

Abstract
5-Guanidino-4-nitroimidazole (NI), derived from guanine oxidation by reactive oxygen and nitrogen species, contains an unusual flexible ring opened structure, with nitro and guanidino groups which possess multiple hydrogen bonding capabilities. In vitro primer extension experiments with bacterial and mammalian polymerases show that NI incorporates C as well as A and G opposite the lesion, depending on the polymerase. In order to elucidate structural and thermodynamic properties of the mutagenic NI lesion, we have investigated the structure of the modified base itself and the NI-containing nucleoside with high level quantum mechanical calculations, and have employed molecular modeling and molecular dynamics simulations in solution for the lesion in B-DNA duplexes with four partner bases opposite the NI. Our results show that the NI adopts a planar structure at the damaged-base level. However, in the nucleoside and in DNA duplexes, steric hindrance between the guanidino group and its linked sugar causes NI to be non-planar. The NI lesion can adopt both syn and anti conformations on the DNA duplex level, with the guanidino group positioned in the DNA major and minor grooves, respectively; the specific preference depends on the partner base. Based on hydrogen bonding and stacking interactions, groove dimensions, and bending, we find that the least distorted NI modified duplex contains partner C, consistent with incorporation of C opposite NI. However, hydrogen bonding interactions between NI with partner G or A are also found, which would be compatible with the observed mismatches.
Reactive oxygen and nitrogen species are products of normal cellular metabolism. In some cases, they are produced specifically to serve essential biological functions, in regulating circulation, energy metabolism, and apoptosis, and they constitute a major defense against pathogens (1, 2). However, these highly reactive species also have the capability of damaging DNA. Base lesions are prominent among the resulting forms of DNA damage (3-9). If not removed by repair enzymes, the processing of the damaged DNA by polymerases may cause mutations, which may in turn initiate cancer (10-14). In addition, aging (15-19), and a variety of diseases such as Alzheimer's disease and cardiovascular disease have been linked to DNA damage caused by oxidative mechanisms (20, 21).
Guanine has the lowest redox potential of the four DNA nucleobases and hence is a primary target of oxidative modification (8, 22). 8-oxo-7,8-dihydroguanine (8-oxoG) (23) is considered to be the most important among over 100 oxidized DNA lesions (12, 24, 25). Other oxidation products of guanine include 5-guanidino-4-nitroimidazole (NI), cyanuric acid (Ca), oxaluric acid (Oa), oxazolone (Oz), imidazolone (Iz), urea (Ua), spiroiminodihydantoin (Sp), and guanidinohydantoin (Gh) (26-32).
5-guanidino-4-nitroimidazole (NI), is derived from guanine oxidation by peroxynitrite anion (ONOO−) (30), or reaction of the nitrogen dioxide radical (·NO2) with guanine radicals in DNA (7, 33). These endogenous active oxidative agents are produced by reactive oxygen and nitrogen species in mitochondria and macrophage/inflammatory cells (12). The chemical change reflects nitration, hydrolytic opening of the 6-membered ring of guanine, and decarboxylation (34). The product contains a guanidino group and a nitroimidazole ring. This DNA lesion lacks a Watson-Crick hydrogen bonding edge, and is characterized by opportunities for torsional flexibility (Figure 1A). Of particular importance are the torsion angles δ (N2-C2-N3-C4) and θ (C2-N3-C4-N9) involving the guanidino group. The rotation of θ governs the co-planarity between the guanidino group and the imidazole ring. The planarity of the guanidino group itself is governed primarily by rotation of δ; rotation between C2 and N2 is also feasible. However, all bond lengths, bond angles and dihedral angles are unrestrained in the QM and MD simulations. The opportunities for conformational flexibility can permit various conformations of the lesion. The lesion is notable for its multiple and unique hydrogen bond donor and acceptor groups. These properties have the potential to produce novel structural features in the lesion-containing DNA, which are of interest in relation to its biological function.
Figure 1.
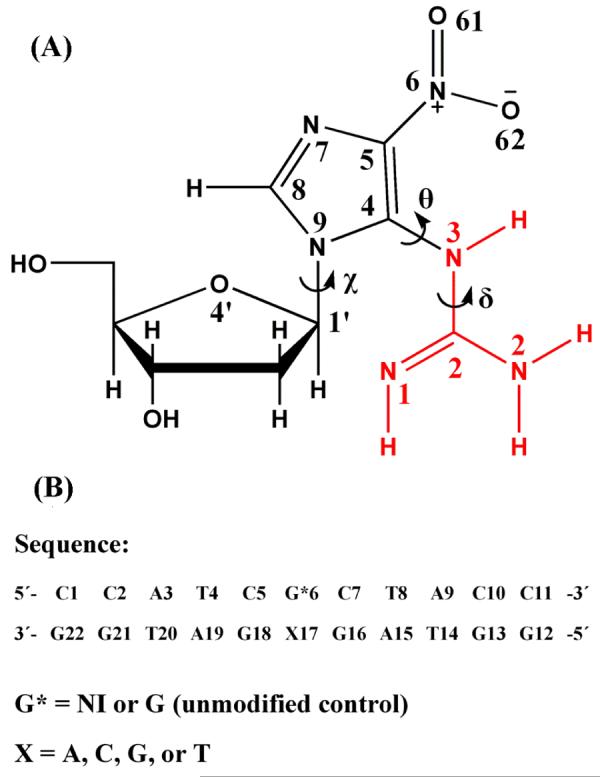
(A) Structure of 5-guanidino-4-nitroimidazole deoxyribonucleoside. Atom numbers and torsion angles are defined as follows: Glycosidic torsion angle χ is O4′-C1-N9-C4 (68) , δ is N2-C2-N3-C4, and θ is C2-N3-C4-N9. The guanidino group is shown in red. (B) Sequences for the molecular dynamics simulations.
It has been demonstrated that the NI lesion blocks polymerases and cause mutations, both in vitro and in vivo (29, 34). In vitro primer extension studies show that NI presents a significant replication block in the case of calf thymus pol α and Escherichia coli pol I (Klenow fragment, exo−), but not in the case of human pol β (34). The normal partner C is primarily incorporated opposite the NI lesion in the case of pol β and pol I, while A and G are also inserted in the case of pol I. In the case of pol α, A and G are chiefly incorporated opposite NI (34). In E. coli, the bypass efficiency of the NI lesion is 7.0 ± 1.6% under normal conditions and 57 ± 1% under SOS-induced conditions where bypass polymerases are likely involved. The order of incorporation preference is C > A ≈ T > G under both conditions (29).
Oxidative DNA base lesions are repaired by the base excision repair (BER) system (35-37). However, a repair pathway has not yet been identified for the NI lesion. Neither E. coli formamidopyrimidine glycosylase (Fpg) nor endonuclease III (Nth) appear to be effective in NI repair (34).
To investigate the biological effects of the NI lesion, a knowledge of the structure of NI itself as well as of NI-damaged DNA duplexes is needed. However, at present, such structural information is not available. We have carried out computational investigations at the levels of the damaged base, nucleoside, and in duplex DNA to elucidate the NI structural preferences. At the base level, quantum mechanical studies provided a geometry-optimized lowest energy planar structure of NI. At the nucleoside level, geometry-optimization reveals the NI base is no longer planar due to steric hindrance to the sugar, consistent with a previous semi-empirical MO calculation (38). At the DNA duplex level, we investigated the NI lesion paired with all four partner bases in a B-form 11mer. Molecular dynamics (MD) simulations in aqueous solution were carried out to obtain ensembles of structures, and trajectories were employed to analyze the structures and to compute free energies. The structural and thermodynamic analyses suggest that the non-planar NI lesion can adopt both syn and anti conformations with the specific preference depending on the partner base, and the guanidino group is positioned in the DNA major and minor grooves, respectively. The unique hydrogen bond properties and the opportunity for flexibility play an important role in the distinct structural features of this unusual lesion.
METHODS
Quantum mechanical geometry- optimization of NI on the base level
NI was built by the program SPARTAN from Wavefunction, Inc., and preliminary geometry-optimization was performed with the MERCK molecular force field (MMFF 94) (39) in SPARTAN. Sixteen starting structures were built by surveying the torsion angles δ (N2-C2-N3-C4) and θ (C2-N3-C4-N9) (Figure 1A) at 90° intervals, in combination. However, two of them θ = 180°, δ = 0° and 180° had collisions between the nitro group and amino/imino group, and were eliminated from further consideration (Table 1). We applied the quantum mechanical density functional theory (DFT) method (B3LYP/6-31G*) (40, 41) in Gaussian 03 (42) from Gaussian, Inc. to perform high level geometry-optimizations. The geometry-optimized structure with the lowest energy was employed for both geometry-optimization on the nucleoside level and molecular dynamics simulation on the DNA duplex level.
Table 1.
NI Base Torsions and Energies, ΔEa
| Initial Structure | Final Structure | |||
|---|---|---|---|---|
| θ( ° ) | δ( ° ) | θ( ° ) | δ( ° ) | ΔE (kcal/ mol) |
| 0 | 0 | 5 | 328 | 8.1 |
| 0 | 90 | −1 | 182 | 0 |
| 0 | 180 | −2 | 182 | 0 |
| 0 | 270 | −1 | 182 | 0 |
| 90 | 0 | 132 | 328 | 15.1 |
| 90 | 90 | 104 | 35 | 13.0 |
| 90 | 180 | −1 | 182 | 0 |
| 90 | 270 | −2 | 182 | 0 |
| 180 | 90 | 104 | 35 | 13.0 |
| 180 | 270 | 1 | 178 | 0 |
| 270 | 0 | 256 | 325 | 13.0 |
| 270 | 90 | −5 | 32 | 8.1 |
| 270 | 180 | −1 | 182 | 0 |
| 270 | 270 | 1 | 178 | 0 |
Energies are relative to 0 kcal/mol for the lowest energy structure.
Quantum mechanical geometry-optimization of NI nucleoside
The planar geometry-optimized NI base was connected to the standard B-DNA C2'-endo sugar (P=162°) (43). The glycosidic torsion χ was systematically rotated to locate structures with minimal collisions between NI and the sugar, since the QM calculations are impossible for structures with severe collisions. Only two starting structures for the QM geometry-optimization could be created in the anti domain. Syn conformers with completely planar NI (Figure 2) were not suitable for the QM calculations, because in this case the guanidino group has massive collisions with the sugar. To investigate the syn domain, we therefore created a representative set of 8 different non-planar syn initial structures by adjusting torsions θ and δ to avoid collisions and at the same time minimize non-planarity (Table 2). The same method as in the base level study was employed to perform QM geometry-optimization. The gas phase geometry-optimized structures produced a non-planar NI amino group, as is usually the case for such computed structures (44, 45). However, in all crystal structures of guanines and adenines, these groups are planar (46-48), and we therefore remodeled them to be planar for the next stage of our studies involving the modified DNA duplexes.
Figure 2.
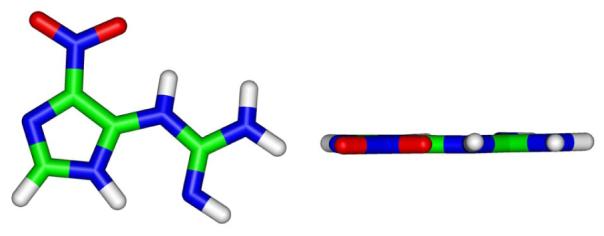
Structures of NI base after QM geometry-optimization. The left view is perpendicular to the NI imidazole ring, and the right view is rotated 90° out of the plane of the paper.
Table 2.
NI Nucleoside Torsions, Sugar Pucker, and Energies, ΔEa
| Initial Structure | Final Structure | |||||||
|---|---|---|---|---|---|---|---|---|
| χ ( ° ) | θ( ° ) | δ( ° ) | P ( ° ) | χ ( ° ) | θ( ° ) | δ( ° ) | P ( ° ) | ΔE(kcal/mol) |
| 200 | 0 | 180 | 162 | 238 | 301 | 196 | 171 | 6.6 |
| 270 | 0 | 180 | 162 | 216 | 55 | 169 | 164 | 8.5 |
| 60 | −145 | 180 | 162 | 51 | −125 | −174 | 155 | 10.6 |
| 60 | 145 | 180 | 162 | 58 | 126 | 180 | 133 | 14.4 |
| 60 | −145 | 0 | 162 | 49 | −118 | −22 | 157 | 0 |
| 60 | 145 | 0 | 162 | 44 | 116 | 31 | 148 | 0.6 |
| 60 | 180 | −135 | 162 | 51 | 125 | −174 | 155 | 10.6 |
| 60 | 180 | 135 | 162 | 59 | 125 | 152 | 149 | 9.9 |
| 60 | 180 | 35 | 162 | 49 | −118 | −23 | 157 | 0 |
| 60 | 180 | −35 | 162 | 44 | 116 | 31 | 148 | 0.6 |
Energy are relative to 0 kcal/mol for the lowest energy structure. Syn is 60°; 200° and 270° are anti.
Molecular dynamics simulation of the NI damaged DNA duplex
Initial models
We created initial DNA models for the molecular dynamics simulations in the sequence shown in Figure 1B by replacing an unmodified guanine with the geometry-optimized NI base in an energy minimized (49) B-DNA of that sequence. This is the same sequence that we employed in our previous study of the Sp R and S stereoisomeric lesions (50) and we retain this sequence here to permit comparison with those results. In addition, mismatches were created by replacing the NI partner C with A, G, or T. Both syn and anti glycosidic bond orientations for the NI, as well as for its partners when these were purines opposite NI(anti) (51, 52), were considered. We did not consider syn-syn pairing or syn pyrimidines since these are rarely observed (46). The B-DNA energy minimized values for χ were employed for the anti partner structures, while the syn structures were obtained by rotation of χ by about 180°, using optimal stacking as the criterion for the actual syn χ values. To avoid collisions between the guanidino and the sugar moieties, torsion δ and θ of the NI were adjusted. Table S1 gives χ, δ, and θ values of NI employed in all starting models. Representative starting structures are shown in Figure S1. In these structures, the NI imidazole ring is stacked within the helix; the guanidino group is also stacked in the anti structures but is in the major groove in the syn ones.
Force field
Computations were carried out with the AMBER 8.0 (53) suite of programs, the Cornell et al. force field (54) and the PARM 99 parameter set (55). The force field was parametrized for the NI consistent with the rest of the force field. Partial charges for the NI were obtained as described by Cieplak et al (56). HF calculations with the 6-31G* basis set (57) were used to calculate the electrostatic potential using Gaussian 03 (42), and the restrained electrostatic potential fitting algorithm RESP (54, 58) was employed to fit the charge to each atom center. Partial charges were separately computed for anti (χ = 240°) and syn (χ = 60°) conformers and averaged. One missing atom type was obtained from the GAFF (General Amber Force Field) parameter set (59). Missing bond length and angle equilibrium values for the NI were taken from the QM optimized structures. Bond length, angle, and dihedral angle force constant parameters not present in the PARM 99 parameter set were obtained from the GAFF parameter set or assigned by analogy with chemically similar atom types already present in the AMBER force field. All of the added force field parameters, atom types and topology assignments are listed in Tables S2 and S3. It is also worth noting that torsion angle δ (Figure 1) in the tautomer A investigated in the present work (Scheme 1) is less rigid than in the guanidino group of arginine because of the proximity to the aromatic imidazole ring with its electron withdrawing nitro group, especially in cases where the lesion is non-planar in which case π electron conjugation is weakened. This is reflected in the relatively low torsion barrier (V0/2 = 2.7 kcal/mol, Table S2) of δ in the GAFF parameter set (59).
Scheme 1.

NI tautomerisma
a: “R” represents a hydrogen atom (on the base level) or the sugar moiety (on the nucleoside level); On the base level, the imidazole ring was protonated at the N9 as the N7 H tautomer is not relevant to DNA.
Molecular dynamics protocol
Details of the MD protocol (53, 60-66) are given in the Supporting Information.
Structural analyses
Snapshots of the DNA structures during the simulation and the average structures, with solvent and counterions stripped away, were obtained with the PTRAJ module of the AMBER 8.0 suite. PTRAJ was also employed to obtain time-dependence of the RMSD, the torsion angles δ, θ, and χ, and the sugar pucker P(43) of the NI residue, and the corresponding dG residue in the unmodified control. Hydrogen bonding analyses were carried out with the CARNAL module of the AMBER 7.0 suite (67). In addition, we employed a hydrogen bond quality index (49), IH, to quantitatively assess the deviation from ideal Watson-Crick hydrogen bonding distances and angles for 5′ and 3′ neighboring base pairs of NI:
where dDA is the instantaneous donor-acceptor distance, d0DA is an ideal donor-acceptor distance (68) (N4 (C) to O6 (G) is 2.91Å, N1 (G) to N3 (C) is 2.95Å, and N2 (G) to O2 (C) is 2.86Å) and γ is the instantaneous D-H⋯A bond angle with ideal value of 180°. The summation is over the three Watson-Crick hydrogen bonds in a G•C base pair and then over the selected trajectory window. IH adopts a value of 0 when ideal Watson-Crick hydrogen bonding is maintained. The DNA groove width was analyzed with MD Toolchest (69, 70). The bend angle of the duplex was analyzed with the program CURVES (71), employing the “UU” option, which yields a bend angle measured between the first and last vectors defining the helical axis. The first and last base pairs were removed prior to this analysis. In addition, we removed the NI and its partner when the partner was syn since CURVES could not recognize these moieties. The computed bend angles were thus based on global helix axes determined by the four base pairs surrounding the lesion in each direction. The stacking interactions were estimated by computing the van der Waals interaction energy between adjacent base pairs, including the NI lesion-containing pair, with the program ANAL from the AMBER 7.0 suite (67). Hydrogen bond occupancies between NI donor or acceptor atoms and water were also computed with CARNAL.
Free energy analyses
The molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) method which has been described in detail (72-77) was employed to perform the free energy analyses. In brief, the free energy (Gtot) was computed from the molecular mechanical energies (EMM), the solvation free energy (Gsolvation), and the solute entropic contributions to the free energy (Gtot = EMM + Gsolvation − TS) (78, 79). The molecular mechanical energies (EMM) were calculated from internal energies (Eint) stemming from deviations of the bonds (Ebonds), angles (Eangles), and dihedral angles (Edihedrals) from their equilibrium values, the van der Waals energies (EvdW), and electrostatic energies (Eelectrostatic). The solvation free energies (Gsolvation) were estimated from the electrostatic solvation energies (GPB) calculated using the DelPhi program (80). and the nonpolar solvation energy (Gnonpolar); the latter was approximated as Gnonpolar = γSA + b (γ= 0.00542 kcal/Å2, b = 0.92 kcal/mol) (81), where SA is the solvent accessible surface area (SASA) (82) computed by Sanner's algorithm in the MSMS program (83). The solute entropic contributions to the free energies were approximated with normal mode calculations (84). Details of the protocol (53-55, 82, 85) are given in the Supporting Information.
INSIGHT II from Accelrys, Inc. was employed for visualization and model building. Computations were carried out on our own cluster of Silicon Graphic Origin and Altix high-performance computers.
RESULTS AND DISCUSSION
The goal of our work was to delineate structural possibilities for NI damaged DNA duplexes in solution through molecular modeling and molecular dynamics simulations. Detailed analyses of the MD generated ensembles were performed to determine the structural and thermodynamic features of these lesion-containing DNA duplexes.
Planar Quantum Mechanical Geometry-Optimized Structures of NI on the Base Level
In the first stage of this study, we created fourteen different conformations for the NI tautomer of Figure 1. We surveyed the torsion angles δ (N2-C2-N3-C4) and θ (C2-N3-C4-N9) (Figure 1A) at 90° intervals, in combination (θ = 180°, δ = 0° and 180°, were excluded since they have collisions between the nitro and amino/imino group), and carried out quantum mechanical geometry-optimization for each structure using the DFT method (B3LYP/6-31G*). From the initial fourteen starting structures, eight converged to one final lowest energy structure. Five additional distinct energy minima were obtained from the other six starting structures (Table 1). These non-planar structures had energies between 8.1 and 15.1 kcal/mol above the lowest energy form. The structure with the lowest energy has a flat five-membered imidazole ring with the C, N, and O atoms in the guanidino and nitro groups in one plane. (Figure 2)
Non-Planar Quantum Mechanical Geometry-Optimized Structures of NI on the nucleoside level
Two anti initial structures (χ = 200° and 270°) with planar NI and eight non-planar syn initial structures employing the QM geometry-optimized structure for the NI base lesion were created, based on minimal collisions between NI and the sugar (see Methods). Due to steric hindrance between the planar NI in the initial structure and the sugar, after the geometry-optimization the NI base itself was never planar. We employ the term “planar” to include co-planarity between the imidazole ring and the guanidino group as well as the planarity within the guanidino group itself. The initial and final torsions and relative energies are shown in Table 2. The eight syn initial structures converged to five different final non-planar structures with the guanidino group above and below the plane of the imidazole ring (Figures 3 and S2). The two initial anti structures provided two different geometry-optimized final structures, also with the guanidino group above and below the plane of the imidazole ring. The anti final structure with lowest energy is 6.6 kcal/mol higher energy than the lowest energy syn one. The syn lowest energy structures (Figure 3) are favored due to a hydrogen bond between the nitro oxygen atom and amino group in the guanidino moiety.
Figure 3.
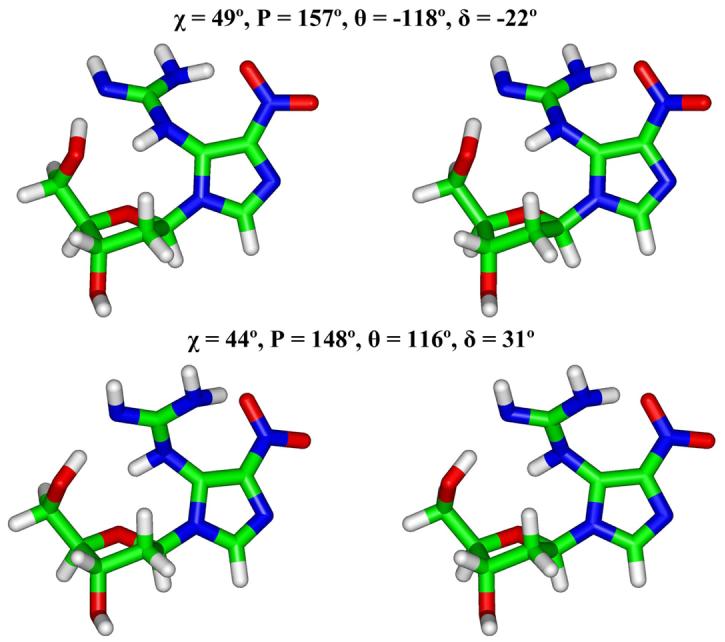
Stereo views of geometry-optimized two lowest energy NI nucleosides (Table 2). All stereo figures are prepared for viewing with a stereoviewer.
Structures in DNA duplexes: major and minor groove NI with flexible and non-planar guanidino group
We carried out 8 ns molecular dynamics simulations to obtain ensembles for structural and thermodynamic analyses. The QM geometry-optimized NI structure with the lowest energy was modeled into four 11-mer B-DNA duplexes in which the partner base opposite the NI was A, C, G, or T. In addition, we investigated an unmodified control duplex containing a normal G·C pair in place of the damaged site. We considered both syn and anti glycosidic bond orientations for the NI modifications, as well as for their partners when these were purines opposite NI(anti). Plots of root-mean-square deviations (RMSD) of the current structure, relative to the starting structure, as a function of time, are shown in Figure S3. The structures generally fluctuate stably after 2 ns and our further analyses employed the most stable 3ns time frame extracted from the relatively stable region of the trajectories (Table S5).
Thermodynamic Analysis
Table 3 shows the results of our thermodynamic analyses. This table reveals relative stabilities of syn and anti conformations for each base partner. Figure 4 shows the structures of each of these conformations. The guanidino group is in the major groove for the syn and minor groove for the anti conformation of NI. The anti conformation is favored when NI is paired with A (regardless of whether the glycosidic torsion of the partner is syn or anti), and the syn conformation is energetically preferred when NI is paired with C and G, while both syn and anti conformations are about equal energy when NI is paired with T.
Table 3.
Relative Free Energies (kcal/mol)a
| NI(anti)•X(anti) | NI(syn)•X(anti) | NI(anti)•X(syn) | |
|---|---|---|---|
| NI•A | 1.4 | 14.8 | 0 |
| NI•C | 7.0 | 0 | |
| NI•G | 7.4 | 0 | 10.3 |
| NI•T | 0 | 0.8 |
Energies are relative to 0 kcal/mol for the lowest energy structure in each sequence.
Figure 4.
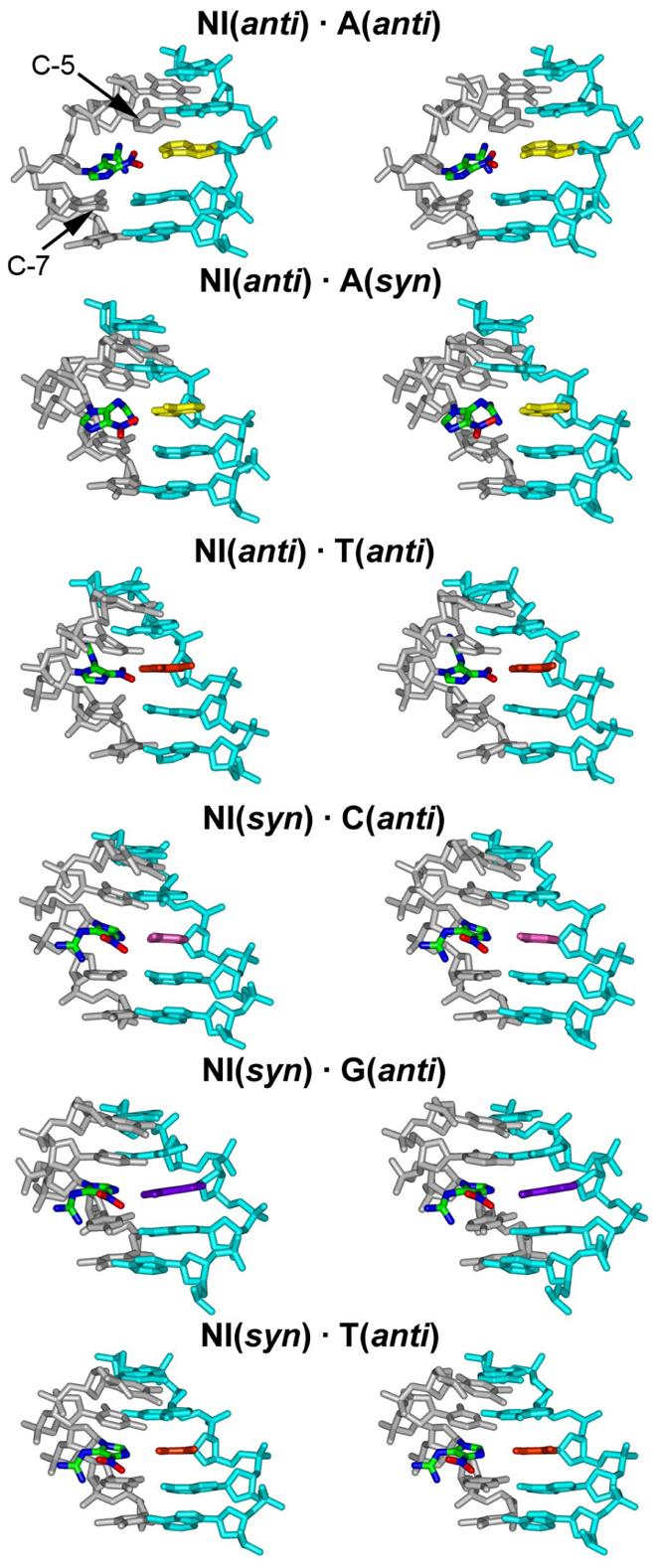
Stereo views of the central 5-mer of the NI trajectory-average duplex structures. The low energy structures of Table 3 are shown. The NI base is colored by atom. The NI base partners are colored as yellow (A), pink (C), purple (G), and orange (T). The NI adjacent bases C5 and C7 are also marked. The view is into the major groove.
To help understand the conformational preferences for each partner suggested from the thermodynamic analyses, we evaluated hydrogen bonding and base stacking properties. Both hydrogen bonding between base partners and base stacking contribute to the stability of the damaged DNA. A partial energetic assessment of stacking interactions can be obtained from van der Waals interactions between base pairs (or the NI residue and partner with each adjacent base pair). These values are given in Table 4. We also analyzed hydrogen bonding at the lesion site and nearest base-pair neighbors (see Methods). The hydrogen bonds in which the NI lesion is involved are shown in Table S6. Watson-Crick hydrogen bond quality indexes of the base pairs adjacent to the lesion are shown in Figure 5.
Table 4.
Sum of van der Waals Interaction Energies Evdwa.
| Evdw (kcal/mol) | |
|---|---|
| NI(anti) · A(anti) | −114.8 |
| NI(anti) · A(syn) | −120.2 |
| NI(syn) · A(anti) | −110.1 |
| NI(anti) · C(anti) | −117.8 |
| NI(syn) · C(anti) | −117.1 |
| NI(anti) · G(anti) | −122.4 |
| NI(anti) · G(syn) | −118.7 |
| NI(syn) · G(anti) | −121.3 |
| NI(anti) · T(anti) | −118.1 |
| NI(syn) · T(anti) | −117.3 |
The sum is over all pairs in the given duplex. The two base pairs at the ends of the duplex are not included. Note that energies may only be compared within the groups containing the same partner base to the NI.
Figure 5.
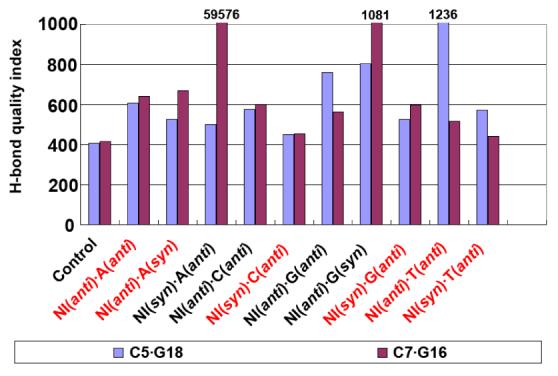
Trajectory summed hydrogen bond quality index for the 5′- (C5•G18) and 3′- (C7•G16) NI neighboring Watson-Crick base pairs. The values for C5•G18 in NI(anti)•T(anti) and C7•G16 in NI(syn)•A(anti) and NI(anti)•G(syn) are not shown to full scale; their values are given at the top of the bar. The energetically favored pairs are in red.
Pairing of NI with A in the complementary strand
In the case of NI paired with A, the NI anti conformation is energetically favored, with A syn or anti, due to better stacking interactions and Watson-Crick hydrogen bonding of base pairs adjacent to the NI lesion. The stacking interaction energies for NI(anti)·A(anti) and NI(anti)·A(syn) are 4.7 and 10.1 kcal/mol lower than the NI(syn)·A(anti) pair, respectively (Table 4); in the latter case the N7 of NI forms a hydrogen bond to H6N6 of partner A, which extrudes the NI imidazole ring out of the duplex (Figures S4 and S5). (This pairing scheme and attendant NI extrusion is not possible for NI(syn)·G(anti): consequently NI is better stacked and hydrogen bonded as described below.) The NI(syn) does not stack well with its base neighbors and hence disturbs their Watson-Crick hydrogen bonding qualities (Figure 5). Specifically, the quality index of the C7·G16 pair is near 60,000 for the NI(syn)·A(anti) sequence, which indicates that this Watson-Crick base pair is completely disrupted. This is consistent with the higher (by 14.6 kcal/mol) free energy of the NI(syn)·A(anti) pair compared to the lowest energy conformation for this sequence, NI(anti)·A(anti) (Table 3).
Pairing of NI with C or G
In the cases of NI paired with C or G, the NI in the syn conformation is energetically favored due to hydrogen bonding within the modified nucleotide and concomitant better Watson-Crick hydrogen bonding of adjacent base pairs, because NI is anchored and avoids disturbing the neighbors. The base stacking energies are nearly the same in the DNA duplexes containing syn and anti NI lesions (Table 4). However, the NI in the syn conformation disturbs its neighboring Watson-Crick base pairs less than when it is in anti conformation. There are 2-3 hydrogen bonds between the guanidino group and the sugar moiety of the NI nucleotide in DNA when NI is syn (Figure 6 and Table S6). These hydrogen bonds, not possible for NI(anti), help hold the flexible guanidino group so that there is little perturbation to the adjacent base pairs when NI(syn) is paired with C or G.. This is shown in the Watson-Crick hydrogen bonding quality indexes of NI(syn)·C(anti) and NI(syn)·G(anti) pairs, which are lower than the NI(anti) cases (Figure 5).
Figure 6.
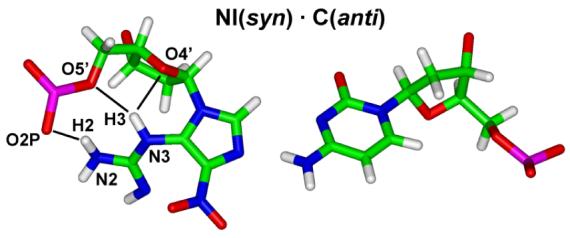
Hydrogen bonds (black lines) between NI and adjacent backbone in NI(syn) structures, illustrated with partner C. Hydrogen bonds (occupancy greater than 50%) within the NI nucleotide are marked.
NI paired with T
When NI is paired with T, both syn and anti conformations are equally favored, with free energy difference less than 1 kcal/mol. The stacking interaction energies are essentially the same for the two conformations (Table 4). In the NI(anti)·T(anti) pair, one bifurcated hydrogen bond forms between the NI and its partner T which stabilizes the pair. In the NI(syn)·T(anti) pair, there is no hydrogen bond formed between NI and its partner T. However, one bifurcated hydrogen bond forms between the N3H3 of the guanidino group and the sugar moiety of the NI nucleotide. This hydrogen bond helps hold the flexible guanidino group, as in the NI(syn)·C/G(anti) cases, so that the Watson-Crick hydrogen bonding of the adjacent base pairs is less disturbed than for the NI(anti) case (Figure 5). Hence, the damaged DNA duplexes with NI(anti) and NI(syn) have interactions overall equally contributing to the free energy and are about equally favored.
Structural Analyses
We confine our discussion of detailed structural analyses to the energetically favored DNA duplexes (Table 3). These show that the NI lesion can adopt both syn and anti conformations. The flexible guanidino group is positioned in the DNA major (syn) and minor (anti) grooves and the specific preference depends on the partner base. As shown in Figure 4, for both syn and anti conformations of NI, the five-membered imidazole ring is stacked into the DNA duplex. The flexible and hydrogen-bond donor-rich guanidino group is located in the minor groove when the NI is in the anti conformation, and in the major groove, stabilized by at least two hydrogen bonds to the sugar moiety, when it is syn. The nitro group on the imidazole ring, which contains multiple hydrogen bond acceptors, is positioned in the DNA duplex, facing the base partner for the NI anti conformation; however, in the syn conformation, this nitro group is placed in the major groove, where it has less opportunity to form hydrogen bonds to the partner.
The NI lesion, both in syn and anti conformation, perturbs the DNA duplexes to various extents, depending on its partner. As detailed below, this can be manifested by opening and closing of the major or minor grooves, with disturbance to the Watson-Crick hydrogen bonding quality in adjacent base pairs and concomitant disturbed base stacking. NI modified duplexes show reduction of thermal stabilities compared to the unmodified ones in the range of 4 – 12 °C, depending on partner base, in 19-mer duplexes (38).
Hydrogen bonding interactions
We analyzed the hydrogen bonds between NI and partner, as shown in Figure 7. Irrespective of partner base and the conformation of NI and its partner, at most one hydrogen bond is observed except for the NI(syn)·G(anti) and NI(anti)·T(anti) pairs, which have two hydrogen bonds and one bifurcated hydrogen bond respectively. There is no hydrogen bond in the NI(syn)·T(anti) pair. Other hydrogen bonding interactions are shown in Table S6.
Figure 7.
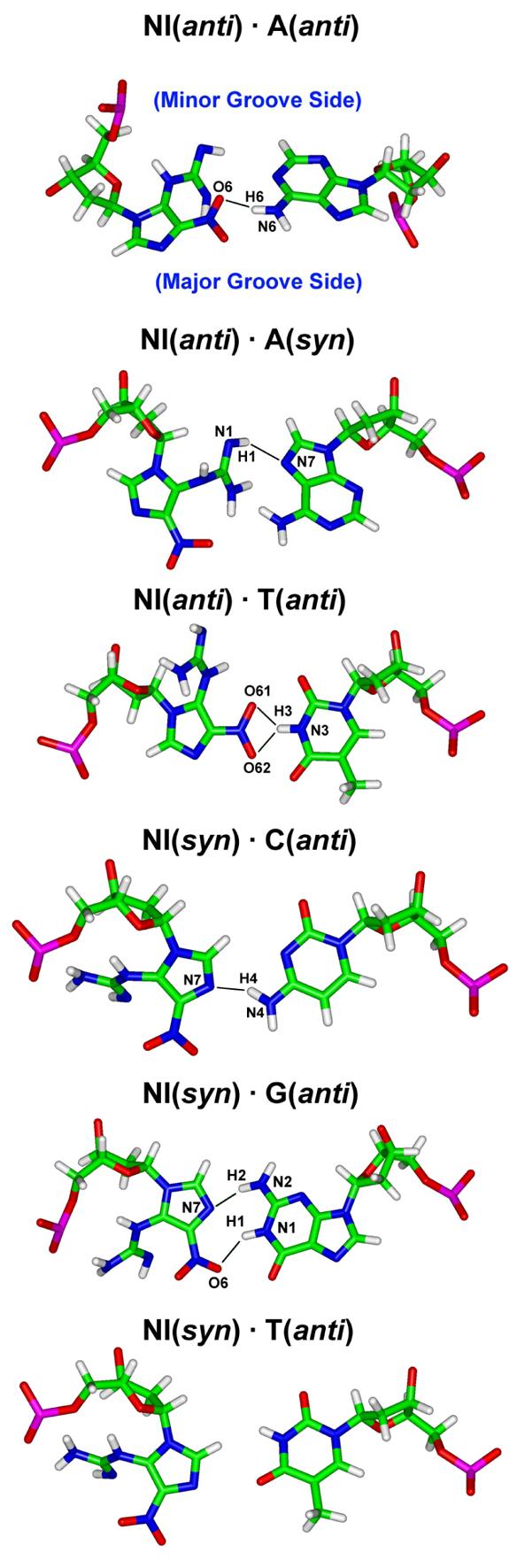
Hydrogen bonds (black lines) between NI and partner base. The low energy structures of Table 3 are shown. The base pairs are obtained from the trajectory average structures of the selected simulation window. Hydrogen bonds shown have occupancy greater than 50%. Major and minor groove sides for all structures are designated in the first pair.
We also analyzed the hydrogen bonding of base pairs adjacent to the lesion. The hydrogen bond quality indexes (Figure 5) of the energetically favored structures show that generally the adjacent base pairs are less disturbed in the NI(syn) damaged DNA duplexes than in the NI(anti) damaged ones. The flexible guanidino group plays a key role in disturbing the base neighbors when NI is anti, in the DNA minor groove. For example, the C5·G18 Watson-Crick pair is quite disturbed in the NI(anti)·T(anti) case (Figure 4), with hydrogen bonding quality index about three times that in the unmodified control (Figure 5). This stems from interaction between the minor groove positioned guanidino group pointing to the 5' direction of the damaged strand, which crowds and repositions the C5 base. However, syn NI places the guanidino group in the DNA major groove so that it does not interact with the base neighbors as much (Figure 4). Among the energetically favored structures, the NI(syn)·C(anti) pair has the least disturbed Watson-Crick hydrogen bonding in both adjacent base pairs compared to the unmodified control. Concomitant with the impaired Watson-Crick hydrogen bonding, stacking is also diminished for NI(anti)·A(anti), NI(anti)·A(syn), NI(anti)·T(anti), and NI(anti)·T(syn) (Figure 4).
Hydrogen bond occupancies between NI donor or acceptor atoms and water were also computed and are shown in Table 5. No such interactions were found for the N6 and N9 atoms of NI (Figure 1). The general principle that emerges is that the more solvent exposed atoms are highly hydrogen bonded to solvent while buried atoms are rarely solvent-hydrogen bonded. (See Figures 4 and 7.) Thus, for the syn NI structures, oxygen atoms (O61 and O62) in the nitro group are most exposed in the major groove and most solvated. Nitrogen atoms (N1 and N2) in the guanidino group are also exposed, but to a lesser extent and are solvated somewhat less. N3 in the guanidino group is not solvated, even though it is on the major groove side, because it is shielded from exposure. N7 in the imidazole ring is either solvated [NI(syn)·T(anti)] or hydrogen bonded to partner base [NI(syn)·C(anti), and NI(syn)·G(anti)]. For two NI(anti) structures [NI(anti)·A(anti), and NI(anti)·T(anti)], the N7 on the major groove side is highly exposed and solvated, while nitrogen atoms (N1, N2, and N3) in the guanidino group are less exposed on the narrower minor groove side and therefore less solvated. Oxygen atoms (O61 and O62) in the nitro group dynamically interchange between hydrogen bonding to solvent or to partner base. For the case of NI(anti)·A(syn), there are unusual structural features: the guanidino group is within the DNA duplex inaccessible to solvent, accounting for the low solvation of atoms N1, N2, and N3. The O6 atoms on the nitro group in the major groove are each only solvated about 25% of the time; at any given instant one is totally exposed and the other is inward directed and shielded, but the nitro group rotates. Also, for this NI(anti)·A(syn) pair, the N7 is less exposed and solvated than the other anti NI structures. The NI syn structures, with the guanidino group in the major groove, appear to be stabilized by the high opportunity for hydrogen bonding to solvent on the major groove side of the lesion. For the anti structures, with the guanidino group in the minor groove or within the DNA duplex, solvation appears to play a lesser role in providing stability, with interactions between NI guanidino, nitro, or imidazole ring with adjacent bases playing a more important part.
Table 5.
Occupancies (%) of Hydrogen Bonds between NI Atoms and H2O in Energetically Favored Structuresa
| Atoms |
N1 | N2 | N3 | N6 | N7 | N9 | O61 | O62 |
|---|---|---|---|---|---|---|---|---|
| Structures | ||||||||
| NI(anti)·A(anti) | 48.1 | 47.9 | 20.4 | 0 | 107.1 | 0 | 119.0 | 51.4 |
| NI(anti)·A(syn) | 4.3 | 14.3 | 4.7 | 0 | 34.2 | 0 | 26.4 | 27.1 |
| NI(syn)·C(anti) | 58.1 | 82.4 | 0 | 0 | 14.6 | 0 | 152.0 | 129.6 |
| NI(syn)·G(anti) | 52.4 | 62.3 | 0 | 0 | 0 | 0 | 117.9 | 60.6 |
| NI(syn)·T(anti) | 67.4 | 90.2 | 0 | 0 | 65.0 | 0 | 155.9 | 115.5 |
| NI(anti)·T(anti) | 46.2 | 67.6 | 51.9 | 0 | 82.5 | 0 | 50.8 | 55.9 |
Occupancy over 100% occurs because the atom is engaged in bifurcated hydrogen bonding to more than one water.
Bending and groove dimensions
We analyzed bending (Figure S6) and groove dimensions (Figure S7) of the NI damaged DNA (see Methods). Among energetically favored structures, increased bending was observed compared to the unmodified control (Figure S6 and Table 6) whose extent depends on the partner. NI(anti)·A(syn) has the highest average bend, ∼ 47°. The NI(syn)·C(anti) and NI(syn)·G(anti) pairs are bent less (∼20° and ∼16°, respectively). Major grooves at the lesion site are all closed for the energetically favored structures, irrespective of whether NI is syn or anti. For the syn case, there are hydrogen bonds between the guanidino group and the sugar moiety which close the groove; in the case of NI in the anti conformation, the guanidino group is in the minor groove, which causes concomitant major groove closing. The minor groove is closed in the NI(syn)·G(anti) case since the guanidino group of the NI(syn) lesion is in the major groove; however it is opened in the NI(anti)·A(anti) case because of the minor groove positioning of the guanidino group of the NI(anti) lesion. The minor groove is normal near the lesion site for the NI(syn)·C(anti), NI(syn/anti)·T(anti), and NI(anti)·A(syn) cases. The pyrimidine partners as well as the A(syn) (which somewhat mimics a pyrimidine) allow more room to accommodate the guanidino group and cause less disturbance to the minor groove.
Table 6.
Structural Analyses for Energetically Favored Structures
| Sequences | HB quality indexa C5G18 C7G16 |
Number of HBs to partnerb |
Bendingc (°) |
Major grooved (Å) |
Minor groovee (Å) |
|
|---|---|---|---|---|---|---|
| Unmodified control |
406 | 417 | 3(100%) | 14.6(8.7)f | 16.4(1.6) | 7.4(1.3) |
| NI(syn)·C(anti) | 451 | 454 | 1(75%) | 19.5(10.0) | 14.1(2.0) | 7.3(1.5) |
| NI(syn)·G(anti) | 526 | 596 | 2(91%) | 16.1(7.7) | 14.4(1.3) | 4.2(1.5) |
| NI(anti)·A(anti) | 606 | 640 | 1(59%) | 16.4(8.9) | 12.1(1.6) | 13.1(4.0) |
| NI(anti)·A(syn) | 527 | 668 | 1(64%) | 46.8(17.7) | 13.0(1.9) | 5.3(1.5) |
| NI(anti)·T(anti) | 1236 | 514 | 1.5g (66%) | 27.5(20.3) | 13.5(2.2) | 6.9(1.4) |
| NI(syn)·T(anti) | 573 | 440 | 0 | 35.5(19.2) | 15.1(1.8) | 6.7(1.3) |
Hydrogen bond quality index (Figure 4)
Number of hydrogen bonds between NI and its partner (Figure 6) with average occupancy shown in parentheses
Trajectory average bending of the DNA duplex (Figure S7)
Trajectory average major groove dimension at the lesion site (Figure S8)
Trajectory average minor groove dimension at the lesion site (Figure S8)
Standard deviation is shown in parentheses
A bifurcated hydrogen bond.
NI torsional flexibility in duplex DNA
An especially interesting feature of the NI lesion is the flexibility of the nitro and guanidino groups. Though the QM geometry-optimized structure with lowest energy for NI itself is planar (Figure 2), the structures from the QM geometry-optimization on the nucleoside level in this work and by Neeley et al. (38), and in our MD simulation for the DNA duplexes are no longer planar. The torsions in the nitro group and guanidino amino/imino group depend on the intrinsic torsional preferences as found in the QM study of the lesion itself, together with steric effects stemming from the presence of adjacent sugar and neighboring bases on the same and opposite strands, as well as possible hydrogen bonding interactions in the DNA duplexes.
The glycosidic torsion χ as well as the torsions θ and δ in the guanidino group during our MD simulations (Figure 1) rotate within certain preferred domains (Figure S8). Among energetically favored structures, there are five clear states for the combination of the three torsions (Table 7). The corresponding NI nucleotide structures are shown in Figure 8. The conformations of the NI base in all five states on the DNA duplex level are different from the planar lowest energy structure obtained by QM geometry-optimization of the lesion itself, and also different from the non-planar QM geometry-optimized nucleoside structures. Due to steric hindrance from the sugar, the conformation of the NI base is no longer planar in the nucleoside. However, the NI conformations on the DNA duplex level differ from those in the nucleoside due to the interactions with the partner base or with adjacent nucleosides on the same strand. The most predominant state is State 1 which is favored in most NI(syn) conformations because of the hydrogen bonds formed between the guanidino group and the sugar moiety. State 2 was found in two trajectories, in which the NI is in the anti conformation paired with pyrimidine or syn (pyrimidine-like) purine. State 3 was observed in one trajectory in which the NI is in the anti conformation paired with syn purine. Two additional states were sampled for 1 – 1.5 ns in the trajectory of NI(anti)·A(anti) pair, anti NI paired with an anti purine. Notably, state 4 shows the NI flipping from the anti conformation to syn. The flexibility of these torsions provides enhanced opportunities for the hydrogen bond donor and acceptor-rich NI lesion to form hydrogen bonds.
Table 7.
Torsions in NI for Energetically Favored DNA Duplexesa.
| State | χ ( ° ) | θ( ° ) | δ( ° ) |
|---|---|---|---|
| 1 | 49 (8) | 127 (14) | 180 (27) |
| 2 | 268 (21) | 71 (15) | 303 (53) |
| 3 | 268 (21) | 264 (11) | 303 (53) |
| 4 | 20 (12) | 238 (15) | 37 (58) |
| 5 | 309 (14) | 116 (22) | 185 (27) |
Average values are shown. The standard deviations are in parentheses. Table 3 gives the energetically favored duplexes.
Figure 8.
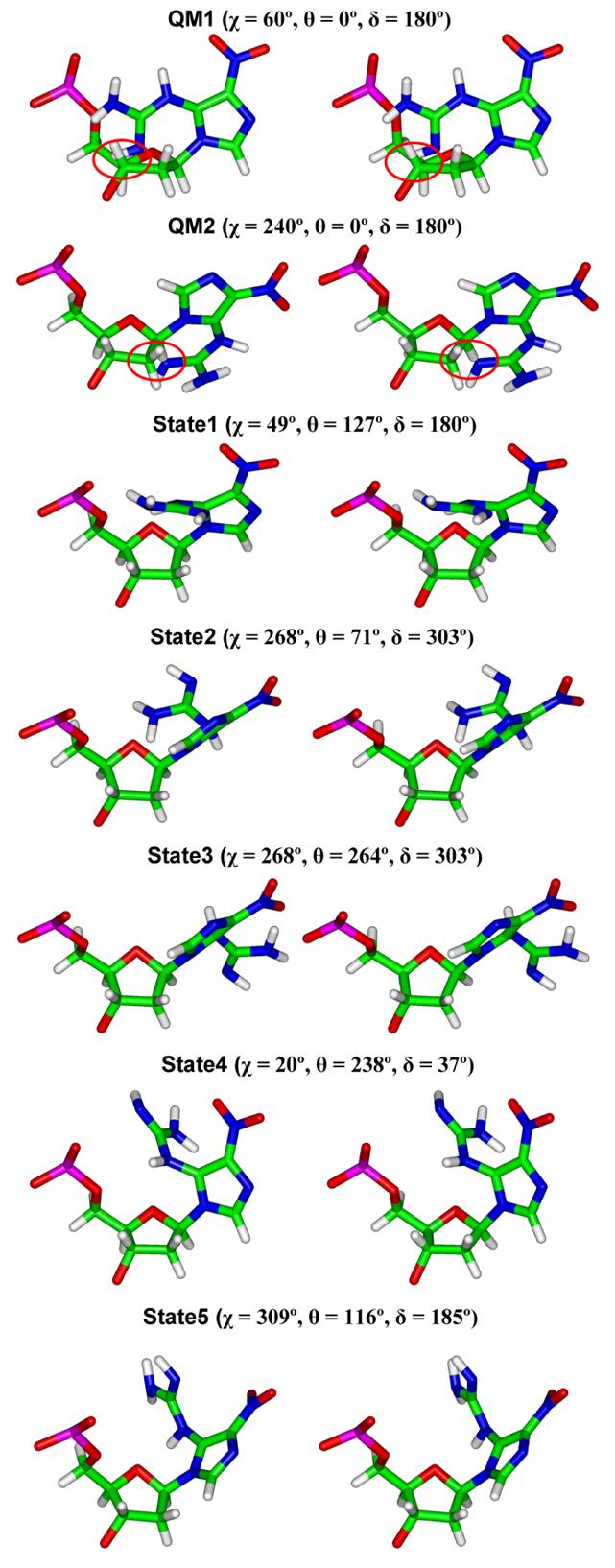
Stereo views of the NI deoxyribonucleotide in five stable states observed during the MD simulations for the DNA duplexes (Table 7). The QM calculated planar base structure with lowest energy (Table 1) is also shown connected to the sugar (C2'-endo P=162°) in syn and anti conformations (χ = 60° and 240° respectively) for comparison; the steric hindrance between NI and sugar moiety is circled.
The non-planar orientation of the guanidino group with respect to the imidazole ring is due to the steric hindrance with its own sugar, and occurs despite the conjugation of the π electrons which favor co-planarity. Such non-planarity due to steric hindrance have been observed in a crystal structure of 2-guanidinium-1,3-dimethyl-benzimidazole (86). Similarly, the guanidino group itself can be non-planar, and has also been observed, in a crystal structure containing Nω-propyl-L-arginine (PDB ID: 1QW6) (87).
Other tautomers may be possible for the NI lesion (Scheme 1). Three tautomeric alternatives involving the guanidino-group are illustrated in Scheme 1. The present study focuses on structure A, which allows maximal coplanarity of the nitroimidazole and guanidino groups in the base as shown in our quantum mechanical studies; this coplanarity promotes resonance interaction between these groups. Other tautomers may be worthy of investigation in future, particularly in relation to the conformational flexibility of the lesion.
Possible Biological Implications
NI does not have a Watson-Crick hydrogen bonding edge. Yet, data from primer extension studies in vitro using Kf (exo−) and human pol β have shown that C is preferentially incorporated opposite the NI lesion, Calf thymus pol α and Kf (exo−) also incorporate A and G.. T is not incorporated with these polymerases (34). In E. coli, both with and without SOS induction the incorporation preference is C > A ≈ T > G (29). Our simulations may provide some insight on these findings.
In the case of partner C, the NI(syn)·C(anti) pair is lower energy (Table 3). In this case, we find that duplex perturbation is the least. Specifically, from Table 6, the Watson-Crick hydrogen bonding quality of the adjacent base pairs and concomitant stacking is maintained well (Figures 4 and 5), the minor groove is not disturbed (Figure S7), and there is little bending (Figure S6), compared to the unmodified control. Also, there is one hydrogen bond in this pair (Figure 7). However, with partner T, the simulations show more disturbances to the duplexes. Both NI(syn) and NI(anti) are about equal in relative energy (Table 3). With the NI(syn)·T(anti) pair there is no hydrogen bond between the lesion and its partner (Figure 7). In the case of the NI(anti)·T(anti) pair, there is one bifurcated hydrogen bond, but the quality of Watson-Crick hydrogen bonding adjacent to the lesion at C5•G18 is the poorest of any low energy structure; the flexible guanidino group distorts this pair when the lesion is anti (Figure 5), resulting also in perturbed stacking of the C5 with T4 (Figure 4). The simulations for partners G and A also show some distortion relative to the unmodified control, but hydrogen bonding interactions between NI and G or A (Figure 5) could support observed G to C and T transversions (34), while least T incorporation may be reflected by the presence of just 1 bifurcated hydrogen bond in the two structures with T partner. However, studies with polymerases are needed for understanding of the NI accommodation within different polymerase active sites, and specific interactions within polymerase active sites could influence the planarity features of the lesion. Repair of the NI lesion may also be affected by its flexibility. Repair data is available for this lesion only for two members of the E. coli BER pathway, Fpg and Nth. However, these do not repair NI (34). There is thus insufficient data to draw conclusions concerning the NI repair susceptibility, but the flexibility and multiple hydrogen bonding opportunities are likely to be important parameters determining the repair enzymology.
Supplementary Material
Abbreviations
- 8-oxoG
8-oxo-7, 8-dihydroguanine
- Ca
cyanuric acid
- Oa
oxaluric acid
- Oz
oxazolone
- Iz
imidazolone
- NI
5-guanidino-4-nitroimidazole
- Ua
urea
- Sp
spiroiminodihydantoin
- Gh
guanidinohydantoin
- G
guanine
- A
adenine
- C
cytosine
- T
thymine
- ONOO−
peroxynitrite anion
- ·NO2
nitrogen dioxide radical
- BER
base excision repair
- Fpg
formamidopyrimidine glycosylase
- Nth
endonuclease III
- QM
quantum mechanics
- DFT
density functional theory
- MD
molecular dynamics
- RESP
restrained electrostatic potential fitting algorithm
- PME
particle mesh Ewald
- RMSD
root-mean-square deviations
- MM-PBSA
molecular mechanics Poisson-Boltzmann surface area
- SASA
solvent accessible surface area
Footnotes
This research is supported by NIH Grant CA-75449 to S. B. and NIH Grant ES-11589 to V. S. and N. E. G.
SUPPORTING INFORMATION AVAILABLE
Figure S1 shows representative starting models for the MD simulations. Figure S2 shows stereo views of geometry-optimized NI nucleosides. Figure S3 shows RMSD vs time plots of each molecular dynamics simulation. Figures S4 and S5 show stereo views of the central 5-mer of the NI trajectory-average duplex structures and hydrogen bonds between NI and partner base of high energy structures. Figures S6-S8 show the average bend angles, groove dimensions of the NI damaged DNA, and trajectory plots of torsion angles of the NI lesion, respectively. Tables S1-S5 show added force field parameters, torsion angles, simulation box sizes, and numbers of added waters of initial models, and time windows chosen for our analyses. Table S6 shows hydrogen bonds involving NI and their occupancies. Table S7 shows MM-PBSA free energy analysis components for each DNA duplex. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Inoue M, Sato EF, Nishikawa M, Park AM, Kira Y, Imada I, Utsumi K. Mitochondrial generation of reactive oxygen species and its role in aerobic life. Curr. Med. Chem. 2003;10:2495–505. doi: 10.2174/0929867033456477. [DOI] [PubMed] [Google Scholar]
- 2.Le Bras M, Clement MV, Pervaiz S, Brenner C. Reactive oxygen species and the mitochondrial signaling pathway of cell death. Histol. Histopathol. 2005;20:205–19. doi: 10.14670/HH-20.205. [DOI] [PubMed] [Google Scholar]
- 3.Cadet J, Berger M, Douki T, Ravanat JL. Oxidative damage to DNA: formation, measurement, and biological significance. Rev. Physiol. Biochem. Pharmacol. 1997;131:1–87. doi: 10.1007/3-540-61992-5_5. [DOI] [PubMed] [Google Scholar]
- 4.Dizdaroglu M. Chemical determination of free radical-induced damage to DNA. Free. Radic. Biol. Med. 1991;10:225–42. doi: 10.1016/0891-5849(91)90080-m. [DOI] [PubMed] [Google Scholar]
- 5.Epe B. DNA damage profiles induced by oxidizing agents. Rev. Physiol. Biochem. Pharmacol. 1996;127:223–49. doi: 10.1007/BFb0048268. [DOI] [PubMed] [Google Scholar]
- 6.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–15. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 7.Misiaszek R, Crean C, Geacintov NE, Shafirovich V. Combination of nitrogen dioxide radicals with 8-oxo-7,8-dihydroguanine and guanine radicals in DNA: oxidation and nitration end-products. J. Am. Chem. Soc. 2005;127:2191–200. doi: 10.1021/ja044390r. [DOI] [PubMed] [Google Scholar]
- 8.Misiaszek R, Crean C, Joffe A, Geacintov NE, Shafirovich V. Oxidative DNA damage associated with combination of guanine and superoxide radicals and repair mechanisms via radical trapping. J. Biol. Chem. 2004;279:32106–15. doi: 10.1074/jbc.M313904200. [DOI] [PubMed] [Google Scholar]
- 9.Valko M, Izakovic M, Mazur M, Rhodes CJ, Telser J. Role of oxygen radicals in DNA damage and cancer incidence. Mol. Cell. Biochem. 2004;266:37–56. doi: 10.1023/b:mcbi.0000049134.69131.89. [DOI] [PubMed] [Google Scholar]
- 10.Greenberg MM. In vitro and in vivo effects of oxidative damage to deoxyguanosine. Biochem. Soc. Trans. 2004;32:46–50. doi: 10.1042/bst0320046. [DOI] [PubMed] [Google Scholar]
- 11.Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat. Rev. Cancer. 2003;3:276–85. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]
- 12.Klaunig JE, Kamendulis LM. The role of oxidative stress in carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 2004;44:239–67. doi: 10.1146/annurev.pharmtox.44.101802.121851. [DOI] [PubMed] [Google Scholar]
- 13.Olinski R, Gackowski D, Foksinski M, Rozalski R, Roszkowski K, Jaruga P. Oxidative DNA damage: assessment of the role in carcinogenesis, atherosclerosis, and acquired immunodeficiency syndrome. Free. Radic. Biol. Med. 2002;33:192–200. doi: 10.1016/s0891-5849(02)00878-x. [DOI] [PubMed] [Google Scholar]
- 14.Weinberg RA. How cancer arises. Sci. Am. 1996;275:62–70. doi: 10.1038/scientificamerican0996-62. [DOI] [PubMed] [Google Scholar]
- 15.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–47. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 16.Mandavilli BS, Santos JH, Van Houten B. Mitochondrial DNA repair and aging. Mutat. Res. 2002;509:127–51. doi: 10.1016/s0027-5107(02)00220-8. [DOI] [PubMed] [Google Scholar]
- 17.Sastre J, Pallardo FV, Vina J. The role of mitochondrial oxidative stress in aging. Free. Radic. Biol. Med. 2003;35:1–8. doi: 10.1016/s0891-5849(03)00184-9. [DOI] [PubMed] [Google Scholar]
- 18.Hamilton ML, Van Remmen H, Drake JA, Yang H, Guo ZM, Kewitt K, Walter CA, Richardson A. Does oxidative damage to DNA increase with age? Proc. Natl. Acad. Sci. U. S. A. 2001;98:10469–74. doi: 10.1073/pnas.171202698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Osterod M, Hollenbach S, Hengstler JG, Barnes DE, Lindahl T, Epe B. Age-related and tissue-specific accumulation of oxidative DNA base damage in 7,8-dihydro-8-oxoguanine-DNA glycosylase (Ogg1) deficient mice. Carcinogenesis. 2001;22:1459–63. doi: 10.1093/carcin/22.9.1459. [DOI] [PubMed] [Google Scholar]
- 20.Aslan M, Ozben T. Reactive oxygen and nitrogen species in Alzheimer's disease. Curr. Alzheimer Res. 2004;1:111–9. doi: 10.2174/1567205043332162. [DOI] [PubMed] [Google Scholar]
- 21.Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. Faseb J. 2003;17:1195–214. doi: 10.1096/fj.02-0752rev. [DOI] [PubMed] [Google Scholar]
- 22.Steenken S, Jovanovic SV. How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution. J. Am. Chem. Soc. 1997;119:617–618. [Google Scholar]
- 23.Brajter-Toth A, Goyal RN, Wrona MZ, Lacava T, Nguyen NT, Dryhurst G. Electrochemical and enzymic oxidation of biological purines. Bioelectrochemistry and Bioenergetics. 1981;8:413–435. [Google Scholar]
- 24.Demple B, Harrison L. Repair of oxidative damage to DNA: enzymology and biology. Annu. Rev. Biochem. 1994;63:915–48. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- 25.Evans MD, Dizdaroglu M, Cooke MS. Oxidative DNA damage and disease: induction, repair and significance. Mutat. Res. 2004;567:1–61. doi: 10.1016/j.mrrev.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 26.Henderson PT, Delaney JC, Gu F, Tannenbaum SR, Essigmann JM. Oxidation of 7,8-dihydro-8-oxoguanine affords lesions that are potent sources of replication errors in vivo. Biochemistry. 2002;41:914–21. doi: 10.1021/bi0156355. [DOI] [PubMed] [Google Scholar]
- 27.Henderson PT, Neeley WL, Delaney JC, Gu F, Niles JC, Hah SS, Tannenbaum SR, Essigmann JM. Urea Lesion Formation in DNA as a Consequence of 7,8-Dihydro-8-oxoguanine Oxidation and Hydrolysis Provides a Potent Source of Point Mutations. Chem. Res. Toxicol. 2005;18:12–18. doi: 10.1021/tx049757k. [DOI] [PubMed] [Google Scholar]
- 28.Luo W, Muller JG, Rachlin EM, Burrows CJ. Characterization of hydantoin products from one-electron oxidation of 8-oxo-7,8-dihydroguanosine in a nucleoside model. Chem. Res. Toxicol. 2001;14:927–38. doi: 10.1021/tx010072j. [DOI] [PubMed] [Google Scholar]
- 29.Neeley WL, Delaney JC, Henderson PT, Essigmann JM. In vivo bypass efficiencies and mutational signatures of the guanine oxidation products 2-aminoimidazolone and 5-guanidino-4-nitroimidazole. J. Biol. Chem. 2004;279:43568–73. doi: 10.1074/jbc.M407117200. [DOI] [PubMed] [Google Scholar]
- 30.Niles JC, Wishnok JS, Tannenbaum SR. A novel nitroimidazole compound formed during the reaction of peroxynitrite with 2',3',5'-tri-O-acetyl-guanosine. J. Am. Chem. Soc. 2001;123:12147–51. doi: 10.1021/ja004296k. [DOI] [PubMed] [Google Scholar]
- 31.Niles JC, Wishnok JS, Tannenbaum SR. Spiroiminodihydantoin is the major product of the 8-oxo-7,8-dihydroguanosine reaction with peroxynitrite in the presence of thiols and guanosine photooxidation by methylene blue. Org. Lett. 2001;3:963–6. [PubMed] [Google Scholar]
- 32.Tretyakova NY, Niles JC, Burney S, Wishnok JS, Tannenbaum SR. Peroxynitrite-induced reactions of synthetic oligonucleotides containing 8-oxoguanine. Chem. Res. Toxicol. 1999;12:459–66. doi: 10.1021/tx980235c. [DOI] [PubMed] [Google Scholar]
- 33.Joffe A, Mock S, Yun BH, Kolbanovskiy A, Geacintov NE, Shafirovich V. Oxidative generation of guanine radicals by carbonate radicals and their reactions with nitrogen dioxide to form site specific 5-guanidino-4-nitroimidazole lesions in oligodeoxynucleotides. Chem. Res. Toxicol. 2003;16:966–73. doi: 10.1021/tx025578w. [DOI] [PubMed] [Google Scholar]
- 34.Gu F, Stillwell WG, Wishnok JS, Shallop AJ, Jones RA, Tannenbaum SR. Peroxynitrite-induced reactions of synthetic oligo 2'-deoxynucleotides and DNA containing guanine: formation and stability of a 5-guanidino-4-nitroimidazole lesion. Biochemistry. 2002;41:7508–18. doi: 10.1021/bi020148q. [DOI] [PubMed] [Google Scholar]
- 35.David SS, Williams SD. Chemistry of Glycosylases and Endonucleases Involved in Base-Excision Repair. Chem. Rev. 1998;98:1221–1262. doi: 10.1021/cr980321h. [DOI] [PubMed] [Google Scholar]
- 36.Michaels ML, Cruz C, Grollman AP, Miller JH. Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA. Proc. Natl. Acad. Sci. U. S. A. 1992;89:7022–5. doi: 10.1073/pnas.89.15.7022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michaels ML, Tchou J, Grollman AP, Miller JH. A repair system for 8-oxo-7,8-dihydrodeoxyguanine. Biochemistry. 1992;31:10964–8. doi: 10.1021/bi00160a004. [DOI] [PubMed] [Google Scholar]
- 38.Neeley WL, Henderson PT, Essigmann JM. Efficient synthesis of DNA containing the guanine oxidation-nitration product 5-guanidino-4-nitroimidazole: generation by a postsynthetic substitution reaction. Org. Lett. 2004;6:245–8. doi: 10.1021/ol036188j. [DOI] [PubMed] [Google Scholar]
- 39.Halgren TA. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996;17:490–519. [Google Scholar]
- 40.Becke AD. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 41.Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 42.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JJA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03. Gaussian, Inc.; Wallingford CT: 2004. Revision C.02. [Google Scholar]
- 43.Altona C, Sundaralingam M. Conformational analysis of the sugar ring in nucleosides and nucleotides. A new description using the concept of pseudorotation. J. Am. Chem. Soc. 1972;94:8205–12. doi: 10.1021/ja00778a043. [DOI] [PubMed] [Google Scholar]
- 44.Hocquet A, Leulliot N, Ghomi M. Ground-state properties of nucleic acid constituents studied by density functional calculations. 3. Role of sugar puckering and base orientation on the energetics and geometry of 2 '-deoxyribonucleosides and ribonucleosides. J. Phys. Chem. B. 2000;104:4560–4568. [Google Scholar]
- 45.Mishra SK, Mishra PC. An ab initio theoretical study of electronic structure and properties of 2'-deoxyguanosine in gas phase and aqueous media. J. Comput. Chem. 2002;23:530–40. doi: 10.1002/jcc.10046. [DOI] [PubMed] [Google Scholar]
- 46.Berman HM, Olson WK, Beveridge DL, Westbrook J, Gelbin A, Demeny T, Hsieh SH, Srinivasan AR, Schneider B. The nucleic acid database. A comprehensive relational database of three-dimensional structures of nucleic acids. Biophys. J. 1992;63:751–9. doi: 10.1016/S0006-3495(92)81649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haschemeyer AE, Sobell HM. The Crystal Structure of a Hydrogen Bonded Complex of Adenosine and 5-Bromouridine. Acta. Crystallogr. 1965;18:525–32. doi: 10.1107/s0365110x65001123. [DOI] [PubMed] [Google Scholar]
- 48.Haschemeyer AE, Sobell HM. The crystal structure of a hydrogen bonded complex of deoxyguanosine and 5-bromodeoxycytidine. Acta. Crystallogr. 1965;19:125–30. doi: 10.1107/s0365110x65002864. [DOI] [PubMed] [Google Scholar]
- 49.Hingerty BE, Figueroa S, Hayden TL, Broyde S. Prediction of DNA-Structure from Sequence - a Buildup Technique. Biopolymers. 1989;28:1195–1222. doi: 10.1002/bip.360280703. [DOI] [PubMed] [Google Scholar]
- 50.Jia L, Shafirovich V, Shapiro R, Geacintov NE, Broyde S. Structural and thermodynamic features of spiroiminodihydantoin damaged DNA duplexes. Biochemistry. 2005;44:13342–53. doi: 10.1021/bi050790v. [DOI] [PubMed] [Google Scholar]
- 51.Brown T, Hunter WN, Kneale G, Kennard O. Molecular structure of the G.A base pair in DNA and its implications for the mechanism of transversion mutations. Proc. Natl. Acad. Sci. U. S. A. 1986;83:2402–6. doi: 10.1073/pnas.83.8.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lane AN, Peck B. Conformational flexibility in DNA duplexes containing single G.G mismatches. Eur. J. Biochem. 1995;230:1073–87. doi: 10.1111/j.1432-1033.1995.tb20658.x. [DOI] [PubMed] [Google Scholar]
- 53.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Wang B, Pearlman DA, Crowley M, Brozell S, Tsui V, Gohlke H, Mongan J, Hornak V, Cui G, Beroza P, Schafmeister C, Caldwell JW, Ross WS, Kollman PA. AMBER. Vol. 8. University of California; San Francisco: 2004. [Google Scholar]
- 54.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 55.Wang JM, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000;21:1049–1074. [Google Scholar]
- 56.Cieplak P, Cornell WD, Bayly C, Kollman PA. Application of the Multimolecule and Multiconformational Resp Methodology to Biopolymers - Charge Derivation for DNA, Rna, and Proteins. J. Comput. Chem. 1995;16:1357–1377. [Google Scholar]
- 57.Hehre WJ, Ditchfie R, Pople JA. Self-Consistent Molecular-Orbital Methods. 12. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular-Orbital Studies of Organic-Molecules. J. Chem. Phys. 1972;56:2257–2261. [Google Scholar]
- 58.Bayly CI, Cieplak P, Cornell W, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 1993;97:10269–10280. [Google Scholar]
- 59.Wang JM, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 60.Berendsen HJC, Postma JPM, Vangunsteren WF, Dinola A, Haak JR. Molecular-Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 61.Darden T, York D, Pedersen L. Particle Mesh Ewald - an N.Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 62.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 63.Harvey SC, Tan RKZ, Cheatham TE. The flying ice cube: Velocity rescaling in molecular dynamics leads to violation of energy equipartition. J. Comput. Chem. 1998;19:726–740. [Google Scholar]
- 64.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 65.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical-Integration of Cartesian Equations of Motion of a System with Constraints - Molecular-Dynamics of N-Alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 66.Yan SX, Shapiro R, Geacintov NE, Broyde S. Stereochemical, structural, and thermodynamic origins of stability differences between stereoisomeric benzo[a]pyrene diol epoxide deoxyadenosine adducts in a DNA mutational hot spot sequence. J. Am. Chem. Soc. 2001;123:7054–7066. doi: 10.1021/ja0043035. [DOI] [PubMed] [Google Scholar]
- 67.Case DA, Pearlman DA, Caldwell JW, Cheatham TE, III, Wang J, Ross WS, Simmerling CL, Darden TA, Merz KM, Stanton RV, Cheng AL, Vincent JJ, Crowley M, Tsui V, Gohlke H, Radmer RJ, Duan Y, Pitera J, Massova I, Seibel GL, Singh UC, Weiner PK, Kollman PA. AMBER. Vol. 7. University of California; San Francisco: 2002. [Google Scholar]
- 68.Saenger W. Principles of nucleic acid structure. Springer-Verlag; New York: 1984. [Google Scholar]
- 69.Ravishanker G, Swaminathan S, Beveridge DL, Lavery R, Sklenar H. Conformational and helicoidal analysis of 30 PS of molecular dynamics on the d(CGCGAATTCGCG) double helix: “curves”, dials and windows. J. Biomol. Struct. Dyn. 1989;6:669–99. doi: 10.1080/07391102.1989.10507729. [DOI] [PubMed] [Google Scholar]
- 70.Ravishanker G, Wang W, Beveridge DL. Molecular Dynamics Analysis Toolchest. Wesleyan University; Middletown, CT: [Google Scholar]
- 71.Lavery R, Sklenar H. The Definition of Generalized Helicoidal Parameters and of Axis Curvature for Irregular Nucleic-Acids. J. Biomol. Struct. Dyn. 1988;6:63–91. doi: 10.1080/07391102.1988.10506483. [DOI] [PubMed] [Google Scholar]
- 72.Huo S, Massova I, Kollman PA. Computational alanine scanning of the 1:1 human growth hormone-receptor complex. J. Comput. Chem. 2002;23:15–27. doi: 10.1002/jcc.1153. [DOI] [PubMed] [Google Scholar]
- 73.Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE., 3rd Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 2000;33:889–97. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 74.Lee MR, Duan Y, Kollman PA. Use of MM-PB/SA in estimating the free energies of proteins: application to native, intermediates, and unfolded villin headpiece. Proteins. 2000;39:309–16. [PubMed] [Google Scholar]
- 75.Reyes CM, Kollman PA. Investigating the binding specificity of U1A-RNA by computational mutagenesis. J. Mol. Biol. 2000;295:1–6. doi: 10.1006/jmbi.1999.3319. [DOI] [PubMed] [Google Scholar]
- 76.Wang J, Morin P, Wang W, Kollman PA. Use of MM-PBSA in reproducing the binding free energies to HIV-1 RT of TIBO derivatives and predicting the binding mode to HIV-1 RT of efavirenz by docking and MM-PBSA. J. Am. Chem. Soc. 2001;123:5221–30. doi: 10.1021/ja003834q. [DOI] [PubMed] [Google Scholar]
- 77.Wang W, Kollman PA. Free energy calculations on dimer stability of the HIV protease using molecular dynamics and a continuum solvent model. J. Mol. Biol. 2000;303:567–82. doi: 10.1006/jmbi.2000.4057. [DOI] [PubMed] [Google Scholar]
- 78.Jayaram B, McConnell KJ, Dixit SB, Beveridge DL. Free energy analysis of protein-DNA binding: The EcoRI endonuclease-DNA complex. J. Comput. Phys. 1999;151:333–357. [Google Scholar]
- 79.Yan S, Wu M, Buterin T, Naegeli H, Geacintov NE, Broyde S. Role of base sequence context in conformational equilibria and nucleotide excision repair of benzo[a]pyrene diol epoxide-adenine adducts. Biochemistry. 2003;42:2339–54. doi: 10.1021/bi0270081. [DOI] [PubMed] [Google Scholar]
- 80.Honig B, Nicholls A. Classical Electrostatics in Biology and Chemistry. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 81.Connolly ML. Analytical Molecular-Surface Calculation. J. Appl. Crystallogr. 1983;16:548–558. [Google Scholar]
- 82.Sitkoff D, Sharp KA, Honig B. Accurate Calculation of Hydration Free-Energies Using Macroscopic Solvent Models. J. Phys. Chem. 1994;98:1978–1988. [Google Scholar]
- 83.Sanner MF, Olson AJ, Spehner JC. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers. 1996;38:305–20. doi: 10.1002/(SICI)1097-0282(199603)38:3%3C305::AID-BIP4%3E3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 84.Srinivasan J, Cheatham TE, Cieplak P, Kollman PA, Case DA. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate - DNA helices. J. Am. Chem. Soc. 1998;120:9401–9409. [Google Scholar]
- 85.Yan S, Wu M, Patel DJ, Geacintov NE, Broyde S. Simulating structural and thermodynamic properties of carcinogen-damaged DNA. Biophys. J. 2003;84:2137–48. doi: 10.1016/S0006-3495(03)75020-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.AndradeLopez N, ArizaCastolo A, Contreras R, VazquezOlmos A, Behrens NB, Tlahuext H. Versatile behavior of 2-guanidinobenzimidazole nitrogen atoms toward protonation, coordination and methylation. Heteroat. Chem. 1997;8:397–410. [Google Scholar]
- 87.Fedorov R, Hartmann E, Ghosh DK, Schlichting I. Structural basis for the specificity of the nitric-oxide synthase inhibitors W1400 and Nomega-propyl-L-Arg for the inducible and neuronal isoforms. J. Biol. Chem. 2003;278:45818–25. doi: 10.1074/jbc.M306030200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.