

Abstract
Cell-signaling peptides have been extensively used to transport functional molecules across the plasma membrane into living cells. These peptides consist of a hydrophobic sequence and a cationic nuclear localization sequence (NLS). It has been assumed that the hydrophobic region penetrates through the hydrophobic lipid bilayer and delivers the NLS inside the cell. To better understand the transport mechanism of these peptides, in this study, we investigated the structure, orientation, tilt of the peptide relative to the bilayer normal, and the membraneinteraction of two cell-signaling peptides, SA and SKP. Results from CD and solid-state NMR experiments combined with molecular dynamics simulations suggest that the hydrophobic region is helical and has a transmembrane orientation with the helical axis tilted away from the bilayer normal. The influence of the hydrophobic mismatch, between the hydrophobic length of the peptide and the hydrophobic thickness of the bilayer, on the tilt angle of the peptides was investigated using thicker POPC and thinner DMPC bilayers. NMR experiments showed that the hydrophobic domain of each peptide has a tilt angle of 15±3° in POPC, while in DMPC 25±3° and 30±3° tilts were observed for SA and SKP peptides respectively. These results are in good agreement with molecular dynamics simulations, which predicts a tilt angle of 13.3° (SA in POPC), 16.4° (SKP in POPC), 22.3° (SA in DMPC) and 31.7° (SKP in POPC). These results and simulations on the hydrophobic fragment of SA or SKP suggest that the tilt of helices increases with a decrease in the bilayer thickness without changing the phase, order, and structure of the lipid bilayers.
Keywords: Cell-permeable peptides, NLS, PISA wheel, tilt, hydrophobic mismatch, transmembrane helix
Noninvasive delivery of peptides, proteins, oligonucleotides and other functional cargo molecules into living cells is highly dependent on their efficient transport across the plasma membrane barrier (1-7). Hydrophobic peptides have been successfully used to transport nuclear localization sequences (NLS) in order to probe intracellular signaling, which involved interactions of thousands of proteins expressed in living cells (3, 6). For example, cellular import of peptides has been used in the agonist-induced translocation of transcription factor NF-kB, FGF-1-stimulated mitogenesis, the function of an intracellular signaling protein SHc whose over-expression leads to cell transformation in NIH 3T3 fibroblasts, and epidermal growth factor (EGF)-stimulated Ras activation in intact cells (1-7). Cellular import is also involved in the regulation of intracellular pathways associated with adhesion, signaling and trafficking to the nucleus. Recent studies have suggested that, in general, cell-signaling peptides translocate a functional nuclear localization sequence directly across the plasma membrane bilayers without this sequence interacting with a receptor or transporter protein in the process (5, 8-10). However, there are no high-resolution studies on the secondary structure, folding, and membrane-interaction of cell-signaling peptides to understand their transport mechanism. In this study, we have therefore investigated the structure and topology of two such peptides, SKP and SA (Figure 1), using solid-state NMR experiments and molecular dynamics simulations of lipid bilayers with varying compositions.
Figure 1.

Primary sequence for 26-residues, SA, 41-residues, SKP, and the transmembrane fragment (TM) peptides. The underlined residues are hydrophobic and identical in all peptides.
Cell-signaling peptides SKP (6) and SA (8, 9) each contain a 16-residue hydrophobic sequence in their N-terminus and different types of nuclear localization sequences at the C-terminus. The hydrophobic region is selected from a signaling peptide sequence. The SKP peptide contains a 25-residue nuclear localization sequence of NF-kB p50 and has been shown to inhibit, in a concentration-dependent manner, nuclear translocation of NF-kB in cultured endothelial and monocytic cells stimulated with lipopolysaccharide or tumor necrosis factor-alpha (6). In the SA peptide, the 16-residue hydrophobic and 7-residue NLS sequences are connected by a spacer region of three Ala residues. This peptide has been shown to stimulate DNA synthesis in NIH 3T3 cells in an FGF-receptor-independent manner and to induce DNA synthesis in bovine hamster kidney-21 cells (8, 9). Studies have shown that these peptides are not cytotoxic, and deletion of the hydrophobic domain or mutation of the NLS sequence abolishes the activity of the peptide (6, 8-10).
Since the above-mentioned functions of these peptides are highly dependent on their ability to interact with cell membranes, structural studies of these peptides could provide insights into their transport mechanism and efficiency of transport across cell membranes. In spite of recent successes in this field, structure determination of membrane-associated peptides and proteins still remains as a big challenge. Recently, it has been demonstrated that solid-state NMR studies can address biological questions related to membrane-bound peptides (11-17) and also determine the backbone conformation (18-27) and tertiary folding of peptides and proteins in lipid bilayers (28-33). Most importantly, solid-state NMR experiments can determine the orientation of these molecules in lipid bilayers, which helps in understanding the function of these systems. A complementary approach is to use molecular dynamics (MD) simulations, which are increasingly used to understand the partitioning of peptides in bilayers (34-36).
In this report, we present results from a combination of solid-state NMR experiments and molecular dynamics simulations. REDOR (Rotational echo double resonance) (37) experiments on multilamellar vesicles (MLVs) under magic angle spinning (MAS) conditions were used to determine the backbone conformation of SKP and SA peptides, while static PISEMA (polarization inversion spin exchange at the magic angle) (38-40) experiments on mechanically aligned bilayers allowed measurement of the orientation of the pep tide in bilayers. Molecular dynamics simulations were performed on lipid bilayers containing SKP or SA peptides. From the simulations, properties such as peptide secondary structure, peptide tilt angle, and peptide-induced lipid perturbations were calculated. Computational and experimental results are in excellent agreement. Our results suggest that the hydrophobic region of these peptides is helical with a kink caused by the Pro residue. The transmembrane orientation of the hydrophobic helix was found to depend on the hydrophobic mismatch between the hydrophobic thickness of the lipid bilayer and the hydrophobic length of the peptide. On the other hand, the backbone conformation of SKP and SA peptides was independent of the bilayer composition.
MATERIALS AND METHODS
Materials
DMPC (1,2-dimyristoyl-phosphatidylcholine) and POPC (1-palmitoyl-2-oleoyl-phosphatidylcholine) were purchased from Avanti Polar Lipids (Alabaster, AL). Chloroform and methanol were procured from Aldrich Chemical Inc. (Milwaukee, WI). Naphthalene was purchased from Fisher Scientific (Pittsburgh, PA). Peptides were synthesized, and cleavage reagents were purchased from Applied Biosystems (Foster City, CA) and Aldrich (Milwaukee, WI). Fmoc-protected amino acids were obtained from Advanced ChemTech (Louisville, KY), and isotopically labeled Fmoc-amino acids from Cambridge Isotope Labs (Cambridge, MA). All chemicals were used without further purification. 15N and 13C labeled peptides were synthesized and purified at the University of Michigan as explained elsewhere (6, 8, 9).
Circular Dichroism
SUVs (small unilamellar vesicles) were prepared by suspending dry lipids in Tris buffer (10 mM Tris•HCl, 100 mM NaCl, 0.1 mM EDTA, pH 7.4) and sonicating the dispersion until a clear solution was obtained. 100mM peptide stock solutions were also prepared using Tris buffer. Each sample was prepared as a 1:1 (vol/vol) mixture of peptide and lipid SUV stock solutions with additional Tris buffer as needed in a 5 mm quartz cuvette. The sample was then equilibrated to 25°C in the CD spectrometer (AVIV, Lake wood, NJ) for 20 minutes and 8 scans were acquired and averaged. The scan rate was 1 nm/minute over the range of 190 to 280 nm. Background contributions from the buffer and SUVs were removed by subtracting the spectrum of a similar sample without peptide.
Solid-state NMR
Mechanically aligned bilayers were prepared using the procedure described by Hallock et al. (41). Briefly, 50 mg of lipids and an appropriate concentration of peptide were dissolved in CHCl3/CH3OH (2:1) mixture. The sample was dried under a stream of nitrogen and re-dissolved in CHCl3/CH3OH (2:1) mixture containing equimolar quantities of naphthalene. An aliquot of the solution (~300 mL) was spread on thin glass plates (11 mm × 22 mm × 50 mm, Paul Marienfeld GmbH and Co., Bad Mergentheim, Germany); 5 to 10 glass plates were used for each sample. The samples were then air-dried and kept under vacuum at 35°C for at least 24 hours to remove naphthalene and any residual organic solvents. After drying, the samples were hydrated at 98% relative humidity using saturated NH4H2PO4 solution (42) for 1-2 days at 37°C, after which approximately 4 μL of H2O was added to the surface of the lipid-peptide film to fully hydrate the samples. The glass plates were stacked, wrapped with parafilm, sealed in plastic bags (Plastic Bagmart, Marietta, GA), and then kept at 4°C for about 12 hours.
Multilamellar vesicles were prepared by mixing the required amounts of lipid and peptide in 2:1 chloroform:methanol. The solution was first dried under N2 gas and then under vacuum overnight to completely remove any residual organic solvents. The mixture was resuspended in 60 weight % water by heating in a water bath at 42°C. The samples were vortexed for 5 min and freeze-thawed using liquid nitrogen several times to obtain a uniform mixture of lipid and peptide.
All of the experiments were performed on a Chemagnetics/Varian Infinity 400 MHz solid-state NMR spectrometer operating at resonance frequencies of 400.138, 161.979, 100.8, and 40.55 MHz for 1H, 31P, 13C, and 15N nuclei, respectively. A Chemagnetics/Varian temperature controller unit was used to maintain the sample temperature, and each sample was equilibrated for at least 45 minutes before starting the experiment. A home-built double resonance probe, which has a four-turn square-coil (12 mm × 12 mm × 4 mm) constructed using a 2 mm wide flat-wire and a spacing of 1 mm between turns, was used for 31P and 15N experiments on aligned samples and a 5mm double-resonance MAS probe was used for experiments on MLVs under static condition. In the case of aligned samples, the lipid bilayers were positioned with the bilayer normal parallel to the external magnetic field of the NMR spectrometer. The 15N chemical shift spectrum of a labeled peptide in oriented bilayers was acquired using a cross-polarization (CP) (43) sequence with a 1H π/2 pulse length of 5 μs, 75 kHz CP RF (radio frequency) power, and a 95 kHz two-phase pulse-modulation (TPPM) (44) decoupling of protons during acquisition. A 1 ms ramp-CP (45) with a 10 kHz ramp on the 1H channel and a recycle delay of 3 s were used. A ramped spin-lock pulse provided better signal-to-noise ratio than a constant amplitude spin-lock pulse in the proton channel. A typical 31P 90°-pulse length of 3.5 μs was used. 31P chemical shift spectra were obtained using a spin echo sequence (90°-τ-180°-τ-acquire with τ=9 μs), 65 kHz RF field for TPPM decoupling of protons, and a recycle delay of 3 seconds. A typical spectrum required the co-addition of about 100 scans for aligned samples and about 1,000 transients for MLVs. The 31P chemical shift spectra are referenced relative to 85% H3PO4 (0 ppm) (41).
2D PISEMA experiments were performed on mechanically aligned lipid bilayers containing a 15N labeled peptide as mentioned in our previous publication (40). A 50 kHz RF field was used in the 1H channel at on-resonance during the preparation 90° pulse and CP, and a 35.355 kHz offset during the Lee-Goldburg (46) sequence in the SEMA (spin exchange at the magic angle) (39,40) sequence (or the t1 period). RF field strengths of 50 and 61.2 kHz were used to spin-lock the 15N magnetization during ramp-CP (45) and SEMA (39) respectively. A 81 kHz RF field was used to decouple protons using the TPPM (44) sequence during the signal acquisition. Due to low signal-to-noise ratio from bilayer samples, the experimental conditions were first optimized on a single crystal of n-acetyl-L-15N-valyl-L-15N-leucine. The optimized experimental conditions were further optimized on a mechanically aligned POPC bilayers containing SA peptide by carrying out several 1D experiments using the regular PISEMA sequence but for different t1 time intervals (39,40). Since the use of a high RF power in the PISEMA experiment could dehydrate the bilayer sample, cold air was circulated in the probe to reduce the effect of RF heat on the sample. In addition, the quality of aligned samples was examined using 1D 31P experiments before and after PISEMA experiments. Data processing was accomplished using the Spinsight software (Varian) on a Sun Sparc workstation.
Magic Angle Spinning solid-state NMR Experiments
A REDOR (37) experiment was used to measure isotropic 13C chemical shifts from peptides selectively labeled with 13C and 15N isotopes. REDOR-filtered experiments as described in the literature (47) were utilized to suppress the 13C background signal from lipids of MLVs. A 5mm triple-resonance Varian/Chemagnetics MAS probe was used and the sample was spun at a speed of 8 kHz spinning speed at −20°C. A 1.0 milliseconds ramp-CP45 was followed by a REDOR dephasing period and then direct 13C detection under proton decoupling with a recycle delay of 3.8s. A single 55 kHz 13C refocusing 180° pulse was placed at the center of the REDOR dephasing time; and 88 kHz TPPM44 decoupling was applied on the proton channel during both dephasing and detection. The 15N and 13C transmitters were set at 115 ppm (relative to liquid ammonia at 25°C) and 175 ppm (relative to tetramethylsilane) respectively. For the S1 (that is with 180° pulses in the 15N channel) acquisition, a 45 kHz 15N 180° pulse at the middle and end of each rotor period in the dephasing time was applied. Other details of the REDOR filtering experiment can be found elsewhere (47).
Molecular dynamics simulations
We performed molecular dynamics simulations on three different sequences (Figure 1) of signaling peptides (SA, SKP and the presumed transmembrane domain, TM, of SA and SKP peptides) embedded in POPC and DMPC bilayers. In all peptides, the presumed transmembrane domain is between residues 1 and 16. We used the GROMACS simulation tool (48), using the GROMOS96 43a2 force field for the protein and an OPLS based force field for the lipids (49). The SPC water model was used. First, ideal helical structures of all three peptides were created using the software SWISSPDB (50). Then, a kink of ~90° was introduced between residues 16 and 17 of the SA and SKP peptides by changing the appropriate torsion angles. This yielded a transmembrane domain roughly perpendicular to the non-transmembrane domain for both SA and SKP peptides. Each of the three peptides was then solvated in a bath of water and the presumed transmembrane segment of each (residues 1-16) was positionally restrained while a 10 ns simulation was performed in an NPT (constant number of atoms, pressure and temperature) ensemble to relax the side chains and the non-transmembrane residues. Short-range interactions used a cut-off of 1.2 nm and the PME algorithm (51) was used for long-range electrostatic interactions. The temperature was coupled to a Berendsen thermostat at 310 K, with the peptide and water molecules coupled separately with coupling constants of 0.1 ps. The pressure was coupled isotropically to a Berendsen barostat at 1 atm pressure with a coupling constant of 5 ps. The peptide conformation at the end of these simulations was used to perform the bilayer simulations.
The initial coordinates of the hydrated POPC and DMPC bilayers were originally obtained from http://moose.bio.ucalgary.ca/files, but were subsequently modified to give sufficient number of water molecules. Each bilayer consisted of 128 lipid molecules (64 lipids per leaflet). After modifications and sufficiently long equilibration runs, the DMPC bilayer had ~3500 water molecules and the POPC bilayer had ~5000 water molecules. Each peptide was inserted into the two bilayers using the “hole” protocol (52), giving us six different simulation systems. The peptides were inserted such that the presumed transmembrane segment was oriented parallel to the bilayer normal and the Ala-8 residue was in the middle of the bilayer. After insertion, the peptide backbone residues were positionally restrained and a 5 ns simulation was performed. The simulation conditions were identical to the “solvated simulations” mentioned above, except that a semi-isotropic pressure coupling was used, with the normal and lateral directions of the bilayer coupled independently to a pressure bath. After 5ns, the restraints on the backbone were released and all six simulations were run for 50 ns. Two simulations (TM peptide in DMPC and POPC) were extended to 100 ns. The coordinates were saved every picosecond and were used in subsequent analysis.
RESULTS
Secondary Structure of SKP and SA peptides
CD Experiments
The amino acid sequences of SA, SKP and the hydrophobic segment (noted as TM) peptides are given in Figure 1. CD experiments were performed on SUVs and SDS micelles containing SA and SKP peptides. The concentration of peptides (up to 3 mole %), pH (4 to 7) and lipid composition were varied. SUVs were prepared using DMPC or POPC lipids. CD spectra of all of these model membrane samples were similar and characterized by the double minima at 208 and 222 nm, attributable to a helical conformation. A representative CD spectrum of SKP in POPC SUVs at 24°C is given in Figure 2. All peptides were assumed to be completely bound to the lipid vesicles at the lipid-peptide ratio of 100:2 used in the present study, as the spectra do not change upon increasing the lipid-peptide ratio. The mean helix contents of vesicle-bound SKP and SA as calculated from the mean residue ellipticity values at l222 nm, are about 45% and 60%, respectively.
Figure 2.

A CD spectrum of SKP peptide in POPC small unilamellar vesicles at 24°C.
Magic Angle Spinning NMR experiments
CD experiments suggest that SKP and SA peptides exist in a helical conformation in membranes. In addition, the dispersion of resonances in 2D 1H-1H NOESY spectra of DPC detergent micelles suggest that these peptides form helices (data not shown); samples were prepared by dissolving a lyophilized peptide in an aqueous solution (90% H2O and 10% D2O) containing 200 mM perdeuterated DPC (Cambridge Isotope Laboratory) and 20 mM phosphate buffer at pH~5 at a final concentration of 3mM. However, determining the conformation of the hydrophobic region of the peptides in lipid bilayers would be useful in understanding the folding of the peptide in membranes. Therefore, we performed MAS experiments on both peptides reconstituted in lipid bilayers. Multilamellar vesicles containing synthetic peptides selectively labeled with 13C and 15N isotopes were used to measure the isotropic 13C chemical shift frequency by performing REDOR experiments (37, 53). Figure 3 shows the difference between the spectra obtained without (S0) and with (S1) 180° pulses in the 15N channel. Both experiments were performed with the same number of scans and a 1.6 milliseconds REDOR dephasing time. The spectra of peptides reconstituted in POPC and DMPC MLVs were similar and therefore only the spectra obtained from POPC are given in Figure 3. The presence of a single peak with a chemical shift frequency of 177.5 ppm (relative to TMS at 0 ppm) suggests that the 13C-labeled residues are in an α-helical conformation (54, 55). The narrowness of the spectral lines suggest that the peptides are in a single conformation, while the observed line width (about 0.5 ppm for SA and 1.5 ppm for SKP peptides) may be attributed to librational motions and/or a small variation in the conformation of the peptides. While more experimental data are needed to solve the high-resolution structure of the peptide in bilayers, these REDOR data show that the hydrophobic domains (residues 1-14) of both peptides form a helical conformation in lipid membranes. These results are in good agreement with the CD data.
Figure 3.
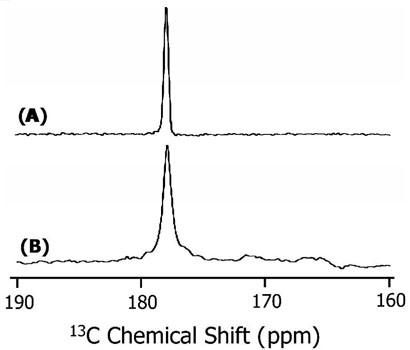
REDOR difference 13C chemical shift spectra of POPC multilamellar vesicles containing 3 mole % of (A) SA and (B) SKP peptides. Both peptides contain 13Carbonyl-labeled Leu11 and 15N labeled Leu12. About 20,000 scans were used to obtain the spectra. Other experimental details are given in the text.
Molecular dynamics simulations
All six simulations showed that the transmembrane segment of all the peptides remains largely helical over the 50-ns duration of the simulation. The secondary structure profiles as a functions of time for TM, SA and SKP peptides are shown in Figure 4. For the TM peptide, at the beginning of the simulation, residues 2-14 are α-helical (blue). This segment remains helical, for almost the entire duration of the simulation, except for the last 15 ns, when slight fraying is observed at the N-terminal. For the SA peptide, as the simulation progresses, the N-terminal region of the transmembrane domain (residues 1-4) and the non-transmembrane region (residues 17-26) both lose their helicity, leading to other motifs such as bend, turn and coil. However, the central region of the presumed transmembrane domain remains helical for the entire duration of the simulation. For the SKP peptide, again, residues 4-16 remain helical for most of the simulation, but, as was observed in the other three simulations in POPC bilayers, near the end of the simulation the transmembrane domain remained helical, but with fraying observed at the ends of the peptide. For the SA peptide, the kink that was introduced near the 17th residue to orient the non-transmembrane domain parallel to the lipid-water interface, seems to induce a non-helical structure to the non-transmembrane domain, as also seen in the experiments. However, for the SKP peptide, some residual helicity remains in the non-transmembrane domain throughout the simulation. Presumably, longer simulations would enable the non-transmembrane domain to attain its preferred secondary structure, which might well be completely devoid of helicity. While the final structure of the whole peptide is not completely obtained over the time scales we can obtain, it is clear that, in stark contrast to the non-transmembrane domain, the transmembrane domain remains mostly helical in all of the simulations, as observed in experiments. We calculated the mean helicity of the peptides in POPC and DMPC lipid bilayers. The data are presented in Table 1. These simulation results are in good agreement with the CD and the MAS experimental results.
Figure 4.

Secondary structure profiles of TM (top), SA (middle) and SKP (bottom) peptides in DMPC bilayers obtained from molecular dynamics simulations. Blue color represents an α-helical structure.
Table 1.
Overall helicity of peptides in phospholipid bilayers measured from molecular dynamics simulations and circular dichroism experiments.
| Method | SA | SKP | TM | |
|---|---|---|---|---|
| Simulations: | POPC | 61% | 42% | 78% |
| DMPC |
57% |
49% |
83% |
|
| Experiments | 60% | 45% | Not determined |
Peptide-induced disorder in bilayers
Since a cell-permeable peptide interacts with membranes and could disrupt the lipid bilayer structure to carry out its function, it is important to examine if SKP and SA induce any changes in the lamellar phase structure of POPC and DMPC bilayers. These results are also essential to interpret the NMR spectra of mechanically aligned lipid samples in terms of the peptide orientation. Therefore, 31P chemical shift spectra of POPC and DMPC MLVs containing various concentrations (up to 5 mole %) of peptides were obtained at 30°C. Representative spectra are given in Figure 5. Spectra of all MLV samples (Figure 5A) suggest that the bilayers were in the lamellar phase. On the other hand, the observation of a single narrow peak near the low-field edge of an unoriented spectrum (~30 ppm) (Figure 5B) suggests that the lipid bilayers were well-hydrated and well-aligned in glass plate samples. These spectra indicate that the interaction of peptides with lipids do not significantly disrupt the lamellar phase bilayer structure of the samples under study. In addition, the 31P data from POPC and DMPC bilayers suggest that the hydrophobic mismatch between the hydrophobic thickness of the lipid bilayer and the hydrophobic length of the peptide had no significant effect on the bilayer structure. Small changes observed in the frequency position of the 31P peak in the aligned spectrum and span of CSA of MLVs with increasing concentration of the peptide can be attributed to the interaction of the peptide, particularly the charged nuclear localization sequence, with the head group region of lipids. Molecular dynamics simulations confirm these experimental results. We calculated the bilayer thickness in the immediate vicinity of the peptide in all six simulations and compared it to the thickness of bilayers free from the peptide. This approach has been used to calculate peptide-induced bilayer disorder in earlier studies (56). We found that the bilayer disruption was minimal, with a small (~ 1Å) difference between the bilayer widths near and far away from the peptide.
Figure 5.
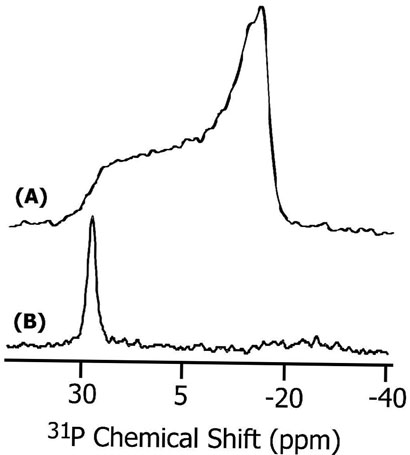
31P chemical shift spectra of (A) multilamellar vesicles and (B) mechanically aligned POPC bilayers containing 3 mole % SKP peptide at 30°C. About 1500 and 150 scans were used to obtain spectra (A) and (B) respectively.
Orientation of SKP and SA peptides in bilayers
Solid State NMR Experiments
The orientations of SKP and SA in phospholipid bilayers were determined using solid-state NMR experiments on mechanically aligned bilayers containing peptides labeled with 15N at a single site. All aligned 15N chemical shift spectra were interpreted with reference to a powder pattern spectrum obtained from MLVs. A spectrum of MLVs containing 3 mole % SKP peptide labeled with 15N-Leu-11 is given in Figure 6. All aligned 15N spectra showed a single narrow peak suggesting that the embedded peptides have a unique orientation in bilayers; some representative spectra are given in Figures 7 and 8. The measured chemical shift values and the line widths are given in Table 2. These spectra contain a single peak (Figures 7 and 8) in a frequency region near to the parallel edge (~160-215 ppm) of an unaligned 15N chemical shift anisotropy (57) spectrum (Figure 6). Since most of the 15N labeled residues are most likely located in the hydrophobic regions of the bilayer, providing considerably less conformational or dynamic disorder, narrow lines are observed in the spectra of aligned samples. On the other hand, some amphipathic peptides oriented near the surface of bilayers could result in broad lines due to heterogeneous orientation (12, 15, 58) or narrow lines due to motion (59). The observed line widths range from 1.8 to 6 ppm and did not depend on the RF power of proton decoupling. The largest line width observed for Ala-15 residue could be attributed to the conformational heterogeneity of this residue as it is near a Pro residue, which is at a junction of the hydrophobic and hydrophilic regions of the peptide. The chemical shift values measured from SKP and SA are, within experimental errors, similar in POPC bilayers while they are different in DMPC bilayers (Table 2). Since the peptide forms a helical structure in lipid bilayers, these data can be interpreted in terms of the orientation of the peptide in bilayers. Based on the reported structural studies on the membrane-associated peptides using solid-state NMR (14, 18, 40), these data reveal the transmembrane orientation of the helical region of these peptides.
Figure 6.

15N chemical shift spectrum of POPC multilamellar vesicles containing 3 mole % 15N-Leu11 SKP at −10°C. A 1 millisecond cross-polarization time, 4000 scans and a recycle delay of 3s were used; all other experimental conditions were as mentioned in the text. The principal components of the amide-15N chemical shift tensor, directly measured from the powder pattern spectrum, are 51±2, 72±2 and 225±2 ppm.
Figure 7.
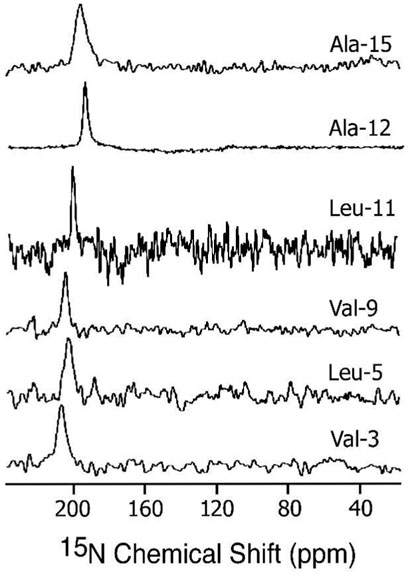
15N chemical shift spectra of mechanically aligned POPC bilayers containing 3 mole % SKP peptides labeled with 15N isotope at 30°C. About 600, 400, 800, 400, 4000, and 2000 scans were used to obtain spectra of peptides labeled with 15N at the amide site Val-3, Leu-5, Val-9, Leu-11, Ala-12, and Ala-15 residues respectively. Other experimental details are given in the text.
Figure 8.

15N chemical shift spectra of mechanically aligned DMPC bilayers containing 3 mole % of (A) SA and (B) SKP peptides labeled with 15N isotope at the amide site of Leu-11. About 4000 scans were used to obtain spectra. Other experimental details are as given in the text.
Table 2.
Experimentally measured 15N chemical shift (Figure 4) and 1H-15N dipolar coupling (Figure 5) values of SKP and SA peptides embedded in mechanically aligned POPC and DMPC lipid bilayers at 30°C.
| Peptide | V3 | L5 | V9 | L11 | L12 | A15 |
|---|---|---|---|---|---|---|
| POPC (SKP or SA): | ||||||
| Chemical Shift (ppm) | 210±2 | 203±3 | 205±1.5 | 200±0.9 | 195±1 | 198±3 |
| Dipolar coupling (kHz) * | 9.5 | 7.2 | 7.0 | 9.3 | 7.7 | 8.2 |
| SKP in DMPC: | ||||||
| Chemical Shift (ppm) | 190±2 | 172±1.7 | 178±2 | 167±1 | 170±1 | 168±2 |
| Dipolar coupling (kHz)* | 8.5 | 3.5 | 3.0 | 8.0 | 5.0 | 7.0 |
| SA in DMPC: | ||||||
| Chemical Shift (ppm) | 200±2 | 185±1 | 192±1 | 181±1.4 | 180±1 | 178±2 |
| Dipolar coupling (kHz)* | 9.0 | 5.0 | 4.8 | 9.0 | 5.5 | 8.5 |
Based on line widths, errors in the reported dipolar couplings are estimated to be about ±0.3 to ±0.4 kHz.
A 2D PISEMA (38-40) experiment that correlates the 15N chemical shift and the 1H-15N dipolar coupling was performed on mechanically aligned bilayer samples. 1H-15N dipolar coupling spectra extracted from 2D PISEMA spectra of different samples are given in Figures 9 and 10. As demonstrated in previous studies, PISEMA provides narrow lines by suppressing 1H-1H dipolar interactions (38-40). Observed line widths range from 0.3 to 0.6 kHz. Since each sample consisted of a single site 15N labeled peptide, an off-resonance dependence of the PISEMA sequence (40) was minimized by setting 1H and 15N carrier frequencies near the amide-1H and 15N resonances respectively. Therefore, errors in the measured parameters due to the variation of the scaling factor of the sequence were minimized.
Figure 9.

1H-15N dipolar coupling spectra of aligned POPC bilayers containing 3 mole % SKP peptides. Since a dipolar coupling spectrum is symmetric with respect to the zero frequency, half of each spectrum is presented. These spectra were extracted from 2D PISEMA spectra (not shown). 2D PISEMA spectra were obtained using a 0.75 millisecond cross-polarization time, 48 t1 experiments, 800 scans and a recycle delay of 3 s.
Figure 10.
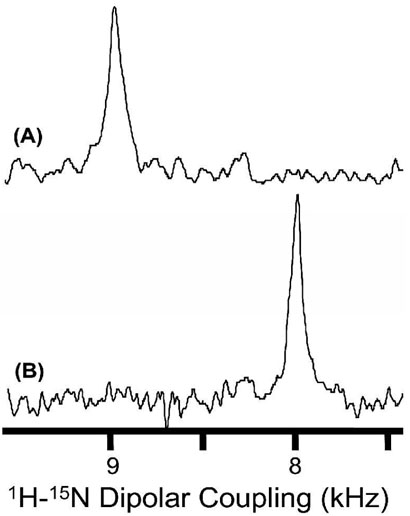
1H-15N dipolar coupling spectra of aligned DMPC bilayers containing 3 mole % of (A) SA and (B) SKP peptides labeled with 15N isotope at the amide site of Leu11. 2D PISEMA spectra were obtained using a 0.75 millisecond cross-polarization time, 64 t1 experiments, 640 scans and a recycle delay of 3 s.
PISA (polarity index slant angle) wheels (60, 61) generated from PISEMA spectra of 15N chemical shifts and 1H-15N dipolar couplings of mechanically aligned samples are given in Figure 11 along with simulated PISA wheel spectra. The simulations were carried out using the SIMMOL program (62, 63). Even though there are not enough experimental points to define the complete shape of the PISA wheel, the simulated and experimental spectra are in good agreement. The comparison of the simulated and experimental results suggests that the helical domain is transmembrane but tilted away from the bilayer normal. The tilt angles are given in Table 3. Interestingly, the tilt angles measured in POPC are smaller but are different in DMPC bilayers. Errors on the reported tilt angles are estimated from the line widths observed in the chemical shift and dipolar coupling spectra. However, there could be additional contributions to the error from the variation of chemical shift tensors (57, 64) from the model tensor used for these calculations and due to peptide dynamics that would have influenced the observed experimental parameters. Nevertheless, the reported tilt angles are in good agreement with results obtained from MD simulations, discussed below.
Figure 11.

Experimental (data points) and simulated (dotted, dashed or solid line) PISA wheels corresponding to hydrophobic transmembrane regions of peptides: SKP or SA in POPC (circles and dotted line), SA in DMPC (filled triangles and a solid line) and SKP in DMPC (stars and a dashed line) bilayers. Experimental points are generated from the data given in Table 2.
Table 3.
Average tilt angles from experiments and molecular dynamics simulations of lipid bilayers containing SA, SKP or TM peptides.
| Tilt Angle | SA (50ns) |
SKP (50ns) |
TM (100ns) |
|---|---|---|---|
| DMPC bilayers : | |||
| Simulations | 22.3±6.3° | 31.7±8.3° | 24.6±4.6° |
| Experimental |
25±3° |
30±3° |
- |
| POPC bilayers : | |||
| Simulations | 13.3±6.7° | 16.4±4.0° | 19.8±1.0° |
| Experimental | 15±3° | 15±3° | - |
Molecular dynamics simulations
We observed tilting of peptides away from their original vertical transmembrane orientation in all six simulations. In Figure 12, we show snapshots at the end of three simulations (TM, SA and SKP in DMPC bilayers). It is clear that the non-transmembrane domain of the SA peptide is fully frayed, with no residual helicity. Residues 30 to 38 of the SKP peptide retain their initial helicity while the residues 18 to 30 seem to lose their helicity quickly. However, the region of the residual helicity is far away from the transmembrane domain and presumably will not affect the tilt behavior of the transmembrane region. We calculated the tilt angle of the transmembrane segment relative to the bilayer normal for all six simulations. The helical axis was defined by selecting two points at the top and bottom of the transmembrane segment. The top point was chosen to be the center of mass of the backbone atoms of residues 3, 4 and 5, while the bottom point was chosen to be the center of mass of the backbone atoms of residues 12, 13 and 14. In Figure 13, we show the tilt angle as a function of time for a representative simulation (TM peptide in POPC bilayers). The average tilt angle was calculated for all simulations and is presented in Table 3. The first 20 ns of each simulation was taken to be an equilibration period and was not used to calculate the average. The standard error was estimated by a block averaging procedure. The peptides show a consistently larger tilt in DMPC bilayers than in the POPC bilayers, which is expected, since POPC bilayers have a larger hydrophobic width than DMPC bilayers, and so the disparity between the hydrophobic length of the peptide and the hydrophobic width is greater for DMPC. It should be noted that the standard errors of tilt angles are rather large, especially for SA and SKP peptides. This is due to the large timescales required for the equilibration of peptide tilt in phospholipid bilayers. In a recent simulation study (56), we found that even 100ns may not be sufficient to fully equilibrate peptide tilt in lipid bilayers. Thus, longer simulations will be required to provide a quantitatively more accurate tilt angle. Nevertheless, qualitatively, the peptides show a consistently larger tilt in DMPC bilayers than in POPC bilayers, in agreement with the experimental results.
Figure 12.

Snapshots at the end of molecular dynamics simulations for TM (left), SA (middle) and SKP (right) peptides in DMPC bilayers. The lipid tails are shown in grey, the water molecules in blue, and the lipid phosphorus atoms as red spheres. The peptide is shown as pink ribbons.
Figure 13.

Tilt angle as a function of simulation time for a representative simulation for the TM peptide in POPC bilayers. The tilt angle is shown as a running average.
DISCUSSION
Cell-signaling peptides such as SA and SKP have been used to carry functional nuclear localization sequences across the cell membrane (6, 8, 9). Previous studies have shown that these peptides are efficient in transporting nuclear localization sequences for intracellular activities (1-10). However, the mechanism of the transport activity and interaction of these peptides with membranes are not clear. In this study, solid-state NMR experiments and molecular dynamics simulations were used to investigate the interaction of SA and SKP peptides with model membranes. In particular, the structure and folding of the hydrophobic domain of these peptides in membrane bilayers were determined and the effect of peptide-membrane interactions on lipid bilayer structure was also examined. The effect of hydrophobic mismatch between the hydrophobic length of the peptide and the thickness of lipid bilayers, on the folding of the peptide was investigated using thicker (POPC) and thinner (DMPC) bilayers.
Secondary structure
Results obtained from CD experiments on SUVs (Figure 2) and solution NMR experiments on detergent micelles (data not given) suggest that the main conformation of both the peptides is helical in membrane environments. CD results suggested that SA (~60%) is more helical than SKP (~45%), which is in agreement with qualitative structure prediction analysis. The difference in the helical content could be due to the difference in the hydrophilic nuclear localization sequences of these peptides. The non-helical NLS sequence of SKP (25 residues) is longer than that of SA (7 residues), and hence its contribution to the non-helicity of the peptide is greater. In addition, the hydrophobic spacer region containing three Ala residues in SA could increase the helicity of the peptide by “insulating” better the non-helical region from the helical transmembrane domain. The CD results are further confirmed using REDOR experiments on MLVs containing peptides labeled with 13C' and 15N isotopes (Figure 3). The observation of similar 13C' chemical shift values from DMPC and POPC bilayers suggests that the conformation is independent of the thickness of the bilayer.
Topology and tilt of peptides in membrane bilayers
PISA wheels (Figure 11) generated from the experimentally measured chemical shift (Figures 7 and 8) and dipolar coupling (Figures 9 and 10) values (Table 2) on mechanically aligned bilayers containing peptides suggest that the hydrophobic domain (residues 1 to 15 in Figure 1) of both the peptides has a transmembrane orientation in DMPC and POPC bilayers. However, the helical axis of this domain is tilted away from the direction of the bilayer normal in both types of bilayers with a larger tilt in thinner DMPC bilayers than in thicker POPC bilayers. These results are in very good agreement with molecular dynamics simulations. Experimental results suggest that the helical axis of both peptides is tilted 15±3° relative to the normal, in POPC bilayers, compared to the MD predicted tilts of 13.3° for SA and 16.4° for SKP. In DMPC bilayers, SA has an experimentally determined tilt of 25±3° while for SKP it is 30±3° compared to predicted values of 22.3° and 31.7°, respectively. The trends and even the magnitudes of the tilts in the simulations are remarkably consistent with the experimental values. The simulations on the hydrophobic sequence (the TM peptide) alone suggest that the tilt angle is 19.8° and 24.6° in POPC and DMPC bilayers respectively.
Solid-state NMR experiments have been successfully used to understand the topology of several membrane-associated peptides and proteins (13, 18-21, 40, 60, 61). Particularly, high resolution rendered by 2D PISEMA experiments has yielded PISA wheel patterns of membrane-bound helices (13, 18, 40, 60, 61). Images of proteins and peptides have been used to address various biological questions related to membrane-peptide and peptide-peptide interactions that determine the function of membrane-associated proteins (11, 40, 65). Our PISEMA results presented in this paper are consistent with the interpretations and explanations provided by the previous studies on different transmembrane helical systems (13, 18, 40, 60, 61, 65). Interestingly, the tilt angles measured from solid-state NMR experiments are also in good agreement with MD simulations. More generally, our results and those of others indicate that a combination of MD simulations and solid-state NMR structural constraints could be a practical way to solve some of the challenging problems involved in studying membranes and membrane-peptide interactions. For example, MD simulations have yielded a unique structure of gramicidin when combined with solid-state NMR structural constraints obtained from mechanically aligned bilayers (66). In addition, a recent study combining MD simulations with NMR determined the orientation and conformational preference of leucine-enkephalin in DMPC bilayers (67). Similar studies are increasingly being carried out in several laboratories. Therefore, we believe that while both NMR and MD simulations will continue to be developed, a suitable combination of them can provide insights into the structure, dynamics, folding, oligomerization, topology, and function of some of the most challenging membrane-associated peptides and proteins.
Effect of hydrophobic mismatch
Recent studies have shown that hydrophobic mismatch between the hydrophobic length of a helical peptide and the hydrophobic thickness of the lipid bilayer can significantly affect the backbone conformation, membrane-orientation, tilt (the angle between the helical axis and the bilayer normal), and oligomerization of the peptide (65, 68-78). It could also lead to changes in the phase, order/disorder, and curvature of lipid bilayers (68). While most previous studies utilized designed peptides (68, 70-72, 74-76), in this study, we considered the effect of hydrophobic mismatch on the folding and tilt of functional peptides. 31P NMR and MD simulation results suggest that the hydrophobic mismatch, in both SA and SKP peptides, did not alter the phase, structure and order of lipid bilayers. CD and REDOR experiments and MD simulations showed that the backbone conformation of SA and SKP peptides is independent of the thickness of the bilayer suggesting that the hydrophobic mismatch does not affect the structure of the peptides, either. However, as mentioned above, 15N NMR and MD simulations results reveal that, rather than altering either the membrane or peptide structure, hydrophobic mismatch is accommodated through tilting of the peptides to keep their hydrophobic domain within the hydrophobic region of the bilayer. The measured and simulated tilt angles for SA and especially SKP are generally larger than observed for model peptides, such as KALP peptides (68). However, the observed qualitative dependence of the tilt of SA and SKP peptides on bilayer thickness of DMPC and POPC is consistent with recent NMR studies on other transmembrane peptide fragments (65, 78). For example, Cross and coworkers used NMR spectra of aligned bilayers containing tetrameric M2 protein of influenza A in DLPC, DMPC and POPC bilayers to understand the effect of hydrophobic mismatch on the tilt of the helical transmembrane domain of the protein (69, 78). Recently, Opella and coworkers used PISA wheels to determine the effect of hydrophobic mismatch on the tilt of a 36-residues N-terminal channel-forming segment of Vpu protein from HIV (65). Interestingly, they reported a large tilt angle of 51° and helix kink in lipid bilayers (65). Therefore, in the absence of oligomerization and disorder in the structure of lipids, a large tilt angle is possible for membrane-associated proteins/peptides while the extent of the tilt depends on the length of the hydrophobic sequence and the membrane-composition (65, 69, 77, 78). The tilted transmembrane orientation of SA and SKP peptides and their avoidance of disruption of cell membrane integrity could be responsible for their efficient transport of nuclear localization sequences across the plasma membrane.
Abbreviations
- CD
circular dichroism
- CP
cross polarization
- CSA
chemical shift anisotropy
- DMPC
1,2-dimyristoyl- sn-glycero-3-phosphatidylcholine
- EGF
epidermal growth factor
- MAS
magic angle spinning
- MD
molecular dynamics
- MLVs
multilamellar vesicles
- NLS
nuclear localization sequence
- NMR
nuclear magnetic resonance
- PISA
polarity index slant angle
- PISEMA
polarization inversion spin exchange at the magic angle
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine
- PBS
phosphate buffered saline
- REDOR
rotational echo double resonance
- RF
radio frequency
- SUV
small unilamellar vesicle
- TM
tansmembrane
- TPPM
two-phase pulse-modulation
- Tris
tris hydroxymethylaminoethane
Footnotes
This research was supported by the research funds from National Institutes of Health (AI054515).
References
- 1.Zorko M, Langel U. Cell-penetrating peptides: mechanism and kinetics of cargo delivery. Adv. Drug Deliv. Rev. 2005;57:529–545. doi: 10.1016/j.addr.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 2.Boon JM, Smith BD. Synthetic membrane transporters. Curr. Opin. Chem. Biol. 2002;6:749–56. doi: 10.1016/s1367-5931(02)00399-x. [DOI] [PubMed] [Google Scholar]
- 3.Hawiger J. Noninvasive intracellular delivery of functional peptides and proteins. Curr. Opin. Chem. Biol. 1999;3:89–94. doi: 10.1016/s1367-5931(99)80016-7. [DOI] [PubMed] [Google Scholar]
- 4.Thoren PE, Persson D, Esbjorner EK, Goksor M, Lincoln P, Norden B. Membrane binding and translocation of cell-penetrating peptides. Biochemistry. 2004;43:3471–89. doi: 10.1021/bi0360049. [DOI] [PubMed] [Google Scholar]
- 5.Veach RA, Liu D, Yao S, Chen Y, Liu XY, Downs S, Hawiger J. Receptor/transporter-independent targeting of functional peptides across the plasma membrane. J. Biol. Chem. 2004;279:11425–31. doi: 10.1074/jbc.M311089200. [DOI] [PubMed] [Google Scholar]
- 6.Lin YZ, Yao SY, Veach RA, Torgerson TR, Hawiger J. Inhibition of nuclear translocation of transcription factor NF-kappa B by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J. Biol. Chem. 1995;270:14255–8. doi: 10.1074/jbc.270.24.14255. [DOI] [PubMed] [Google Scholar]
- 7.Christiaens B, Grooten J, Reusens M, Joliot A, Goethals M, Vandekerckhove J, Prochiantz A, Rosseneu M. Membrane interaction and cellular internalization of penetratin peptides. Eur. J. Biochem. 2004;271:1187–97. doi: 10.1111/j.1432-1033.2004.04022.x. [DOI] [PubMed] [Google Scholar]
- 8.Lin YZ, Tao SY, Hawiger J. Role of the nuclear localization sequence in fibroblast growth factor-1-stimulated mitogenic pathways. J. Biol. Chem. 1996;271:5305–5308. doi: 10.1074/jbc.271.10.5305. [DOI] [PubMed] [Google Scholar]
- 9.Rojas M, Yao SY, Lin YZ. Controlling epidermal growth factor (EFG)-stimulated Ras activation in intact cells by a cell-permeable peptide mimicking phosphorylated EGF receptor. J. Biol. Chem. 1996;271:27456–27461. doi: 10.1074/jbc.271.44.27456. [DOI] [PubMed] [Google Scholar]
- 10.Liu XY, Timmons S, Lin YZ, Hawiger J. Identification of a functionally important sequence in the cytoplasmic tail of integrin b3 by using cell-permeable peptide analog. Proc. Natl. Acd. Sci. USA. 1996;93:11819–11824. doi: 10.1073/pnas.93.21.11819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu J, Fu R, Nishimura K, Zhang L, Zhou HX, Busath DD, Vijayvergiya V, Cross TA. Histidines, heart of the hydrogen ion channel from influenza A virus: Toward an understanding of conductance and proton selectivity. Proc. Natl. Acd. Sci. USA. 2006;103:6865–6870. doi: 10.1073/pnas.0601944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henzler-Wildman KA, Lee DK, Ramamoorthy A. Mechanism of lipid bilayer disruption by the human antimicrobial peptide, LL-37. Biochemistry. 2003;42:6545. doi: 10.1021/bi0273563. [DOI] [PubMed] [Google Scholar]
- 13.Marassi FM. A simple approach to membrane protein secondary structure and topology based on NMR spectroscopy. Biophys. J. 2001;80:994–1003. doi: 10.1016/S0006-3495(01)76078-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strandberg E, Wadhwani P, Tremouilhac O, Durr UHN, Ulrich AS. Solid-state NMR analysis of the PGLa peptide orientation in DMPC bilayers: Structural fidelity of H-2-labels versus high sensitivity of F-19-NMR. Biophys. J. 2006;90:1676–1686. doi: 10.1529/biophysj.105.073858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hallock KJ, Lee DK, Ramamoorthy A. MSI-78, an analogue of the magainin antimicrobial peptides, disrupts lipid bilayer structure via positive curvature strain. Biophys. J. 2003;84:3052. doi: 10.1016/S0006-3495(03)70031-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang J, Prorok M, Castellino FJ, Weliky DP. Oligomeric beta-structure of the membrane-bound HIV-1 fusion peptide formed from soluble monomers. Biophys. J. 2004;87:1951–1963. doi: 10.1529/biophysj.103.028530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu JX, Damodaran K, Blazyk J, Lorigan GA. Solid-state nuclear magnetic resonance relaxation studies of the interaction mechanism of antimicrobial peptides with phospholipid bilayer membranes. Biochemistry. 2005;44:10208–10217. doi: 10.1021/bi050730p. [DOI] [PubMed] [Google Scholar]
- 18.Opella SJ, Marassi FM. Structure Determination of Membrane Proteins by NMR Spectroscopy. Chem. Rev. 2004;104:3587–3606. doi: 10.1021/cr0304121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tian CL, Gao PF, Pinto LH, Lamb RA, Cross TA. Initial structural and dynamic characterization of the M2 protein transmembrane and amphipathic helices in lipid bilayers. Prot. Sci. 2004;12:2597–2605. doi: 10.1110/ps.03168503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quine JR, Cross TA. Protein structure in anisotropic environments: Unique structural fold from orientational constraints. Concep. Magn. Reson. 2000;12:71–82. [Google Scholar]
- 21.Gong XM, Choi JY, Franzin CM, Zhai DY, Reed JC, Marassi FM. Conformation of membrane-associated proapoptotic tBid. J. Biol. Chem. 2004;279:28954–28960. doi: 10.1074/jbc.M403490200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Porcelli F, Buck-Koehntop B, Thennarasu S, Ramamoorthy A, Veglia G. Structures of the dimeric and monomeric variants of magainin antimicrobial peptides (MSI-78 and MSI-594) in micelles and bilayers by NMR spectroscopy. Biochemistry. 2006;45:5793–5799. doi: 10.1021/bi0601813. [DOI] [PubMed] [Google Scholar]
- 23.McDowell LM, Schaefer J. High-resolution NMR of biological solids. Curr. Opin. Struct. Biol. 1996;6:624–629. doi: 10.1016/s0959-440x(96)80028-5. [DOI] [PubMed] [Google Scholar]
- 24.Kamihira M, Vosegaard T, Mason AJ, Straus SK, Nielsen NC, Watts A. Structural and orientational constraints of bacteriorhodopsin in purple membranes determined by oriented-sample solid-state NMR spectroscopy. J. Struct. Biol. 2005;149:7–16. doi: 10.1016/j.jsb.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 25.Crocker E, Eilers M, Ahuja S, Hornak V, Hirshfeld A, Sheves M, Smith SO. Location of Trp265 in metarhodopsin II: implications for the activation mechanism of the visual receptor rhodopsin. J. Mol. Biol. 2006;357:163–172. doi: 10.1016/j.jmb.2005.12.046. [DOI] [PubMed] [Google Scholar]
- 26.Sharpe S, Yau WM, Tycko R. Structure and dynamics of the HIV-1 Vpu transmembrane domain revealed by solid-state NMR with magic-angle spinning. Biochemistry. 2006;45:918–933. doi: 10.1021/bi051766k. [DOI] [PubMed] [Google Scholar]
- 27.Thonpson LK. Solid-state NMR studies of the structure and mechanisms of proteins. Curr. Opin. Struct. Biol. 2002;12:661–669. doi: 10.1016/s0959-440x(02)00374-3. [DOI] [PubMed] [Google Scholar]
- 28.Lange A, Giller K, Hornig S, Martin-Eauclaire MF, Pongs O, Becker S, Baldus M. Toxin-induced conformational changes in a potassium channel revealed by solid-state NMR. Nature. 2006;440:959–962. doi: 10.1038/nature04649. [DOI] [PubMed] [Google Scholar]
- 29.Andronesi OC, Becker S, Seidel K, Heise H, Young HS, Baldus M. Determination of membrane protein structure and dynamics by magic-angle-spinning solid-state NMR spectroscopy. J. Am. Chem. Soc. 2005;127:12965–12974. doi: 10.1021/ja0530164. [DOI] [PubMed] [Google Scholar]
- 30.Baldus M. Spectral Assignment of (Membrane) Proteins under Magic-Angle Spinning. In: Ramamoorthy A, editor. NMR spectroscopy of biological solids. Taylor and Francis; New York: 2006. pp. 39–56. [Google Scholar]
- 31.Todokoro Y, Yumen I, Fukushima K, Kang SW, Park JS, Kohno T, Wakamatsu K, Akutsu H, Fujiwara T. Structure of tightly membrane-bound mastoparan-X, a G-protein-activating Peptide, determined by solid-state NMR. Biophys J. 2006;91:1368–1379. doi: 10.1529/biophysj.106.082735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luca S, Heise H, Baldus M. High-Resolution Solid-State NMR Applied to Polypeptides and Membrane Proteins. Acc. Chem. Res. 2003;36:858–865. doi: 10.1021/ar020232y. [DOI] [PubMed] [Google Scholar]
- 33.Porcelli F, Buck B, Lee DK, Hallock KJ, Ramamoorthy A, Veglia G. Structure and Orientation of Pardaxin Determined by NMR Experiments in Model Membranes. J. Biol. Chem. 2004;279:46815–46823. doi: 10.1074/jbc.M405454200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tieleman DP, Berendsen HJC, Sansom MSP. Surface Binding of Alamethicin Stabilizes its Helical Structure: Molecular Dynamics Simulations. Biophys. J. 1999;76:3186–3191. doi: 10.1016/S0006-3495(99)77470-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kandasamy SK, Larson RG. Binding and insertion of alpha-helical anti-microbial peptides in POPC bilayers studied by molecular dynamics simulations. Chem. Phys. Lipids. 2004;132:113–132. doi: 10.1016/j.chemphyslip.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 36.Lensink MF, Christiaens B, Vandekerckhove J, Prochiantz A, Rosseneu M. Penetratin-Membrane Association: W48/R52/W56 Shield the Peptide from the Aqueous Phase. Biophys. J. 2005;88:939–952. doi: 10.1529/biophysj.104.052787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gullion T, Schaefer J. Rotational-echo double-resonance NMR. J. Magn. Reson. 1987;81:196–200. doi: 10.1016/j.jmr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 38.Wu CH, Ramamoorthy A, Opella SJ. High Resolution Heteronuclear Dipolar Solid-state NMR spectroscopy. J. Magn. Reson. 1994;A109:270. [Google Scholar]
- 39.Ramamoorthy A, Wu CH, Opella SJ. Experimental aspects of multidimensional solid-state NMR correlation spectroscopy. J. Magn. Reson. 1999;140:131. doi: 10.1006/jmre.1999.1827. [DOI] [PubMed] [Google Scholar]
- 40.Ramamoorthy A, Wei Y, Lee DK. PISEMA Solid-State NMR Spectroscopy. Ann. Rep. NMR Spectrosc. 2004;52:1–52. [Google Scholar]
- 41.Hallock KJ, Henzler Wildman KA, Lee DK, Ramamoorthy A. Sublimable Solids can be Used to Mechanically Align Lipid Bilayers for Solid-State NMR Studies. Biophys. J. 2002;82:2499. doi: 10.1016/S0006-3495(02)75592-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Washburn E, West CJ, Hull C. McGraw-Hill; New York: 1926. International Critical, Tables of Numerical Data, Physics, Chemistry, and Technology. [Google Scholar]
- 43.Pines A, Gibby MG, Waugh J. Proton-enhanced NMR of dilute spins in solids. J. Chem. Phys. 1973;59:569–590. [Google Scholar]
- 44.Bennet AE, Rienstra CM, Auger M, Lakshmi KV, Griffin RG. Heteronuclear Decoupling in Rotating Solids. J. Chem. Phys. 1995;103:6951–6958. [Google Scholar]
- 45.Metz G, Wu X, Smith SO. Ramped-amplitude cross polarization in magic-angle-spinning NMR. J. Magn. Reson. Ser., A. 1994;110:219–227. [Google Scholar]
- 46.Lee M, Goldburg WI. Nuclear-Magnetic-Resonance Line Narrowing By a Rotating Rf Field. Phys. Rev. 1965;140:1261. [Google Scholar]
- 47.Yang J, Parkanzky PD, Bodner ML, Duskin CA, Weliky DP. Application of REDOR Subtraction for Filtered MAS Observation of Labeled Backbone Carbons of Membrane-Bound Fusion Peptides. J. Magn. Res. 2002;159:101–110. doi: 10.1016/s1090-7807(02)00033-2. [DOI] [PubMed] [Google Scholar]
- 48.Lindahl E, Hess B, van der Spoel. D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 2001;7:306–317. [Google Scholar]
- 49.Berger O, Edholm O, Jahnig F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 1997;72:2002–2013. doi: 10.1016/S0006-3495(97)78845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 51.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 52.Faraldo-Gomez JD, Smith GR, Sansom MSP. Setting up and optimization of membrane protein simulations. Eur. Biophys. J. 2002;31:217–227. doi: 10.1007/s00249-002-0207-5. [DOI] [PubMed] [Google Scholar]
- 53.Toke O, O'Connor RD, Weldeghiorghis TK, Maloy WL, Glaser RW, Ulrich AS, Schaefer J. Structure of (KIAGKIA)(3) aggregates in phospholipid bilayers by solid-state NMR. Biophys. J. 2004;87:675–687. doi: 10.1529/biophysj.103.032714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wei Y, Lee DK, Ramamoorthy A. Solid-State 13C chemical shift anisotropy tensors of polypeptides. J. Am. Chem. Soc. 2001;123:6118–6126. doi: 10.1021/ja010145l. [DOI] [PubMed] [Google Scholar]
- 55.Tuzi S, Komoto T, Ando I, Saito H, Shoji A, Ozaki T. C-13-NMR chemical-shift and conformation of l-alanine residues incorporated into poly(beta-benzyl l-aspartate) in the solid-state. Biopolymers. 1983;26:1983–1992. [Google Scholar]
- 56.Kandasamy SK, Larson RG. Molecular Dynamics Simulations of Model Trans-Membrane Peptides in Lipid Bilayers: A Systematic Investigation of Hydrophobic Mismatch. Biophys. J. 2006;90:2326–234357. doi: 10.1529/biophysj.105.073395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Poon A, Birn J, Ramamoorthy A. Variation of Amide-15N Chemical Shift Tensors in Proteins. J. Phys. Chem. B. 2004;108:16577. doi: 10.1021/jp0471913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ramamoorthy A, Marassi FM, Zasloff. Opella SJ. 3-Dimensional solid-state NMR spectroscopy of a peptide oriented in membrane bilayers. J. Biomol. NMR. 1995;6:329–334. doi: 10.1007/BF00197814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aisenbrey C, Bechinger B. Investigations of polypeptide rotational diffusion in aligned membranes by 2H and 15N solid-State NMR spectroscopy. J. Am. Chem. Soc. 2004;126:16676–16683. doi: 10.1021/ja0468675. [DOI] [PubMed] [Google Scholar]; Chekmenev EY, Vollmar BS, Forseth KT, Manion MN, Jones SM, Wagner TJ, Endicott RM, Kyriss BP, Homem LM, Pate M, He J, Raines J, Gor'kov P, Brey WW, Mitchell DJ, Auman AJ, Ellard-Ivey MJ, Blazyk J, Cotton M. Investigating molecular recognition and biological function at interfaces using piscidins, antimicrobial peptides from fish. Biochim. Biophys. Acta. 2006;1758:1359–1372. doi: 10.1016/j.bbamem.2006.03.034. [DOI] [PubMed] [Google Scholar]
- 60.Marassi FM, Opella SJ. A solid-state NMR index of helical membrane protein structure and topology. J. Magn. Reson. 2000;144:150–155. doi: 10.1006/jmre.2000.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang J, Denny J, Tian C, Kim S, Mo Y, Kovacs F, Song Z, Nishimura K, Gan Z, Fu R, Quine JR, Cross TA. Imaging membrane protein helical wheels. J. Magn. Reson. 2000;144:162–167. doi: 10.1006/jmre.2000.2037. [DOI] [PubMed] [Google Scholar]
- 62.Bak M, Rasmussen JT, Nielsen NC. SIMPSON: A general simulation program for solid-state NMR spectroscopy. J. Magn. Reson. 2000;147:296–330. doi: 10.1006/jmre.2000.2179. [DOI] [PubMed] [Google Scholar]
- 63.Bak M, Schultz R, Vosegaad T, Nielsen NC. Specification and visualization of anisotropic interaction tensors in polypeptides and numerical simulations in biological solid-state NMR. J. Magn. Reson. 2002;154:28–45. doi: 10.1006/jmre.2001.2454. [DOI] [PubMed] [Google Scholar]
- 64.Lee DK, Wittebort RJ, Ramamoorthy A. Characterization of 15N Chemical Shift and 1H-15N Dipolar Coupling Interactions in a Peptide Bond of Uniaxially Oriented and Polycrystalline Samples by One-Dimensional Dipolar - Chemical Shift Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 1998;120:8868. [Google Scholar]
- 65.Park SH, Opella SJ. Tilt angle of a trans-membrane helix is determined by hydrophobic mismatch. J. Mol. Biol. 2005;350:310–318. doi: 10.1016/j.jmb.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 66.Allen TW, Andersen OS, Roux B. Structure of Gramicidin A in a Lipid Bilayer Environment Determined using Molecular Dynamics Simulations and Solid-State NMR Data. J. Am. Chem. Soc. 2003;125:9868–9877. doi: 10.1021/ja029317k. [DOI] [PubMed] [Google Scholar]
- 67.Chandrasekar I, van Gunsteren WF, Zandomeneghi G, Williamson PTF, Meier BH. Orientation and conformational preference of leucine-enkephalin at the surface of a hydrated dimyristoylphosphatidylcholine bilayer: NMR and MD simulation. J. Am. Chem. Soc. 2005;128:159–170. doi: 10.1021/ja054785q. [DOI] [PubMed] [Google Scholar]
- 68.Killian JA, Nyholm TKM. Peptides in lipid bilayers: the power of simple models. Curr. Opin. Str. Biol. 2006;16:1–7. doi: 10.1016/j.sbi.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 69.Kovacs FA, Cross TA. Transmembrane four-helix bundle of influenza A M2 protein channel: structural implications from helix tilt and orientation. Biophys. J. 1997;73:2511–2517. doi: 10.1016/S0006-3495(97)78279-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.White SH, Wimley WC. Hydrophobic interactions of peptides with membrane interfaces. Biochim. Biophys. Acta. 1998;1376:339–352. doi: 10.1016/s0304-4157(98)00021-5. [DOI] [PubMed] [Google Scholar]
- 71.Killian JA. Hydrophobic mismatch between proteins and lipids in membranes. Biochim. Biophys. Acta. 1998;1376:401–415. doi: 10.1016/s0304-4157(98)00017-3. [DOI] [PubMed] [Google Scholar]
- 72.Zhang YP, Lewis RN, Hodges RS, McElhaney RN. Interaction of a peptide model of a hydrophobic transmembrane alpha-helical segment of a membrane protein with phosphatidylcholine bilayers: differential scanning calorimetric and FTIR spectroscopic studies. Biochemistry. 1992;31:11579–11588. doi: 10.1021/bi00161a042. [DOI] [PubMed] [Google Scholar]
- 73.de Planque MR, Greathouse DV, Koeppe RE, II, Schafer H, Marsh D, Killian JA. Influence of lipid/peptide hydrophobic mismatch on the thickness of diacylphosphatidylcholine bilayers. A 2H NMR and ESR study using designed transmembrane alpha-helical peptides and gramicidin A. Biochemistry. 1998;37:9333–9345. doi: 10.1021/bi980233r. [DOI] [PubMed] [Google Scholar]
- 74.Harzer U, Bechinger B. Alignment of lysine-anchored membrane peptides under conditions of hydrophobic mismatch: a CD, 15N and 31P solid-state NMR spectroscopy investigation. Biochemistry. 2000;39:13106–13114. doi: 10.1021/bi000770n. [DOI] [PubMed] [Google Scholar]
- 75.Weiss TM, van der Wel PC, Killian JA, Koeppe RE, Huang HW. Hydrophobic mismatch between helices and lipid bilayers. Biophys. J. 2003;84:379–385. doi: 10.1016/S0006-3495(03)74858-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Strandberg E, Ozdirekcan S, Rijkers DTS, val der Wel CA, Koepee RE, II, Liskamp RMJ, Killian JA. Tilt angles of transmembrane model peptides in oriented and non-orientated lipid bilayers as determined by 2H solid-state NMR. Biophys. J. 2004;86:3709–3721. doi: 10.1529/biophysj.103.035402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Duong-ly KC, Nanda V, Degrado WF, Howard KP. The conformation of the pore region of the M2 proton channel depends on lipid bilayer environment. Protein Sci. 2005;14:856–861. doi: 10.1110/ps.041185805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kovacs FA, Denny JF, Song Z, Quine JR, Cross JA. Helix tilt of the M2 transmembrane peptide from influenza A virus: An intrinsic property. J. Mol. Biol. 2000;295:117–125. doi: 10.1006/jmbi.1999.3322. [DOI] [PubMed] [Google Scholar]