

Abstract
Objective
Posttraumatic stress disorder (PTSD) involves deficits in information processing that may reflect hypervigilence and deficient inhibitory control. To date, however, no PTSD neuroimaging study has directly examined PTSD-related changes in executive inhibition. Our objective was to investigate the hypothesis that executive inhibitory control networks are compromised in PTSD.
Methods
Functional magnetic resonance imaging (fMRI) was used during a Go/No-Go inhibition task completed by a sample of patients with PTSD (n = 23), a matched sample of healthy (i.e. without trauma exposure) control participants (n = 23) and a sample of control participants with trauma exposure who did not meet criteria for PTSD (n = 17).
Results
Participants with PTSD showed more inhibition-related errors than did individuals without trauma exposure. During inhibition, control participants activated a right-lateralized cortical inhibitory network, whereas patients with PTSD activated only the left lateral frontal cortex. PTSD was associated with a reduction in right cortical activation and increased activation of striatal and somatosensory regions.
Conclusion
The increased inhibitory error and reduced right frontal cortical activation are consistent with compromised inhibitory control in PTSD, while the increased activation of brain regions associated with sensory processing and a greater demand on inhibitory control may reflect enhanced stimulus processing in PTSD, which may undermine cortical control mechanisms.
Medical subject headings: inhibition; stress disorders, posttraumatic; motor activity; neurophysiology
Abstract
Objectif
Le syndrome de stress post-traumatique (SSPT) met en cause des déficits du traitement de l'information qui peuvent traduire une hypervigilance et un déficit du contrôle inhibiteur. Jusqu'à maintenant, toutefois, aucune étude de neuro-imagerie portant sur le SSPT n'a examiné directement les changements de l'inhibition de l'exécution reliés au SSPT. Notre objectif consistait à étudier l'hypothèse selon laquelle les réseaux de contrôle inhibiteur de l'exécution sont affaiblis dans les cas de SSPT.
Méthodes
On a utilisé l'imagerie par résonance magnétique fonctionnelle (IMRf) au cours d'une tâche d'inhibition oui/non exécutée par un échantillon de patients atteints de SSPT (n = 23), un échantillon jumelé de participants témoins (n = 23) en bonne santé (c.-à-d. sans exposition à un traumatisme) et un échantillon de participants témoins exposés à un traumatisme mais ne satisfaisant aux critères du SSPT (n = 17).
Résultats
Les participants atteints de SSPT ont commis plus d'erreurs reliées à l'inhibition que les individus sans exposition à un traumatisme. Au cours de l'inhibition, les participants témoins ont activé un réseau inhibiteur cortical latéralisé à droite tandis que les patients atteints de SSPT ont activé seulement le cortex frontal latéral gauche. Le SSPT était associé à une réduction de l'activation du cortex droit et une augmentation de l'activation des aires striées et somatosensorielles.
Conclusion
L'erreur accrue de l'inhibition et la réduction de l'activité du cortex frontal droit concordent avec le contrôle affaibli de l'inhibition dans les cas de SSPT, tandis que l'activation accrue des aires cérébrales associées au traitement sensoriel et une plus grande demande imposée au contrôle inhibiteur peuvent refléter le traitement accru des stimulis dans les cas de SSPT, ce qui peut miner les mécanismes de contrôle cortical.
Introduction
Posttraumatic stress disorder (PTSD) is associated with deficits in information processing and with hypervigilence to salient and threat-related stimuli.1 It has been proposed that these deficits are related to increased “bottom–up” hyperarousal coupled with a breakdown of the inhibitory functions required for attentional control and working memory.2 Consistent with this view, neuroimaging studies have characterized PTSD as a disorder involving excessive arousal-related subcortical activity in response to emotional or threat-related stimuli, as well as impaired inhibition over arousal/fear networks in the medial prefrontal cortex (mPFC) and ventral-rostral anterior cingulate cortex (rACC).3–6 A reduction in the capacity to regulate emotion-and arousal-related processing may affect other forms of information processing by increasing the demand on cognitive and attentional control networks.6 However, although many PTSD neuroimaging studies have examined response to emotional stimuli such as fear faces,5,7 trauma symptom provocation or emotional recall,4,8–17 relatively few PTSD neuroimaging studies have examined response to cognitive and attentional tasks that engage control networks by using affectively neutral stimuli.
Neuroimaging findings using generic stimulus processing tasks in PTSD patients have shown significant changes in blood flow in the anterior cingulate cortext (ACC)/mPFC, orbitofrontal cortex, amygdala, hippocampus and parahippocampal gyrus, relative to control participants, in continuous performance tasks,18 as well as significant changes in memory networks,19–22 including reduced dorsolateral prefrontal cortex (DLPFC) activation during working memory.21 Attentional difficulties in PTSD have also been demonstrated through the use of Stroop interference paradigms that require participants to name a word in an incongruent semantic context, which results in a delay in naming relative to a semantically congruent context; these have also shown a reduction in mPFC activation during exposure to emotional but not to nonemotional words.3,6
In contrast to neuroimaging studies that have found a PTSD-related reduction in rACC/mPFC activation in the presence of emotional stimuli,5–17 it has been found that attending to targets in an auditory oddball task is related to increased activation of the mPFC, rostral and dorsal regions of the ACC, the DLPFC and the inferior frontal cortex (IFC) in PTSD patients relative to control participants.2 The findings in the oddball task may reflect a general enhancement of stimulus processing in PTSD that may be related to generalized hypervigilence2 and that may explain electrophysiologic and attentional disturbances in PTSD, including abnormal frontal-and parietal-related electrophysiologic potentials,22 alterations in cortical arousal23 and changes in cortical excitability.24 These disturbances may have an impact on various forms of cognitive control, such as inhibition. To date, no neuroimaging study has studied the executive control of generic stimulus processing in PTSD.
The auditory oddball task used previously2 involves target detection and attentional allocation and requires the passive inhibition of auditory distractors. In contrast, motor response inhibition paradigms are commonly used to study the active, volitional inhibition of stimulus processing.25–30 In these inhibition tasks, a stimulus that requires the participant to respond (“Go”) is presented frequently until a dominant (or prepotent) tendency to respond is established, and then, an infrequent (“No-Go”) stimulus is presented that requires the participant to refrain from responding. This places a demand on executive control because the subject is required to make a decision to inhibit a tendency to respond. Successful inhibitory control is readily operationalized in this paradigm by the presence or absence of motor behaviour. Although attentional allocation tasks such as the oddball have been shown to activate the ACC/mPFC, DLPFC, left ventrolateral prefrontal cortex (VLPFC) and supramarginal gyrus,31 studies in normal populations suggest that inhibitory processing during response inhibition activates a network that is mostly right-lateralized, particularly the right IFC (as well as the right parietal and temporal cortex, ACC, cerebellum and striatum25–30). A change in inhibitory task demand dissociates the neural networks involved in response inhibition; specifically, tasks requiring reduced preparation and a higher demand on inhibitory control have been shown to recruit additional areas of the right VLPFC, right DLPFC, bilateral parietal cortex, insula and striatum.29,30
Converging evidence indicates that in PTSD there may be an enhanced motor readiness, or an increased prepotency to respond, that includes enhanced motor cortical activation in response to fear-and trauma-related stimuli13,20 and increased excitability of the motor cortex.24 PTSD may therefore involve an enhanced demand on inhibitory control during response inhibition. The current study investigated PTSD-related changes in executive control networks with a response inhibition task that uses emotionally neutral stimuli to engage inhibitory control. If PTSD involves enhanced stimulus processing, we would predict that PTSD involves a higher demand on inhibitory control and a recruitment of neural systems involved in higher-demand inhibitory processing (such as the DLPFC, parietal cortex, insula and striatum). Consistent with the suggestion that executive inhibition is compromised in PTSD, we would also predict a reduction in cortical inhibitory control by the right VLPFC/IFC, along with associated inhibitory behavioural deficits.
Method
Participants
Participants included 23 patients with PTSD (10 men, 13 women), 23 healthy individuals without trauma exposure (10 men, 13 women) and 17 trauma-exposed participants (11 men, 6 women) who did not meet the criteria for PTSD. These participants were recruited from the community in collaboration with the Brain Resource International Database32 and the Traumatic Stress Unit, Westmead Hospital, Westmead, Australia. Participant groups were matched for age and handedness. The mean age of participants in the PTSD group was 38.3 (standard deviation [SD] 12.16, range 19–65) years; 21 were right-handed and 2 were left-handed. The mean age of healthy control participants was 39.3 (SD 12.6, range 21–68) years; 21 were right-handed and 2 were left-handed. The mean age of trauma-exposed control participants was 32.4 (SD 15, range 19–64) years; 15 were right-handed and 2 were left-handed. An analysis was also performed with left-handed participants excluded, and this did not significantly alter the main pattern of findings. Exclusion criteria included the following: any current substance abuse or alcohol abuse or dependence (one patient with PTSD met criteria for alcohol abuse 3 months before testing but did not meet criteria for abuse at testing; all other participants did not meet criteria for any substance or alcohol abuse within at least 6 months of testing), any history of traumatic brain injury or neurologic condition, any significant medical condition, a history of psychosis or borderline personality disorder or any loss of consciousness. All participants were screened with the SPHERE-1233for axis I psychiatric disorder. Trauma-exposed control participants had experienced a criterion A stressor but did not develop PTSD in their lifetime (meeting no more than a single lifetime PTSD cluster criterion). Both healthy and trauma-exposed control participants had no current or lifetime psychiatric disorder. Trauma-exposed participants were screened for PTSD according to the Composite International Diagnostic Interview (CIDI).34 The CIDI assesses disorder according to International Classification of Diseases, tenth edition,35 and Diagnostic and Statistical Manual of Mental Disorders, fourth edition (DSM-IV),36 criteria. Trauma-exposed control participants were considered asymptomatic if they endorsed no more than a single DSM-IV PTSD symptom cluster (out of 3 possible PTSD symptom clusters including reexperiencing, avoidance/numbing and arousal). Clusters encompass the 17 PTSD symptoms described by DSM-IV PTSD criteria. The mean total number of PTSD symptoms in trauma-exposed control participants was 3.25 (SD 3.24), whereas the mean total number of PTSD symptoms in patients with PTSD was 12.16 (SD 2.48). To index PTSD severity and specific comorbidity within the PTSD group, these patients were also given the Clinician-Administered PTSD Scale (CAPS),37 the Structured Clinical Interview for DSM-IV Axis I Disorders38 and the Beck Depression Inventory (BDI).39 The CAPS indexes the 17 symptoms described by DSM-IV PTSD criteria. Each symptom is rated on a 5-point scale in terms of the severity and frequency of the symptom in the past week. We adopted a conservative estimate of good end-state functioning for PTSD by following an established cut-off total CAPS score of 19 (combining frequency and intensity scores) as a measure of the absence of PTSD.40 The mean total CAPS score in the PTSD group was 76.1 (SD 17.4, range 44–111), and the mean BDI score was 27.8 (SD 13.2, range 6–52). Trauma in the PTSD group was the result of a physical assault (n = 14) or motor vehicle or industrial accident (n = 9). Trauma in the trauma-exposed control group was a result of physical assault (n = 2), motor vehicle or industrial accident (n = 4), witnessing a bad injury or a killing (n = 4), being involved in a fire or natural disaster (n = 4) or being threatened with a weapon, held captive or kidnapped (n = 3). Comorbid disorders in the PTSD group included major depression (n = 15), obsessive–compulsive disorder (n = 1), panic disorder (n = 3) and bulimia (n = 1). Eleven patients with PTSD were taking medication (citalopram n = 1, paroxetine n = 2, venlafaxine n = 1, bupivacaine n = 1, mirtazepine n = 2, St. John's wort n = 1, contraceptive n = 1 and diarrhea tablets n = 1). Average time posttrauma did not significantly differ between the PTSD group (56.7 mo, SD 74.7) and the trauma-exposed control group (80 mo, SD 57.1); p = 0.17. The Western Sydney Area Health Service Human Research Ethics Committee approved the study, and all participants gave written informed consent before participating.
Behavioural procedure and functional magnetic resonance image acquisition
A Go/No-Go behavioural procedure was employed during the functional magnetic resonance imaging (fMRI) scanning. The Go/No-Go paradigm requires participants to respond during “Go” trials and then to withhold this prepotent response during “No-Go” trials, with commission error (the inability to withhold a response) reflecting a deficit in response inhibition. Participants were placed on the MR scanner table and fitted with MRI-compatible headphones; a mirror fitted into the head coil projected a visual display from an external projector (Sanyo Pro-X, Multiverse, Tokyo, Japan), maximum 60 Hz. Go stimuli (“PRESS” in green-coloured type in the centre of a black screen) and No-Go stimuli (“PRESS” in red-coloured type in the centre of a black screen) were presented to participants by the projector system. Each Go and No-Go stimulus was presented for 500 milliseconds, with an interstimulus interval of 1143 milliseconds. To create a tendency to respond, Go stimuli were presented 75% of the time (total Go stimuli = 126), and No-Go stimuli were randomly presented 25% of the time (total No-Go stimuli = 42). Participants received standardized visual and audio instructions to tap a response box as quickly as possible when the Go stimulus appeared and to withhold response to No-Go stimuli (each subject was instructed to tap the response box with both their left and right thumbs simultaneously to counterbalance for motor activity). Ninety-three T2-weighted volumes (including 3 initial “dummy” scans) depicting blood oxygen level dependent (BOLD) contrast were acquired with a VISION Plus 1.5 Tesla scanner (Siemens Magnetom) fitted with a standard quadrature head coil. T2-weighted images were obtained by using a gradient echoplanar sequence, and 15 axial noncontiguous slices of 6-mm thickness (0.6 interslice gap) were measured, positioned in parallel to the intercommissural (AC-PC) line (repetition time 3.2 s [3 s, 200 ms = 3.278 with delay]; time to echo 40 ms, matrix 128 × 128; field of view 24 cm × 24 cm2, flip angle 90°). These slices covered the whole cerebrum.
Behavioural and fMRI analysis
For behavioural analysis, we determined significant differences between groups for the number of commission (failure to inhibit) errors. Total CAPS scores in the PTSD group were also correlated with behavioural performance. fMRI analysis was conducted with Statistical Parametric Mapping (SPM2, Wellcome Department of Neurology, London, UK). All T2-weighted volumes were realigned, unwarped, spatially normalized into standardized Montréal Neurological Institute space and smoothed with a Gaussian kernel (full width at half maximum 8 mm). A hemodynamic response function–convolved boxcar model with temporal derivative was created to correspond to Go and No-Go stimuli, and a high-pass filter was applied to remove low-frequency fluctuations in the BOLD signal. To examine voxel-wise effects of signal changes, BOLD signal change was analyzed for the contrast of No-Go minus Go stimuli in each subject. This contrast determines activation related to No-Go compared with the Go responding, which better isolates the activation responsible for the decision to inhibit and inhibitory response components of the task (and minimizes the measurement of activation due to selective attention and response selection processes). The No-Go minus Go contrast was then used for all within-group and between-group random effects analyses. We undertook whole brain t test analyses with a statistical threshold of p < 0.005 (uncorrected) and an extent threshold of greater than or equal to 5 contiguous voxels per cluster to determine significant areas of activation due to No-Go minus Go responding within each group. To control for the contribution of inhibitory ability to the between-group neural activation differences, we used total commission error as a covariate in our between-group analyses. Within the PTSD group, we determined the relation between inhibitory ability, PTSD severity and neural activation by using a multiple regression analysis in SPM2 for the contrast of No-Go minus Go, with CAPS score and total commission error as within-group regressors of interest. This allowed us to determine how level of PTSD severity (i.e., higher or lower total CAPS scores in the PTSD participants) may predict changes in BOLD activation during No-Go minus Go responding, as well as how differences in inhibitory ability (measured by total number of commission errors) may predict changes in No-Go minus Go–related activation. Considering that the PTSD group exhibited some comorbid depression, we also determined the impact of depressive symptoms in PTSD on No-Go minus Go responding by performing a separate partial regression analysis (using SPM2) within the PTSD group, with total BDI score as a regressor of interest (taking into account differences in PTSD severity, i.e., differences in total CAPS score, and commission error).
Results
Behavioural data
Patients with PTSD performed more commission errors than healthy control participants (Mann–Whitney U = 122, p = 0.004; PTSD mean number of commission errors 3.5, SD 2.7; healthy control participants mean number of commission errors 1.38, SD 1.2) but not trauma-exposed control participants (U = 137, p = 0.33; trauma-exposed control mean number of commission errors 2.3, SD 1.6). There was a significant positive correlation between total CAPS score and commission error (Spearman's r = 0.48, p = 0.04).
fMRI data
Within-group findings
With regard to areas recruited during inhibitory processing, Table 1 and Figure 1 indicate the significant activations within PTSD and control participants during No-Go related processing. Both healthy and trauma-exposed control participants activated a network that included the right IFC and right parietal cortex, whereas participants with PTSD activated the left VLPFC (Brodmann area [BA] 10) and right temporal cortex (BA 21 and 22).
Table 1
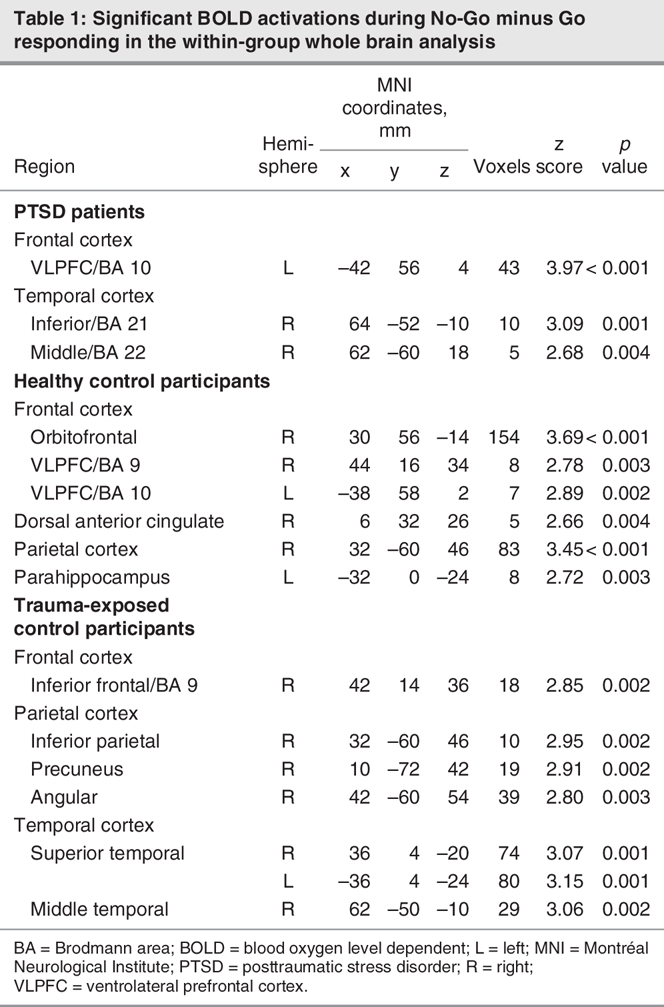

Fig. 1: Significant activation (increased blood oxygen level dependent signal) elicited during inhibitory responding (No-Go relative to Go) within patients with PTSD and healthy control participants. Control participants (top) activated mostly right ventrolateral prefrontal cortex/orbitofrontal cortex, whereas patients with PTSD (bottom) showed a lack of activation in right frontal areas. Those with PTSD instead activated the left ventrolateral frontal cortex. L = left; PTSD = posttraumatic stress disorder; R = right.
With regard to the effects of PTSD severity, comorbid depression and inhibitory ability on inhibitory processing. Table 2 outlines the independent effects of PTSD severity (total CAPS score), inhibitory ability (total commission error) and depression comorbidity (total BDI score) on neural activation during No-Go–related responding in patients with PTSD. Better inhibitory performance (i.e., a diminished total number of commission errors) in PTSD predicted increased activation in the putamen, ventral ACC and VLPFC/IFC, whereas poorer inhibitory performance (increased number of commission errors) predicted greater activation in the mPFC and motor cortex. Diminished PTSD severity predicted greater activation of inhibition-relevant areas such as the right IFC, DLPFC, mPFC and cerebellum (Fig. 2). Comorbid depression in the PTSD participants did not affect activation in these inhibition-relevant areas during No-Go minus Go responding.
Table 2
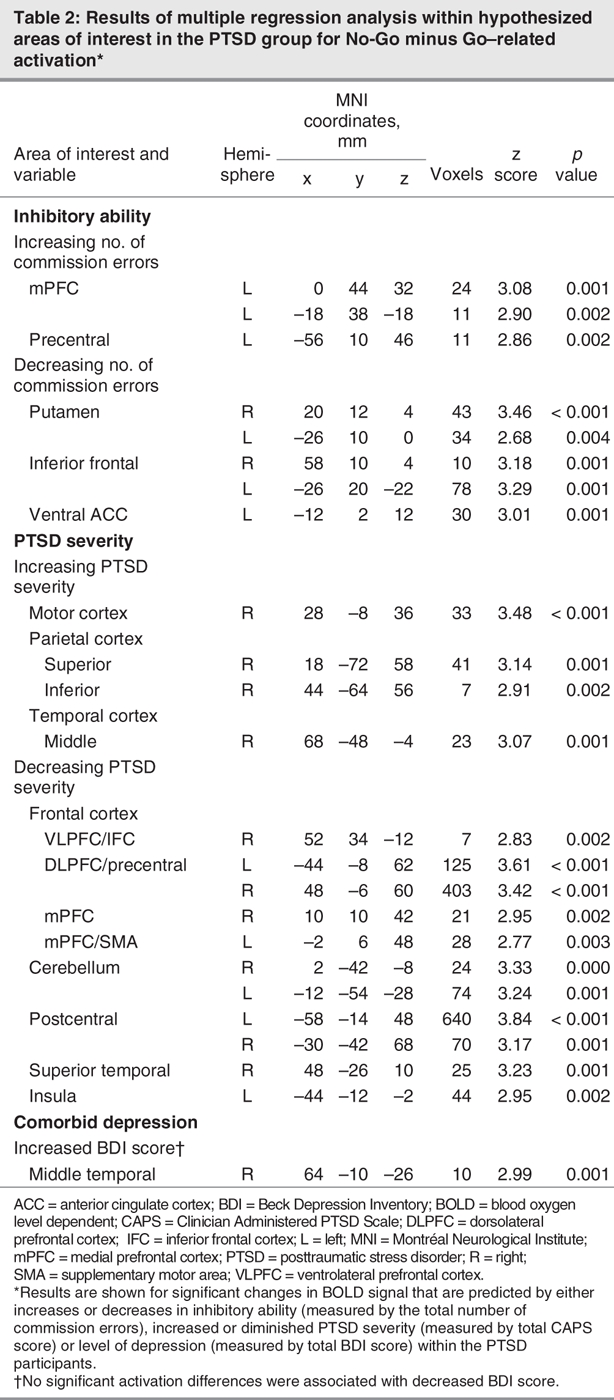

Fig. 2: Significant differences observed during No-Go–related processing between healthy control participants and subjects with posttraumatic stress disorder (PTSD) (puncorrected < 0.005). Increased blood oxygen level dependent (BOLD) signal elicited by No-Go stimuli minus Go stimuli and changes in BOLD activation due to PTSD severity within PTSD patients. DLPFC = dorsolateral prefrontal cortex; mPFC = medial prefrontal cortex; VLPFC = ventrolateral prefrontal cortex.
Between-group comparisons
Table 3 and Table 4 and Figure 2 summarize the between-group differences during No-Go related processing in hypothesized brain regions (cortex, basal ganglia and cerebellum). Both groups of control participants showed significantly greater activation of the right VLPFC/IFC, mPFC, DLPFC and temporoparietal cortex relative to the PTSD group. In contrast, patients with PTSD activated the postcentral gyrus and cuneus to a greater extent than both groups of control participants. Patients with PTSD also activated the putamen, parahippocampal area and cerebellum to a greater extent than healthy control participants. Trauma-exposed and healthy control participants did not differ in inhibition-related activation in regions of interest.
Table 3
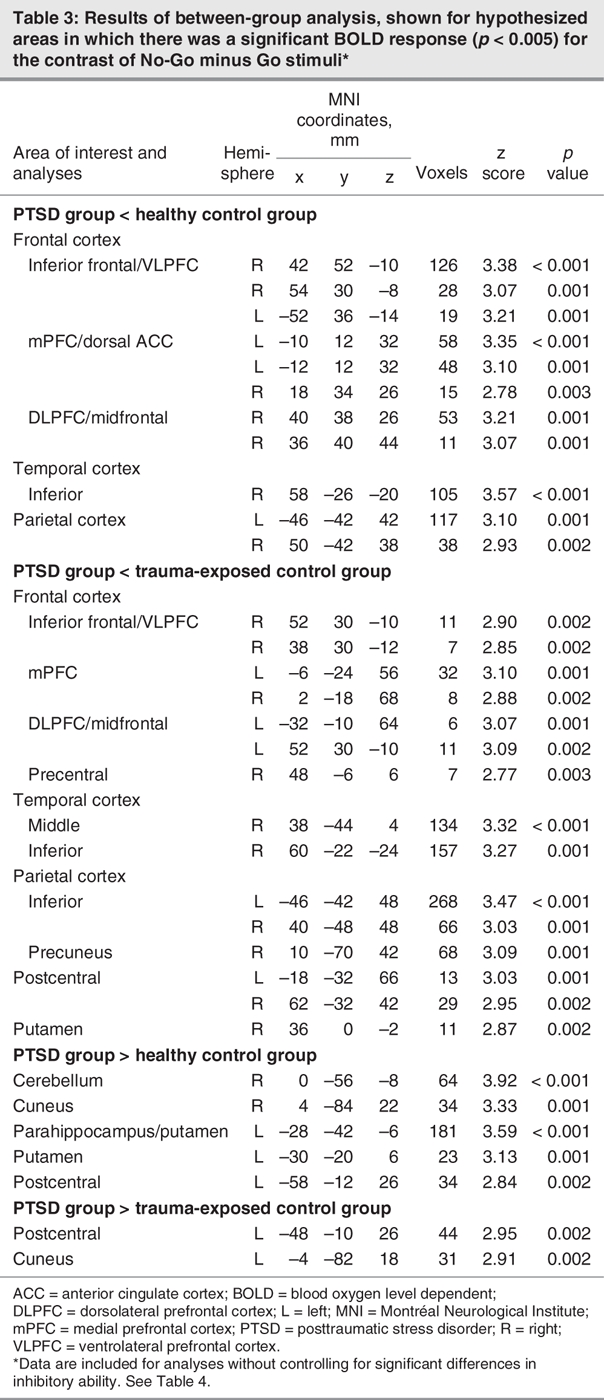
Table 4
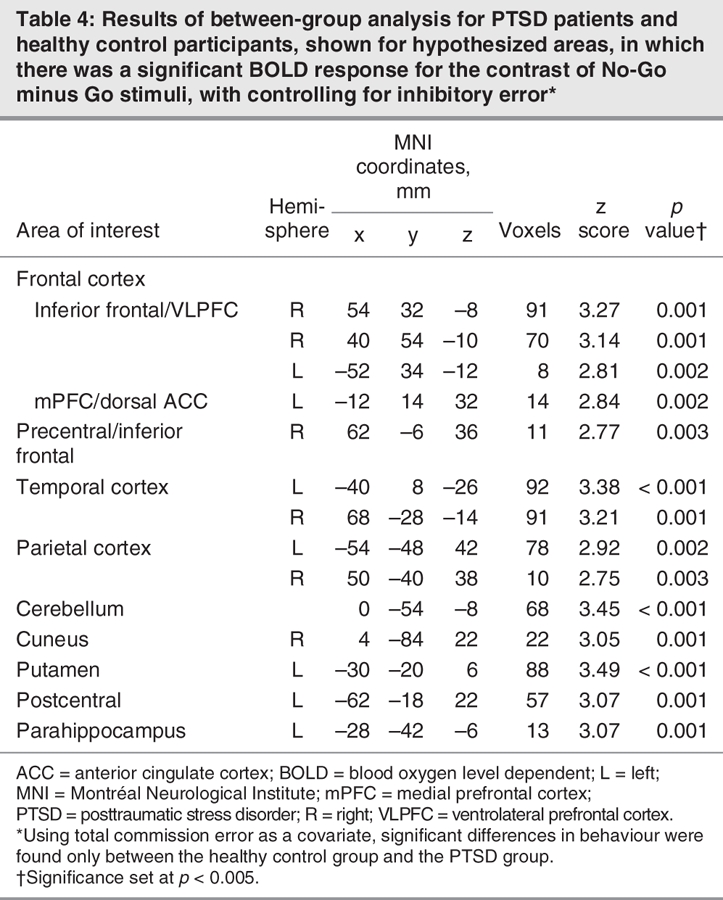
Considering the significant difference in inhibitory ability in PTSD relative to healthy control participants (i.e., increased total commission error), we determined significant between-group differences during No-Go responding while controlling for inhibitory error (using commission error as a covariate in analysis of covariance; see Table 4). Even after taking commission error into account, we found that control participants activated the right inferior frontal/VLPFC cortex to a greater extent than did patients with PTSD, and those with PTSD activated the cuneus, cerebellum, putamen, parahippocampus and postcentral cortex relative to healthy control participants.
Discussion
This study represents the first demonstration of neural networks associated with executive inhibitory control of generic information in PTSD. We found that PTSD involves both inhibitory deficits and reduced activation of brain areas normally associated with successful inhibition (particularly the right IFC, which may be an important locus of inhibitory control27–29). Consistent with previous studies in healthy individuals, control participants activated a mostly right-lateralized fronto-parietal cortical network during inhibitory processing.27–30 In contrast, patients with PTSD activated mostly the left VLPFC and showed reduced activation of a right-lateralized frontotemporoparietal cortical inhibitory network, relative to control participants. We also found that inhibitory responding in PTSD was associated with the recruitment of areas related to sensory processing (somatosensory cortex, parahippocampus and visual cortex) and higher inhibitory task demand (striatal regions30).
We found that there was an increase in inhibitory error with increased PTSD severity. Appropriately, our findings suggest not only that there is an increase in inhibitory error with increased PTSD severity but also that more severe PTSD may be related to the diminished recruitment of inhibitory control networks. Successful inhibition (reduced commission error) in patients with PTSD recruited a bilateral inferior frontal and striatal (putamen) inhibition network. We found that decreased PTSD severity related to the enhanced recruitment of inhibitory control areas (right IFC and DLPFC) and to additional recruitment of the mPFC/left supplementary motor area (which has been implicated in motor response planning and inhibition27). That is, patients with less severe PTSD recruited inhibitory control areas to a greater extent during inhibitory responding than did those with more severe PTSD. One possible explanation for this reduction in executive control with increased PTSD severity is that PTSD disrupts cortical control systems; participants with PTSD are able to recruit inhibitory control networks, but these control systems may be deficient or “overwhelmed” with increasing PTSD severity. Alternatively, it is possible that individuals with a diminished ability to recruit these executive areas are at greater risk for developing more severe PTSD symptoms. Importantly, our findings argue against the possibility that the diminished cortical recruitment observed in PTSD is adaptive or reflects an enhanced cortical efficiency because increased PTSD severity was associated with both increased inhibitory error and a diminished recruitment of inhibitory control systems.
PTSD involves generalized hypervigilence, excessive arousal-related processing and enhanced response to salient stimuli2 as well as cortical hyperexcitability.24 Adrenergic, arousal-related processing stimulates the cortex and contributes to deficits in prefrontal function and executive control.41 As such, inhibitory deficits in PTSD may be explained by increased stimulus processing that stimulates and places a demand on the cortex. Our finding of increased somatosensory cortical (postcentral gyrus), parahippocampal and visual cortical activation in patients with PTSD relative to control participants is consistent with a state of enhanced sensory processing during inhibitory control.2,42 The increased activation in the putamen in patients with PTSD relative to control participants during inhibitory processing suggests that there may be an increased demand placed on inhibitory control systems in PTSD. The striatum is recruited during Go/No-Go performance when the task requires more urgent inhibition and therefore involves an increased demand on inhibitory control.30 Animal models also show that differential activation of the frontal cortex and striatal network is involved in behavioural flexibility, with the frontal cortex supporting the inhibition of a previous choice to generate a new behavioural choice, whereas the striatum maintains a behavioural strategy once it is selected.43 Disruptions in this corticostriatal pathway possibly underlie the lack of both cognitive and behavioural flexibility and the repetitive, stereotyped behaviour that is characteristic of certain neuropsychiatric conditions (such as obsessive–compulsive disorder44). The striatum is also important for learning to predict aversive and rewarding outcomes,45 and it is suggested that the striatum plays a role in regulating attention to predict danger.46 Considering this role of the striatum, an increase in striatal activation in PTSD is particularly relevant to the enhanced attentional bias to threat and novelty shown to be characteristic of PTSD.2 Taken together, the increased activation of sensory-related areas and the striatum in PTSD during No-Go responding may reflect enhanced sensory processing, an increased demand on control systems and a possible dysregulation in the substrates of behavioural flexibility.
Notably, we found diminished mPFC and dorsal ACC (dACC) activation in patients with PTSD relative to control participants during inhibitory control in our Go/No-Go task. This stands in contrast to previous studies showing increased dACC/mPFC activation in patients with PTSD relative to control participants during target-related responding in an auditory oddball task.2 Similar to the auditory oddball task, the Go/No-Go task involves both selective attention and response selection, but in addition, it involves executive inhibitory control. These convergent findings suggest that processing related to attention and response selection may be heightened in PTSD, whereas an additional demand on executive inhibition may involve a breakdown in cortical control. That is, our findings of reduced mPFC/dACC in patients with PTSD during inhibitory responding may indicate that our emotionally neutral inhibitory control task involves a sufficient demand on cortical control to overwhelm this regulatory structure.6 This suggestion is further supported by our observation that decreased PTSD severity was related to increased activation of the mPFC during inhibitory control. The mPFC, ACC and orbitofrontal cortex may have a critical role in coordinating the networks involved in alerting and processing significant stimuli with those involved in more detailed, controlled contextual processing (hippocampus and lateral prefrontal cortex).47 Our findings of reduced dACC/mPFC and orbitofrontal cortex activation in PTSD during inhibitory control may suggest a disruption of “significance processing” networks in PTSD.
Previous trauma exposure in the control participants did not significantly influence our main findings; both trauma-exposed and healthy control participants activated the right orbitofrontal/VLPFC during inhibitory control and also activated this area to a greater extent than participants with PTSD. However, whereas those with PTSD differed in their inhibitory performance relative to healthy control participants, our behavioural findings indicated that participants with PTSD did not differ in inhibition-related performance relative to trauma-exposed control participants. This may reflect a possible floor effect, since our Go/No-Go task was relatively simple (commission errors: PTSD mean 3.5; trauma-exposed control mean 2.3; healthy control mean 1.4). It is interesting, however, that even this simple inhibitory task was associated with a significant reduction in cortical activation in participants with PTSD.
Limitations of this study include the possible effects on our findings of comorbid diagnoses, possible axis II disorder (DSM-IV36) or the use of medication. Comorbid depressive symptoms did not significantly affect inhibition-related frontal cortical activation in patients with PTSD; those having PTSD both with and without depression showed diminished activation of the orbitofrontal/VLPFC and mPFC inhibition areas as well as increased activation of somatosensory areas (postcentral cortex) and areas related to increased inhibitory demand (striatum), relative to control participants. Further, depression severity did not significantly influence frontal cortical activation in participants with PTSD during No-Go inhibition. Another limitation of the current study is the possible effect of sex or handedness on our findings (considering that inhibitory control networks showed right lateralization). This possibility should be explored in future studies.
In conclusion, our findings not only support previous models of PTSD suggesting that PTSD is accompanied by enhanced stimulus processing2 and reduced cortical control,3–22 they also extend these models to show that PTSD involves unique neural alterations during the executive inhibitory control of emotionally neutral information processing. Our findings indicate that increasing PTSD severity may be related to a greater disruption of cortical control networks. Additional research should be undertaken to explore further the mechanisms associated with these PTSD-related changes in inhibitory processing, and particularly the ways in which autonomic arousal may modulate these changes.
Acknowledgments
This study was funded by an Australian Research Council Linkage Grant (LP0212045) and a National Health and Medical Research Council (NHMRC) program grant (300304). We would like to acknowledge the contribution of the Brain Resource International Database (BRID). L.M.W. is a small equity holder and E.G. holds significant equity and stock options in the Brain Resource Company Ltd. We acknowledge the support of the Brain Resource International Database (under the auspices of the Brain Resource Company) in data acquisition and methodology.
Footnotes
Contributors: Ms. Falconer and Drs. Bryant, Gordon and Williams designed the study. Ms. Olivieri and Dr. Perduto acquired the data, which Ms. Falconer and Drs. Bryant, Felmingham, Kemp and Williams analyzed. Ms. Falconer and Drs. Williams, Bryant and Felmingham wrote the article, which Mses. Falconer and Olivieri and Drs. Bryant, Felmingham, Kemp, Gordon, Peduto and Williams reviewed. All authors gave final approval for publication.
Competing interests: None declared for Drs. Bryant, Felmingham, Peduto and Ms. Olivieri. Ms. Falconer has been employed part-time by the Brain Resource Company Ltd. to conduct work unrelated to the study. Dr. Kemp is currently employed by the Brain Resource Company Ltd. in work unrelated to this study. Dr. Gordon is CEO of the Brain Resource Company Ltd. Dr. Williams owns stock in the Brain Resource Company Ltd. and has received paid contract work from them unrelated to the study.
Correspondence to: Ms. E. Falconer, Brain Dynamics Centre, Westmead Hospital, Westmead, NSW 2145, Australia; fax 61-2-96357734; efalconer@psy.unsw.edu.au
References
- 1.Buckley TC, Blanchard EB, Neill WT. Information processing and PTSD: a review of the empirical literature. Clin Psychol Rev 2000;20:1041-65. [DOI] [PubMed]
- 2.Bryant RA, Felmingham KL, Kemp AH, et al. Neural networks of information processing in posttraumatic stress disorder: a functional magnetic resonance imaging study. Biol Psychiatry 2005;58: 11-8. [DOI] [PubMed]
- 3.Bremner JD, Vermetten E, Vythilingam M, et al. Neural correlates of the classic color and emotional stroop in women with abuse-related posttraumatic stress disorder. Biol Psychiatry 2004;55:612-20. [DOI] [PubMed]
- 4.Lanius RA, Williamson PC, Densmore M, et al. Neural correlates of traumatic memories in posttraumatic stress disorder: a functional MRI investigation. Am J Psychiatry 2001;158:1920-2. [DOI] [PubMed]
- 5.Shin LM, Wright CI, Cannistraro PA, et al. A functional magnetic resonance imaging study of amygdala and medial prefrontal cortex responses to overtly presented fearful faces in posttraumatic stress disorder. Arch Gen Psychiatry 2005;62:273-81. [DOI] [PubMed]
- 6.Shin LM, Whalen PJ, Pitman RK, et al. An fMRI study of anterior cingulate function in posttraumatic stress disorder. Biol Psychiatry 2001;50:932-42. [DOI] [PubMed]
- 7.Rauch SL, Whalen PJ, Shin LM, et al. Exaggerated amygdala response to masked facial stimuli in posttraumatic stress disorder: a functional MRI study. Biol Psychiatry 2000;47:769-76. [DOI] [PubMed]
- 8.Rauch SL, Van der Kolk BA, Fisler RE, et al. A symptom provocation study of posttraumatic stress disorder using positron emission tomography and script-driven imagery. Arch Gen Psychiatry 1996;53:380-7. [DOI] [PubMed]
- 9.Liberzon I, Taylor SF, Amdur R, et al. Brain activation in PTSD in response to trauma-related stimuli. Biol Psychiatry 1999;45:817-26. [DOI] [PubMed]
- 10.Shin LM, McNally RJ, Kosslyn SM, et al. Regional cerebral blood flow during script-driven imagery in childhood sexual abuse-related PTSD: a PET investigation. Am J Psychiatry 1999;156:575-84. [DOI] [PubMed]
- 11.Shin LM, Orr SP, Carson MA, et al. Regional cerebral blood flow in amygdala and medial prefrontal cortex during traumatic imagery in male and female Vietnam veterans with PTSD. Arch Gen Psychiatry 2004;61:168-76. [DOI] [PubMed]
- 12.Liberzon I, Britton JC, Phan KL. Neural correlates of traumatic recall in posttraumatic stress disorder. Stress 2003;6:151-6. [DOI] [PubMed]
- 13.Bremner JD, Narayan M, Staib LH, et al. Neural correlates of memories of childhood sexual abuse in women with and without posttraumatic stress disorder. Am J Psychiatry 1999;156:1787-95. [DOI] [PMC free article] [PubMed]
- 14.Shin LM, Kosslyn SM, McNally RJ, et al. Visual imagery and perception in posttraumatic stress disorder. A positron emission tomographic investigation. Arch Gen Psychiatry 1997;54:233-41. [DOI] [PubMed]
- 15.Lanius RA, Williamson PC, Hopper J, et al. Recall of emotional states in posttraumatic stress disorder: an fMRI investigation. Biol Psychiatry 2003;53:204-10. [DOI] [PubMed]
- 16.Pissiota A, Frans O, Fernandez M, et al. Neurofunctional correlates of posttraumatic stress disorder: a PET symptom provocation study. Eur Arch Psychiatry Clin Neurosci 2002;252:68-75. [DOI] [PubMed]
- 17.Gilboa A, Shalev AY, Laor L, et al. Functional connectivity of the prefrontal cortex and the amygdale in posttraumatic stress disorder. Biol Psychiatry 2004;55:263-72. [DOI] [PubMed]
- 18.Semple WE, Goyer PF, McCormick R, et al. Higher brain blood flow at amygdala and lower frontal cortex blood flow in PTSD patients with comorbid cocaine and alcohol abuse compared to normals. Psychiatry 2000;63:65-74. [DOI] [PubMed]
- 19.Shaw ME, Strother SC, McFarlane AC, et al. Abnormal functional connectivity in posttraumatic stress disorder. Neuroimage 2002;15:661-74. [DOI] [PubMed]
- 20.Bremner JD, Vythilingam M, Vermetten E, et al. MRI and PET study of deficits in hippocampal structure and function in women with childhood sexual abuse and posttraumatic stress disorder. Am J Psychiatry 2003;160:924-32. [DOI] [PubMed]
- 21.Clark CR, McFarlane AC, Morris P, et al. Cerebral function in posttraumatic stress disorder during verbal working memory updating: a positron emission tomography study. Biol Psychiatry 2003;53:474-81. [DOI] [PubMed]
- 22.Weber DL, Clark CR, McFarlane AC, et al. Abnormal frontal and parietal activity during working memory updating in post-traumatic stress disorder. Psychiatry Res 2005;140:27-44. [DOI] [PubMed]
- 23.Veltmeyer MD, McFarlane AC, Bryant RA, et al. Integrative assessment of brain function in PTSD: brain stability and working memory. J Integr Neurosci 2006;5:123-38. [DOI] [PubMed]
- 24.Centonze D, Palmieri MG, Boffa L, et al. Cortical hyperexcitability in posttraumatic stress disorder secondary to minor accidental head trauma: a neurophysiologic study. J Psychiatry Neurosci 2005;30:127-32. [PMC free article] [PubMed]
- 25.Badcock JC, Michie PT, Johnson L, et al. Acts of control in schizophrenia: dissociating the components of inhibition. Psychol Med 2002;32:287-97. [DOI] [PubMed]
- 26.Bannon S, Gonsalvez CJ, Croft RJ, et al. Response inhibition deficits in obsessive-compulsive disorder. Psychiatry Res 2002;110:165-74. [DOI] [PubMed]
- 27.Garavan H, Ross TJ, Stein EA. Right hemispheric dominance of inhibitory control: an event-related functional MRI study. Proc Natl Acad Sci U S A 1999;96:8301-6. [DOI] [PMC free article] [PubMed]
- 28.Rubia K, Smith AB, Brammer MJ, et al. Right inferior prefrontal cortex mediates response inhibition while mesial prefrontal cortex is responsible for error detection. Neuroimage 2003;20:351-8. [DOI] [PubMed]
- 29.de Zubicaray GI, Andrew C, Zelaya FO, et al. Motor response suppression and the prepotent tendency to respond: a parametric fMRI study. Neuropsychologia 2000;38:1280-91. [DOI] [PubMed]
- 30.Kelly AM, Hester R, Murphy K, et al. Prefrontal-subcortical dissociations underlying inhibitory control revealed by event-related fMRI. Eur J Neurosci 2004;19:3105-12. [DOI] [PubMed]
- 31.Yoshiura T, Zhong J, Shibata DK, et al. Functional MRI study of auditory and visual oddball tasks. Neuroreport 1999;10:1683-8. [DOI] [PubMed]
- 32.Gordon E. Integrative neuroscience. Neuropsychopharmacology 2003;28 (Suppl 1):S2-8. [DOI] [PubMed]
- 33.Hickie IB, Davenport TA, Hadzi-Pavlovic D, et al. Development of a simple screening tool for common mental disorders in general practice. Med J Aust 2001;(Suppl 175):S10-7. [DOI] [PubMed]
- 34.World Health Organization (WHO). The Composite International Diagnostic Interview (CIDI). Geneva: WHO; 1993.
- 35.World Health Organization (WHO). International Classification of Diseases (ICD). 10th rev. Geneva: WHO; 2007.
- 36.American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4th ed. Washington: The Association; 1994.
- 37.Blake DD, Weathers FW, Nagy LM, et al. The development of a Clinician-Administered PTSD Scale. J Trauma Stress 1995;8:75-90. [DOI] [PubMed]
- 38.First MB, Spitzer RL, Gibbon M, et al. Structured clinical interview for DSM-IV axis 1 disorders - clinician version (SCID_CV). New York: New York State Psychiatric Institute; 1998.
- 39.Beck AT, Ward CH, Mendelson M, et al. An inventory for measuring depression. Arch Gen Psychiatry 1961;4:53-63. [DOI] [PubMed]
- 40.Weathers FW, Keane TM, Davidson JRT. Clinician-administered PTSD scale: a review of the first ten years of research. Depress Anxiety 2001;13:132-56. [DOI] [PubMed]
- 41.Arnsten AF, Steere JC, Jentsch DJ, et al. Noradrenergic influences on prefrontal cortical cognitive function: opposing actions at postjunctional alpha 1 versus alpha 2-adrenergic receptors. Adv Pharmacol 1998;42:764-7. [DOI] [PubMed]
- 42.Posner MI, Peterson SE. The attention system of the human brain. Annu Rev Neurosci 1990;13:25-42. [DOI] [PubMed]
- 43.Ragozzino M. The contribution of the medial prefrontal cortex, orbitofrontal cortex and dorsomedial striatum to behavioral flexibility. Ann N Y Acad Sci 2007;1121:355-75. [DOI] [PubMed]
- 44.Graybiel AM, Rauch SL. Toward a neurobiology of obsessive-compulsive disorder. Neuron 2000;28:343-7. [DOI] [PubMed]
- 45.O'Doherty J, Dayan P, Schultz J, et al. Dissociable roles of ventral and dorsal striatum in instrumental conditioning. Science 2004;304:452-4. [DOI] [PubMed]
- 46.McNally GP, Westbrook RF. Predicting danger: the nature, consequences, and neural mechanisms of predictive fear learning. Learn Mem 2006;13:245-53. [DOI] [PMC free article] [PubMed]
- 47.Williams LM. An integrative neuroscience model of “significance” processing. J Integr Neurosci 2006;5:1-47. [DOI] [PubMed]