

Abstract
Ultra-high-throughput sequencing is emerging as an attractive alternative to microarrays for genotyping, analysis of methylation patterns, and identification of transcription factor binding sites. Here, we describe an application of the Illumina sequencing (formerly Solexa sequencing) platform to study mRNA expression levels. Our goals were to estimate technical variance associated with Illumina sequencing in this context and to compare its ability to identify differentially expressed genes with existing array technologies. To do so, we estimated gene expression differences between liver and kidney RNA samples using multiple sequencing replicates, and compared the sequencing data to results obtained from Affymetrix arrays using the same RNA samples. We find that the Illumina sequencing data are highly replicable, with relatively little technical variation, and thus, for many purposes, it may suffice to sequence each mRNA sample only once (i.e., using one lane). The information in a single lane of Illumina sequencing data appears comparable to that in a single array in enabling identification of differentially expressed genes, while allowing for additional analyses such as detection of low-expressed genes, alternative splice variants, and novel transcripts. Based on our observations, we propose an empirical protocol and a statistical framework for the analysis of gene expression using ultra-high-throughput sequencing technology.
Since the mid-1990s, DNA microarrays have been the technology of choice for large-scale studies of gene expression levels. The ability of these arrays to simultaneously interrogate thousands of transcripts has led to important advances in a wide range of biological problems, including the identification of gene expression differences among diseased and healthy tissues, and new insights into developmental processes, pharmacogenomic responses, and the evolution of gene regulation (Scherf et al. 2000; White 2001; Rifkin et al. 2003; Passador-Gurgel et al. 2007). Nonetheless, array technology has several limitations. For example, background levels of hybridization (i.e., hybridization to a probe that occurs irrespective of the corresponding transcript’s expression level) limit the accuracy of expression measurements, particularly for transcripts present in low abundance. Furthermore, probes differ considerably in their hybridization properties (Gautier et al. 2004). Thus, although comparing hybridization results across arrays can identify gene expression differences among samples (Allison et al. 2006), hybridization results from a single sample may not provide a reliable measure of the relative expression of different transcripts. Finally, arrays are limited to interrogating transcripts with relevant probes on the array.
Sequencing-based approaches to measuring gene expression levels have the potential to overcome these limitations. New, ultra-high-throughput sequencing techniques enable thousands of megabases of DNA to be sequenced in a matter of days. Several technologies, including those developed by 454 Life Sciences (Roche) (Margulies et al. 2005) and Illumina (formerly Solexa sequencing) (Bennett et al. 2005), are currently available and have been used to investigate genetic variation (Korbel et al. 2007), transcription factor binding sites (Mikkelsen et al. 2007), and DNA methylation (Cokus et al. 2008). Applications to the measurement of mRNA expression levels have proceeded more slowly, partly because of difficulties in developing appropriate experimental protocols, but also because expression studies aim to identify (perhaps subtle) quantitative differences between samples, while other applications have, thus far, focused on detecting the absence or presence of an event, such as a transcription factor binding.
In this study, we describe the results of a pilot project to assess the potential of Illumina sequencing for detecting and measuring mRNA expression levels and comparing expression levels across samples. Specifically, we applied Illumina sequencing to a liver RNA sample and a kidney RNA sample, sequencing each sample seven times, and compared the results with Affymetrix array data on the same samples. Although several papers have described the use of 454 sequencing to examine mRNA expression levels (Weber et al. 2007; Sugarbaker et al. 2008; Torres et al. 2008), we chose the Illumina sequencing platform because currently, for a fixed cost, its coverage and depth are far greater than other sequencing technologies, making it particularly attractive for expression studies. Our study also differs from previous reports in its assessment of variability across technical replicates for a single sample, and direct comparison of the sequence-based results with those from a state-of-the-art array platform.
We find that the sequencing data are highly reproducible, with few systematic differences among technical replicates. Statistically, we find that the variation across technical replicates can be captured using a Poisson model, with only a small proportion (∼0.5%) of genes showing clear deviations from this model. This Poisson model can be used to identify differentially expressed genes, and using this approach, the sequence data identified 30% more differentially expressed genes than were obtained from a standard analysis of the array data at the same false discovery rate. We also illustrate the potential for sequence-based approaches to identify alternative-spliced forms.
Results
Experimental design
Illumina’s sequencing technology uses massively parallel Sanger sequencing to simultaneously sequence millions of short fragments of DNA. Each time a machine is run, DNA samples can be independently sequenced in one of eight lanes, although one lane is normally used to sequence a control sample. Typically, each lane generates many millions of short reads (e.g., 32 bp in the data considered here). To assess the ability of Illumina sequencing to measure gene expression differences between samples, we used the following study design (Fig. 1A): We extracted total RNA from liver and kidney samples of a single human male, purified the poly(A) mRNA, and sheared it prior to cDNA synthesis. The cDNA was then processed into a library of template molecules suitable for sequencing on the Illumina Genome Analyzer (see Methods). To assess technical variance within and between runs, we sequenced each sample seven times, split across two runs of the machine (Fig. 1B). To investigate the effects of cDNA concentration, two different cDNA concentrations were used: 3 pM (five lanes per sample) and 1.5 pM (two lanes per sample).
Figure 1.

Graphical representation of the study design. (A) Summary of the experimental design. (B) The lanes in which each sample was sequenced across the two runs. In each run, the control sample was sequenced in lane 5. Samples were sequenced at two concentrations: 1.5 pM (indicated by an asterisk) and 3 pM (no asterisk).
To allow comparisons with an array-based technology, we hybridized the same RNA samples to Affymetrix U133 Plus 2 arrays (www.affymetrix.com/products/arrays/specific/hgu133plus.affx). We used three arrays (technical replicates) for each RNA sample, and the sample preparation and data analysis were designed to be as similar to the sequence-based approach as possible (Methods). To facilitate a direct comparison between the sequence and array data, we mapped the array probe sets to annotated genes in the Ensembl database v.48 (Flicek et al. 2008). In total, 70% of probe sets mapped to an Ensembl gene, and, after accounting for multiple probe sets mapping to the same gene and probe sets that did not map uniquely, we identified a set of 17,708 probe sets, mapping uniquely to 17,708 genes, which were used in subsequent analyses (see Methods).
Illumina sequencing data processing
Each RNA sample was sequenced in seven lanes, producing 12.9–14.7 million reads per lane at the 3 pM concentration and 8.4–9.3 million reads at the 1.5 pM concentration (Supplemental Table 1). We aligned all reads against the whole genome using the Illumina-supplied algorithm ELAND, which is designed to be particularly efficient for 32-bp reads. Tolerances were set to allow at most two mismatches in each alignment, and reads that aligned to multiple genomic locations were ignored. By these criteria, 40% of reads mapped uniquely to a genomic location, and of these, 65% mapped to autosomal or sex chromosomes (the remainder mapped almost exclusively to mitochondrial DNA). These percentages were similar for 3 pM and 1.5 pM concentrations and are comparable to results from other studies that have used Illumina sequencing (Nagalakshmi et al. 2008). Possible reasons for reads not mapping uniquely to the genome include the presence of sequencing errors or polymorphisms, reads that come from repetitive sequence, and reads from exon–exon junctions (which can potentially be recovered by a more sophisticated alignment strategy; see below).
As expected, the distribution of the locations of mapped reads showed a strong bias toward annotated genic regions based on the Ensembl database: 83% of mapped reads fell in such regions; of these, 68% fell in annotated exons. Furthermore, reads mapping to intergenic locations (i.e., reads mapping outside the furthest 5′ and 3′ exons for every gene) tended to fall near an annotated gene (Supplemental Fig. 1), suggesting that many genes in the Ensembl annotation may require extension or revision. Nonetheless, a sizable minority (10.6%) of intergenic reads was mapped to locations at least 100 kb from a known gene, supporting other published data (The ENCODE Project Consortium 2007), suggesting that many transcriptionally active regions (TARs) are currently unannotated.
We obtained, for each lane, a measure of the “overall” expression of each gene in the Ensembl database by summing the number of reads mapping to exons within each gene (Supplemental Table 2). For genes with multiple transcripts, we took the median across transcripts. Within each lane, under idealized assumptions (e.g., no alignment errors, and no sequence-context sequencing bias), these “gene counts” would, in expectation, be proportional to the transcript length times the mRNA expression level. Of the genes in the Ensembl database, 22,925 (72%) were mapped to by at least one read. Among these, the distribution of the number of reads was very skewed across genes (Supplemental Fig. 2), with many genes having relatively few reads (median = 46 for liver, 101 for kidney).
A first (albeit rather rough) indication that sequence data are highly replicable is that, for each sample, the gene counts are highly correlated across lanes (average Spearman correlation = 0.96) (Supplemental Fig. 3).
An issue of particular importance is to what extent the data exhibit a “lane effect,” by which we mean systematic differences between results for the same sample, sequenced at the same concentration in different lanes, over and above those expected from sampling error. We examined this issue in two ways, first by considering each pair of lanes in turn (which allows any outlying lanes to be identified), and then by considering multiple lanes simultaneously (which should increase the power to detect lane effects if they consistently affect the same genes).
When comparing a pair of lanes, we computed, for each gene, a P-value testing the null hypothesis that the gene counts in one lane resembled a random sample from the reads in both lanes (this is done using the fact that, in the absence of a lane effect, after accounting for the different total gene counts in each lane, the individual gene counts in each lane should follow a hypergeometric distribution). In the absence of a lane effect, the distribution of these P-values across genes should be uniform, whereas deviations from uniformity (which we assessed using a qq-plot) indicate a lane effect. Among the 22 total two-way comparisons between lanes in which the same sample was sequenced at the same concentration, we found that only a small proportion of genes (consistently <0.5%) had very small P-values that indicated clear evidence for a lane effect (Fig. 2A; Supplemental Fig. 4). This was true for comparisons both within and across the two different runs, although comparisons across different runs seemed to show slightly larger proportions of genes with small P-values (larger experiments will be required to assess comprehensively run-to-run variability). In contrast, using the same procedure to compare results from the same sample sequenced at different concentrations produced P-values that showed much greater deviations from uniformity (Fig. 2B; Supplemental Fig. 5).
Figure 2.

Plots to assess lane effects. Each panel shows a qq-plot comparing the distribution of a statistic (Y-axis) against its theoretical distribution in the absence of a lane effect (X-axis). Deviations from the line y = x indicate the presence of a lane effect. (Points in red) Those above the 95th percentile; (points in blue) those above the 99.5th percentile. (A) A typical result when using P-values derived from a hypergeometric test statistic to compare two lanes used to sequence the same sample at the same concentration. (In this panel, data generated when the kidney sample was sequenced in Run 1, lane 1 and Run 2, lane 2 were used; see Supplemental Fig. 4 for all pairwise comparisons.) (B) Analogous results when comparing two lanes used to sequence the same sample at different concentrations. (In this panel, data generated when the kidney sample was sequenced in Run 1, lane 1 and Run 2, lane 4 were used; see Supplemental Fig. 5 for all pairwise comparisons.) (C,D) Results (on two different scales) when the goodness-of-fit statistic is used to assess the fit of the Poisson model to the kidney data sequenced at a concentration of 3 pM. The liver sample showed a similar pattern (Supplemental Fig. 6).
To compare multiple lanes for a lane effect, we took a closely related approach based on the following Poisson model. If xijk represents the number of reads mapped to gene j for the kth lane of data from sample i, xijk can be modeled as independent Poisson random variables with mean μijk = cikλijk, where the λijk are constrained to sum to 1 across genes j. The parameter cik represents the total rate at which lane k of sample i produces reads, and the parameter λijk represents the rate at which reads map to gene j (in lane k of sample i) relative to other genes. The hypothesis of no lane effect corresponds to λijk being constant across lanes k. For each gene, we compute a goodness-of-fit statistic across L lanes to test this hypothesis: if there is no lane effect, then this statistic should be χ2 distributed on L − 1 degrees of freedom. A qq-plot of these values (Fig. 2C,D; Supplemental Fig. 6) shows that, in each case, only a small proportion of genes (∼0.5%) show strong evidence for a lane effect (i.e., extra-Poisson variation).
In summary, for lanes sequencing the same sample at the same concentration, only a small proportion of genes show evidence for differences among lanes over those expected from sampling error. For sequences sampled at different concentrations, the differences were more appreciable. Thus, for the remainder of this paper, we consider only the data sequenced at a concentration of 3 pM (five lanes for each sample).
Identifying differentially expressed genes
The Poisson model described above provides a natural framework for identifying differentially expressed genes. Indeed, the model can be cast as a generalized linear model (McCullagh and Nelder 1989), and standard methods exist to estimate parameters, and to compute P-values for each gene testing the null hypothesis that it is not differentially expressed between two groups (see Methods).
The results from the goodness-of-fit test above suggest that a small proportion of genes show deviations from the Poisson assumption (extra-Poisson variation). To check whether this aspect of the data will lead to false-positive identifications of differentially expressed genes, we applied the Poisson model to identify differentially expressed genes between groups of lanes used to sequence the same sample. We observed that even for the pair of lanes that displayed the strongest evidence of a lane effect, only 14 genes were identified as differentially expressed at a false discovery rate (FDR) of 0.1% (Supplemental Fig. 7). Similarly, when we applied this model to groups that each contained two lanes used to sequence the same sample, the worst comparison yielded only 24 genes that were incorrectly identified as differentially expressed. We conclude that, in this context, at this stringent FDR, deviations from the Poisson model do not lead to the identification of an appreciable number of false-positive differentially expressed genes.
We next used this approach to identify differentially expressed genes from the Illumina sequencing data, by comparing five lanes each of liver-versus-kidney samples. At an FDR of 0.1%, we identified 11,493 genes as differentially expressed between the samples (94% of these had an estimated absolute log2-fold change > 0.5; 71% > 1).
Comparison of results across technologies
As a first step to comparing the sequence and array data, we compared the number of sequence reads mapped to each gene with the corresponding (normalized) absolute intensities from the array (Fig. 3). Reassuringly, these two independent measures of transcript abundance are highly correlated (Spearman correlation = 0.73 for liver, 0.75 for kidney). Interestingly, where results from the two technologies differ, it is generally where the array intensities are large and the sequence counts small; a pattern that might be explained by probe-specific background hybridization on the array.
Figure 3.

Comparing counts from Illumina sequencing with normalized intensities from the array, for kidney (left) and liver (right). In each panel, the average (log2) counts for each gene are plotted on the X-axis, and the corresponding normalized intensities from the array are shown on the Y-axis. To avoid taking the log of 0, we added 1 to each of the average counts prior to taking logs.
We next compared differentially expressed genes called from the Illumina sequencing data with those identified from the array. By applying a widely used empirical Bayes approach (Smyth 2004) to the array data, we identified 8113 genes as differentially expressed at an FDR of 0.1% (83% with an estimated absolute log2-fold change > 0.5, 43% > 1). Of these, 81% of genes were also identified as differentially expressed from the Illumina sequencing data, providing strong evidence that the majority of genes called from the sequence data are genuinely differentially expressed between the two samples. Furthermore, estimates of the log2-fold changes of gene expression levels between the samples across the two technologies are correlated (Spearman correlation = 0.73) (Fig. 4). The correlation is greater for genes that are mapped to by large numbers of sequence reads. For example, for genes mapped to by (on average) more than 32 reads in both tissues (≥5 on the log scale in Fig. 3), the Spearman correlation of the fold changes across technologies is 0.79 compared with 0.60 for genes mapped to by at least one but fewer than 32 reads. These comparisons with the array data demonstrate that the Illumina sequencing technology and our analysis approach are performing well. A complete comparison of genewise results from both technologies is available in Supplemental Table 3.
Figure 4.

Comparison of estimated log2 fold changes (liver/kidney) from Illumina (Y-axis) and Affymetrix (X-axis). We consider only genes that were interrogated using both platforms and genes where the mean number of counts across lanes was greater than 0 for both the liver and kidney samples. (Red and green dots) Genes called as differentially expressed based on the Illumina sequencing data at an FDR of 0.1%, with a mean number of counts greater than (red) or less than (green) 250 reads in both tissues. (Black dots) Genes not called as differentially expressed based on the Illumina sequencing data. The set of differentially expressed genes that show the strongest correlation between the two technologies seems to be those that are mapped to by many reads (red), while the correlation is weaker for differentially expressed genes mapped to by fewer reads (green).
Considered together, 6538 genes were identified as differentially expressed using either the sequencing or the array data but not by both (Fig. 5). To further examine these discrepancies, we used a third technology, quantitative PCR (qPCR), to test for differences in expression between the liver and kidney samples for five genes called differentially expressed from the sequence data but not the array (MMP25, SLC5A1, MDK, ZNF570, GPR64) and for six genes that were found to be differentially expressed using the array, but not the sequencing data (C16orf68, CD38, LSM7, S100P, PEX11A, GLOD5). We designed primers for the qPCR within 1 kb upstream of the annotated 3′-end of the genes (Methods). The qPCR results confirmed as differentially expressed (t-test, P < 0.01) four of the first set of genes (all but ZNF570), but only two of the second set (CD38 and GLOD5). Thus, overall, the qPCR results agreed more closely with the Illumina sequencing results than with the array.
Figure 5.

A Venn diagram summarizing the overlap between genes called as differentially expressed from the (left circle) sequence data and from the (right circle) array. The number of genes called by both technologies is indicated by the overlap between the two circles.
Beyond the analysis of differences in gene expression
In addition to identifying gene expression differences, sequencing data can be used to identify novel exons and transcripts andto study alternative splicing. For example, to find novel exons or transcripts, the distribution of intergenic reads (i.e., reads mapped between currently annotated genes) across the genome could be examined. If a large number of reads were mapped to a particular genomic region, it would suggest that this region might provide a good target for follow-up work. Additionally, identifying sequence reads that span exon–exon junctions should help reconstruct the composition of alternative splice variants (although reconstructing entire transcripts will be challenging, particularly with short reads). A comprehensive analysis of both these topics is beyond the scope of this study. Nevertheless, to illustrate the potential of these data, we performed a preliminary analysis to identify reads that span exon–exon junctions.
Since reads that cover exons that have been spliced together will not map directly back to the reference human genome, we developed a splicing detection algorithm (see Methods) to examine all of the reads that did not align to (at least one location in) the genome. In kidney, we identified more than 200,000 reads that mapped to possible exon–exon junctions within a gene. Of the junctions mapped to, more than 30,000 showed twofold or greater coverage. As expected, we also found evidence for alternative splicing (i.e., splice junctions that skip one or more of the exons). An example of a specific gene for which putative alternative splice variants are present is C17orf45 on chromosome 17 (Fig. 6). We observed similar proportions of splicing isoforms for the liver. The number of reads supporting alternative splicing (Supplemental Table 4) should be taken as an estimate of the order of magnitude at this point, because a more careful analysis is needed to resolve possible exon-annotation conflicts in the databases. A comprehensive examination of these data and their reliability is therefore still necessary, but these preliminary data show the potential for short-sequence reads to detect splicing variation.
Figure 6.

An example of alternative splicing. The full exon structure of C17orf45 (ENSG00000175061) is shown for kidney (top) and liver (bottom), with exons plotted to scale. (Black) The number of reads mapping to each exon and to each exon junction. (Gray) The number of reads mapping to alternative splice exon junctions (i.e., junctions between non-consecutive exons). (The black lines below the exon) The location of reads mapped to this gene in Run 2, lane 2 (kidney) and Run 2, lane 3 (liver).
Discussion
Our results demonstrate the efficacy of high-throughput sequencing for measuring gene expression levels. Using the Illumina sequencing platform, we detected differential expression for 81% of genes called significantly differentially expressed from the array data, and the correlation of fold change ratios between the two technologies (Spearman correlation = 0.73) is similar or higher than observed in comparisons across different microarray platforms (Shi et al. 2006). Furthermore, our analysis suggests that a large proportion of genes called differentially expressed from the sequencing data but not from the array may be true positives: First, comparisons of lanes from the same sample identified at most 14 genes as differentially expressed, and secondly, results from qPCR on five genes identified as differentially expressed in Illumina sequencing but not on the array confirmed four of them. The remaining gene (ZNF570) may represent a false positive in the Illumina sequencing data. Alternatively, it may reflect differences in the genic regions surveyed by the two technologies.
Alternative analysis strategies
The approach we took here to identifying differentially expressed genes in the sequence data was based on a Poisson model. Goodness-of-fit tests indicate that a small proportion of genes show clear deviations from this model (extra-Poisson variation), and although we found that these deviations did not lead to false-positive identification of differentially expressed genes at a stringent FDR, there is nevertheless room for improved models that account for the extra-Poisson variation. One natural strategy would be to replace the Poisson distribution with another distribution, such as the quasi-Poisson distribution (Venables and Ripley 2002) or the negative binomial distribution (Robinson and Smyth 2007), which have an additional parameter that estimates over- (or under-) dispersion relative to a Poisson model. Alternatively, with appropriate transformations of the count data, existing approaches for microarray experiments (Allison et al. 2006) may also work well. For example, a natural approach would be to first convert the count data in each lane to proportions, and then to apply an arcsin-root transformation, which is the standard variance-stabilizing transformation for a proportion. More precisely, we suggest transforming each count x to √n arcsin (√x/n), where n is the total number of counts in the lane. These transformed data could then be used as input to the Empirical Bayes approach of Smyth (2004). Although this approach lacks the elegance of working directly with the count data, the hierarchical structure of the Empirical Bayes method may provide more accurate estimates of the gene-specific variability than a simple quasi-Poisson approach, potentially improving power.
RNA-seq study design
An important aspect of our study is the investigation of technical variance associated with Illumina sequencing. Our analyses suggest that the Illumina sequencing data are highly replicable, with relatively few genes showing evidence for a “lane effect”: the reads in one lane represent an approximately random sample from those obtained across multiple lanes. Note that this does not require, or imply, that sequence data exhibit few systematic biases [e.g., effects of GC content, or poly(N)s]. However, it does suggest that any such biases are largely consistent across lanes, both within and between runs of the same concentration (although to fully assess variation among runs, data from many more runs will be necessary).
We note that our study design did not include replicates of the processing step of the Illumina sequencing library. As equivalent processing steps for microarrays (i.e., fragmentation and amplification) result in the introduction of very little technical variance, we expected that rather than the processing of the library, sequencing over different lanes and flow-cells would introduce most of the technical variance associated with Illumina sequencing. However, given our observation that very little technical variance is associated with sequencing in different lanes or plates, the variance introduced in the library processing step may contribute a nontrivial proportion of the total technical variance associated with the sequencing technology.
The relatively small proportion of genes exhibiting a lane effect, combined with the count nature of the data, makes it possible to perform meaningful comparisons between samples sequenced in just one lane each. This does not, in itself, imply that sequencing each sample in just one lane will necessarily suffice: fewer lanes of data will inevitably reduce the number of genes whose expression can be assessed and reduce the power to detect differences in expression. However, in our data, additional lanes provided only modest gains in the number of genes detected. For example, increasing the number of lanes per sample from one to two, and hence doubling the experiment’s cost, increased the number of genes mapped to by at least one read by only 7%–8%; adding further lanes produces smaller additional increases (Supplemental Table 5). Similarly, while the power to detect differentially expressed genes increases more substantially with additional lanes of data (Table 1), even using only one lane identifies 5670 differentially expressed genes, 70% as many as were found using three technical microarray replicates. Furthermore, using three lanes of sequence data detects more than the three microarrays.
Table 1.
Power to detect differentially expressed genes depends on the number of lanes used for each sample
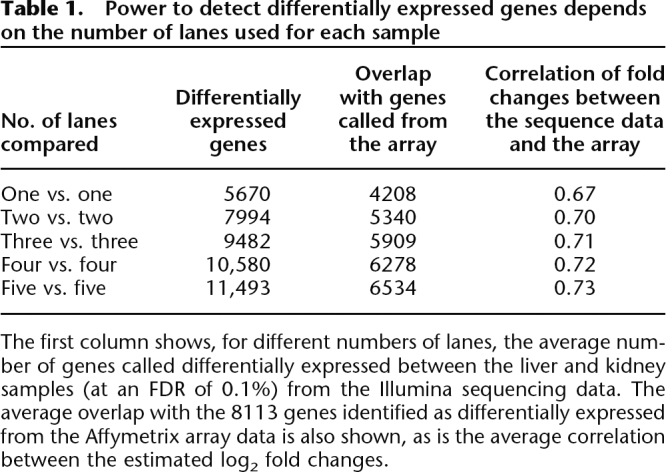
The first column shows, for different numbers of lanes, the average number of genes called differentially expressed between the liver and kidney samples (at an FDR of 0.1%) from the Illumina sequencing data. The average overlap with the 8113 genes identified as differentially expressed from the Affymetrix array data is also shown, as is the average correlation between the estimated log2 fold changes.
Typical gene expression experiments compare expression levels among many RNA samples (e.g., technical and biological replicates), hybridizing each sample only once to a microarray. Our data suggest that, in this setting, with current sequencing protocols, replacing each array with a single lane of Illumina sequencing data (randomizing samples appropriately across runs) is already an attractive alternative, and one that will become increasingly popular as improvements in experimental protocol and alignment methods lead to more usable data being generated in each lane.
Finally, while the primary focus of this work was establishing whether Illumina sequencing could be used to characterize gene expression differences between samples, sequence data may help answer other questions that are difficult to address using arrays. In particular, array technologies can measure expression only of genes that have corresponding probes on the array, and, in most cases, probes are designed only to cover a very small portion of a gene (in the case of the Affymetrix U133 Plus 2 array, most of the probes are at the 3′-end of the transcripts). Consequently, it is not possible to detect novel transcribed regions or (in general) the presence of alternative splice forms of a gene. Both of these problems can potentially be tackled using the Illumina sequencing data, and we have developed approaches to begin addressing these issues (Methods). However, identifying splice variants using a sequencing approach assumes that a sufficient number of reads span exon–exon junctions. This may not be the case if a sample is sequenced in only a single lane, and additional data will probably be required to solve this problem.
In summary, Illumina sequencing appears to be an extremely promising technology for measuring mRNA expression and identifying differentially expressed genes, comparable, and in some ways superior, to existing array-based approaches. Given the rapidly falling costs of sequencing, it seems only a matter of time before sequence-based approaches are widely adopted for this purpose.
Methods
Processing samples for Illumina sequencing
Tissue samples from liver and kidney from one human male were collected for us by the National Development and Research Institute (NDRI; http://www.ndri.org/), within 6 h post mortem. The tissue samples were snap-frozen and kept on dry ice until processing. We extracted total RNA from each tissue using TRIzol (Invitrogen). Total RNA quality from both tissues was high and comparable, based on analysis with a Bioanalyzer 2100 (Agilent).
Aliquots from the total RNA samples were subjected to Illumina sequencing, following the protocol offered by Illumina for sequencing of cDNA samples. Briefly, we used Dynal oligo(dT) beads (Invitrogen) to isolate poly(A) mRNA from the total RNA samples. We then fragmented the mRNA by using the RNA fragmentation kit from Ambion, followed by first- and second-strand cDNA synthesis using random hexamer primers. We complemented the cDNA synthesis by an “end repair” reaction using T4 DNA polymerase and Klenow DNA polymerase for 30 min at 20°C. We then added a single A base to the cDNA molecules by using 3′-to-5′ exo-nuclease, and ligated the Illumina adapter. The detailed laboratory protocol is available upon request. Images taken during the sequencing reactions were processed in three stages by Illumina’s software (v.192): Firecrest performs image analysis, base-calling is done by Bustard, and the sequence analysis is performed with Gerald.
Microarray processing and low-level analysis
Aliquots from the same total RNA samples used for the Illumina sequencing were hybridized to Affymetrix HG-U133 Plus 2.0 arrays in three technical replicates (5 μg of total RNA were used for each hybridization). To minimize sources of variation and to make the comparison between the sequencing and array-based approaches as fair as possible, the RNA samples hybridized to the array were subjected to a single labeling reaction. Hybridizations and scanning were performed at the University of Chicago Functional Genomics Facility.
We obtained background-corrected, normalized, summary raw values for all probe sets from each array using the RMA algorithm (Gautier et al. 2004). We used MA plots (hybridization intensity plotted against the fold change in expression) (Smyth and Speed 2003) to assess the quality of the data and the consistency across technical replicates (see Supplemental Fig. 8).
Subsequently, we considered the subset of probe sets that were mapped to Ensembl genes. We used mapping information from the NetAffx Analysis Center (v24; www.affymetrix.com/analysis/index.affx) and BioMart (Flicek et al. 2008) to map as many probe sets as possible. Hence, of a total of 54,675 probe sets on the array, we were able to map 38,059 to Ensembl genes. Where only a single probe set was mapped to a gene, we used the corresponding intensities in all future analyses. Where multiple probe sets mapped to the same gene, we considered the probe set that was most often called as present by the Affymetrix software across the six hybridizations. If two or more probe sets were called present in the same number of hybridizations, we chose a probe set at random and used it in all further analyses. To identify differentially expressed genes between the two tissues, we used an empirical Bayes modified t-statistic (Smyth 2004). The false discovery rate (FDR) for these P-values was calculated using the approach of Storey and Tibshirani (2003).
Quantitative PCR
We designed qPCR primers for gene regions within 1 kb upstream of the 3′-end of each gene. Primer sequences are available upon request. As templates, we used the same liver and kidney total RNA that was used for the Illumina sequencing and microarray experiments. Quantitative RT-PCR was performed in a 25-μL reaction containing 2× SYBR master mix (Sigma), 0.2 pM each primer, and 1 μL of cDNA template. PCR was performed in a 7900HT Fast Real-Time PCR System (Applied Biosystem, Inc.), in three technical replicates for each sample. The detection threshold cycle for each reaction was determined using a standard curve, after normalization of the results using quantitative RT-PCR with primers for the RPS7 gene, which was shown to have constant expression levels in many tissues (de Jonge et al. 2007); this was also the case for both technologies used in our experiments. The significance of differences in transcript levels between tissues was assessed by a t-test. We note that although we report results for 11 qPCRs, we originally chose 12 genes (six genes that were called differentially expressed from Illumina sequencing but not the array and six genes that were found to be differentially expressed using the array, but not Illumina sequencing). However, we erroneously included the FBXL6 gene, which was only marginally significant in the sequence data (Q-value = 0.001003) and not significant in the array data (Q-value = 0.078); this gene was also found to be differentially expressed using qPCR.
Assessing lane effects, within and between runs
For each gene mapped to by at least one read, we used a test based on the hypergeometric distribution to compute a P-value testing whether the number of counts in each lane differed over and above what would be expected under random sampling. Specifically, let xt1 and xt2 denote the number of counts for gene t in two lanes, and C1 and C2 denote the total number of reads in these lanes. In the absence of a lane effect, the reads in one lane will be a random sample from the reads in both lanes. This results in xt1 having a hypergeometric distribution (conditional on xt1 + xt2). Specifically, under the null hypothesis of no lane effect, the probability that xt1 = x is given by
where
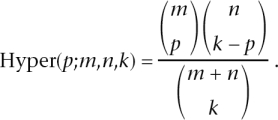 |
Based on this null distribution, we compute a one-sided P-value for the observed value of xt1, as
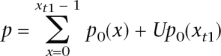 |
where U is a random number generated uniformly on [0, 1). (The use of this randomized U ensures that the P-values are genuinely uniform under the null; without this step, the P-values would have a discrete distribution under the null that is only approximately uniform.) We then converted these one-sided P-values into two-sided P-values: if the original P-value was <0.5, we multiplied it by 2 and, if the original value was >0.5, we subtracted it from 1 and multiplied this value by 2.
Under the null hypothesis of no lane effect, these hypergeometric P-values are uniformly distributed on [0, 1). We assessed deviations from uniformity visually via qq-plots (Fig. 2; Supplemental Figs. 4, 5).
Likelihood ratio test
For each gene, we separated the L lanes of interest into two groups (e.g., group A might be all lanes used to sequence the kidney mRNA, and group B might be all lanes used to sequence the liver mRNA). We then fitted the Poisson model described in the main text, computing the maximum likelihood estimates both under the null hypothesis that, for gene j, λijk = λj, and under the alternative that λijk = 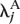 for samples/lanes in group A, and λijk =
for samples/lanes in group A, and λijk = 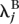 for samples/lanes in group B. The standard likelihood ratio statistic, being twice the log-likelihood ratio, was computed, and P-values were obtained using the fact that, under the null hypothesis, this statistic has a χ2 distribution with 1 degree of freedom.
for samples/lanes in group B. The standard likelihood ratio statistic, being twice the log-likelihood ratio, was computed, and P-values were obtained using the fact that, under the null hypothesis, this statistic has a χ2 distribution with 1 degree of freedom.
This procedure leads to a P-value for each gene. The significance threshold to control the FDR at a given value was calculated using the method of Storey and Tibshirani (2003), using the package qvalue within the R statistical package.
Fold changes were estimated by fitting the Poisson model under the alternative hypothesis to the lanes of interest. The estimated fold change for gene j was therefore calculated as 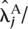 /
/ 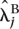 where and denote the maximum likelihood estimates of and .
where and denote the maximum likelihood estimates of and .
χ2 goodness-of-fit test
The χ2 goodness-of-fit statistic, Xij, is calculated for gene j and sample i as
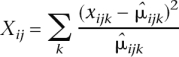 |
where the sum is over all lanes for sample i. Here 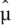 ijk denotes the maximum likelihood estimate of the Poisson mean μijk = cikλijk, under the constraint that λijk is constant across lanes k.
ijk denotes the maximum likelihood estimate of the Poisson mean μijk = cikλijk, under the constraint that λijk is constant across lanes k.
If the counts xijk are independent Poissons with mean μijk, then these statistics should follow a χ2 distribution with L − 1 degrees of freedom, where L is the number of lanes for sample i. To assess whether this is the case for a given sample i, the values of Xij can be plotted against the quantiles of the appropriate χ2 distribution using a qq-plot.
Identifying novel alternative splice forms
To examine splicing and alternative splicing, we used a two-stage approach. First, we removed from consideration all reads that were aligned by ELAND to a unique position or multiple positions in the human genome (Build 36, hg18), allowing for up to two mis-matches. To discover reads mapping to exon–exon junctions in novel splice forms, we used the following alignment strategy:
First, we extracted all exon sequences from the Ensembl gene annotations (v. 48) and used a Perl program to create a database (an exon-edge database, EEDB) consisting of two parts: a 3′ database containing the 32 bp from the 3′-end of every exon, and a 5′ database containing the 32 bp from the 5′-end of every exon.
We then repeatedly partitioned each unmapped read into two segments, A and B, where A has increasing size n (9 < n < 23), and B has decreasing size (32 − n). We tested A for alignment with the last n bp of each entry in the 3′ database, and B for alignment with the first 32 − n bp of each entry in the 5′ database. In these tests, we required exact matches (no mismatches). For each n, if we found an alignment for both A and B, then this pair of alignments was noted as an exon–exon junction to which the read maps. This search was conducted for both forward and reverse strand orientations.
This process maps reads to exon junctions, some reads mapping to one or more junctions, others mapping to no junctions. Of the junctions that are mapped to, some span multiple exons that are in the same gene, whereas others span exons in different genes. In this study, we report numbers of matches only for those junctions that span exons in the same gene, leaving investigation and verification of other junctions for future study.
Acknowledgments
We thank Terry Speed, Tony Long, Alicia Oshlack, and the members of the Przeworski, Pritchard, and Stephens groups at the University of Chicago for helpful discussions. We also acknowledge the support and input of Matthew W. State from the Yale Program on Neurogenetics, Nicholas Carriero from the Yale High Performance Computing Cluster, and the Keck Microarray Facility, where the sequencing experiments were performed. J.C.M. and M.S. were supported by NIH grant HG002585, S.M.M. was supported by grant U24 NS051869, and Y.G. was supported by NIH grant GM077959 and the Sloan Foundation.
Footnotes
[Supplemental material is available online at www.genome.org. Raw microarray CEL files have been deposited in the GEO database with accession number GSE11045. The raw Illumina sequencing data are available in the NCBI short read archive (http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi) with accession number SRA000299. A summary of the mapped reads and of the processed microarray data is available at http://giladlab.uchicago.edu/data.html.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.079558.108.
References
- Allison D., Cui X., Page G., Sabripour M., Cui X., Page G., Sabripour M., Page G., Sabripour M., Sabripour M. Microarray data analysis: From disarray to consolidation and consensus. Nat. Rev. Genet. 2006;7:55–65. doi: 10.1038/nrg1749. [DOI] [PubMed] [Google Scholar]
- Bennett S., Barnes C., Cox A., Davies L., Brown C., Barnes C., Cox A., Davies L., Brown C., Cox A., Davies L., Brown C., Davies L., Brown C., Brown C. Toward the 1,000 dollars human genome. Pharmacogenomics. 2005;6:373–382. doi: 10.1517/14622416.6.4.373. [DOI] [PubMed] [Google Scholar]
- Cokus S., Feng S., Zhang X., Chen Z., Merriman B., Haudenschild C., Pradhan S., Nelson S., Pellegrini M., Jacobsen S., Feng S., Zhang X., Chen Z., Merriman B., Haudenschild C., Pradhan S., Nelson S., Pellegrini M., Jacobsen S., Zhang X., Chen Z., Merriman B., Haudenschild C., Pradhan S., Nelson S., Pellegrini M., Jacobsen S., Chen Z., Merriman B., Haudenschild C., Pradhan S., Nelson S., Pellegrini M., Jacobsen S., Merriman B., Haudenschild C., Pradhan S., Nelson S., Pellegrini M., Jacobsen S., Haudenschild C., Pradhan S., Nelson S., Pellegrini M., Jacobsen S., Pradhan S., Nelson S., Pellegrini M., Jacobsen S., Nelson S., Pellegrini M., Jacobsen S., Pellegrini M., Jacobsen S., Jacobsen S. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452:215–219. doi: 10.1038/nature06745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jonge H., Fehrmann R., de Bont E., Hofstra R., Gerbens F., Kamps W., de Vries E., van der Zee A., te Meerman G., ter Elst A., Fehrmann R., de Bont E., Hofstra R., Gerbens F., Kamps W., de Vries E., van der Zee A., te Meerman G., ter Elst A., de Bont E., Hofstra R., Gerbens F., Kamps W., de Vries E., van der Zee A., te Meerman G., ter Elst A., Hofstra R., Gerbens F., Kamps W., de Vries E., van der Zee A., te Meerman G., ter Elst A., Gerbens F., Kamps W., de Vries E., van der Zee A., te Meerman G., ter Elst A., Kamps W., de Vries E., van der Zee A., te Meerman G., ter Elst A., de Vries E., van der Zee A., te Meerman G., ter Elst A., van der Zee A., te Meerman G., ter Elst A., te Meerman G., ter Elst A., ter Elst A. Evidence based selection of housekeeping genes. PLoS One. 2007;2:e898. doi: 10.1371/journal.pone.0000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium Identification and analysis of functional elements in 1% of the human genome by the encode pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P., Aken B., Beal K., Ballester B., Caccamo M., Chen Y., Clarke L., Coates G., Cunningham F., Cutts T., Aken B., Beal K., Ballester B., Caccamo M., Chen Y., Clarke L., Coates G., Cunningham F., Cutts T., Beal K., Ballester B., Caccamo M., Chen Y., Clarke L., Coates G., Cunningham F., Cutts T., Ballester B., Caccamo M., Chen Y., Clarke L., Coates G., Cunningham F., Cutts T., Caccamo M., Chen Y., Clarke L., Coates G., Cunningham F., Cutts T., Chen Y., Clarke L., Coates G., Cunningham F., Cutts T., Clarke L., Coates G., Cunningham F., Cutts T., Coates G., Cunningham F., Cutts T., Cunningham F., Cutts T., Cutts T., et al. Ensembl 2008. Nucleic Acids Res. 2008;36:D707–D714. doi: 10.1093/nar/gkm988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier L., Cope L., Bolstad B., Irizarry R., Cope L., Bolstad B., Irizarry R., Bolstad B., Irizarry R., Irizarry R. affy—Analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- Korbel J., Urban A., Affourtit J., Godwin B., Grubert F., Simons J., Kim P., Palejev D., Carriero N., Du L., Urban A., Affourtit J., Godwin B., Grubert F., Simons J., Kim P., Palejev D., Carriero N., Du L., Affourtit J., Godwin B., Grubert F., Simons J., Kim P., Palejev D., Carriero N., Du L., Godwin B., Grubert F., Simons J., Kim P., Palejev D., Carriero N., Du L., Grubert F., Simons J., Kim P., Palejev D., Carriero N., Du L., Simons J., Kim P., Palejev D., Carriero N., Du L., Kim P., Palejev D., Carriero N., Du L., Palejev D., Carriero N., Du L., Carriero N., Du L., Du L., et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies M., Egholm M., Altman W., Attiya S., Bader J., Bemben L., Berka J., Braverman M., Chen Y., Chen Z., Egholm M., Altman W., Attiya S., Bader J., Bemben L., Berka J., Braverman M., Chen Y., Chen Z., Altman W., Attiya S., Bader J., Bemben L., Berka J., Braverman M., Chen Y., Chen Z., Attiya S., Bader J., Bemben L., Berka J., Braverman M., Chen Y., Chen Z., Bader J., Bemben L., Berka J., Braverman M., Chen Y., Chen Z., Bemben L., Berka J., Braverman M., Chen Y., Chen Z., Berka J., Braverman M., Chen Y., Chen Z., Braverman M., Chen Y., Chen Z., Chen Y., Chen Z., Chen Z., et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullagh P., Nelder J., Nelder J. Generalized linear models. Chapman & Hall; Boca Raton, FL: 1989. [Google Scholar]
- Mikkelsen T., Ku M., Jaffe D., Issac B., Lieberman E., Giannoukos G., Alvarez P., Brockman W., Kim T., Koche R., Ku M., Jaffe D., Issac B., Lieberman E., Giannoukos G., Alvarez P., Brockman W., Kim T., Koche R., Jaffe D., Issac B., Lieberman E., Giannoukos G., Alvarez P., Brockman W., Kim T., Koche R., Issac B., Lieberman E., Giannoukos G., Alvarez P., Brockman W., Kim T., Koche R., Lieberman E., Giannoukos G., Alvarez P., Brockman W., Kim T., Koche R., Giannoukos G., Alvarez P., Brockman W., Kim T., Koche R., Alvarez P., Brockman W., Kim T., Koche R., Brockman W., Kim T., Koche R., Kim T., Koche R., Koche R., et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagalakshmi U., Wang Z., Waern K., Shou C., Raha D., Gerstein M., Snyder M., Wang Z., Waern K., Shou C., Raha D., Gerstein M., Snyder M., Waern K., Shou C., Raha D., Gerstein M., Snyder M., Shou C., Raha D., Gerstein M., Snyder M., Raha D., Gerstein M., Snyder M., Gerstein M., Snyder M., Snyder M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passador-Gurgel G., Hsieh W., Hunt P., Deighton N., Gibson G., Hsieh W., Hunt P., Deighton N., Gibson G., Hunt P., Deighton N., Gibson G., Deighton N., Gibson G., Gibson G. Quantitative trait transcripts for nicotine resistance in Drosophila melanogaster. Nat. Genet. 2007;39:264–268. doi: 10.1038/ng1944. [DOI] [PubMed] [Google Scholar]
- Rifkin S., Kim J., White K., Kim J., White K., White K. Evolution of gene expression in the Drosophila melanogaster subgroup. Nat. Genet. 2003;33:138–144. doi: 10.1038/ng1086. [DOI] [PubMed] [Google Scholar]
- Robinson M., Smyth G., Smyth G. Moderated statistical tests for assessing differences in tag abundance. Bioinformatics. 2007;23:2881–2887. doi: 10.1093/bioinformatics/btm453. [DOI] [PubMed] [Google Scholar]
- Scherf U., Ross D.T., Waltham M., Smith L.H., Lee J.K., Tanabe L., Kohn K.W., Reinhold W.C., Myers T.G., Andrews D.T., Ross D.T., Waltham M., Smith L.H., Lee J.K., Tanabe L., Kohn K.W., Reinhold W.C., Myers T.G., Andrews D.T., Waltham M., Smith L.H., Lee J.K., Tanabe L., Kohn K.W., Reinhold W.C., Myers T.G., Andrews D.T., Smith L.H., Lee J.K., Tanabe L., Kohn K.W., Reinhold W.C., Myers T.G., Andrews D.T., Lee J.K., Tanabe L., Kohn K.W., Reinhold W.C., Myers T.G., Andrews D.T., Tanabe L., Kohn K.W., Reinhold W.C., Myers T.G., Andrews D.T., Kohn K.W., Reinhold W.C., Myers T.G., Andrews D.T., Reinhold W.C., Myers T.G., Andrews D.T., Myers T.G., Andrews D.T., Andrews D.T. A gene expression database for the molecular pharmacology of cancer. Nat. Genet. 2000;24:236–244. doi: 10.1038/73439. [DOI] [PubMed] [Google Scholar]
- Shi L., Reid L., Jones W., Shippy R., Warrington J., Baker S., Collines P., de Longueville F., Kawasaki E., Lee K., Reid L., Jones W., Shippy R., Warrington J., Baker S., Collines P., de Longueville F., Kawasaki E., Lee K., Jones W., Shippy R., Warrington J., Baker S., Collines P., de Longueville F., Kawasaki E., Lee K., Shippy R., Warrington J., Baker S., Collines P., de Longueville F., Kawasaki E., Lee K., Warrington J., Baker S., Collines P., de Longueville F., Kawasaki E., Lee K., Baker S., Collines P., de Longueville F., Kawasaki E., Lee K., Collines P., de Longueville F., Kawasaki E., Lee K., de Longueville F., Kawasaki E., Lee K., Kawasaki E., Lee K., Lee K., et al. The MicroArry Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat. Biotechnol. 2006;24:1151–1161. doi: 10.1038/nbt1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth G. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;3:Article3.. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- Smyth G., Speed T., Speed T. Normalization of cDNA microarray data. Methods. 2003;31:265–273. doi: 10.1016/s1046-2023(03)00155-5. [DOI] [PubMed] [Google Scholar]
- Storey J., Tibshirani R., Tibshirani R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugarbaker D., Richards W., Gordon G., Dong L., De Rienzo A., Maulik G., Glickman J., Chirieac L., Hartman M., Taillon B., Richards W., Gordon G., Dong L., De Rienzo A., Maulik G., Glickman J., Chirieac L., Hartman M., Taillon B., Gordon G., Dong L., De Rienzo A., Maulik G., Glickman J., Chirieac L., Hartman M., Taillon B., Dong L., De Rienzo A., Maulik G., Glickman J., Chirieac L., Hartman M., Taillon B., De Rienzo A., Maulik G., Glickman J., Chirieac L., Hartman M., Taillon B., Maulik G., Glickman J., Chirieac L., Hartman M., Taillon B., Glickman J., Chirieac L., Hartman M., Taillon B., Chirieac L., Hartman M., Taillon B., Hartman M., Taillon B., Taillon B., et al. Transcriptome sequencing of malignant pleural mesothelioma tumors. Proc. Natl. Acad. Sci. 2008;105:3521–3526. doi: 10.1073/pnas.0712399105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres T., Metta M., Ottenwälder B., Schoötterer C., Metta M., Ottenwälder B., Schoötterer C., Ottenwälder B., Schoötterer C., Schoötterer C. Gene expression profiling by massively parallel sequencing. Genome Res. 2008;1:172–177. doi: 10.1101/gr.6984908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables W., Ripley B., Ripley B. Modern applied statistics with S. Springer; New York: 2002. [Google Scholar]
- Weber A., Weber K., Carr K., Wilkerson C., Ohlrogge J., Weber K., Carr K., Wilkerson C., Ohlrogge J., Carr K., Wilkerson C., Ohlrogge J., Wilkerson C., Ohlrogge J., Ohlrogge J. Sampling the Arabidopsis transcriptome with massively parallel pyrosequencing. Plant Physiol. 2007;144:32–42. doi: 10.1104/pp.107.096677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White K. Functional genomics and the study of development, variation and evolution. Nat. Rev. Genet. 2001;2:528–537. doi: 10.1038/35080565. [DOI] [PubMed] [Google Scholar]