sizes were modest. The largest OR observed was 1.69 for a SNP in SORCS2, although most of the ORs were smaller than 1.4. Three SNPs in SORCS2 and three SNPs in DGKH were significant at the p<0.05 level in both samples. The most significant result was observed for the DGKH SNP, rs1012053 (p = 1.5 × 10−8, OR= 1.59 in combined samples). This SNP is significant at the experiment-wide p<0.01 after Bonferroni correction of the combined results for the total number of SNPs studied.
Multi-locus analysis
Are particular loci more influential than others in conferring risk? To explore this question, we tested all 2- and 3- locus combinations of the 37 individually-genotyped SNPs in the combined sample. There were 95 combinations that were globally significant at p<0.05 after Bonferroni correction for the 1072 combinations tested. Of these, 23% contained a SNP in SORCS2 (rs10937823) and 22% contained a SNP in DGKH (rs1170191). The most significant combination (p=1.2 × 10−8) was a 2-marker model containing both of these SNPs. No other SNPs were present in over 20% of significant combinations. This suggests that no one SNP was necessary to produce bipolar disorder, but the SNPs we studied in SORCS2 and DGKH may be particularly influential in conferring risk in the samples we studied.
If multiple different loci, each of small effect, contribute to the risk of bipolar disorder as our data suggest, this raises the question of how the risks of individual loci accumulate to influence risk of disease. To explore this question, we analyzed the 10 SNPs that were supported by the individual genotyping in the combined sample at the p<0.001 level (representing DGKH, DFNB31, SORCS2, GANTL4, NXN, and VGCNL1). We classified alleles as “risk” alleles if they were more common in cases than in controls. We then counted the number of risk alleles carried by each case and each control (homozygotes were counted as 2) and summed these across loci. The results are illustrated in Figure 2.
Figure 2.
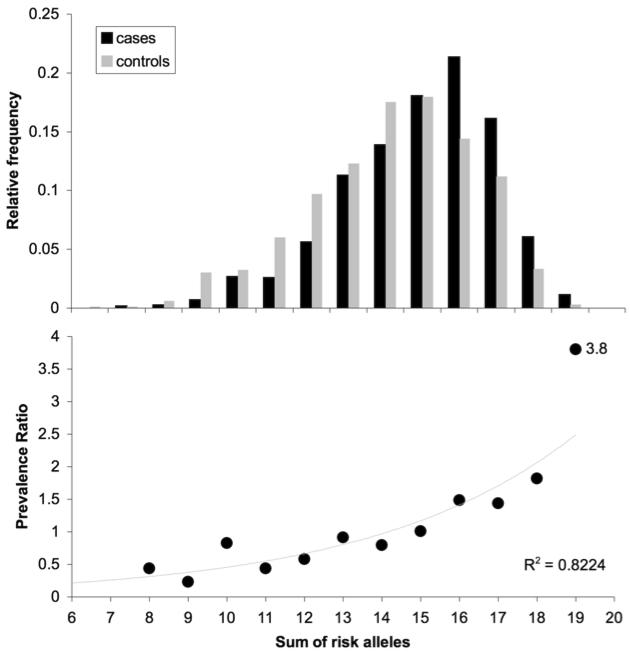
Distribution of risk alleles among cases and controls in the combined sample (a), and relationship of prevalence ratio to risk allele burden (b). Data from ten individually genotyped SNPs (rs4411993, rs7683874, rs10937823, rs942518, rs11021955, rs10120953, rs1170191, rs9315885, rs9513877, rs2360111) are plotted. Cases are indicated in black, controls in gray. An exponential function provides a good fit to the data (R2 = 0.82).
As expected, the proportion of cases rises with the number of risk alleles carried. The case and control distributions are significantly different (Kolmogorov-Smirnov D=0.155, p<0.0001). Many controls also carry substantial burdens of risk alleles. This implies that the identified alleles are not sufficient to cause bipolar disorder. Only 5 cases (out of 1152 scored) carried fewer than 9 risk alleles, suggesting that multiple risk alleles are a necessary part of the vulnerability to bipolar disorder. Persons carrying 19 or more risk alleles were almost 4 times more likely to be classified as cases than controls (Figure 2; prevalence ratio = 3.8, 95% CI 1.24-11.6 , χ2 = 20.25, p = 6.8 × 10−6).
Discussion
This first genome-wide association study of bipolar disorder has detected and replicated evidence of genetic association at several distinct loci. While the contribution of each locus to risk of disease is modest, cases carry significantly more risk alleles than controls, and disease risk increases substantially with the total burden of risk alleles carried. Many of the identified genes lie within previously-reported regions of genetic linkage to bipolar disorder or schizophrenia. Some loci, primarily DGKH, stand out for their biological relevance and consistency between samples, and such loci may be more influential than the others in determining risk for bipolar disorder. These data suggest that bipolar disorder is a polygenic disease influenced by many genes, each of small effect.
This study has several limitations. Our strategy of pooling DNA samples at the initial screen reduced power to detect true associations. Still, the study design had sufficient power to detect common loci of modest effect (OR > 1.4) that were present in both samples. (While odds ratios were often lower in the individual genotyping than the pooled genotyping results, this would be expected in any screening method where the primary screen has a larger variance than the secondary screen; the so-called “winner's curse.”) The Illumina HumanHap550 chip gives very good, but incomplete, coverage of the European genome, and we restricted our replication testing to SNPs in and near genes. This strategy cannot detect uncommon alleles and SNPs in unannotated genes or regulatory regions. Despite these limitations, we detected and replicated multiple alleles that confer modest risk for bipolar disorder. Our data therefore imply that high-risk loci for bipolar disorder, if they exist, are probably very uncommon.
Population stratification is always a risk in case-control studies, even where groups have been carefully matched for ancestry (26), as in the present study. Probability plots of the distribution p-values in the pooled results showed no evidence of deviation from the expected distribution, except at the extreme, where the German sample actually returned fewer highly-significant results than would be expected by chance. Thus we conclude that population stratification is very unlikely to account for our findings.
We have not corrected p-values in the pooled DNA results for the number of SNPs evaluated. This is because we used the pooling results only as a means for identifying SNPs that warrant individual genotyping. While p-value adjustment methods such as false discovery rates may be considered, the ranking of the SNPs is invariant to such adjustments, and we considered other criteria, such as presence in a linkage region, in prioritizing SNPs for further study, as recommended by some authorities (27, 28). On the other hand, the p-values obtained for SNPs that were individually genotyped should, arguably, be corrected for multiple testing. One reasonable approach would be to correct for the 1877 SNPs that were eligible for replication testing. We have not individually genotyped all of these SNPs, but we do know their p-value ranking in the pooled NIMH sample. Thus, we can use a Bonferroni-Holm step-down procedure to correct the p-values of the SNPs that were individually genotyped. When we apply this correction to our results, one SNP in DGKH (rs1012053) remains significant at the experiment-wise p=0.002 level. This same SNP would remain significant at the experiment-wise p<0.01 level after Bonferroni-correction of the combined p-value for the 555,055 SNPs we studied. Thus we can reject the null hypothesis of no association between bipolar disorder and at least this SNP.
Given that we did not take functional relevance into account when selecting SNPs to individually genotype, it is remarkable that the strongest result in this study, replicated by individual genotyping in both samples, involves a SNP in the gene DGKH. A total of four SNPs in DGKH (rs1170195, rs9525570, rs1170191, in a 7kb region in the 1st intron, and rs9315897 in the 7th intron) showed evidence of association at the p<0.05 level in both pooled samples (Figure 3). Three SNPs (rs1170191, rs9315885, and rs1012053) showed strong evidence of association in both samples after individual genotyping. DGKH is located within the bipolar disorder linkage region on 13q14 (29, 30). DGKH encodes diacylglycerol kinase eta, a key protein in the lithium-sensitive phosphatidyl inositol pathway (31) (Figure 4). We do not have lithium-response data available in either of the samples we studied, but it may be of particular interest to test this finding in a sample where lithium response data is available.
Figure 3.

Pooling-derived association results for SNPs in the gene DGKH (181 kb). The graphic was produced by UCSC Genome Browser, using the May 2004 build and the custom tracks option. SNPs associated at the p<0.05 level are shown in the NIMH and German tracks, and SNPs genotyped by the HumanHap550 are shown directly above the gene track. Red SNPs were significant at p < 0.05 in both samples, blue SNPs had p < 0.05 in only one sample. There is substantial linkage disequilibrium across the region, according to the HapMap CEPH-European data (not shown).
Figure 4.
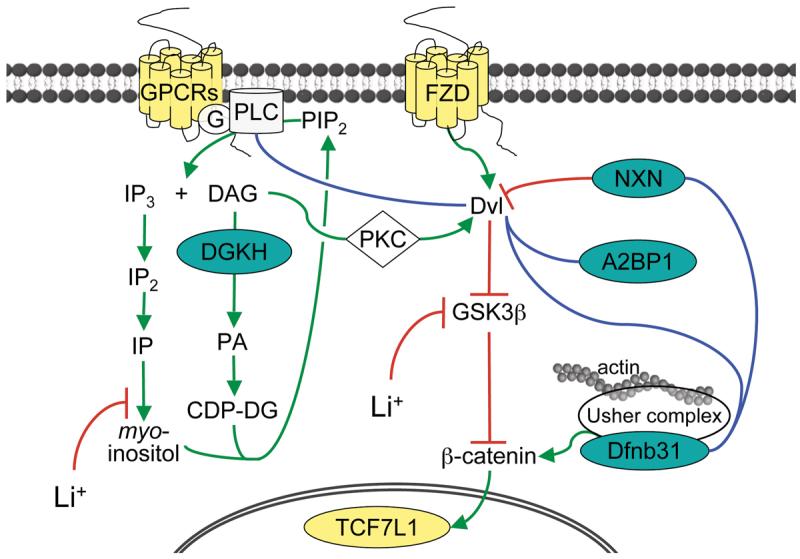
Graphic illustrating the roles of 8 genes implicated in the present study in lithium-sensitive signaling pathways. Green lines denote enzymatic transformations or cofactor activations, red lines confirmed inhibitory actions, blue lines protein interactions of unknown nature from Lim et al (54) or hypothetical interactions. All colored genes contain at least one replicated SNP based on individual genotyping (teal) or pooled (yellow) data. G-protein coupled receptors (such as GPC51) activate the phosphatidyl inositol signaling pathway via G-protein (G) activation of phospholipase C (PLC, such as PLCG2). PLC cleaves PIP2 into IP3 and diacylglycerol (DAG). DAG is metabolized by DAG kinases (such as DGKH). DAG is a necessary cofactor for activation of most protein kinase C (PKC) isoforms. PKC cross-talks via disheveled (Dvl) with the Wnt/B-catenin signaling pathway. Dvl is activated by Wnt receptors (such as FZD2) and directly inhibited by nucleoredoxin (NXN) by binding the PDZ domain. A2BP1 also binds Dvl. Dvl inhibits glycogen synthase kinase-3 beta (GSK3B), which itself inhibits β-catenin. β-catenin is activitated by whirlin (DFNB31) via the Usher protein complex. Since it also contains PDZ domains, DFNB31 may also bind nucleoredoxin and Dvl. In the nucleus, β-catenin modulates the activity of TCF/LEF transcription factors (such as TCF7L1), which ultimately effects the expression of a large number of target genes. Lithium (Li+) lowers intracellular myo-inositol levels, reducing production of PIP2, and increases β–catenin signaling through inhibition of GSK3B.
Another replicated signal is in the gene NXN, which encodes the protein nucleoredoxin, which inhibits Wnt-β-catenin signaling by binding to the PDZ domain of the protein disheveled (Figure 4) (32). The phosphitidylinositol and Wnt pathways, which connect via protein kinase C, have been hypothesized to play an important role in the mechanism of action of mood stabilizing medications (33). These results illustrate how a genome-wide association study can highlight the potential etiologic importance of specific components of even well-understood pathways.
Our results also implicate novel proteins and pathways. The association signals we detected in VGCNL1, DFNB31, and SORCS2 are good examples of this. VGCNL1 encodes a voltage-gated ion channel highly expressed in the brain, and lies within a region on chromosome 13q that has shown linkage to bipolar disorder in several studies (29). Its Drosophila homolog, narrow abdomen (na), regulates circadian rhythms (34), suggesting that this gene may play a role in the circadian disruption that is a hallmark of bipolar disorder.
DFNB31 is located within a bipolar linkage region (35). The gene encodes the neuronally-expressed protein whirlin, which binds to most members of the Usher protein complex (Figure 4). Whirlin contains three highly-conserved PDZ domains, mutations in which cause the “whirler” phenotype in mice and prelingual-onset non-syndromic deafness in humans (36), including some types of Usher syndrome (37). Like nucleoredoxin, the Usher protein complex is an effector of β-catenin, and also affects neuronal morphogenesis and structural plasticity (38). Usher syndrome can cause CNS structural changes (39) and psychosis (40), and there is one report of increased incidence of bipolar disorder among adults with prelingual-onset non-syndromic deafness (41), but it is not known if any of those patients carried the “whirler” mutation. Finally, SORCS2 maps to a region on chromosome 4p that has been widely linked to bipolar disorder (42, Supplemental References). SORCS2 encodes a VPS10 domain-containing receptor prominently expressed in the developing brain (43).
These results lead us to hypothesize that most bipolar disorder is best explained by the polygenic threshold model, a classical model of the relationship between genes and disease, based in quantitative genetic theory (44). Many risk alleles, each of small effect size relative to the total risk, are assumed to exist in a population. Each person's disease risk is influenced by the total burden of risk alleles they carry. Fewer alleles confer a lower risk, while more alleles confer greater risk. Disease occurs when the allele burden crosses some threshold, although the exact disease threshold for a given person may be influenced by non-genetic factors.
The polygenic threshold model provides a good fit to the evidence to date in the field of bipolar disorder genetics. The model predicts that no single locus or small set of loci will be necessary or sufficient to develop disease. Thus there is expected to be a high degree of genetic heterogeneity, since different combinations of loci can create the same disease state in different people. Such heterogeneity would lead to many weak and inconsistent genetic linkage findings, and linkage peaks that are broad and ill-defined, because the magnitude of linkage scores would be reduced by locus heterogeneity and any linkage region might harbor more than one risk locus. Both phenomena have consistently characterized the genetic linkage findings in bipolar disorder (45). Similarly, a number of weak genetic associations have been reported, but the effect sizes have been small and difficult to replicate (2). Again, this is consistent with a polygenic model. Many of the replicated loci in this study lie within regions previously linked to schizophrenia. The overlap of risk alleles between bipolar disorder, schizophrenia, and other mental illnesses is also consistent with a polygenic model, since different constellations of risk alleles might lead to different disease outcomes.
A polygenic threshold model may seem inconsistent with the apparent vertical transmission of bipolar disorder that has been long observed (46), but not generally supported by segregation analyses (reviewed in 47). However, many of the risk alleles that we have identified are the most common allele in the population, with frequencies over 80% even in controls. Such common alleles could create a pseudo-dominant transmission pattern (48), especially in conjunction with the assortative mating that is common among people with mood disorders (49).
The replication design we employed made it possible to separate out some robust association signals from among the large set of statistically significant results in this genome-wide study. However, some of the most significant results in the NIMH sample did not replicate in the German sample, and by design we did not pursue association signals present only in the German sample. Association signals that did not replicate under our criteria were observed in several genes that have been implicated by previous studies (50-53), including NPAS3, GRIK2, GRM3, GRM4, KCNQ3, and GRIN2B (data not shown). Since others may wish to pursue these findings, we will make our genome-wide set of results available at http://mapgenetics.nimh.nih.gov/bp_pooling.
In conclusion, this first genome-wide association study of bipolar disorder has detected weak but reproducible association with markers in several genes. None of the replicated genes confers a large risk of disease, but the gene DGKH contains a SNP that is significantly associated with bipolar disorder even after conservative experiment-wise correction. Several implicated genes reside in well-known regions of genetic linkage to bipolar disorder, and several lie within schizophrenia linkage regions, but no one gene appears to be necessary or sufficient for disease. These findings imply that major gene effects in bipolar disorder are very uncommon, if they exist at all. Future studies may benefit from multi-locus approaches that embrace the genetic heterogeneity of bipolar disorder.
Supplementary Material
Acknowledgements
We thank the study participants who make this research possible. Layla Kassem and Jo Steele provided technical assistance; Michael O'Donovan and Paul Boyce gave advice on the pooling methods; Husseini Manji provided helpful comments on the manuscript; and the Rutgers University Cell & DNA Repository managed DNA samples.
Data and biomaterials for the NIMH sample were collected as part of 10 projects that participated in the NIMH Bipolar Disorder Genetics Initiative. From 1991-98, the Principal Investigators and Co-Investigators were: Indiana Univ., Indianapolis, IN, U01 MH46282, John Nurnberger, MD , PhD, Marvin Miller, MD , and Elizabeth Bowman, MD ; Washington Univ., St. Louis, MO, U01 MH46280, Theodore Reich, MD , Allison Goate, PhD, and John Rice, PhD; Johns Hopkins Univ., Baltimore, MD U01 MH46274, J. Raymond DePaulo, Jr MD Sylvia Simpson, MD , MPH, and Colin Stine, PhD; NIMH Intramural Research Program, Clinical Neurogenetics Branch, Bethesda, MD, Elliot Gershon, MD , Diane Kazuba, BA, and Elizabeth Maxwell, M.S.W. From 1999-03, the Principal Investigators and Co-Investigators were: Indiana Univ., Indianapolis, IN, R01 MH59545, John Nurnberger, MD , PhD, Marvin J. Miller, MD , Elizabeth S. Bowman, MD , N. Leela Rau, MD , P. Ryan Moe, MD , Nalini Samavedy, MD , Rif El-Mallakh, MD (at Univ. of Louisville), Husseini Manji, MD (at Wayne State Univ.), Debra A. Glitz, MD (at Wayne State Univ.), Eric T. Meyer, M.S., Carrie Smiley, RN, Tatiana Foroud, PhD, Leah Flury, M.S., Danielle M. Dick, PhD, Howard Edenberg, PhD; Washington Univ., St. Louis, MO, R01 MH059534, John Rice, Ph.D,Theodore Reich, MD , Allison Goate, PhD, Laura Bierut, MD ; Johns Hopkins Univ., Baltimore, MD, R01 MH59533, Melvin McInnis MD , J. Raymond DePaulo, Jr MD Dean F. MacKinnon, MD , Francis M. Mondimore, MD , James B. Potash, MD , Peter P. Zandi, Ph.D, Dimitrios Avramopoulos, and Jennifer Payne; Univ. of Pennsylvania, PA, R01 MH59553, Wade Berrettini MD ,PhD ; Univ. of California at Irvine, CA, R01 MH60068, William Byerley MD , and Mark Vawter MD ; Univ. of Iowa, IA, R01 MH059548, William Coryell MD , and Raymond Crowe MD ; Univ. of Chicago, IL, R01 MH59535, Elliot Gershon, MD , Judith Badner PhD , Francis McMahon MD , Chunyu Liu PhD, Alan Sanders MD , Maria Caserta, Steven Dinwiddie MD , Tu Nguyen, Donna Harakal; Univ. of California at San Diego, CA, R01 MH59567, John Kelsoe, MD , Rebecca McKinney, BA; Rush Univ., IL, R01 MH059556, William Scheftner MD , Howard M. Kravitz, D.O., MPH, Diana Marta, BA, Annette Vaughn-Brown, MSN, RN, and Laurie Bederow, MA; NIMH Intramural Research Program, Bethesda, MD, 1Z01MH002810-01, Francis J. McMahon, MD , Layla Kassem, PsyD, Sevilla Detera-Wadleigh, Ph.D, Lisa Austin, Ph.D, Dennis L. Murphy, MD.
The NIMH control subjects were collected by the NIMH Schizophrenia Genetics Initiative “Molecular Genetics of Schizophrenia II” (MGS-2) collaboration. The investigators and coinvestigators are: ENH/Northwestern Univ., Evanston, IL, MH059571, Pablo V. Gejman, MD (Collaboration Coordinator; PI), Alan R. Sanders, MD ; Emory Univ. School of Medicine, Atlanta, GA,MH59587, Farooq Amin, MD (PI); Louisiana State Univ. Health Sciences Center; New Orleans, Louisiana, MH067257, Nancy Buccola APRN, BC, MSN (PI); Univ. of California-Irvine, Irvine, CA,MH60870, William Byerley, MD (PI); Washington Univ., St. Louis, MO, U01, MH060879, C. Robert Cloninger, MD (PI); Univ. of Iowa, Iowa, IA,MH59566, Raymond Crowe, MD (PI), Donald Black, MD ; Univ. of Colorado, Denver, CO, MH059565, Robert Freedman, MD (PI); Univ. of Pennsylvania, Philadelphia, PA, MH061675, Douglas Levinson MD (PI); Univ. of Queensland, Queensland, Australia, MH059588, Bryan Mowry, MD (PI); Mt. Sinai School of Medicine, New York, NY,MH59586, Jeremy Silverman, PhD (PI).
We thank the following clinician colleagues for help in collecting German patients: Margot Albus, Margitta Borrmann-Hassenbach, Ernst Franzek, Jürgen Fritze, Magdalena Gross, Thilo Held, Roland Kreiner, Mario Lanczik, Dirk Lichtermann,Wolfgang Maier, Jürgen Minges, Stephanie Ohlraun, Ulrike Reuner, Monja Tullius, Bettina Weigelt.
Supported by the NIMH Intramural Research Program, Deutsche Forschungsgemeinschaft, the National German Genome Research Network of the Federal Ministry of Education and Research, the National Alliance for Research on Schizophrenia and Depression, the Alfried Krupp von Bohlen und Halbach-Stiftung, and the National Institute on Aging. JS is supported by NIH grants R01GM60457 and R01CA098438.
REFERENCES
- 1.Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1516. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 2.Detera-Wadleigh SD, McMahon FJ. Genetic association studies in mood disorders: issues and promise. Int Rev Psychiatry. 2004;16:301–301. doi: 10.1080/09540260400014377. [DOI] [PubMed] [Google Scholar]
- 3.Jorgenson E, Witte JS. A gene-centric approach to genome-wide association studies. Nat Rev Genet. 2006;7:885–885. doi: 10.1038/nrg1962. [DOI] [PubMed] [Google Scholar]
- 4.Clark AG, Boerwinkle E, Hixson J, Sing CF. Determinants of the success of whole-genome association testing. Genome Res. 2005;15:1463–1463. doi: 10.1101/gr.4244005. [DOI] [PubMed] [Google Scholar]
- 5.Sham P, Bader JS, Craig I, O'Donovan M, Owen M. DNA Pooling: a tool for large-scale association studies. Nat Rev Genet. 2002;3:862–862. doi: 10.1038/nrg930. [DOI] [PubMed] [Google Scholar]
- 6.Steer S, Abkevich V, Gutin A, Cordell HJ, Gendall KL, Merriman ME, et al. Genomic DNA pooling for whole-genome association scans in complex disease: empirical demonstration of efficacy in rheumatoid arthritis. Genes Immun. 2007;8:57–57. doi: 10.1038/sj.gene.6364359. [DOI] [PubMed] [Google Scholar]
- 7.Pearson JV, Huentelman MJ, Halperin RF, Tembe WD, Melquist S, Homer N, et al. Identification of the genetic basis for complex disorders by use of pooling-based genomewide single-nucleotide-polymorphism association studies. Am J Hum Genet. 2007;80:126–126. doi: 10.1086/510686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nurnberger JI, Jr., Blehar MC, Kaufmann CA, York-Cooler C, Simpson SG, Harkavy-Friedman J, et al. Diagnostic interview for genetic studies. Rationale, unique features, and training. NIMH Genetics Initiative. Arch Gen Psychiatry. 1994;51:849–849. doi: 10.1001/archpsyc.1994.03950110009002. [DOI] [PubMed] [Google Scholar]
- 9.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. GRR: graphical representation of relationship errors. Bioinformatics. 2001;17:742–742. doi: 10.1093/bioinformatics/17.8.742. [DOI] [PubMed] [Google Scholar]
- 10.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–945. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cichon S, Schumacher J, Muller DJ, Hurter M, Windemuth C, Strauch K, et al. A genome screen for genes predisposing to bipolar affective disorder detects a new susceptibility locus on 8q. Hum Mol Genet. 10:2933–2933. doi: 10.1093/hmg/10.25.2933. [DOI] [PubMed] [Google Scholar]
- 12.Leckman JF, Sholomskas D, Thompson WD, Belanger A, Weissman MM. Best estimate of lifetime psychiatric diagnosis: a methodological study. Arch Gen Psychiatry. 1982;39:879–879. doi: 10.1001/archpsyc.1982.04290080001001. [DOI] [PubMed] [Google Scholar]
- 13.Hoefgen B, Schulze TG, Ohlraun S, von Widdern O, Hofels S, Gross M, et al. The power of sample size and homogenous sampling: association between the 5-HTTLPR serotonin transporter polymorphism and major depressive disorder. Biol Psychiatry. 2005;57:247–247. doi: 10.1016/j.biopsych.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 14.Fangerau H, Ohlraun S, Granath RO, Nothen MM, Rietschel M, Schulze TG. Computer-assisted phenotype characterization for genetic research in psychiatry. Hum Hered. 2004;58:122–122. doi: 10.1159/000083538. [DOI] [PubMed] [Google Scholar]
- 15.Purcell S, Cherny SS, Sham PC. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics. 2003;19:149–149. doi: 10.1093/bioinformatics/19.1.149. [DOI] [PubMed] [Google Scholar]
- 16.Barratt BJ, Payne F, Rance HE, Nutland S, Todd JA, Clayton DG. Identification of the sources of error in allele frequency estimations from pooled DNA indicates an optimal experimental design. Ann Hum Genet. 2002;66:393–393. doi: 10.1017/S0003480002001252. [DOI] [PubMed] [Google Scholar]
- 17.Norton N, Williams NM, O'Donovan MC, Owen MJ. DNA pooling as a tool for large-scale association studies in complex traits. Ann Med. 2004;36:146–146. doi: 10.1080/07853890310021724. [DOI] [PubMed] [Google Scholar]
- 18.Steemers FJ, Gunderson KL. Whole genome genotyping technologies on the BeadArray platform. Biotechnol J. 2007;2:41–41. doi: 10.1002/biot.200600213. [DOI] [PubMed] [Google Scholar]
- 19.Klemens B. Modeling with Data: Modern Statistical Computing with C. Princeton University Press; Princeton, NJ: forthcoming. [Google Scholar]
- 20.Riva A, Kohane IS. SNPper: retrieval and analysis of human SNPs. Bioinformatics. 2002;18:1681–1681. doi: 10.1093/bioinformatics/18.12.1681. [DOI] [PubMed] [Google Scholar]
- 21.Wigginton JE, Abecasis GR. PEDSTATS: descriptive statistics, graphics and quality assessment for gene mapping data. Bioinformatics. 2005;21:3445–3445. doi: 10.1093/bioinformatics/bti529. [DOI] [PubMed] [Google Scholar]
- 22.Dudbridge F. Pedigree disequilibrium tests for multilocus haplotypes. Genet Epidemiol. 2003;25:115–115. doi: 10.1002/gepi.10252. [DOI] [PubMed] [Google Scholar]
- 23.Dudbridge F. Technical Report 2006/5. MRC Biostatistics Unit; Cambridge, UK: 2006. UNPHASED user guide. [Google Scholar]
- 24.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–997. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 25.Balding DJ. A tutorial on statistical methods for population association studies. Nat Rev Genet. 2006;7:781–781. doi: 10.1038/nrg1916. [DOI] [PubMed] [Google Scholar]
- 26.Steffens M, Lamina C, Illig T, Bettecken T, Vogler R, Entz P, et al. SNP-based analysis of genetic substructure in the German population. Hum Hered. 2006;62:20–20. doi: 10.1159/000095850. [DOI] [PubMed] [Google Scholar]
- 27.Roeder K, Bacanu SA, Wasserman L, Devlin B. Using linkage genome scans to improve power of association in genome scans. Am J Hum Genet. 2006;78:243–243. doi: 10.1086/500026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun L, Craiu RV, Paterson AD, Bull SB. Stratified false discovery control for large-scale hypothesis testing with application to genome-wide association studies. Genet Epidemiol. 2006;30:519–519. doi: 10.1002/gepi.20164. [DOI] [PubMed] [Google Scholar]
- 29.Detera-Wadleigh SD, McMahon FJ. G72/G30 in schizophrenia and bipolar disorder: review and meta analysis. Biol Psychiatry. 2006;60:106–106. doi: 10.1016/j.biopsych.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 30.Badner JA, Gershon ES. Meta-analysis of whole-genome linkage scans of bipolar disorder and schizophrenia. Mol Psychiatry. 2002;7:405–405. doi: 10.1038/sj.mp.4001012. [DOI] [PubMed] [Google Scholar]; Berridge MJ. The Albert Lasker Medical Awards. Inositol trisphosphate, calcium, lithium, and cell signaling. JAMA. 1989;262:1834–1834. [PubMed] [Google Scholar]
- 31.Funato Y, Michiue T, Asashima M, Miki H. The thioredoxin-related redox-regulating protein nucleoredoxin inhibits Wnt-beta-catenin signalling through dishevelled. Nat Cell Biol. 2006;8:501–501. doi: 10.1038/ncb1405. [DOI] [PubMed] [Google Scholar]
- 32.Coyle JT, Manji HK. Getting balance: drugs for bipolar disorder share target. Nat Med. 2002;8:557–557. doi: 10.1038/nm0602-557. [DOI] [PubMed] [Google Scholar]
- 33.Nash HA, Scott RL, Lear BC, Allada R. An unusual cation channel mediates photic control of locomotion in Drosophila. Curr Biol. 2002;12:2152–2152. doi: 10.1016/s0960-9822(02)01358-1. [DOI] [PubMed] [Google Scholar]
- 34.Venken T, Claes S, Slyjis S, Paterson AD, van Duijn C, Adolfsson R, et al. Genomewide scan for affective disorder susceptibility locs in families of a northern Swedish isolated population. Am J Hum Genet. 2005;76:237–237. doi: 10.1086/427836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mburu P, Mustapha M, Varela A, Weil D, El-Amraoui A, Holme RH, et al. Defects in whirlin, a PDZ domain molecule involved in stereocilia elongation, cause deafness in the whirler mouse and families with DFNB31. Nat Genet. 2003;34:421–421. doi: 10.1038/ng1208. [DOI] [PubMed] [Google Scholar]
- 36.Ebermann I, Scholl HP, Charbel Issa P, Becirovic E, Lamprecht J, Jurklies B, et al. A novel gene for Usher syndrome type 2: mutations in the long isoform of whirlin are associated with retinitis pigmentosa and sensorineural hearing loss. Hum Genet. doi: 10.1007/s00439-006-0304-0. in press. [DOI] [PubMed] [Google Scholar]
- 37.Kremer H, van Wijk E, Marker T, Wolfrum U, Roepman R. Usher syndrome: molecular links of pathogenesis, proteins and pathways. Hum Mol Genet. 2006;15(Spec No 2):R262–262. doi: 10.1093/hmg/ddl205. [DOI] [PubMed] [Google Scholar]
- 38.Piazza L, Fishman GA, Kaplan RD, Horowitz AL, Hindo WA, Mafee MF. Magnetic resonance imaging of central nervous system defects in Usher's syndrome. Retina. 1987;7:241–241. doi: 10.1097/00006982-198707040-00009. [DOI] [PubMed] [Google Scholar]
- 39.Koizumi J, Ofuku K, Sakuma K, Shiraishi H, Iio M, Nawano S. CNS changes in Usher's syndrome with mental disorder: CT, MRI and PET findings. J Neurol Neurosurg Psychiatry. 1988;51:987–987. doi: 10.1136/jnnp.51.7.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shapira NA, DelBello MP, Goldsmith TD, Rosenberger BM, Keck PE., Jr. Evaluation of bipolar disorder in inpatients with prelingual deafness. Am J Psychiatry. 1999;156:1267–1267. doi: 10.1176/ajp.156.8.1267. [DOI] [PubMed] [Google Scholar]
- 41.Blackwood DH, He L, Morris SW, McLean A, Whitton C, Thomson M, et al. A locus for bipolar affective disorder on chrosmosome 4p. Nat Genet. 1996;12:427–427. doi: 10.1038/ng0496-427. [DOI] [PubMed] [Google Scholar]
- 42.Rezgaoui M, Hermey G, Riedel IB, Hampe W, Schaller HC, Hermans-Borgmeyer I. Identification of SorCS2, a novel member of the VPS10 domain containing receptor family, prominently expressed in the developing mouse brain. Mech Dev. 2001;100:335–335. doi: 10.1016/s0925-4773(00)00523-2. [DOI] [PubMed] [Google Scholar]
- 43.Falconer DS. The inheritance of liability to diseases with variable age of onset, with particular reference to diabetes mellitus. Ann Hum Genet. 1967;31:1–1. doi: 10.1111/j.1469-1809.1967.tb01249.x. [DOI] [PubMed] [Google Scholar]
- 44.Segurado R, Detera-Wadleigh SD, Levinson DF, Lewis CM, Gill M, Nurnberger JI, Jr., et al. Genome scan meta-analysis of schizophrenia and bipolar disorder, part III: Bipolar disorder. Am J Hum Genet. 2003;73:49–49. doi: 10.1086/376548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Slater E. The inheritance of manic-depressive insanity. Proc R Soc Med. 1936;29:39–39. doi: 10.1177/003591573602900846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Faraone SV, Kremen WS, Tsuang MT. Genetic transmission of major affective disorders: quantitative models and linkage analyses. Psychol Bull. 1990;108:109–109. doi: 10.1037/0033-2909.108.1.109. [DOI] [PubMed] [Google Scholar]
- 47.Pyeritz RE. Formal genetics in humans: mendelian and nonmendelian inheritance. Res Publ Assoc Res Nerv Ment Dis. 1991;69:47–47. [PubMed] [Google Scholar]
- 48.Negri F, Melica AM, Zuliani R, Smeraldi E. Assortative mating and affective disorders. J Affect Disord. 1979;1:247–247. doi: 10.1016/0165-0327(79)90011-9. [DOI] [PubMed] [Google Scholar]
- 49.Pickard BS, Pieper AA, Porteous DJ, Blackwood DH, Muir WJ. The NPAS3 gene--emerging evidence for a role in psychiatric illness. Ann Med. 2006;38:439–439. doi: 10.1080/07853890600946500. [DOI] [PubMed] [Google Scholar]
- 50.Fallin MD, Lasseter VK, Avramopoulos D, Nicodemus KK, Wolyniec PS, McGrath JA, et al. Bipolar I disorder and schizophrenia: a 440-single-nucleotide polymorphism screen of 64 candidate genes among Ashkenazi Jewish case-parent trios. Am J Hum Genet. 2005;77:918–918. doi: 10.1086/497703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buervenich S, Detera-Wadleigh SD, Akula N, Thomas CJM, Kassem L, Rezvani A, et al. Fine mapping on chromosome 6q in the NIMH Genetics Initiative bipolar pedigrees; Annual Meeting of The American Society of Human Genetics; Los Angeles CA. 2003; [Abstract 1882] [Google Scholar]
- 52.Avramopoulos D, Willour VL, Zandi PP, Huo Y, MacKinnon DF, Potash JB, et al. Linkage of bipolar affective disorder on chromosome 8q24: follow-up and parametric analysis. Mol Psychiatry. 2004;9:191–191. doi: 10.1038/sj.mp.4001388. [DOI] [PubMed] [Google Scholar]
- 53.Lim J, Hao T, Shaw C, Patel AJ, Szabo G, Rual JF, Fisk CJ, Li N, Smolyar A, Hill DE, Barabasi AL, Vidal M, Zoghbi HY. A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell. 2006;125:801–801. doi: 10.1016/j.cell.2006.03.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.