

Abstract
Tissue transglutaminase (TGase-2), which binds GTP and catalyzes the crosslinking of proteins (transamidation), has been implicated both in the promotion of cell death and in the protection of cells against apoptotic insults. However, a novel transcript originally identified from the brains of Alzheimer’s patients, encoding a truncated form of TGase-2 (called TGase-S), shows strong apoptotic activity. TGase-S exhibits no detectable GTP-binding capability, suggesting that its ability to induce cell death might be due to its inability to bind GTP. Thus, we have examined whether eliminating the GTP-binding capability of full-length human TGase-2 would prevent it from conferring protection against apoptotic challenges and instead convert it into a protein that causes cell death. A number of point mutants of human TGase-2 defective for binding GTP, as well as a mutant that shows impaired GTP-hydrolytic activity, were generated. Similar to what we had found for TGase-S, there was a time-dependent decrease in the expression of the GTP-binding-defective TGase-2 mutants in different cell lines, whereas the expression of wild-type TGase-2 and the GTP hydrolysis-defective mutant was sustained. Moreover, the GTP-binding-defective TGase-2 mutants induced cell death. The cell death responses triggered by these mutants were not due to their transdamidation activity, because double-mutants that were both GTP-binding- and transamidation-defective also stimulated cell death. Therefore, these results point to the inability to bind GTP as being sufficient for the apoptotic activity exhibited by the TGase-S protein. They also highlight a novel example of how the loss of GTP-binding activity can convert a protein that provides protection against apoptotic stimuli into a cell death-promoting factor.
Keywords: GTP, signaling, transglutaminase, apoptosis, Alzheimer’s, transamidation
Tissue transglutaminase (TGase-2) belongs to a family of enzymes (transglutaminases) that catalyze the Ca2+-dependent, post-translational modification of proteins through the incorporation of polyamines or the formation of covalent crosslinks (1–4). The underlying chemical reaction involves the generation of an isopeptide bond between the γ-carboxamide group of a peptide-bound glutamine residue and either the ε-amino group of a lysine residue or the primary amino group of a polyamine. The expression of TGase-2 and its accompanying transamidation activity are stimulated when cells are exposed to different stresses, differentiation agents, and growth factors (5–11). A number of reports have suggested that TGase-2 is able to both promote and prevent apoptosis (4,12). For example, studies performed in U937 cells and in neuroblastoma cell lines have suggested that the over-expression of TGase-2 is correlated with increased cell death (13). The de-regulated expression and/or transamidation activity of TGase-2 have also been implicated in various neurodegenerative disorders characterized by proteinacious aggregates including Alzheimer’s, Huntington’s, and Parkinson’s disease (6,7,14–22). On the other hand, the regulation of TGase-2 by growth and differentiation factors has also been shown to be important in conferring a survival advantage to cells challenged with apoptotic stimuli like the chemotherapeutic drug doxorubicin (8,23–27).
We have been interested in trying to better understand how TGase-2 can give rise to contradictory cellular outcomes such as cell survival versus cell death. Recently, we have found that one possible explanation for these apparent discrepancies may arise from the alternative processing of the TGase-2 transcript (28). In fact, an alternative TGase-2 transcript was shown to be expressed in cells upon their treatment with cytokines, and interestingly, was also detected in the brains of Alzheimer’s patients (29–31). This transcript encodes a carboxyl-terminal truncated form of TGase-2 (lacking the last 138 amino acids) and has been designated TGase-S (for TGase-2-short-form). We showed that in various cell lines, TGase-S is capable of inducing the exact opposite effects on cell viability compared to its longer TGase-2 counterpart. In particular, while TGase-2 confers a survival advantage to different cells challenged with apoptotic agents, the shorter TGase-S protein caused these same cells to undergo cell death.
These findings lead to the obvious question of how the two forms of TGase-2 give rise to such diametrically opposed cellular effects? The carboxyl-terminal truncation that generates the TGase-S protein removes a portion of its guanine nucleotide-binding domain, as well as its binding site for phospholipase-Cδ1, and results in enhanced protein aggregation (28,32,33). Thus, in the present study, we have set out to determine whether the selective abrogation of TGase-2’s GTP-binding capability might be sufficient to give rise to apoptotic activity. In doing so, we have taken advantage of the detailed structural information provided by the X-ray crystal structure of GDP-bound TGase-2 (33), as well as other studies (34) that have suggested point mutations that would lead to the generation of GTP-binding-defective TGase-2 molecules. We have carefully characterized the ability of a number of different TGase-2 mutants to bind guanine nucleotides by using a convenient fluorescent spectroscopic assay (27). We also examined the abilities of the different TGase-2 mutants to exhibit constitutive, guanine nucleotide-independent, transamidation activity and their capabilities for hydrolyzing GTP. We then went on to examine specific GTP-binding-defective mutants, as well as a mutant that showed impaired GTP-hydrolytic activity, for their abilities to influence cell viability. Similar to the case for TGase-S, the expression of TGase-2 molecules that were defective for binding GTP could not be sustained in NIH 3T3 fibroblasts nor in a human cancer (Hela) cell line. Moreover, these same mutants induced cell death in a manner that was independent of their transamidation activity and solely attributable to their inability to bind GTP. These and other results described below lead us to conclude that the apoptotic activity exhibited by the TGase-S transcript is likely a result of its defective GTP-binding capability. They also point to an interesting example of how a dual function GTP-binding/acyl transferase (TGase-2) can be converted from a protective agent against apoptotic challenges into factor that induces cell death through the selective elimination of one of these activities.
EXPERIMENTAL PROCEDURES
Site-directed mutagenesis
A PCR-based method (Quik-change mutation kit, Stratagene) was used to generate the different TGase-2 mutants using a plasmid encoding wild-type TGase-2 as the template. A pair of oligonucleotide primers containing the desired mutation were used for the PCR reactions. The template plasmid DNA was linearized by DpnI digestion before transformation into Escherichia coli strain DH5α. Mutations were verified by DNA sequence analysis.
Expression and purification of recombinant wild-type TGase-2 and the different TGase-2 mutants
Wild-type TGase-2 and the individual TGase-2 mutant constructs were transformed into E. coli BL21(DE3) cells which were eventually harvested to purify the recombinant TGase-2 proteins using previously described methods (27).
Fluorescence measurements
Fluorescence measurements were performed using a Varian Eclipse Spectrofluorimeter. All experiments were carried out in 200 mM MOPS, pH 7.3, containing 2 mM DTT and 1 mM EDTA. BODIPY-FL-GTPγS was used as a GTP-analog for the fluorescence-binding assays. The excitation and emission wavelengths for BODIPY fluorescence were set at 504 nm and 520 nm, respectively.
GTP hydrolysis assays
The GTP-hydrolytic activity of wild-type TGase-2 or the TGase-2 mutants was assayed at room temperature using [γ32P]GTP in a buffer containing 200 mM MOPS, pH 7.3, 2 mM MgCl2, 100 M EDTA, and 2 mM DTT. The hydrolysis reaction was initiated by the addition of [γ32P]GTP (final concentration of 10 μM; specific activity = 5 Ci/mmol). Aliquots (50 μl) were removed at specific time points and added to 750 μL of 5% activated charcoal (neutralized) in 50 mM NaH2PO4. The samples were centrifuged at high speed and then aliquots from the supernatant (40 μL) were removed and counted.
Spectroscopic assay for transamidation activity
The transamidation activities of recombinant wild-type TGase-2 and the different TGase-2 mutant proteins were measured using a spectrophotometric assay originally developed by Day and Keillor (35) and as previously described (27).
In some cases, the transamidation activity of wild-type TGase-2 or the TGase-2 mutants expressed in mammalian cells was measured in cellular lysates. For these assays, whole cell extracts were incubated with transamidation reaction buffer (50 mM Tris-HCl, pH 7.5, 20 mM NaCl and 10 mM DTT) containing 1 mM 5-(biotinamido)pentylamine and 10 mM CaCl2 for 30 minutes and then the reaction mixtures were subjected to SDS-PAGE. The proteins were transferred to a PVDF membrane and blocked in BBST buffer (100 mM boric acid, 20 mM sodium borate, 0.01% SDS, 0.02% Tween 20, and 80 mM NaCl) containing 10% bovine serum albumin. The proteins that incorporated 5-(biotinamido)pentylamine were detected using horseradish peroxidase-conjugated streptavidin followed by exposure to ECL (Amersham/GE Health Care).
Cell culture and preparation of cell lysates
NIH 3T3 cells were grown in DMEM medium (Invitrogen) containing 10% calf serum. Hela cells were grown in MEM medium (Eagle) containing 10% fetal bovine serum. To express the different TGase-2 proteins in either of the cell lines, Myc-tagged pCDNA3 constructs encoding wild-type TGase-2 and the various TGase-2 mutants were generated and transfected into cells using Lipofectamine (Invitrogen). In order to prepare cell lysates, the cells were rinsed with phosphate-buffered saline (PBS) and lysed with cell lysis buffer (25 mM Tris-HCl, pH 7.2, 100 mM NaCl, 1 mM EDTA, 1 mM DTT, 1 mM PMSF, 1 mM Na3VO4, and 1 μg/mL each of aprotinin and leupeptin). To assess the expression of different TGase-2 constructs, equivalent concentrations of protein from the cell lysates were subjected to SDS-PAGE, and then the proteins were transferred to a PVDF membrane. The filters were blocked with TBST [20 mM Tris, pH 7.4, 137 mM NaCl, and 0.02% Tween 20] containing 10% non-fat dry milk, and then incubated with anti-Myc (Covance) or anti-actin (Sigma) antibodies diluted in TBST. The primary antibodies were detected with horseradish peroxidase-conjugated secondary antibodies followed by exposure to ECL (Amersham Biosciences).
Immunofluorescence and cell death assays
NIH 3T3 or Hela cells were plated on dual-chamber microscope slides (Nalge-Nunc) and cultured until they were 60% confluent. The cells were then transfected with the different Myc-TGase-2 constructs. Twenty-four or 36 hours following transfection, the NIH 3T3 cells were fixed with 3.7% formaldehyde. Similarly, Hela cells were fixed either 36 or 60 hours post-transfection. The transfectants were permeabilized with PBS containing 0.2% Triton X-100, blocked with PBS containing 5% bovine serum albumin, and then probed with either anti-Myc antibody or anti-AIF antibody (Upstate). The primary antibodies were detected with Oregon green 488- or Texas Red-conjugated secondary antibodies (Molecular Probes). The slides were also incubated with DAPI (Sigma) to stain nuclei, and the cells were then visualized by fluorescence microscopy. Cells undergoing apoptosis were identified by nuclear condensation or blebbing, and the percentage of cell death was determined by calculating the ratio of apoptotic to non-apoptotic cells for each condition.
RESULTS
Generation of GTP-binding-defective mutants of human TGase-2
The goal of this study was to gain insight into the molecular basis for the cell death caused by TGase-S, and in particular, to establish whether the loss of GTP-binding capability was sufficient to enable the human TGase-2 protein to mimic the actions of TGase-S. Thus, the first step was to generate point-mutants of TGase-2 that were defective for binding GTP. Figure 1A shows the guanine nucleotide-binding site for TGase-2 based on the X-ray crystal structure for the GDP-bound form of the protein (33). Although the guanine nucleotide-binding domain of TGase-2 is conserved among other transglutaminase family members that are capable of binding GTP (Figure 1B), it is clearly distinct from that of the more traditional (large and small) G-proteins that function in cellular signal transduction. For example, while both large and small G-proteins share a highly conserved P-loop that binds to the phosphate moiety of the guanine nucleotide, TGase-2 has a series of positively charged residues that appear to perform this function. Three residues in particular, Arg 476, Arg 478, and Arg 580, are positioned to make critical contacts with the phosphate groups of GDP through their side chains (Figure 1A), and indeed, they have been suggested to be important for the ability of the rat TGase-2 protein to bind guanine nucleotides (34). Therefore, we made amino acid substitutions for these residues in the human TGase-2 protein (denoted in orange in Figure 1B).
Figure 1.
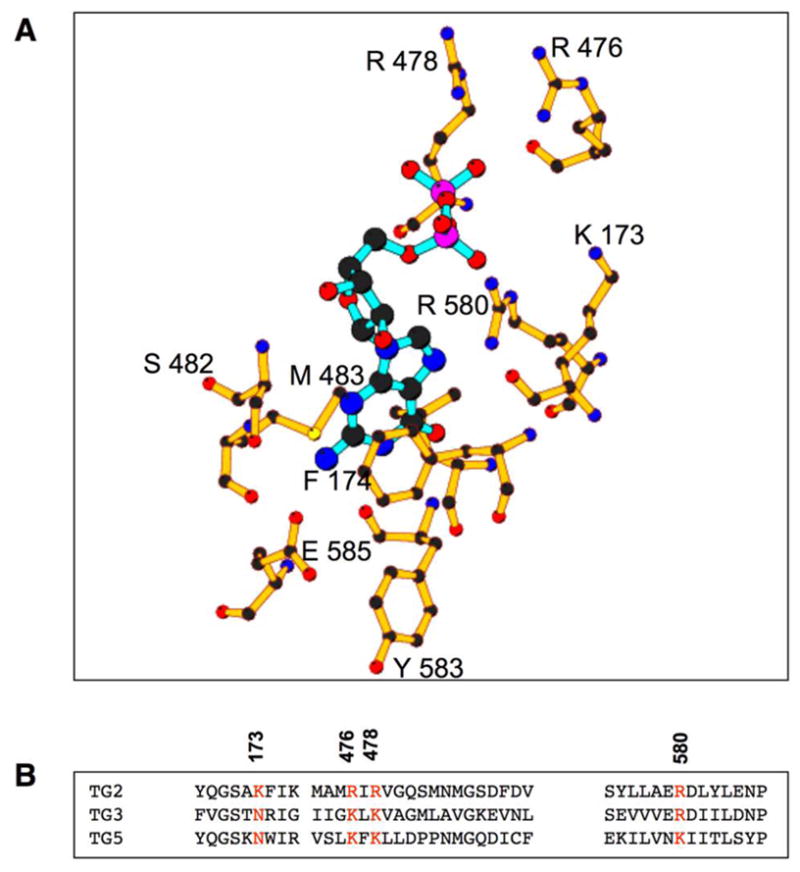
The guanine-nucleotide binding site on human TGase-2. (A) TGase-2 residues which interact with the guanine nucleotide (GDP-shown in blue) are represented by ball and stick. (B) Sequence alignments using ClustalW (59) of the guanine nucleotide-binding sites of TGase-2 and other members of the transglutaminase family that bind guanine nucleotides. The numbered residues in orange represent the sites mutated in TGase-2.
In order to verify that these different amino acid substitutions in fact compromised the GTP-binding activity of human TGase-2, we expressed histidine (His)-tagged forms of wild-type TGase-2 and the different point-mutated TGase-2 proteins in E. coli and purified them through a series of chromatographic steps beginning with nickel affinity chromatography and then followed by anion-exchange and size exclusion chromatography, as previously described (27). Figure 2A shows the Coomassie blue-stained SDS gel profile for wild-type TGase-2 and some representative examples of the mutants used in this study. Unlike the case for the TGase-S protein (28), none of the TGase-2 point-mutants showed detectable aggregation during their purification (data not shown). We then examined their GTP-binding capability by using a spectroscopic assay that takes advantage of the fluorescence properties of the GTP-analog, BODIPY-FL GTPγS (BOD-GTPγS). Previously, we demonstrated that BOD-GTPγS was capable of undergoing a rapid binding interaction with wild-type human TGase-2, resulting in an ~ 2-fold increase in the fluorescence emission from the BODIPY fluorophore (27). Figure 2B shows a plot of the relative change in the fluorescence emission of BOD-GTPγS, which accompanies its binding to the recombinant wild-type TGase-2 protein, as a function of the concentration of the fluorescent nucleotide-analog. The binding of BOD-GTPγS to wild-type TGase-2 is dose-dependent and can be completely reversed upon the addition of GTPγS (Figure 2C). This confirms our earlier finding that BOD-GTPγS binds to the guanine nucleotide-binding site on the protein (27).
Figure 2.
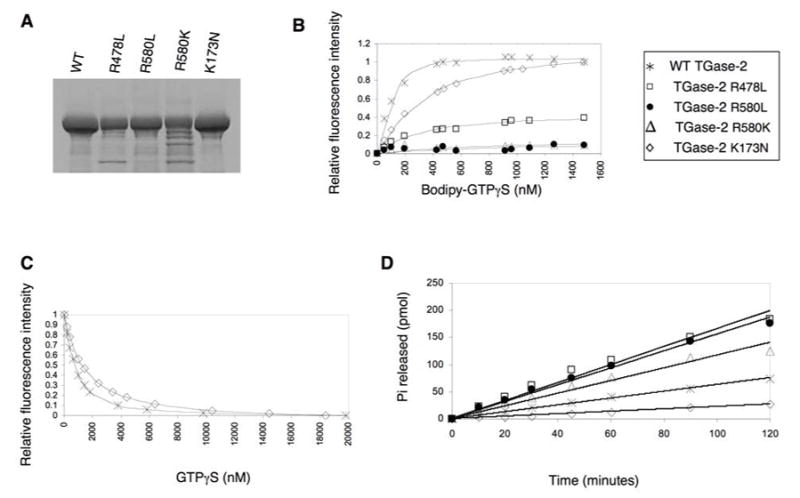
Assays comparing the guanine nucleotide-binding and GTP-hydrolytic activities of wild-type TGase-2 and the different TGase-2 mutants. (A) SDS-PAGE analysis of purified E. coli recombinant, His-tagged forms of human wild-type TGase-2 (WT) and the indicated TGase-2 mutants. The gel was stained with Coomassie-blue to visualize the recombinant proteins. (B) The binding of BOD-GTPγS to the various recombinant forms of TGase-2. The recombinant TGase-2 proteins (300 nM) were dissolved in a buffer containing 200 mM MOPS, pH 7.3, 1 mM EDTA, and 2 mM DTT. Increasing concentrations of BOD-GTPγS were incubated with the proteins at room temperature for 1 hour and then the changes in the BOD-GTPγS fluorescence emission at 520 nm (excitation = 504 nm) were determined. (C) Competition between BOD-GTPγS and GTPγS as read-out by changes in BODIPY fluorescence. Wild-type TGase-2 (300 nM) or the TGase-2(K173N) mutant (300 nM) was incubated with 1.8 μM BOD-GTPγS alone or in the presence of increasing concentrations of GTPγS. Each addition of GTPγS was followed by a 2 minute incubation and then changes in fluorescence were measured. The changes in fluorescence were plotted against different GTPγS concentrations and fit to a competitive inhibition curve (27). (D) Wild-type TGase-2 (9 μM) and the indicated TGase-2 mutants (9 μM, each) were assayed for GTP-hydrolytic activity in a buffer containing 200 mM MOPS (pH 7.3), 2 mM MgCl2, 100 μM EDTA, and 2 mM DTT. Assays were initiated by the addition of [γ32P]GTP, and the production of 32Pi (product) was measured as described in “Experimental Procedures”.
While the TGase-2(R476L) mutant was able to bind guanine nucleotides with affinities similar to, although slightly weaker than, those of the wild-type protein (Table 1), the TGase-2(R478L) mutant bound guanine nucleotides much more weakly than wild-type TGase-2, whereas the TGase-2(R580L) mutant showed little detectable binding of either the fluorescent nucleotide-analog or GTPγS (Figure 2B and Table 1). Even the conservative substitution of a lysine residue for the normal arginine at position 580 [TGase-2(R580K)] resulted in a marked reduction in guanine nucleotide-binding capability. [35S]GTPγS-binding assays were performed in parallel with the fluorescence assays and yielded essentially identical results (data not shown).
Table 1.
Ability of Wild-type TGase-2 and Different TGase-2 Mutants to Bind BODIPY-GTPγS and GTPγS
| Protein | Apparent Dissociation Constant for BOD-GTPγS (nM)a | Apparent Dissociation Constant for GTPγS (nM) |
|---|---|---|
| Wild-type | 400 ± 13 | 80 ± 6 |
| R476L | 850 ± 30 | 200 ± 10 |
| R476K | 500 ± 15 | 50 ± 4 |
| R478L | ND | ND |
| R478K | 500 ± 25 | 55 ± 6 |
| R580L | ND | ND |
| R580K | ND | ND |
| K173L | 388 ± 20 | 250 ± 20 |
| K173R | 345 ± 12 | 70 ± 5 |
| K173N | 388 ± 20 | 120 ± 9 |
The apparent dissociation constants for the binding of BOD-GTPγS to wild-type TGase-2 and the listed mutants were determined from the best fits to the titration data (Figure 2B) for a simple bimolecular interaction model. ND indicates that the mutant showed little or no detectable binding of BOD-GTPγS.
The ability of varying concentrations of GTPγS to compete with BOD-GTPγS for binding to wild-type TGase-2 and the indicated TGase-2 mutants was assessed as described in the legend of Figure 2C. The apparent dissociation constants were determined from the best fits to the titration data assuming a simple competition between BOD-GTPγS and GTPγS, as previously described (27).
Identifying a residue in TGase-2 that is necessary for maximal GTP-hydrolytic activity
In addition to generating GTP-binding-defective forms of TGase-2, we also identified a TGase-2 mutant that was fully capable of binding guanine nucleotides, while being impaired in its ability to hydrolyze GTP. Although TGase-2 lacks the classical “arginine-finger motif” that is found within the α subunits of large G-proteins and plays a critical role in their ability to hydrolyze GTP, it exhibits measurable GTP-hydrolytic activity (27,36–38). The X-ray structure for TGase-2 (Figure 1A) shows that Lys173 might be in position to help catalyze GTP hydrolysis. Thus, we changed this lysine residue either to alanine, asparagine or arginine. Each of these TGase-2 mutants was able to bind guanine nucleotides with affinities similar to that measured for wild-type TGase-2 (Figures 2B and 2C, and Table 1). The GTP-hydrolytic activities for the recombinant wild-type TGase-2 and the different Lys173 mutants were then compared by assaying the rate of [32Pi] release from each of the recombinant proteins, following their loading with [γ32P]GTP. While the TGase-2(K173A) and TGase-2(K173R) mutants exhibited rates of GTP hydrolysis that were similar to or even slightly faster (~2 fold) than that measured for the wild-type protein, the TGase-2(K173N) mutant showed significantly reduced GTP-hydrolytic activity (Figure 2D). Likewise, earlier studies using rat TGase-2 showed that the corresponding K173N mutant was significantly impaired in its ability to hydrolyze GTP (39). Interestingly, while the TGase-2(R478L), TGase-2(R580L) and TGase-2(R580K) mutants showed very low affinity for GTP, they were still able to hydrolyze GTP at rates that were comparable to, if not faster than, that of the wild-type protein. A similar observation was recently reported when examining two isoforms of TGase-2 that either lacked 13 or 43 amino acids from the carboxyl-terminus, as each of these TGase-2 isoforms showed very weak affinity for guanine nucleotides but nonetheless exhibited enhanced GTP-hydrolytic activity (40).
The guanine nucleotide-binding-defective and GTP hydrolysis-defective TGase-2 mutants exhibit full transamidation capability
Figure 3A shows that each of the guanine nucleotide-binding-defective mutants were capable of catalyzing crosslinking activity, ranging from ~80% to ~125% of that measured for wild-type TGase-2. Consistent with previous reports (27,38,41,42), the crosslinking activity of wild-type TGase-2 was progressively inhibited by increasing levels of GTPγS (Figure 3B). However, the TGase-2(R478L) mutant, which binds guanine nucleotides with weak affinity (see Figure 2A), showed only a subtle decrease in its transamidation activity when incubated with GTPγS. Moreover, the transamidation activities of those TGase-2 mutants which were most ineffective in binding guanine nucleotides, namely TGase-2(R580L) and TGase-2(R580K), showed no detectable sensitivity to GTPγS (Figure 3B) nor to other guanine nucleotides and, therefore, were insensitive to the usual nucleotide-regulation exhibited by wild-type TGase-2. The GTP hydrolysis-defective TGase-2(K173N) mutant was also still capable of catalyzing transamidation and in fact typically yielded activities that were even higher than those measured for the wild-type protein (Figure 3A; also see Figure 4B, below). However, unlike the GTP-binding-defective mutants, the crosslinking activity for the GTP hydrolysis-defective K173N mutant was susceptible to negative regulation by guanine nucleotides (e.g. GTPγS, see Figure 3B).
Figure 3.
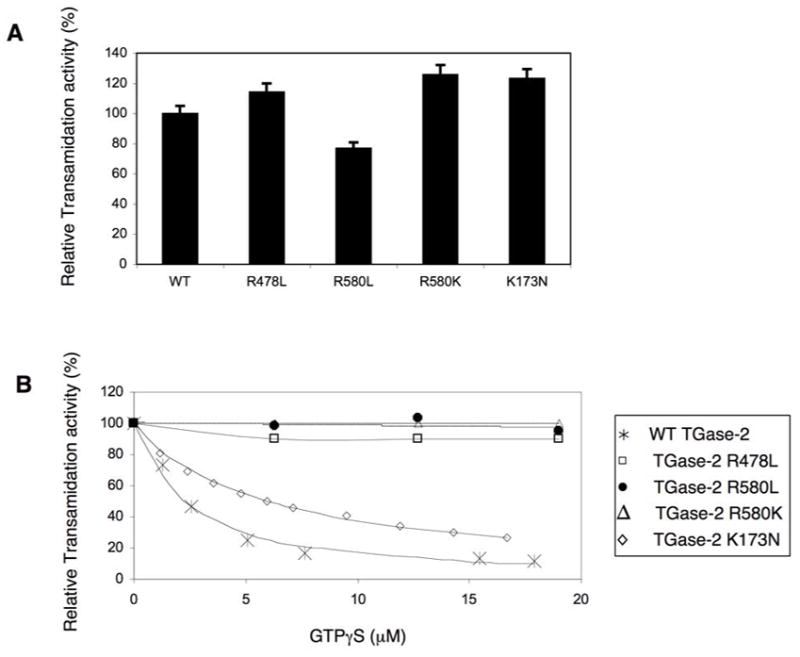
All of the TGase-2 mutants retain transamidation activity. (A) Relative transamidation activities of the recombinant wild-type TGase-2 protein (WT) and different GTP-binding- and GTP hydrolysis-defective forms of TGase-2 (600 nM, each) were measured for 30 minutes at room temperature using a colorimetric assay as previously described (27). (B) Increasing concentrations of GTPγS were incubated with wild-type TGase-2 or the indicated TGase-2 mutants (600 nM, each) prior to assaying transamidation activity.
Figure 4.
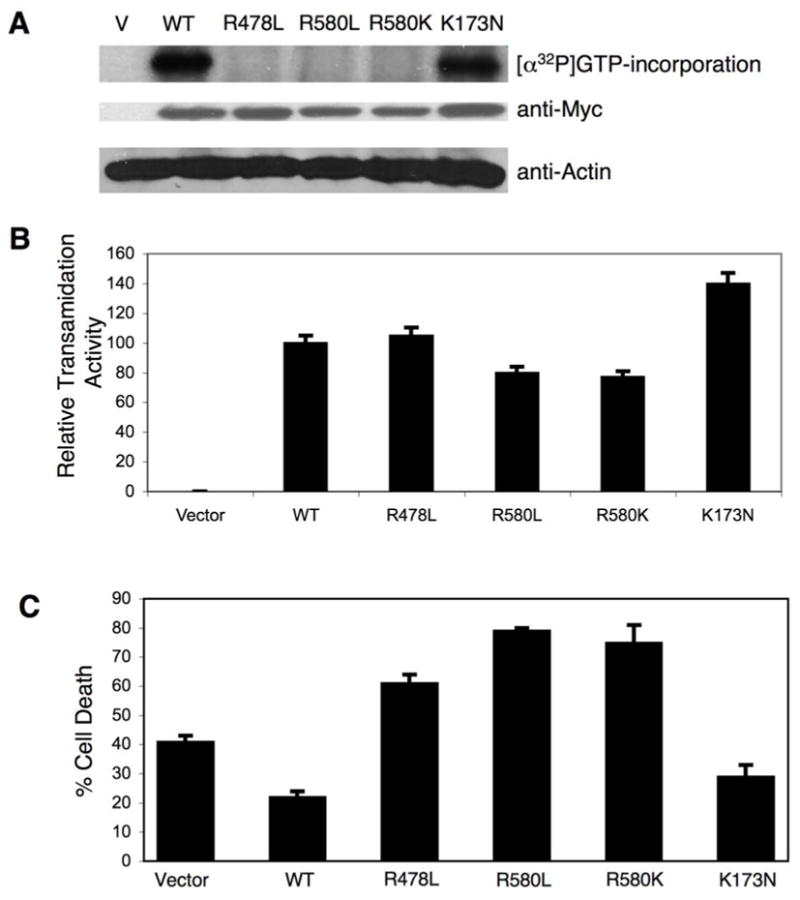
Functional activities of wild-type TGase-2 and different TGase-2 mutants expressed in cells. (A) NIH 3T3 cells expressing Myc-tagged forms of wild-type TGase-2 (WT) and the indicated TGase-2 mutants, and cells transfected with vector (V) alone, were grown for 12 hours and then lysed. Each lysate (~50 μg protein) was then assayed for photo-incorporation of [α32P]GTP as previously described (8,38). Western blot analysis using an anti-Myc antibody was also performed on cell extracts to assess the relative expression levels of each of the TGase-2 constructs. Actin served as a loading control. (B) Lysates from cells transfected with vector alone, and from cells expressing wild-type TGase-2 and the different TGase-2 mutants, were assayed for transamidation activity as determined by the incorporation of 5-biotinamido pentylamine into the lysate proteins as described in “Experimental Procedures”. (C) Comparisons of the protective effects conferred by wild-type TGase-2 and different TGase-2 mutants against serum starvation-induced cell death. NIH 3T3 cells expressing Myc-tagged forms of wild-type TGase-2 (WT) and the indicated TGase-2 mutants, or cells transfected with vector alone, were allowed to grow in medium containing 10% serum for 12 hours, and then the cells were serum-starved for 24 hours. The cells were then fixed and prepared for immunofluorescence microscopy to assay for apoptosis as described in “Experimental Procedures”. The average percentage of cell death is plotted for each of the conditions and the error bars indicate standard deviations.
The GTP-binding-defective and GTP hydrolysis-defective forms of TGase-2 are capable of catalyzing transamidation activity when expressed in cells
We next wanted to verify that the different GTP-binding-defective TGase-2 mutants described above, when expressed in cells, still lacked the ability to bind guanine nucleotides but were capable of catalyzing transamidation reactions. Myc-tagged forms of wild-type TGase-2 and the different TGase-2 mutants were ectopically expressed in NIH 3T3 cells and then twelve hours post-transfection, we examined their GTP-binding capability in cell lysates as assessed by the photo-incorporation of [α32P]GTP (11,38), as well as their transamidation activity. The expression of the different mutants during this time period was detected by Western blot analysis of the cell lysates with anti-Myc antibody (Figure 4A). As expected, both wild-type TGase-2 and the GTP-hydrolysis-defective TGase-2(K173N) mutant were capable of incorporating [α32P]GTP (Figure 4A), whereas the TGase-2(580L) and TGase-2(580K) mutants showed no detectable incorporation of the radiolabeled GTP, consistent with their inability to show measurable guanine nucleotide-binding activity in the fluorescence assays. The TGase-2(R478L) mutant also showed no detectable incorporation of [α32P]GTP following its expression in cells, even though the purified recombinant protein exhibited weak binding activity in the fluorescence assays (Figure 2A).
Each of the mutants showed crosslinking activity in cell lysates, as read-out by their ability to catalyze the incorporation of biotinylated pentylamine into lysate proteins (Figure 4B). Interestingly, we again found that the GTP hydrolysis-defective TGase-2(K173N) mutant gave rise to increased crosslinking activity, suggesting that this substitution in some way confers a positive influence on its ability to catalyze transamidation reactions.
We had earlier shown that wild-type TGase-2 protects NIH 3T3 cells from cell death caused by serum starvation (43). Thus, we compared the abilities of the different mutants to provide a similar protective effect. The wild-type TGase-2 and the different TGase-2 mutants were expressed in NIH 3T3 cells for 12 hours and then the cells were cultured in the absence of serum for another 24 hours. The ratio of dead to healthy cells was determined by immunofluorescence microscopy (see “Experimental Procedures”) and then plotted as histograms (Figure 4C). It is clear from the data that while wild-type TGase-2 and the GTP hydrolysis-defective TGase-2(K173N) mutant were able to provide protection against cell death caused by serum starvation, each of the GTP-binding-defective mutants was unable to provide a protective effect and in fact significantly enhanced apoptosis under these conditions.
The expression levels of the GTP-binding-defective TGase-2 mutants decrease with time
In our studies of the TGase-S protein, we found that there was a decrease in its expression over time (12–48 hours) in NIH 3T3 (28). The decreased expression of TGase-S was shown to be due to a selective pressure as an outcome of its apoptotic activity. Interestingly, we found that the guanine nucleotide-binding-defective TGase-2 mutants exhibited similar time-dependent changes in their expression levels. Figure 5A shows the results of experiments performed in NIH 3T3 cells, where we transiently transfected the indicated TGase-2 constructs and then lysed the cells and examined their expression by Western blot analysis using anti-Myc antibody after different periods of time. Both wild-type TGase-2 and the GTP hydrolysis-defective TGase-2(K173N) mutant were robustly expressed 12 hours post-transfection, with their expression levels then being maintained through 36 hours post-transfection. In contrast, the GTP-binding-defective TGase-2 mutants showed very different expression profiles. Specifically, the expression levels of Myc-TGase-2(R478L), Myc-TGase-2(R580L), and Myc-TGase-2(R580K) were highest at 12–24 hours post-transfection and then underwent a time-dependent reduction. These effects were not limited to NIH 3T3 cells, as the GTP-binding-defective forms of TGase-2 also showed a time-dependent decrease in their expression levels in the human cervical carcinoma (Hela) cell line. However, the time-courses were shifted such that the expression of the GTP-binding-defective mutants in Hela cells typically began to decline between 36 and 60 hours post-transfection (Figure 5B), compared to a decline between 24 and 36 hours in NIH 3T3 cells.
Figure 5.
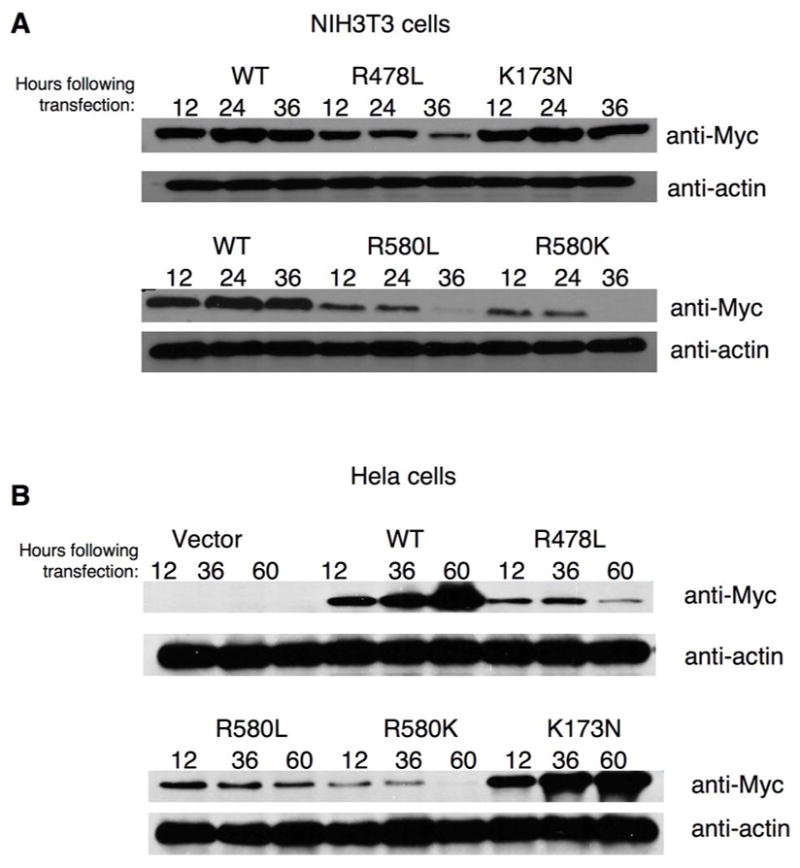
Time-dependent expression profiles for wild-type TGase-2 and the different TGase-2 mutants in cells. (A) NIH 3T3 cells and (B) Hela cells transiently expressing either vector alone, Myc-tagged wild-type TGase-2 (WT), or the different Myc-tagged TGase-2 mutants for the indicated times, were lysed and then equivalent amounts of protein from each of the cell lysates were subjected to Western blot analysis.
The GTP-binding-defective forms of TGase-2 induce cell death
We then examined whether the selective pressure against the expression of the GTP-binding-defective forms of TGase-2 in cells was due to these mutants having deleterious effects on cell viability. We first examined NIH 3T3 cells for cell death, as indicated by nuclear condensation and blebbing (also, see Figure 9C, below), under conditions where the cells still exhibited significant expression of the GTP-binding-defective mutants (i.e. 24 hours following their transient expression). Figure 6A shows that the cells expressing the GTP-binding-defective mutants at this time point were already showing significant cell death, compared to vector control cells and cells expressing wild-type TGase-2 or the GTP hydrolysis-defective TGase-2(K173N) mutant. The GTP-binding-defective mutants had very similar effects in Hela cells (Figure 6B), although we examined them 36 hours after transfection because they showed a shifted time-course for the decline in the expression of the GTP-binding-defective mutants, compared to NIH 3T3 cells (Figures 5A and 5B).
Figure 9.
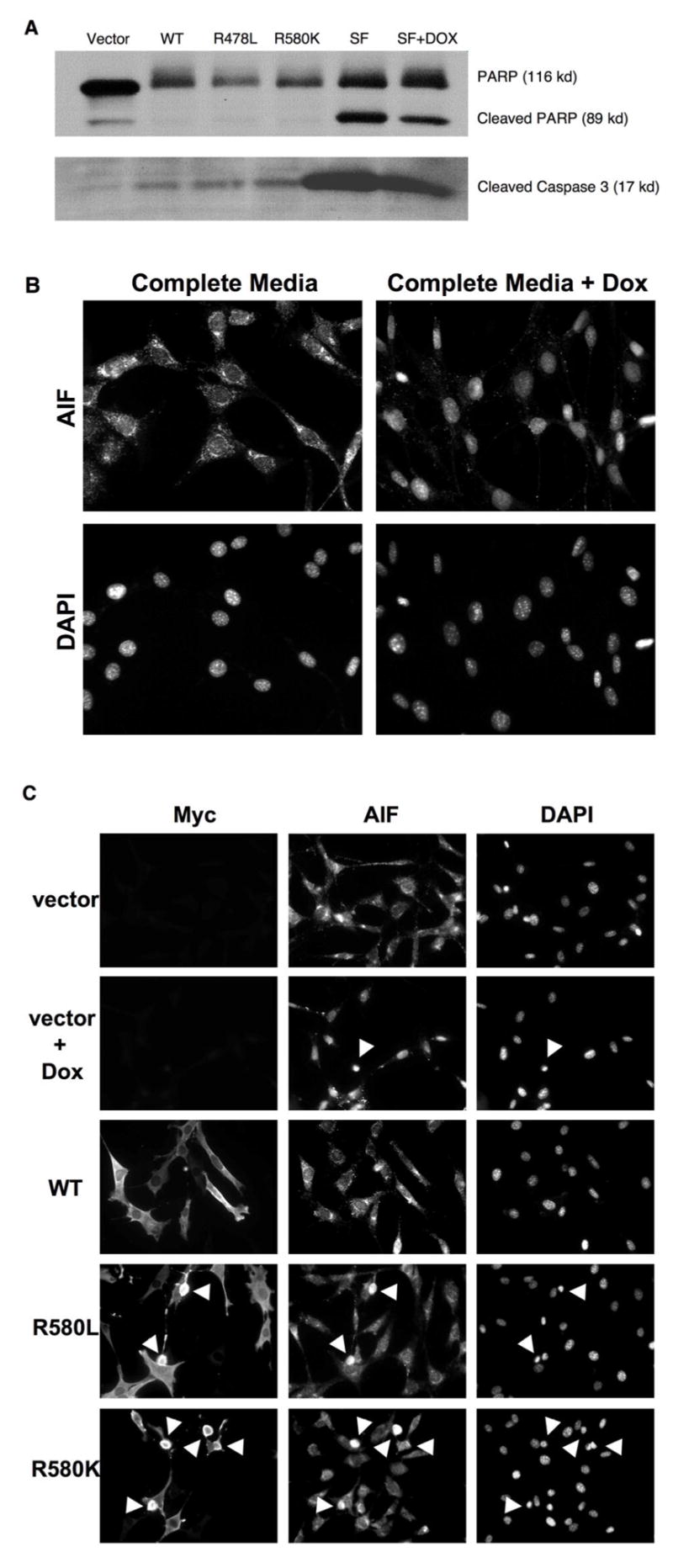
GTP-binding-defection TGase-2 mutants induce cell death via a caspase-independent mechanism. (A) NIH 3T3 cells transfected with vector alone, or cells expressing wild-type Myc-TGase-2 and the indicated TGase-2 mutants, were grown in complete medium (medium containing 10% serum) for 24 hours post-transfection and then lysed. Additionally, cultures of NIH 3T3 cells were either maintained in just serum-free (SF) media or serum-free medium plus 1.0 μM doxorubicin (SF + DOX) for about 30 hours and then lysed. Caspase activity in the different cell lysates was assayed using an antibody that detects activated caspase-3 and by the proteolytic cleavage of PARP. (B) NIH 3T3 cells were cultured in complete media or complete media plus 1.0 μM doxorubicin (DOX) for 24 hours and then fixed. Immunofluorescence using anti-AIF antibody and DAPI was performed on the cells. Note the appearance of AIF in the nuclei of cells treated with doxorubicin. (C) Cells transfected with the vector alone, cells transfected with the vector and then exposed to 1.0 μM doxorubicin (DOX), or cells transfected with the indicated Myc-tagged TGase-2 constructs were maintained in complete medium (medium containing 10% serum) for 24 hours and then were fixed. Immunofluorescence was performed on the cells using anti-Myc and anti-AIF antibodies and DAPI was used to stain the nuclei. Cells containing nuclei which are condensed/blebbed are indicated with arrows.
Figure 6.
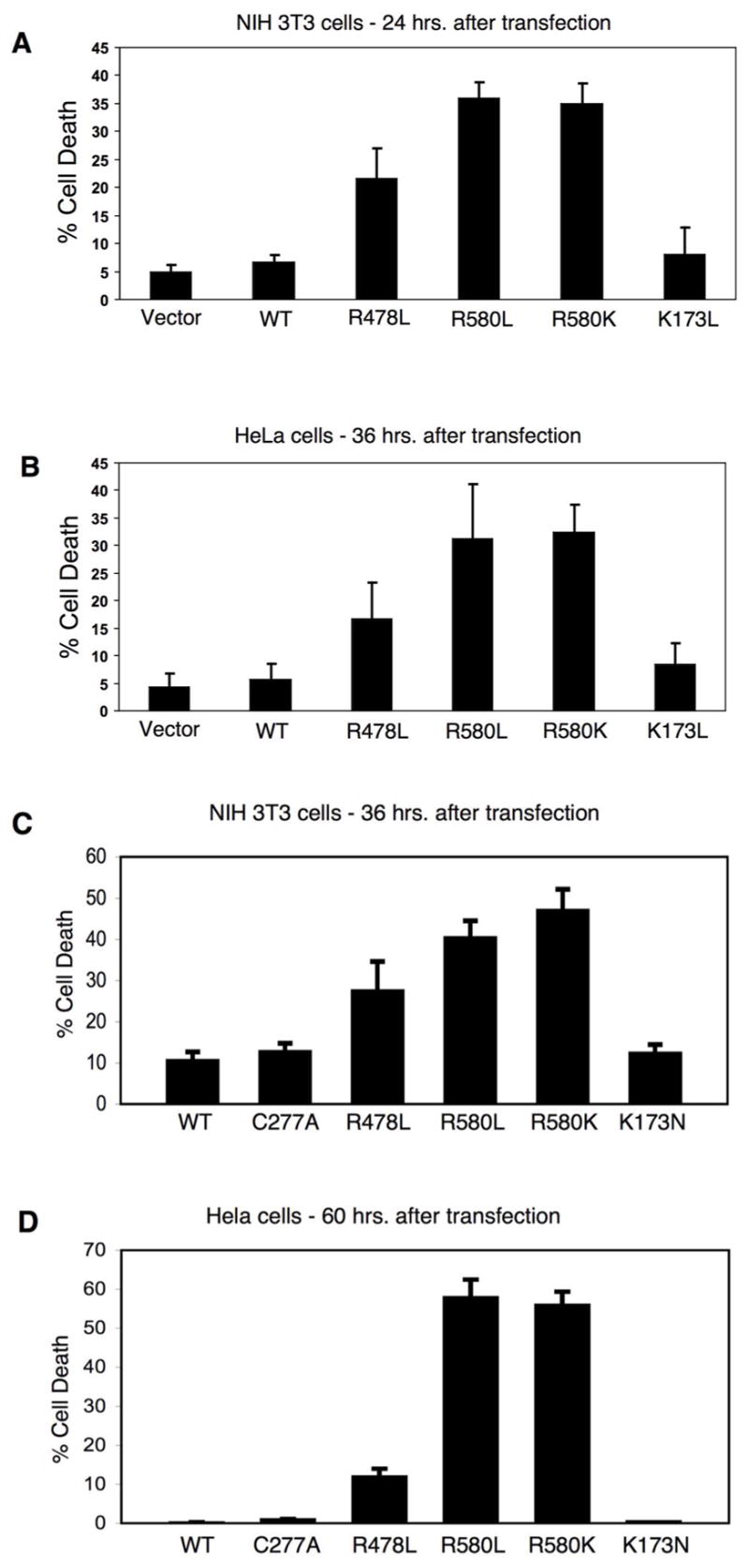
GTP-binding-defective TGase-2 mutants induce cell death. (A, C) NIH 3T3 cells and (B, D) Hela cells were transiently transfected with vector alone or with one of the indicated Myc-tagged TGase-2 constructs and maintained in complete medium (medium containing 10% serum). NIH 3T3 cells were fixed either 24 hours (A) or 36 hours (C) after transfection, while the Hela cells were fixed 36 hours (B) or 60 hours (D) post-transfection. Immunofluorescence using anti-Myc antibody and DAPI (to stain nuclei) was performed on the cells, and nuclear condensation or blebbing was used to detect cell death. The average percentage of cell death was plotted for each of the conditions and the error bars indicate standard deviations.
We have also performed a similar analysis at longer time points where we had observed a pronounced decline in the levels of expression of the GTP-binding-defective mutants (i.e. after 36 hours in NIH 3T3 cells and 60 hours in Hela cells). In those cells where we were still able to detect the expression of the Myc-tagged GTP-binding-defective TGase-2 mutants based on immunofluorescence, there was a greater degree of cell death compared to what we quantified after 24 hours, whereas wild-type TGase-2 and the TGase-2(K173N) mutant again caused little or no cell death under these conditions (Figures 6C and 6D).
Cell death caused by the GTP-binding-defective TGase-2 mutants is not due to uncontrolled crosslinking of cellular proteins nor to caspase activation
We wanted to make certain that the apoptotic activity of the mutants can be attributed to their inability to bind GTP rather than being due to their un-regulated transamidation activity. Thus, we introduced transamidation-defective substitutions into the background of the nucleotide-binding-defective TGase-2(R580L) and R580K mutants. These double-mutants were then expressed in both normal fibroblasts and Hela cells. Figures 7A and 7B show the expression profiles for these double-mutants along with wild-type TGase-2 and the different point-mutants. Both double-mutants showed expression profiles like those for the TGase-2(R580L) and TGase-2(R580K) mutants, as well as caused similar extents of cell death in NIH 3T3 and Hela cells (Figures 8A and 8B, respectively). Thus, the cell death caused by the GTP-binding-defective TGase-2 mutants cannot be attributed to uncontrolled transamidation reactions.
Figure 7.
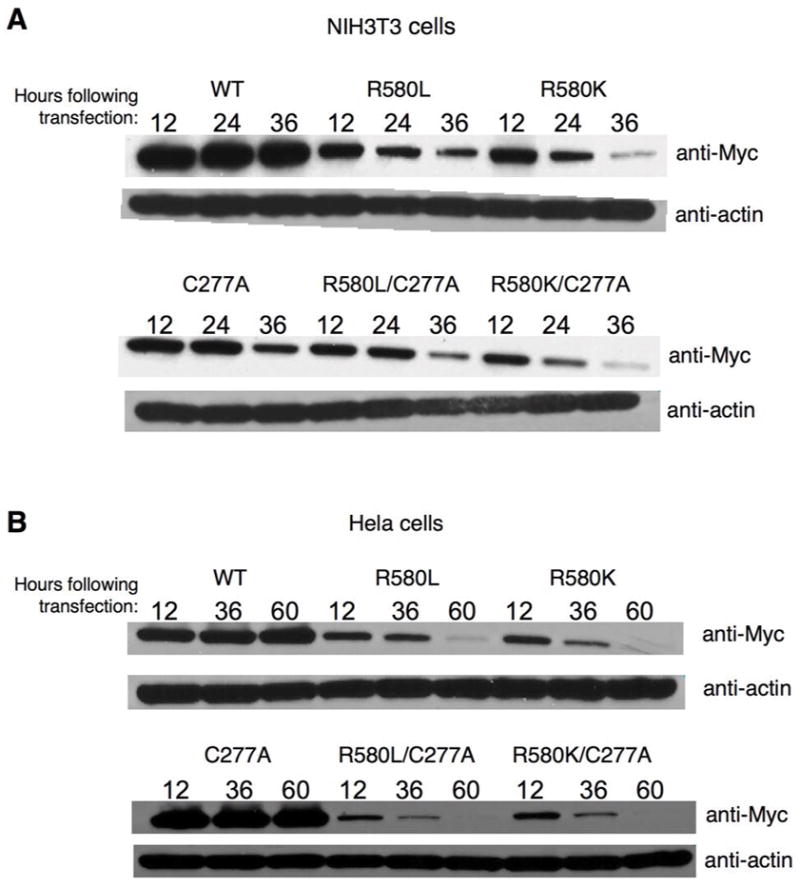
Time-dependent expression profiles for TGase-2 double-mutants defective for both GTP-binding and transamidation activity. Myc-tagged forms of wild-type TGase-2 (WT), the transamidation-defective TGase-2(C277A) mutant, and the various single- and double-mutants of TGase-2 were transiently expressed in (A) NIH 3T3 cells and (B) in Hela cells for the indicated times and then lysed. Equivalent amounts of protein from each of the cell lysates were used for Western blot analysis with anti-Myc and anti-actin antibodies.
Figure 8.
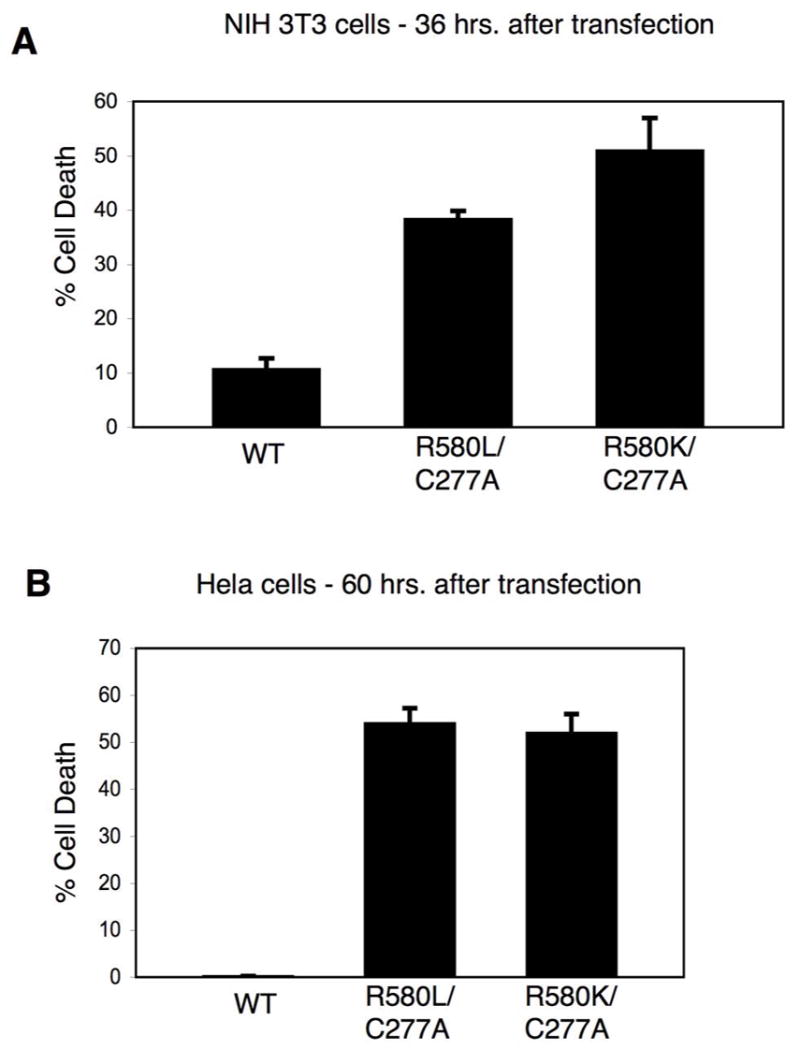
The apoptotic activity of the GTP-binding-defective TGase-2 mutants is not due to their constitutive transamidation activity nor to the activation of caspases. (A) NIH 3T3 cells and (B) Hela cells transiently expressing Myc-tagged wild-type TGase-2 (WT) and the indicated Myc-TGase-2 double-mutants were grown for 36 and 60 hours, respectively, and then fixed and assayed for cell death as described in Figures 6C and 6D. The average percentage of cell death is plotted for each of the conditions and the error bars indicate standard deviations.
We wondered whether the GTP-binding-defective TGase-2 mutants might stimulate apoptotic-signaling pathways that culminate in caspase activation. However, this does not appear to be the case. Figure 9A shows that the expression of GTP-binding-defective TGase-2 mutants in cells, under conditions where they gave rise to cell death, was not accompanied by caspase activation, either as read-out by Western blotting using an antibody that recognizes activated caspase-3 or as detected by degradation of the caspase-substrate PARP. This contrasts with the apoptotic responses of cells exposed to different serum-free conditions (i.e. in the presence and absence of doxorubicin), which result in caspase-activation and the cleavage of PARP.
We have also compared the ability of doxorubicin, versus the expression of the GTP-binding-defective TGase-2 mutants, to cause the accumulation of the apoptosis-inducing factor protein (AIF) in the nucleus, i.e. as an outcome of its release from mitochondria (44). Figure 9B provides an example of this response following the treatment of NIH 3T3 cells with doxorubicin for 24 hours. There is a clear change in the cellular distribution of AIF upon treatment with the drug, such that it is primarily detected in the nuclei of the cells. This change in AIF localization can be seen in virtually all of the cells treated with doxorubicin, even though only a percentage of these cells are starting to show blebbing and condensation (see Figure 9B, the top panels, and Figure 9C, the top row and the second row from the top). However, we did not detect the same nuclear accumulation of AIF in cells expressing the GTP-binding-defective TGase-2 mutants. Figure 9C (the bottom row, and the row second from the bottom) shows that the cellular localization of AIF in the cells expressing the TGase-2 mutants was indistinguishable from cells expressing wild-type TGase-2 (Figure 9C, middle row), even though the cells that are expressing the mutants are starting to undergo nuclear blebbing and condensation, similar to those treated with doxorubicin. Taken together, these findings show that the GTP-binding-defective TGase-2 mutants do not use the same mechanism to induce cell death as used by doxorubicin or serum starvation.
DISCUSSION
A number of lines of evidence now show that TGase-2 plays an important role in ensuring the survival of different cells, including various types of cancer cells, against cellular stresses and chemotherapeutic challenges (8,11,23–37). Most of the work done thus far seems to indicate that the transamidation activity of TGase-2 is essential for its ability to confer cell survival (11,24,27). However, there also have been a number of reports suggesting that TGase-2 may play a direct role in cell death, dependent upon its cellular localization or under conditions where it is aberrantly expressed and/or its transamidation activity is de-regulated (4,6,7,12,16–21,45). These contradictory results have led to a good deal of confusion regarding the roles played by TGase-2 in cell survival versus apoptosis. Recently, we found that some of the conflicting actions of TGase-2 might be attributed to the existence of multiple transcripts that exhibit distinct functional properties (28). In particular, we showed that expressing wild-type TGase-2 in various cells, versus a carboxyl-terminal truncated form called TGase-S, had exactly the opposite effects on cell viability. While the full-length TGase-2 protein conferred protection against different apoptotic stimuli, its shorter counterpart gave rise to a strong apoptotic response. There was an apparent selective pressure against the expression of TGase-S in cells, and we showed that the apoptotic factor TNF-α selectively up-regulated its expression, while knocking down the expression of TGase-S inhibited TNF-α-induced apoptotic activity. Interestingly, TGase-S expression was originally detected in Alzheimer’s patients (29–31), raising the intriguing possibility that it might directly contribute to the neurotoxicity that accompanies this disease.
These findings lead to the obvious question of how the shorter form of TGase-2 exhibits such markedly deleterious effects on cell viability. At the time of our original study, there seemed to be at least two plausible possibilities. One was that the ability of TGase-S to induce cell death might be related to its unusual propensity to undergo higher order oligomerization (28), although this is difficult to test directly because it requires being able to manipulate the oligomerization of the protein in a highly defined manner, which is not yet possible to do. A second possibility was that the actions of TGase-S were the outcome of its inability to bind GTP, because the carboxyl-terminal truncation disrupts the guanine nucleotide-binding site (33). We were in position to test this possibility by seeing whether the selective abrogation of the GTP-binding activity of TGase-2 gives rise to a similar capability to induce cell death as seen with its shorter isoform. Indeed, we were able to take advantage of the structural information available for the guanine nucleotide-binding site of TGase-2, together with methods for directly assaying guanine nucleotide-binding and GTP hydrolysis, to generate mutants that were selectively defective for GTP-binding or hydrolytic activity.
We first showed that unlike the wild-type TGase-2 protein, when TGase-2 mutants significantly compromised in their ability to bind GTP were transiently expressed in NIH 3T3 cells, they were unable to protect against cell death caused by serum-deprivation. We then demonstrated that like the TGase-S protein, the GTP-binding-defective mutants of TGase-2 exhibited a time-dependent decrease in their expression, and in a corresponding fashion, these mutants also induced cell death. The effects of the GTP-binding-defective TGase-2 mutants cannot be trivially explained by the mis-folding of these proteins. Each of these mutants is soluble and does not show detectable aggregation either when examined in vitro or in cells; moreover, they are capable of relatively strong transamidation activity. In fact, we wondered whether the guanine nucleotide-binding-defective TGase-2 mutants caused cell death as an outcome of their uncontrolled crosslinking (transamidation) activity. Under normal cellular conditions, a significant percentage of the endogenous (wild-type) TGase-2 molecules contains either bound GTP or GDP, and consequently would exhibit a suppressed crosslinking activity. On the other hand, the nucleotide-binding-defective TGase-2 mutants should be capable of constitutive transamidation activity. However, we have been able to clearly establish that the ability of these mutants to induce cell death is not dependent on their capability to catalyze transamidation reactions, because double-mutants that were defective for both guanine nucleotide-binding and crosslinking activity gave rise to equally strong responses.
These findings argue that the selective impairment of the GTP-binding activity of TGase-2 is sufficient to convert this protein from an agent that protects cells against apoptotic challenges into a cell death-inducing factor. They also suggest that the ability of TGase-S to induce cell death can likely be attributed to its inability to bind GTP. We cannot completely rule out the possibility that the increased tendency of TGase-S to form higher oliogomers also contributes to cell death. However, because we have not detected higher-order oligomerization with any of the GTP-binding-defective mutants of TGase-2, either by non-denaturing gel electrophoresis, gel filtration chromatography, or immunofluorescence, it does not appear to be necessary to invoke oligomerization in the cellular actions of TGase-S. Indeed, it would seem more likely that this represents an independent phenomenon that occurs as an outcome of the carboxyl-terminal truncation.
Clearly, an important question concerns how the loss of GTP-binding activity enables TGase-2, as well as TGase-S, to induce cell death. At present, the only known target/effector for GTP-bound TGase-2 is phospholipase C-δ1 (32,46). It has been suggested that certain subtypes of the α1-adrenergic receptor family, together with some other cell surface receptors, stimulate nucleotide exchange on TGase-2 (46–51), enabling it to then activate phospholipase C-δ1. However, thus far, we have not been able to implicate this interaction in the survival of cells. It also seems unlikely that the GTP-binding-defective TGase-2 mutants are acting as classical dominant-negative inhibitors of a function provided by endogenous TGase-2 that is essential for the fundamental viability of cells. Indeed, if TGase-S or the GTP-binding-defective mutants of TGase-2 were acting as dominant-negative mutants to block such a function imparted by TGase-2, then cells lacking TGase-2 would not be expected to be viable. However, TGase-2−/− mice survive and do not exhibit excessive apoptosis (52,53).
All of this seems to point to a rather interesting and novel mechanism by which TGase-S and the guanine nucleotide-binding-defective TGase-2 mutants cause cell death. In particular, the results seem to suggest that these mutants are either able to selectively bind and disable a cellular protein whose function is important for cell survival and/or they activate a protein that is necessary for promoting apoptosis. Thus far, we have not been able to see any indication that caspases are activated upon the over-expression of the GTP-binding-defective TGase-2 mutants in various cell types, nor does it appear that AIF is released from the mitochondria and enters the nucleus, as is the case when cells are stimulated to undergo apoptosis by doxorubicin. The same is true when examining cells that over-express TGase-S (i.e. no indication of caspase activation). Thus, apparently, both the GTP-binding-defective mutants, as well as TGase-2, trigger a caspase-independent cell death event (54,55). At the present time, we favor the idea that GTP-binding-defective TGase-2 mutants are able to selectively bind to and compromise the actions of a protein that is essential for cell viability. Wild-type TGase-2 might either be less effective at binding the same protein, or does not exert the same type of disabling effect on its function. This might also explain why the TGase-2(S171E) mutant, which was originally reported to be GTP-binding-defective (39), but was subsequently found to also be transamidation-defective (8), fails to induce cell death. The single S171E substitution, which knocks-out two different activities, may also preclude binding to the target protein that is disabled by the GTP-binding-defective point mutants. Therefore, a major emphasis of future efforts will be to identify unique binding partners for these TGase-2 mutants.
The importance attributed to TGase-2 in maintaining the survival of different cancer cell lines, as well as in promoting their invasive activity, has led to suggestions that this protein represents a viable target for cancer therapy (56–58). Much of the emphasis toward developing a therapeutic strategy has been directed toward disabling the transamidation activity of TGase-2. However, the results reported here now suggest that another potential option for the future may be to develop strategies that will impair the GTP-binding capability of this protein.
Acknowledgments
We would like to acknowledge Cindy Westmiller for expert secretarial assistance.
Abbreviations
- BOD-GTPγS
BODIPY-FL-modified form of GTPγS
- DTT
dithiothreitol
- G-protein
GTP-binding protein
- PARP
poly(ADP-ribose)polymerase
- PMSF
phenylmethylsulfonyl fluoride
- TGase-2
transglutaminase-2
- TGase-S
transglutaminase-short form
Footnotes
This work was supported by GM61762.
References
- 1.Folk JE. Transglutaminases. Annu Rev Biochem. 1980;49:517–531. doi: 10.1146/annurev.bi.49.070180.002505. [DOI] [PubMed] [Google Scholar]
- 2.Griffin M, Casadio R, Bergamini CM. Transglutaminases: nature’s biological glues. Biochem J. 2002;368:377–396. doi: 10.1042/BJ20021234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol. 2003;4:140–156. doi: 10.1038/nrm1014. [DOI] [PubMed] [Google Scholar]
- 4.Fesus L, Szondy Z. Transglutaminase 2 in the balance of cell death and survival. FEBS Letters. 2005;579:3297–3302. doi: 10.1016/j.febslet.2005.03.063. [DOI] [PubMed] [Google Scholar]
- 5.Monsonego A, Shani Y, Friedmann I, Paas Y, Eizenberg O, Schwartz M. Expression of GTP-dependent and GTP-independent tissue-type transglutaminase in cytokine-treated rat brain astrocytes. J Biol Chem. 1997;272:3724–3732. doi: 10.1074/jbc.272.6.3724. [DOI] [PubMed] [Google Scholar]
- 6.Lesort M, Chun W, Johnson GV, Ferrante RJ. Tissue transglutaminase is increased in Huntington’s disease brain. J Neurochem. 1999;73:2018–2027. [PubMed] [Google Scholar]
- 7.Karpuj MV, Garren H, Slunt H, Price DL, Gusella J, Becher MW, Steinman L. Transglutaminase aggregates huntingtin into nonamyloidogenic polymers, and its enzymatic activity increases in Huntington’s disease brain nuclei. Proc Natl Acad Sci USA. 1999;96:7388–7393. doi: 10.1073/pnas.96.13.7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Antonyak MA, Singh US, Lee DA, Boehm JE, Combs C, Zgola MM, Page RL, Cerione RA. Effects of tissue transglutaminase on retinoic acid-induced cellular differentiation and protection against apoptosis. J Biol Chem. 2001;276:33582–33587. doi: 10.1074/jbc.M105318200. [DOI] [PubMed] [Google Scholar]
- 9.Tucholski J, Lesort M, Johnson GV. Tissue transglutaminase is essential for neurite outgrowth in human neuroblastoma SH-SY5Y cells. Neuroscience. 2001;102:481–491. doi: 10.1016/s0306-4522(00)00482-6. [DOI] [PubMed] [Google Scholar]
- 10.Tucholski J, Johnson GV. Tissue transglutaminase differentially modulates apoptosis in a stimuli-dependent manner. J Neurochem. 2002;81:780–791. doi: 10.1046/j.1471-4159.2002.00859.x. [DOI] [PubMed] [Google Scholar]
- 11.Antonyak MA, Miller AM, Jansen JM, Boehm JE, Balkman CE, Wakshlag JJ, Page RL, Cerione RA. Augmentation of tissue transglutaminase expression and activation by epidermal growth factor inhibit doxorubicin-induced apoptosis in human breast cancer cells. J Biol Chem. 2004;279:41461–41467. doi: 10.1074/jbc.M404976200. [DOI] [PubMed] [Google Scholar]
- 12.Lesort M, Tucholski J, Miller ML, Johnson GV. Tissue transglutaminase: a possible role in neurodegenerative diseases. Prog Neurobiol. 2000;61:439–463. doi: 10.1016/s0301-0082(99)00052-0. [DOI] [PubMed] [Google Scholar]
- 13.Olivero S, Amendola A, Rodolfo C, Spinedi A, Piacentini M. Inhibition of “tissue” transglutaminase increases cell survival by preventing apoptosis. J Biol Chem. 1999;274:34123–34128. doi: 10.1074/jbc.274.48.34123. [DOI] [PubMed] [Google Scholar]
- 14.Piacentini M, Martinet N, Beninati S, Folk JE. Free and protein-conjugated polyamines in mouse epidermal cells. Effect of high calcium and retinoic acid. J Biol Chem. 1988;263:3790–3794. [PubMed] [Google Scholar]
- 15.Piacentini M, Fesus L, Farrace MG, Ghibelli L, Piredda L, Melino G. The expression of “tissue” transglutaminase in two human cancer cell lines is related with the programmed cell death (apoptosis) Eur J Cell Biol. 1991;54:246–254. [PubMed] [Google Scholar]
- 16.Johnson GV, Cox TM, Lockhart JP, Zinnerman MD, Miller ML, Powers RE. Transglutaminase activity is increased in Alzheimer’s disease brain. Brain Res. 1997;751:323–329. doi: 10.1016/s0006-8993(96)01431-x. [DOI] [PubMed] [Google Scholar]
- 17.Kim SY, Grant P, Lee JH, Pant HC, Steinert PM. Differential expression of multiple transglutaminases in human brain. Increased expression and cross-linking by transglutaminases 1 and 2 in Alzheimer’s disease. J Biol Chem. 1999;274:30715–30721. doi: 10.1074/jbc.274.43.30715. [DOI] [PubMed] [Google Scholar]
- 18.Junn E, Ronchetti RD, Quezado MM, Kim SY, Mouradian MM. Tissue transglutaminase-induced aggregation of alpha-synuclein: Implications for Lewy body formation in Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci USA. 2003;100:2047–2052. doi: 10.1073/pnas.0438021100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kahlem P, Green H, Djian P. Transglutaminase action imitates Huntington’s disease: selective polymerization of Huntingtin containing expanded polyglutamine. Mol Cell. 1998;1:595–601. doi: 10.1016/s1097-2765(00)80059-3. [DOI] [PubMed] [Google Scholar]
- 20.Mastroberardino PG, Iannicola C, Nardacci R, Bernassola F, De Laurenzi V, Melino G, Moreno S, Pavone F, Oliverio S, Fesus L, Piacentini M. ‘Tissue’ transglutaminase ablation reduces neuronal death and prolongs survival in a mouse model of Huntington’s disease. Cell Death Differ. 2002;9:873–880. doi: 10.1038/sj.cdd.4401093. [DOI] [PubMed] [Google Scholar]
- 21.Dedeoglu A, Kubilus JK, Jeitner TM, Matson SA, Bogdanov M, Kowall NW, Matson WR, Cooper AJ, Ratan RR, Beal MF, Hersch SM, Ferrante RJ. Therapeutic effects of cystamine in a murine model of Huntington’s disease. J Neurosci. 2002;22:8942–8950. doi: 10.1523/JNEUROSCI.22-20-08942.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karpuj MV, Becher MW, Springer JE, Chabas D, Youssef S, Pedotti R, Mitchell D, Steinman L. Prolonged survival and decreased abnormal movements in transgenic model of Huntington disease, with administration of the transglutaminase inhibitor cystamine. Nat Med. 2002;8:143–149. doi: 10.1038/nm0202-143. [DOI] [PubMed] [Google Scholar]
- 23.Boehm JE, Singh U, Combs C, Antonyak MA, Cerione RA. Tissue transglutaminase protects against apoptosis by modifying the tumor suppressor protein p110 Rb. J Biol Chem. 2002;277:20127–20130. doi: 10.1074/jbc.C200147200. [DOI] [PubMed] [Google Scholar]
- 24.Herman JF, Mangala LS, Mehta K. Implications of increased tissue transglutaminase (TG2) expression in drug-resistant breast cancer (MCF-7) cells. Oncogene. 2006;25:3049–3058. doi: 10.1038/sj.onc.1209324. [DOI] [PubMed] [Google Scholar]
- 25.Zhang R, Tremblay TL, McDermid A, Thibault P, Stanimirovic D. Identification of differentially expressed proteins in human glioblastoma cell lines and tumors. Glia. 2003;42:194–208. doi: 10.1002/glia.10222. [DOI] [PubMed] [Google Scholar]
- 26.Yuan L, Choi K, Khosla C, Zheng X, Higashikubo R, Chicoine MR, Rich KM. Tissue transglutaminase 2 inhibition promotes cell death and chemosensitivity in glioblastomas. Mol Cancer Ther. 2005;4:1293–1302. doi: 10.1158/1535-7163.MCT-04-0328. [DOI] [PubMed] [Google Scholar]
- 27.Datta S, Antonyak MA, Cerione RA. Importance of Ca(2+)-dependent transamidation activity in the protection afforded by tissue transglutaminase against doxorubicin-induced apoptosis. Biochemistry. 2006;45:13163–13174. doi: 10.1021/bi0606795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Antonyak MA, Jansen JM, Miller AM, Ly TK, Endo M, Cerione RA. Two isoforms of tissue transglutaminase mediate opposing cellular fates. PNAS. 2006;103:18609–18614. doi: 10.1073/pnas.0604844103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Citron BA, Suo Z, SantaCruz K, Davies PJ, Qin F, Festoff BW. Protein crosslinking, tissue transglutaminase, alternative splicing and neurodegeneration. Neurochem Int. 2002;40:69–78. doi: 10.1016/s0197-0186(01)00062-6. [DOI] [PubMed] [Google Scholar]
- 30.Festoff BW, SantaCruz K, Arnold PM, Sebastian CT, Davies PJ, Citron BA. Injury-induced “switch” from GTP-regulated to novel GTP-independent isoform of tissue transglutaminase in the rat spinal cord. J Neurochem. 2002;81:708–718. doi: 10.1046/j.1471-4159.2002.00850.x. [DOI] [PubMed] [Google Scholar]
- 31.Fraij BM, Birckbichler PJ, Patterson MK, Jr, Lee KN, Gonzales RA. A retinoic acid-inducible mRNA from human erythroleukemia cells encodes a novel tissue transglutaminase homologue. J Biol Chem. 1992;267:22616–22623. [PubMed] [Google Scholar]
- 32.Im MJ, Russell MA, Feng JF. Transglutaminase II: a new class of GTP-binding protein with new biological functions. Cell Signal. 1997;9:477–482. doi: 10.1016/s0898-6568(97)00049-1. [DOI] [PubMed] [Google Scholar]
- 33.Liu S, Cerione RA, Clardy JC. Structural basis for the guanine nucleotide-binding activity of tissue transglutaminase and its regulation of transamidation activity. Proc Natl Acad Sci. 2002;99:2743–2747. doi: 10.1073/pnas.042454899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Begg GE, Holman SR, Stokes PH, Matthews JM, Graham RM, Iismaa SE. Mutation of a critical arginine in the GTP-binding site of transglutaminase 2 disinhibits intracellular cross-linking activity. J Biol Chem. 2006;281:12603–12609. doi: 10.1074/jbc.M600146200. [DOI] [PubMed] [Google Scholar]
- 35.Day N, Keillor JW. A continuous spectrophotometric linked enzyme assay for transglutaminase activity. Anal Biochem. 1999;274:141–144. doi: 10.1006/abio.1999.4255. [DOI] [PubMed] [Google Scholar]
- 36.Fraij BM. GTP hydrolysis by human tissue transglutaminase homologue. Biochem Biophys Res Commun. 1996;218:45–49. doi: 10.1006/bbrc.1996.0009. [DOI] [PubMed] [Google Scholar]
- 37.Lee KN, Birckbichler PJ, Patterson MK. GTP hydrolysis by guinea pig liver transglutaminase. Biochem Biophys Res Commun. 1989;162:1370–1375. doi: 10.1016/0006-291x(89)90825-5. [DOI] [PubMed] [Google Scholar]
- 38.Singh US, Erickson JW, Cerione RA. Identification and biochemical characterization of an 80 kilodalton GTP-binding/transglutaminase from rabbit liver nuclei. Biochemistry. 1995;34:15863–15871. doi: 10.1021/bi00048a032. [DOI] [PubMed] [Google Scholar]
- 39.Iismma SE, Wu MJ, Nanda N, Church WB, Graham RM. GTP binding and signaling by Gh/transglutaminase II involves distinct residues in a unique GTP-binding pocket. J Biol Chem. 2000;275:18259–18265. doi: 10.1074/jbc.M000583200. [DOI] [PubMed] [Google Scholar]
- 40.Lai T-S, Liu Y, Li W, Greenberg CS. Identification of two GTP-independent alternatively spliced forms of tissue transglutaminase in human leukocytes, vascular smooth muscle, and endothelial cells. FASEB J. 2007 doi: 10.1096/fj.06-7598com. online communication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Achyuthan KE, Greenberg CS. Identification of a guanosine triphosphate-binding site on guinea pig liver transglutaminase. Role of GTP and calcium ions in modulating activity. J Biol Chem. 1987;262:1901–1906. [PubMed] [Google Scholar]
- 42.Lai TS, Slaughter TF, Peoples KA, Hettasch JM, Greenberg CS. Regulation of human tissue transglutaminase function by magnesium-nucleotide complexes. Identification of distinct binding sites for Mg-GTP and Mg-ATP. J Biol Chem. 1998;273:1776–1781. doi: 10.1074/jbc.273.3.1776. [DOI] [PubMed] [Google Scholar]
- 43.Antonyak MA, McNeill CJ, Wakshlag JJ, Boehm JE, Cerione RA. Activation of the Ras-ERK pathway inhibits retinoic acid-induced stimulation of tissue transglutaminase expression in NIH3T3 cells. J Biol Chem. 2003;278:15859–15866. doi: 10.1074/jbc.M300037200. [DOI] [PubMed] [Google Scholar]
- 44.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Constantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-induced factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 45.Milakovic T, Tucholski J, McCoy E, Johnson GV. Intracellular localization and activity state of tissue transglutaminase differentially impacts cell death. J Biol Chem. 2004;279:8715–8722. doi: 10.1074/jbc.M308479200. [DOI] [PubMed] [Google Scholar]
- 46.Feng JF, Rhee SG, Im MJ. Evidence that phospholipase delta1 is the effector in the Gh (transglutaminase II)-mediated signaling. J Biol Chem. 1996;271:16451–16454. doi: 10.1074/jbc.271.28.16451. [DOI] [PubMed] [Google Scholar]
- 47.Nakaoka H, Perez DM, Baek KJ, Das T, Husain AM, Misono K, Im MJ, Graham RM. Gh: a GTP-binding protein with transglutaminase activity and receptor signaling function. Science. 1994;264:1593–1596. doi: 10.1126/science.7911253. [DOI] [PubMed] [Google Scholar]
- 48.Chen S, Lin F, Iismaa S, Lee KN, Birckbichler PJ, Graham RM. Alpha1-adrenergic receptor signaling via Gh is subtype specific and independent of its transglutaminase activity. J Biol Chem. 1996;271:32385–32391. doi: 10.1074/jbc.271.50.32385. [DOI] [PubMed] [Google Scholar]
- 49.Vezza R, Habib A, FitzGerald GA. Differential signaling by the thromboxane receptor isoforms via the novel GTP-binding protein, Gh. J Biol Chem. 1999;274:12774–12779. doi: 10.1074/jbc.274.18.12774. [DOI] [PubMed] [Google Scholar]
- 50.Zhang Z, Vezza R, Plappert T, McNamara P, Lawson JA, Austin S, Pratico D, Sutton MS, FitzGerald GA. COX-2-dependent cardiac failure in Gh/tTG transgenic mice. Circ Res. 2003;92:1153–1161. doi: 10.1161/01.RES.0000071749.22027.45. [DOI] [PubMed] [Google Scholar]
- 51.Park ES, Won JH, Han KJ, Suh PG, Ryu SH, Lee HS, Yun HY, Kwon NS, Baek KJ. Phospholipase C-delta1 and oxytocin receptor signalling: evidence of its role as an effector. Biochem J. 1998;331:283–289. doi: 10.1042/bj3310283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nanda N, Iismaa SE, Owens WA, Husain A, Mackay F, Graham RM. Targeted inactivation of Gh/tissue transglutaminase II. J Biol Chem. 2001;276:20673–20678. doi: 10.1074/jbc.M010846200. [DOI] [PubMed] [Google Scholar]
- 53.De Laurenzi V, Melino G. Gene disruption of tissue transglutaminase. Mol Cell Biol. 2001;21:148–155. doi: 10.1128/MCB.21.1.148-155.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shinzawa K, Tsujimoto Y. PLA2 activity is required for nuclear shrinkage in caspase-independent cell death. J Cell Biol. 2003;163:1219–1230. doi: 10.1083/jcb.200306159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kroemer G, Martin SJ. Caspase-independent cell death. Nat Med. 2005;11:725–730. doi: 10.1038/nm1263. [DOI] [PubMed] [Google Scholar]
- 56.Yuan L, Choi K, Khosla C, Zheng X, Higashikubo R, Chicoine MR, Rich KM. Tissue transglutaminase 2 inhibition promotes cell death and chemosensitivity in glioblastomas. Mol Cancer Ther. 2005;4:1293–1302. doi: 10.1158/1535-7163.MCT-04-0328. [DOI] [PubMed] [Google Scholar]
- 57.Choi K, Siegel M, Piper JL, Yuan L, Cho E, Strnad P, Omary B, Rich KM, Khosla C. Chemistry and biology of dihydroisoxazole derivatives: selective inhibitors of human transglutaminase 2. Chem Biol. 2005;12:469–475. doi: 10.1016/j.chembiol.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 58.Keillor JW. Chem Biol. 2005;12:469–475. doi: 10.1016/j.chembiol.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 59.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]