

Abstract
Understanding the mechanism of protein-DNA interactions at the molecular level is one of the main focuses in structural and molecular biological investigations. Currently, NMR spectroscopy is the only approach that can provide atomic details of protein-DNA recognition in solution. However, solving the structures of protein-DNA complexes using NMR spectroscopy has been dependent on the observation of intermolecular nuclear Overhauser effects (NOE) and their assignments, which are difficult to obtain in many cases. In this study, we have shown that intermolecular distance constraints derived from a single spin-label in combination with docking calculations have defined many specific contacts of the complex between the AT-rich interaction domain (ARID) of Mrf2 and its target DNA. Mrf2 contacts DNA mainly using the two flexible loops, L1 and L2. While the L1 loop contacts the phosphate backbone, L2 and several residues in the adjacent helices interact with AT base pairs in the major groove of DNA. Despite the structural diversity in the ARID family of DNA-binding proteins, Mrf2 maintains similar contacts with DNA as those observed in the homologous Dri-DNA complex.
Understanding the mechanism of protein-DNA interactions at the molecular level is one of the main focuses in structural and molecular biological investigations. Currently, NMR spectroscopy is one of the two main approaches to provide atomic details of protein-DNA recognition. Solving the structures of protein-DNA complexes using NMR spectroscopy is usually a labor-intensive process, involving the complete resonance assignments of the proteins and DNA molecules and the assignments of intra and inter-molecular nuclear Overhauser effects (NOE). In many cases, costly heteronuclear enriched DNA samples are needed in order to resolve ambiguities in the assignments.
Paramagnetic spin labeling has a long history in NMR structural studies (1–4). Dipolar interactions between unpaired electrons and the nearby nuclei can either induce NMR relaxation effects or chemical shift changes, depending on the relaxation property and the paramagnetic tensor of a spin-label. Nitroxide free radical has been frequently used to provide structural information, because it induces strong relaxation effects on the nearby nuclei that are strictly distance-dependent (1). Therefore, it is straightforward to generate distance restraints from the paramagnetic relaxation effect. However, structure determination based solely on these loose distance constraints has been nearly impossible. Recently, significant progress has been made in docking calculations of macromolecular complexes using ambiguously assigned distance restraints derived from NMR chemical shift perturbation (5–7). In this study, we have explored the use of paramagnetic relaxation effect in combination with docking calculations based on NMR chemical shift perturbation for solving the structure of the AT-rich interaction domain (ARID) of the protein Mrf2 in complex with its target DNA sequence.
ARID is a family of homologous DNA-binding domains, and is named as such because the initial ARID proteins to be characterized interacted with specific AT-rich sequences (8). ARID domains occur in a wide variety of species ranging from yeast to nematodes, insects, mammals and one species of plant, and have diverse cellular functions. The ARID domains have conserved central regions, but the N- and C-termini are not conserved. The structures of three ARID domains from the proteins Mrf2, Dri, and SWI1 have been solved (9–11). These DNA-binding domains form a structurally similar central region that contains six α-helices, known as H1 to H6, and two long loops, termed L1 and L2 (11). The diverse sequences in the N- and C-termini of these domains correlate to their structural differences in these regions. The Dri ARID contains two extra helices, located at each of the N- and C-termini. However, both helices do not exist in the Mrf2 ARID. The SWI1 ARID contains a short 310 helix at the N-terminus, but lacks the C-terminal helix. Preliminary NMR studies have shown that the structurally conserved central regions of ARIDs interact with DNA using homologous segments of the proteins, suggesting that the conserved central regions bind DNA using a similar mechanism (9, 11, 12). However, differences exist in how the structurally different termini contact DNA. To date, only the high-resolution structure of the Dri-DNA complex has been determined using the well-established approach with NOEs and coupling constants. Consequently, it is not clear on how the structural differences in the ARID family influences their DNA binding mechanisms.
In this study, we used distance constraints derived from paramagnetic line-broadening effects in combination with docking calculations based on NMR chemical shift perturbation to determine the structure of the Mrf2 ARID in complex with its target DNA sequence. Well-defined structure of the protein-DNA complex has been obtained. This approach is efficient, and the assignments of intermolecular distance constraints are unambiguous without the need of any expensive isotope-labeling scheme. Well-converged structures of the protein-DNA complex obtained provide information on how the Mrf2 ARID recognizes DNA and on how the structural diversity of the ARID family affects their DNA-binding activities.
Materials and Methods
Sample Preparation
The 15N-labeled Mrf2 ARID domain was expressed and purified as described previously (9, 13). Mrf2 binds to a specific AT-rich 14 basepair oligo with the following sequence
5′-ACAATATAACGTCG -3′
3′-TGTTATATTGCAGC -5′
In order to incorporate paramagnetic spin-labels to derive distance constraints between the protein and DNA, deoxy-4-thiouracil was incorporated into each end of the oligonucleotide through standard solid phase synthesis. Such oligonucleotides are shown as DNA-1 and DNA-2 in Figure 1. Deoxy-4-thiouracil reacts with 3-(2-Iodoacetamido)-proxyl (14) and thus allows the covalent attachment of the spin-label to one end of the DNA molecule (Fig. 1B). A third shortened double-stranded DNA (DNA-3, Fig. 1A) was also made where the 3′-end of the upper strand is a dT-phosphothioate (Fig. 1B). It was made by a thioation step following standard DNA synthesis at the DNA/RNA Synthesis Core Facility at the City of Hope. Phosphothioate can also form a covalent bond with 3-(2-Iodoacetamido)-proxyl. Each of the DNA samples was labeled with proxyl, prior to complexing with Mrf2. The spin-label was reduced by directly adding excess sodium hydrosulfite to the complex.
Figure 1. Summary of paramagnetic labeling strategies used in this study.

(a) The sequences of three DNA molecules that were synthesized and used in this study. “u” denotes deoxy-4-thiouracil and the red “T” denotes the 3′-end of phosphothioate. (b) The spin-labeling strategies using both modified base and phosphothiate are employed as depicted here. Both deoxy-4-thiouracil and the 3′-end of phosphothioate reacts with 3-(2-Iodoacetamido)-proxyl and thus allowed the covalent attachment of the spin-label to one end of a DNA molecule.
Distance Restraint Determination and Structural Calculations
The spin labeling approach is based on the fact that the unpaired electron of the spin label can induce line broadening effects of nuclei within 25 Å. The magnitude of the line broadening effects depends on the distances between the spin label and the nuclear spins. In this study, 1H-15N HSQC spectra were acquired before and after proxyl was reduced. Intermolecular distance constraints (between the spin label on DNA and amide protons of the protein) were calculated from the paramagnetic line broadening effects by comparing peak heights in the 1H-15N HSQC spectra before and after the reduction of the spin label (1). Specifically, proton paramagnetic relaxation rate enhancement, denoted as R2SP in Equation 1, is the difference in proton transverse relaxation rates between the oxidized and reduced spectra. This can be solved from the equation
| (1) |
where Iox and Ired are the peak intensities (heights) of oxidized and reduced resonances, respectively, t is the total INEPT evolution time of HSQC (~10 ms), R2 is the transverse relaxation rate of amide protons in the reduced spectrum that was estimated from the line-widths of cross peaks in the proton dimension of the reduced spectrum in this study. Then the R2SP values were converted to distance constraints using (15, 16)
| (2) |
where r is the distance between the electron and nuclear spins (protons), S is the electron spin number (S=1/2 for the proxyl), γ is the nuclear gyromagnetic ratio, g is the electronic g factor, β is the Bohr magneton, τc is the protein rotational correlation time, which has been determined in our previous study (13), ωh is the Larmor frequency of proton.
Residues that are affected by the spin-label so severely that their cross peaks completely disappeared in the spectrum before spin-label reduction are assumed to be within 12 Å of the paramagnetic spin label. For those residues that are broadened by the spin-label with the intensity ratio 0< Iox/Ired < 0.6, intermolecular distance constraints were calculated using Equations (1) and (2). To avoid the extra steps required to create the coordinates and potential function terms associated with the proxyl group, the distance constraints used in structural calculation are between the sulfur atom of thiouracyl and protons of the protein. Then, in order to compensate for the difference in the distances between the sulfur group and the unpaired electron on proxyl, an additional 8 Å was added to all distance constraints. For residues that are too broad to be detectable in the spectra with oxidized spin-label, lower limits were not used, and 4 Å was added as the upper limit of the distance constraints. The residues that are broadened (0< Iox/Ired < 0.6) but detectable in the oxidized spectrum are constrained with ±4 Å bounds. We decided to only use information derived from peaks with 0< Iox/Ired < 0.6 because of their clear paramagnetic relaxation effects. Excluding the overlapped peaks, a total of 13 distance constraints (Table 1) were used in the structural calculation.
Table 1.
Unambiguous distance constraints calculated from paramagnetic line broadening effects and used in the structural determination
| Atom | Iox/Ired | R2SP (Hz) | ra (Å) |
|---|---|---|---|
| E35 (NH) | N/Ab | N/Ab | 20 + 4Å |
| I37 (NH) | N/Ab | N/Ab | 20 + 4Å |
| Y24 (NH) | 0.209 | 80.8 | 22.4 ± 4Å |
| Q44 (HE21) | 0.330 | 54.9 | 24.7 ± 4Å |
| Q44 (HE22) | 0.330 | 54.9 | 24.7 ± 4Å |
| N46 (HD21) | 0.330 | 54.9 | 24.7 ± 4Å |
| N46 (HD22) | 0.330 | 54.9 | 24.7 ± 4Å |
| A22 (NH) | 0.376 | 45.6 | 24.3 ± 4Å |
| A86 (NH) | 0.504 | 30.4 | 25.4 ± 4Å |
| W48 (NH) | 0.533 | 31.5 | 25.3 ± 4Å |
| W69 (HE1) | 0.541 | 25.3 | 26.0 ± 4Å |
| A85 (NH) | 0.574 | 26.2 | 25.9 ± 4Å |
| T89 (NH) | 0.593 | 27.6 | 25.7 ± 4Å |
Notes: Distance constraints between the spin label and the amide or sidechain protons of individual residues.
The peaks in the oxidized spectrum are too broad to be observed. In these cases, the distance constraints were set with upper bounds 20 Å plus 4 Å and no lower bound was used. Details are described in the Materials and Methods section.
The structure of the protein-DNA complex was calculated using the program HADDOCK (5) with the intermolecular distance constraints, dihedral angle constraints on Mrf2, and chemical shift perturbation constraints. The dihedral angle constraints were derived from Cα chemical shifts of Mrf2 in the complex with DNA using the program TALOS (17). The NMR structure of free Mrf2 (13) and a B-form double-stranded DNA structure, manually built, were used as the starting structures. The two loops of Mrf2 at the binding interface (L1 and L2), the C-terminus of Mrf2 and the spin-labeled thiouracil of DNA were defined as “fully flexible”, because these regions have high conformational flexibility in the free form (13). As such, both backbones and side chains of these segments are allowed to move in the calculation. Other residues of Mrf2 that showed large chemical shift perturbation but were located in helical regions, were defined as “semi-flexible”. Only side chains of these residues were allowed to move in the structural calculation. All other residues on Mrf2 and DNA were fixed in the calculation. To ensure that Mrf2 does not change its overall structure, several artificial distance restraints between helices were introduced. A total of 1000 structures were initially generated. The top 300 structures were subjected to simulated annealing calculations, and the best 17 structures were analyzed and shown here.
Results and Discussion
The need of a spin-label
As indicated by localized chemical shift perturbations induced by DNA binding, Mrf2, like many DNA-binding proteins, does not undergo an overall conformational change upon forming the complex with DNA (13). NMR data of the free DNA molecule indicates that the Mrf2 target DNA sequence has a standard B-form conformation (13). A circular permutation assay (18) indicated that binding of Mrf2 to DNA did not induce severe DNA bending (data not shown), which suggests a lack of a major conformational change in DNA upon the complex formation. Taken together, these data demonstrate that both the Mrf2 ARID and the DNA molecule do not undergo global changes of their structures upon the protein-DNA complex formation.
We investigated whether calculation using ambiguous distance restraints derived from NMR chemical shift changes can result in converged structures of the complex, as reported for some protein-protein complexes (5). Since neither Mrf2 nor the DNA undergo major conformational changes, the high-resolution solution structure of Mrf2 and its target DNA sequence (built as a standard B-form) were used for the calculations utilizing ambiguous constraints derived from NMR chemical shift perturbation data. The residues in Mrf2 that showed chemical shift changes upon the complex formation were defined as the “active residues” in HADDOCK calculations. All base pairs in the DNA were defined as “active residues”, because it is not known which specific base pairs contact the protein. The initial calculation of the protein-DNA complex using HADDOCK did not yield converged structures. Particularly, in some of these structures the protein reverses its bound orientation on DNA, likely due to the two-fold symmetry of the DNA structure.
In order to determine the bound orientation of the protein relative to the DNA, we introduced a spin-label at the end of the DNA molecule as described in the Materials and Methods section, as illustrated in Fig. 1. The unpaired electron of proxyl can induce line-broadening effects of the resonances of residues within 25 Å of the spin-label. The magnitude of the line broadening effect depends on the distance between the spin label and the nuclear spins. In this study, 1H-15N HSQC spectra were acquired before and after the proxyl group was reduced. Spin labeling did not change the protein-DNA interaction, because the 1H-15N HSQC spectrum of Mrf2 in complex with the spin-labeled DNA after the reduction of the proxyl group is very similar to that of Mrf2 in complex with its target DNA (Fig. 2, and (13)).
Figure 2. Paramagnetic line-broadening effects used in the structural calculation.
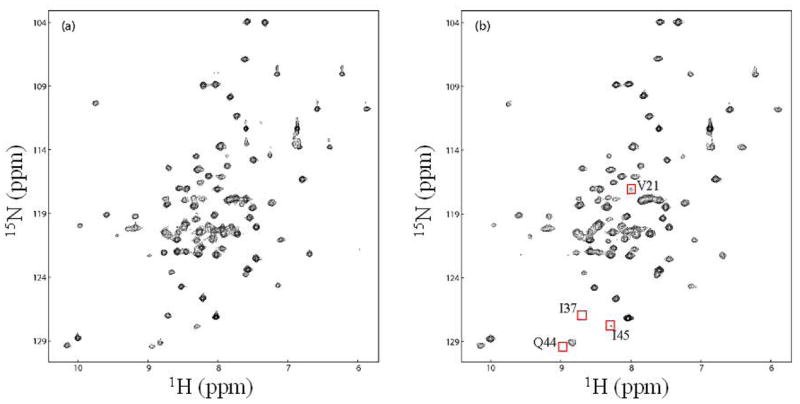
1H-15N HSQC spectra of Mrf2 in complex with a proxyl-labeled DNA (DNA-1 in Figure 1) (a) after and (b) before reduction of the proxyl group. A few severely broadened residues close to the spin label were very weak or not detectable in (b). These residues are marked with a red square in (b), and their assignments are indicated.
Dramatic NMR line broadening effects in Mrf2 were observed in the complex with the spin-labeled DNA-1 molecule (Fig. 1). Specifically, the amide resonances of residues 35 and 37 are completely “bleached out” by the spin-label, indicating that they are very close to the labeled end of the DNA (Fig. 2). This data immediately provided information on the relative orientation of the protein on the DNA. In addition, the cross-peaks of the backbone amide groups of several other residues, as well as the sidechain NH2 groups of an Asn and a Gln, and the sidechain NH groups of two Trp residues in the protein, were significantly broadened (Table 1). These data provided inter-molecular distance constraints for structure calculation. In order to generate additional distance constraints for structure determination, the proxyl spin-label was introduced to the other end of the target sequence in DNA-2 and DNA-3 molecules (Fig. 1A). However, the spin-label introduced to DNA-2 did not produce dramatic line-broadening effects. In order to move the spin-label on this end of the DNA closer to the protein, two basepairs were removed from this end in the DNA-3 molecule. Unfortunately, DNA-3 has significantly altered ability to interact with the protein, as indicated by overall line-broadening effects in the HSQC spectra that are independent of whether the spin-label is reduced or oxidized (data not shown). This effect may due to aggregation of the sample, since complexes with shorter DNA molecules have lower solubility. The effect may also be due to reduced affinity that results in intermediate exchange rate of the free and bound forms of the protein. Therefore, only the data from the spin-labeled DNA-1 was used for structure calculation of the protein-DNA complex.
Structure calculation of the protein-DNA complex
The intermolecular distance constraints obtained from spin-labels were incorporated into structure calculation using the program HADDOCK (5). The “active” and “passive” residues in the protein were defined based on chemical shift perturbation for HADDOCK calculation (Table 2). Since it is not clear which residues in the DNA were involved in the interaction, all bases of the DNA-1 molecule were defined as active residues (Table 2). The calculation led to a well-converged family of structures (Fig. 3A) with favorable covalent geometries and minimal bond length and angle violations (Table 3). In addition, the structures have favorable Ramachandran statistics. Despite the loose constraints, the structures are well converged with a root-mean-square-deviation (RMSD) of 1.31 Å among backbone atoms of both the protein and DNA (Table 3). The structure is consistent with all available experimental data. There is no distance constraint violated by more than 0.5 Å. In addition, the structures explain the lack of line broadening effects when Mrf2 forms a complex with the spin-labeled DNA-2 molecules; the closest residue of Mrf2 to the spin-labeled end of DNA-2 is more than 22 Å away, so the spin-label would not have a dramatic line-broadening effect to induce the disappearance of any cross peaks.
Table 2.
Lists of “active” and “passive” residues used in the definition of the ambiguous interaction restraints (AIRs) in HADDOCK calculations
| Mrf2 | Active residues | 38, 39, 41–44, 70, 71, 77–84, 87–90, 116–119 |
| Passive residues | 32, 35, 36, 46, 60, 64, 66–68, 73, 76, 91, 92, 94, 115 | |
| Flexible segments | 38–48, 77–84, 109–119 | |
| DNA | Active residues | All 28 bases |
| Passive residues | None | |
| Flexible segments | Thy1 (the residue with the spin label) |
Figure 3. The calculated structures of the Mrf2-DNA complex.

(a) Superposition of 20 structures of the Mrf2-DNA complex. (b) Side-by-side comparison of the structures of the Mrf2-DNA complex (left) and Dri-DNA complex (right). All helices, loops, N- and C-termini are indicated in the structures.
Table 3.
Structural statistics of 17 final structures
| Dihedral angle restraints derived from chemical shift | |
|
| |
| All | 150 |
| Φ | 75 |
| Ψ | 75 |
|
| |
| RMSD from the average structure | Å |
| Backbonea excluding the C-terminus | 1.31±0.83 |
| All heavy atoms for residues at the protein-DNA interface | 1.29±0.78 |
|
| |
| Distance violations | |
| Unambigious distance restraints (>0.5 Å)b | 0 |
|
| |
| RMSD from idealized covalent geometry | |
| Bonds (Å) | 0.009±0.00004 |
| Angles (°) | 1.12±0.005 |
| Impropers (°) | 1.84±0.01 |
|
| |
| Ramachandran analysis (excluding the C-terminus) | |
| Residues in most favored regions (%) | 83.0 |
| Residues in additional allowed regions (%) | 16.5 |
| Residues in generously allowed regions (%) | 0.5 |
| Residues in disallowed allowed regions (%) | 0.0 |
Notes: The protein backbone atoms include C, Cα, N. The DNA backbone atoms include P, C3′, C4′, C5′, O3′ and O5′.
Violations of the AIR restraints were not analyzed, because they are based on loosely defined “active” and “passive” residues. Violations of the dihedral angle constraints generated by TOLAS were not analyzed either, as the upper and lower bounds were artificially defined by model molecules.
The structure of the Mrf2 ARID in complex with DNA is compared with that of the homologous Dri ARID in complex with DNA (12). The Mrf2-DNA and Dri-DNA complexes have major structural similarities (Fig. 3B). In particular, the conserved central regions of Mrf2 and Dri ARIDs have similar DNA binding modes. Loop L2 and the adjacent helix H5 insert into the major groove of DNA, whereas loop L1 makes contact with the phosphate backbone. The similarity is quite remarkable, given that the Dri-DNA complex was determined using thousands of NOE constraints, while the Mrf2-DNA complex was determined using chemical shift perturbation constraints and 13 intermolecular distance constraints. The two flexible loops of the protein rigidified upon binding DNA (13). The “folding upon binding” phenomenon has been proposed to be generally important for specificity in protein-DNA recognition (19), since non-specific interactions are not likely to induce a specific structure formation.
The main difference in DNA binding modes between Mrf2 and Dri are in the structurally different C-termini. The C-terminal segment of the Dri ARID is 4 residues longer than that of Mrf2, forms a helix that is absent in Mrf2, and interacts with the minor groove of DNA (12). In contrast, the Mrf2 C-terminus is disordered and appears to form transient non-specific contacts with DNA. The ensemble of structures indicates that the C-terminus of Mrf2 can bind to a position on the DNA analogous to that bound by the Dri ARID C-terminus. The C-terminus of the SWI1 ARID, which also lacks the helix, does not interact with DNA at all, as indicated by the lack of significant chemical shift perturbation upon the complex formation (11). The conformational flexibility of the Mrf2 C-terminus in the structural ensemble is consistent with 15N relaxation measurements (13). The C-terminal residues of Mrf2 retain significant conformational flexibility in the complex with DNA, as indicated by 1H-15N NOE values close to zero. The reduction in conformational flexibility of the C-terminal residues, as signified by the increase in 1H-15N NOE values from −0.7 in the free form to approximately 0 in the complex with DNA, is likely due to electrostatic interactions between the highly positively charged residues at the C-terminus and the negatively charged DNA backbone.
Mechanism of Mrf2-DNA recognition
Mrf2 contacts DNA mainly using the two flexible loops, L1 and L2. Despite the modest resolution of the structures, consistent protein-DNA interactions can be defined in the family of structures. The side chains of residue Arg36 and Pro38, which are located in loop L1, interact with the sugars of Cyt3, Ade4, and Ade5 in the minor groove (Figure 4). Lys70, Ser82, Thr83, Ser84, and Cys88 in helices H4, H5, and loop L2 form base-specific contacts with A-T base pairs in the major groove. Most of these base-specific contacts in the major groove appear to be van der Waals interactions, except for the hydrogen bond between Ser84 and the base of Thy8. In addition, His92 forms intermolecular hydrogen bonds with the phosphate backbone, and Arg67 from helix H4 makes contact with the sugar-phosphate backbone. Furthermore, Arg36, Arg67 and Lys70 also form electrostatic interactions with the negatively charged phosphate backbone of the DNA. Since the structural studies were carried out at pH 6.0, His92 is likely to be protonated and also contributes to the electrostatic interaction between Mrf2 and DNA. The combination of the interactions mentioned above is present in more than 35% of the structures.
Figure 4. The intermolecular contacts between the Mrf2 ARID domain and DNA.
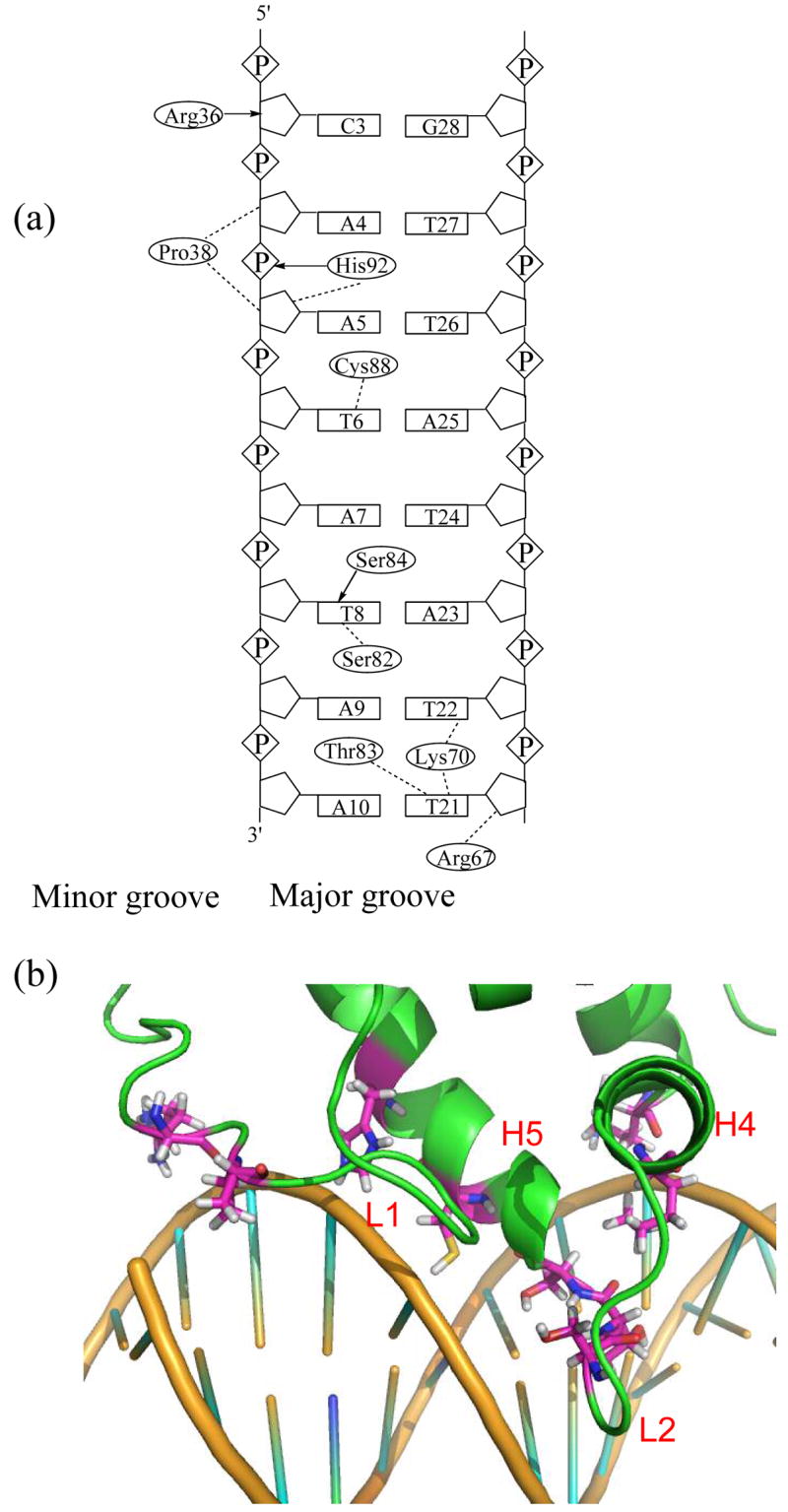
(a) Summary of the contacts observed between the protein and DNA. Dashed lines indicate intermolecular van der Waals interactions. Arrows indicate intermolecular hydrogen-bond interactions with arrowheads denoting the hydrogen bond acceptors. (b) Detailed view of the protein-DNA interface. The amino acid residues involved in DNA-binding are from loops L1 and L2, and helices H4 and H5.
The intermolecular contacts between Mrf2 and DNA are very similar to those observed in the homologous Dri-DNA complex (12). Arg304, Ala309 and Lys310 in Dri loop L1 interact with the sugar phosphate backbone in the minor groove of the DNA, analogous to the aforementioned interaction of Arg36 and Pro38 of Mrf2 with DNA. Residues Ile350–Ser352 of Dri, corresponding to Ser82–Ser84 of Mrf2 in sequence alignment, are located in the major groove and make base-specific contacts with the DNA. In particular, in the Dri-DNA complex, many intermolecular NOEs between Thr351/Ser352 and Ade10/Thy21/Ade20 of DNA were observed. Thr356 of Dri, corresponding to Cys88 of Mrf2 in sequence alignment, makes similar base-specific interaction as Cys88. The extensive intermolecular contacts between the polar residues of the proteins, such as Thr and Ser, and the Ade and Thy basepairs of the DNA are seen in both complexes. Therefore, these major groove interactions are likely critical for sequence-specific DNA-recognition. Despite the significant difference in their overall folds, Mrf2 and Dri ARIDs afford similar base-specific contacts through the highly conserved sequences in their L2 loops and adjacent helices. Therefore, the length and sequence of the L2 loop appear to correlate well with the sequence-specific DNA-binding activity of an ARID domain, and thus are good indicators of ARID DNA-binding activity. In contrast, the comparison between Mrf2 and Dri (Fig. 3B) shows that the overall folds of ARIDs are not correlated with their DNA-binding mechanism and activity. This finding is consistent with a previous study on the DNA-binding properties of the human SWI1 homologue p270 (20).
An efficient approach for structure determination of protein-DNA complexes
In this study, we demonstrated the success of an alternative approach to determine the structure of a protein-DNA complex. Proteins in the ARID family, like many other DNA-binding proteins, do not readily produce NMR spectra of workable quality for structure determination. The residues at the binding interfaces of both Dri and Mrf2 have broad resonances that have prevented the identification of intermolecular NOEs. The quality of NMR spectra of the Dri ARID-DNA complex was improved dramatically by a site-directed mutation at the binding interface (12). We have endeavored to improve the quality of NMR spectra of the Mrf2–DNA complex, and tested different lengths of DNA and buffer conditions. Helix H5 in Mrf2 contains a Cys residue, which is not conserved in the ARID family. We mutated the Cys to a Ser in an effort to avoid spectral complications due to oxidation. This improved the spectral quality of the protein-DNA complex to some extent, but was not sufficient to allow us to obtain intermolecular NOEs.
As shown here, intermolecular distance constraints derived from a single spin-label in combination with docking calculations are sufficient to define many of the interactions important for the Mrf2-DNA complex formation. It is usually much easier to observed backbone resonances in HSQC or TROSY spectra than detecting intermolecular NOEs in NOESY spectra. First, an HSQC spectrum has much higher sensitivity than a 3D NOESY spectrum. Second, proteins’ backbones may not engage in as extensive conformational exchange and fast motions as their sidechains. Thus the signal intensities of backbone resonances are generally much higher in HSQC spectra that those of sidechain resonances in NOESY spectra. This approach is also less labor intensive, and the assignments of intermolecular distances are unambiguous. In cases where intermolecular NOEs can be readily obtained, a global fold generated by spin-label constraints can be used to verify or guide NOE assignments when some of the NOE assignments are ambiguous. Similar approaches can also be applied to the structure determination of RNA-protein complexes.
Acknowledgments
This work is supported by NIH grant CA94595 to Y. Chen.
Abbreviations
- NMR
nuclear magnetic resonance
- NOE
nuclear overhauser effect
- Mrf2
modulator recognition factor 2
- ARID
AT-rich interaction domain
References
- 1.Battiste JL, Wagner G. Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry. 2000;39:5355–65. doi: 10.1021/bi000060h. [DOI] [PubMed] [Google Scholar]
- 2.Folmer RH, Nilges M, Folkers PJ, Konings RN, Hilbers CW. A model of the complex between single-stranded DNA and the single- stranded DNA binding protein encoded by gene V of filamentous bacteriophage M13. J Mol Biol. 1994;240:341–57. doi: 10.1006/jmbi.1994.1449. [DOI] [PubMed] [Google Scholar]
- 3.Gillespie JR, Shortle D. Characterization of long-range structure in the denatured state of staphylococcal nuclease. II. Distance restraints from paramagnetic relaxation and calculation of an ensemble of structures. J Mol Biol. 1997;268:170–84. doi: 10.1006/jmbi.1997.0953. [DOI] [PubMed] [Google Scholar]
- 4.Folkers PJ, van Duynhoven JP, van Lieshout HT, Harmsen BJ, van Boom JH, Tesser GI, Konings RN, Hilbers CW. Exploring the DNA binding domain of gene V protein encoded by bacteriophage M13 with the aid of spin-labeled oligonucleotides in combination with 1H-NMR. Biochemistry. 1993;32:9407–16. doi: 10.1021/bi00087a020. [DOI] [PubMed] [Google Scholar]
- 5.Dominguez C, Boelens R, Bonvin AMJJ. HADDOCK: A Protein-Protein Docking Approach Based on Biochemical or Biophysical Information. Journal of American Chemical Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 6.Arnesano F, Banci L, Bertini I, Bonvin AM. A docking approach to the study of copper trafficking proteins; interaction between metallochaperones and soluble domains of copper ATPases. Structure (Camb) 2004;12:669–76. doi: 10.1016/j.str.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 7.Dominguez C, Bonvin AM, Winkler GS, van Schaik FM, Timmers HT, Boelens R. Structural model of the UbcH5B/CNOT4 complex revealed by combining NMR, mutagenesis, and docking approaches. Structure (Camb) 2004;12:633–44. doi: 10.1016/j.str.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 8.Kortschak RD, Tucker PW, Saint R. ARID proteins come in from the desert. Trends Biochem Sci. 2000;25:294–9. doi: 10.1016/s0968-0004(00)01597-8. [DOI] [PubMed] [Google Scholar]
- 9.Yuan YC, Whitson RH, Liu Q, Itakura K, Chen Y. A novel DNA-binding motif shares structural homology to DNA replication and repair nucleases and polymerases. Nat Struct Biol. 1998;5:959–64. doi: 10.1038/2934. [DOI] [PubMed] [Google Scholar]
- 10.Iwahara J, Clubb RT. Solution structure of the DNA binding domain from Dead ringer, a sequence-specific AT-rich interaction domain (ARID) Embo J. 1999;18:6084–94. doi: 10.1093/emboj/18.21.6084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim S, Zhang Z, Upchurch S, Isern N, Chen Y. Structure and DNA-binding sites of the SWI1 AT-rich interaction domain (ARID) suggest determinants for sequence-specific DNA recognition. J Biol Chem. 2004;279:16670–6. doi: 10.1074/jbc.M312115200. [DOI] [PubMed] [Google Scholar]
- 12.Iwahara J, Iwahara M, Daughdrill GW, Ford J, Clubb RT. The structure of the Dead ringer-DNA complex reveals how AT-rich interaction domains (ARIDs) recognize DNA. Embo J. 2002;21:1197–209. doi: 10.1093/emboj/21.5.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu L, Hu J, Lin D, Whitson RH, Itakura K, Chen Y. Dynamics of the Mrf-2 Domain Free and in Complex with DNA. Biochemistry. 2001;40:9142–50. doi: 10.1021/bi010476a. [DOI] [PubMed] [Google Scholar]
- 14.Ramos A, Varani G. A New Method to Detect Long-Range Protein-RNA Contacts: NMR Detection of Electron-Proton Relaxation Induced by Nitroxide Spin-Labled RNA. J Am Chem Soc. 1998;120:10992–10993. [Google Scholar]
- 15.Krugh TR. Spin Labeling: Theory and Applications. Academic Press; New York: 1976. pp. 339–372. [Google Scholar]
- 16.Solomon I, Bloembergen N. J Chem Phys. 1956;25:261–266. [Google Scholar]
- 17.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 18.Levene SD, Wu HM, Crothers DM. Bending and flexibility of kinetoplast DNA. Biochemistry. 1986;25:3988–95. doi: 10.1021/bi00362a003. [DOI] [PubMed] [Google Scholar]
- 19.Spolar RS, Record MT., Jr Coupling of local folding to site-specific binding of proteins to DNA. Science. 1994;263:777–84. doi: 10.1126/science.8303294. [DOI] [PubMed] [Google Scholar]
- 20.Wilsker D, Patsialou A, Zumbrun SD, Kim S, Chen Y, Dallas PB, Moran E. The DNA-binding properties of the ARID-containing subunits of yeast and mammalian SWI/SNF complexes. Nucleic Acids Res. 2004;32:1345–53. doi: 10.1093/nar/gkh277. [DOI] [PMC free article] [PubMed] [Google Scholar]