

SUMMARY
The aged heart sustains greater injury during ischemia (ISC) and reperfusion (REP) compared to the adult heart. In the Fischer 344 (F344) rat, aging decreases oxidative phosphorylation and complex III activity increasing the production of reactive oxygen species in interfibrillar mitochondria (IFM) located among the myofibrils. In the isolated, perfused 24 month old elderly F344 rat heart 25 min. of stop-flow ISC causes additional damage to complex III, further decreasing the rate of OXPHOS. We did not observe further progressive mitochondrial damage during REP. We next asked if ISC or REP increased oxidative damage within mitochondria of the aged heart. Cardiolipin (CL) is a phospholipid unique to mitochondria consisting predominantly of four linoleic acid residues (C18:2). Following ISC and REP in the aged heart, there is a new CL species containing three oxygen atoms added to one linoleic residue. ISC alone was sufficient to generate this new oxidized molecular species of CL. Based upon oxidative damage to CL, complex III activity, and oxidative phosphorylation, mitochondrial damage thus occurs in the aged heart mainly during ISC, rather than during REP. Mitochondrial damage during ischemia sets the stage for mitochondrial-driven cardiomyocyte injury during reperfusion in the aged heart.
Keywords: ischemia, reperfusion, heart, ubiquinone:cytochrome c reductase (complex III), cardiolipin, mitochondria, aging
INTRODUCTION
Despite timely and successful reperfusion elderly patients sustain greater mortality and cardiac injury with acute myocardial infarction [1]. The Fischer 344 rat model of aging (F344) was used to investigate mechanisms of the enhanced susceptibility to myocardial damage. Isolated, buffer-perfused hearts from 24 mo. F344 rats (aged) exhibit decreased hemodynamic recovery and greater myocardial cell death following 25 min. of 37°C global stop-flow ischemia and 30 min. reperfusion compared to hearts from 6 mo. adult controls [2], providing an appropriate cardiac model for study. Other laboratories also have observed enhanced cardiac damage in the aged Fischer 344 rat heart [3–5] as well as age-related increases in other rat strains [6] and other species [7, 8]. Ischemic preconditioning, where antecedent periods of brief ischemia limit injury from a subsequent longer period of ischemia [5, 9, 10], is ineffective in the aged rat [5, 11, 12] and human hearts [13, 14]. Thus, additional understanding of the mechanisms that underlie the age-enhanced susceptibility to ischemic damage are needed in order to develop strategies to protect the elderly heart during ischemia and reperfusion.
Mitochondrial dysfunction contributes to aging in the heart by increasing the production of reactive oxygen species (ROS) [15] and by favoring the release of cytochrome c to activate cell death pathways [16]. Mitochondrial defects predispose to an increase in cardiomyocyte death that leads to age-related decreases in cardiomyocyte number and an increase in areas of fibrosis [17]. Cardiac mitochondria exist in two functionally distinct populations within the myocyte. Subsarcolemmal mitochondria (SSM) are located underneath the plasma membrane and interfibrillar mitochondria (IFM) are situated among the myofibrils [18]. The content of IFM is decreased in the aged heart [19]. The rate of oxidative phosphorylation (OXPHOS) is decreased in IFM from hearts in 24 mo. aged F334 rats, whereas SSM remain unaffected [19]. OXPHOS is tightly coupled in IFM from aged rats [19]. Dinitrophenol-uncoupled respiration is decreased, localizing the defect to the electron transport chain (ETC) [19]. IFM exhibit a decrease in OXPHOS with age using TMPD-ascorbate, an electron donor to complex IV, as a substrate [19]. Complex IV enzyme activity decreases with aging and is reversed by the addition of exogenous phospholipid liposomes [19, 20], localizing the defect to the lipid environment of the inner mitochondrial membrane, rather than to the peptide subunits of complex IV [19–21].
Aging decreases the maximally expressed activity of complex III measured in detergent-solubilized mitochondria in IFM from aged hearts, but SSM are unaffected [22]. Complex III catalyzes electron transfer from ubiquinol to cytochrome c coupled to proton translocation [23–25]. The complex is composed of two 11 subunit monomers. Each monomer contains three subunits, cytochrome b, cytochrome c1, and the iron-sulfur protein (ISP) that participate in electron transfer [23, 24, 26, 27]. The content of subunit peptides is not altered by age [22]. Functional studies using partial reactions within complex III localized the aging defect to the ubiquinone oxidation site of cytochrome b in complex III (Qo site) in IFM whereas SSM were unaffected [28]. The aging defect in the Qo site of complex III in IFM increases ROS production [28]. The net release of H2O2 was increased in IFM isolated from aged Fischer 344 rat hearts compared to adult controls, whereas SSM, without the aging defect in complex III, showed no increase in ROS production [15].
Mitochondria sustain progressive damage during ischemia in adult and aged hearts [29–37]. Twenty-five min. of global ischemia decreases the activity of complex III, the content of cytochrome c, and respiration through complex IV [38]. In contrast to the selectivity of the aging defect to IFM, ischemia damages both SSM and IFM to a similar extent in 6 and 24 mo. Fischer 344 rats [38]. In IFM from the aged heart, ischemic defects are superimposed upon the pre-existing aging defects [38]. Ischemia decreased complex III activity in both SSM and IFM in adult and aged hearts via a functional inactivation of the iron-sulfur peptide [38]. The iron-sulfur peptide is central to electron transfer within complex III [24], in line with the observed additional decrease in complex III activity. Based upon studies in the adult heart ischemic damage to complex III also favors an increase in the production and net release of ROS from mitochondria[39].
Cytochrome c transfers electrons from complex III to complex IV. Following 25 min. ischemia, cytochrome c content decreases in SSM and IFM in the adult and aged heart [38]. Cardiolipin, enriched in oxidatively-sensitive linoleic acyl-groups [40], interacts with cytochrome c via non-ionic [41–43] and electrostatic [44] mechanisms to localize cytochrome c at the inner mitochondrial membrane [44]. A decrease in cardiolipin content or the oxidative modification of cardiolipin diminishes the affinity of cytochrome c for the inner membrane [41–43], delocalizing cytochrome c into the intermembrane space [44, 45], the first step leading to cytochrome c loss from mitochondria [44]. Release of cytochrome c from mitochondria in turn activates caspases and cell death programs [46]. An increase in the frequency of apoptotic myocytes is observed following ischemia and reperfusion in the aged heart [3, 8].
Mitochondria are both targets and sources of damage during ischemia and reperfusion [46–48]. Ischemia damages the electron transport chain [33] and decreases the rate of OXPHOS [37, 38]. The aged heart sustains greater myocardial injury during ischemia and reperfusion compared to the adult heart[2]. We asked if mitochondria in the aged heart sustain damage mainly during ischemia or sustain progressive injury during reperfusion. We next asked if the mitochondrial injury occurred at least in part as a result of oxidative damage. Preservation of mitochondrial function is critical in order to diminish myocardial injury during ischemia and reperfusion.
METHODS
The Animal Care and Use Committees of the Louis Stokes Cleveland Department of Veterans Affairs Medical Center and Case Western Reserve University approved the protocol. Male Fisher 344 rats (adult: 6–8 months of age and aged: 24 months of age) were anesthetized with pentobarbital sodium (100 mg/kg i.p.) and anti-coagulated with heparin (1000 IU/kg i.p.). Hearts were excised and perfused retrograde via the aorta with modified Krebs-Henseleit (K-H) buffer oxygenated with 95% O2/5% CO2, as previously described [49]. Isolated hearts were perfused for 15 min. followed by 25 min. global stop-flow ischemia at 37°C with or without 30 min. of reperfusion. Cardiac mitochondria were isolated using the procedure of Palmer [18] except that trypsin was used as the protease [49, 50]. Mitochondrial protein concentration was measured by the Lowry method, using bovine serum album as a standard. Mitochondrial respiration was measured using a Clark-type oxygen electrode at 30°C as previously described [37, 51]. Maximally expressed complex III activity was measured by following the increase in absorbance of reduced cytochrome c at 550 nm in mitochondria solubilized with 2% sodium cholate in 25 mM KH2PO4, pH 7.4, using 100 μM decylubiquinol as the electron donor [22]. The separation, quantification, and characterization of cardiolipin was performed as previously described [50, 52]. Phospholipids were extracted from SSM and IFM by the Folch method with 50μM butylated hydroxytoluene added as an antioxidant [52], the phospholipid fraction isolated using silica gel chromatography [53], and cardiolipin isolated from the phospholipid fraction using normal phase HPLC [52]. Cardiolipin was quantified by organic phosphate measurement [52] and composition assessed by separation into component molecular species using reversed phase HPLC followed by electrospray ionization/mass spectrometry as previously described using a Finnegan LCQ-Deca (San Jose, CA, USA) in the negative ion mode [52]. Data-dependent MS3 fragmentation analysis from reverse phase-HPLC separation was utilized to identify and characterize individual cardiolipin molecular species. Data are expressed as the mean ± standard error of the mean (SEM). Differences among groups were compared by two-tailed Student t-test. A difference of p<0.05 was considered significant.
RESULTS
Ischemia, not reperfusion, leads to mitochondrial damage in the aged heart
We studied if damage to the ETC and OXPHOS occurred mainly during ischemia or during reperfusion in aged hearts. Ischemia markedly decreased the rate of state 3 and increased the rate of state 4 respiration with glutamate as substrate compared to mitochondria isolated from time control rat hearts (Figure 1A). The ADP/O ratio was not changed by ischemia (data not shown). There were no differences in the rate of state 3, state 4, and 2 mM ADP stimulated respiration in SSM or IFM with glutamate as complex I substrate following ischemia and reperfusion compared to ischemia alone (Figure 1A). Ischemia decreased the rate of oxidative phosphorylation using substrates that donate electrons to complex III (duroquinol), and to complex IV via cytochrome c (TMPD-ascorbate) in both SSM and IFM (Figures 1B, 1C). Reperfusion failed to further reduce the rate of oxidative phosphorylation in SSM and IFM in aged hearts (Figures 1B, 1C). State 4 rates, respiratory control ratios (RCR), and ADP:O ratios also remained unchanged (data not shown). Similar rates were observed for uncoupled respiration, localizing the defect to the ETC (data not shown). Reperfusion did not cause an additional decrease in oxidation rate of duroquinol or TMPD-ascorbate, indicating that reperfusion did not lead to additional damage to the distal electron transport chain.
Figure 1.



Ischemia markedly decreases the maximal ADP-stimulated rate of oxidative phosphorylation in both subsarcolemmal (SSM) and interfibrillar (IFM) mitochondria obtained from the 24 month old Fischer 344 rat heart. Reperfusion does not result in additional decreases in the rate of oxidative phosphorylation. Decreases are observed with glutamate (A), duroquinol (B), and TMPD-ascorbate (C) as substrates. The decrease in the rate of respiration are similar in the mitochondria isolated both following 25′ ischemia and following 25′ ischemia plus 30′ reperfusion. (Mean ± SD; * p<0.05; p=NS Ischemia vs. Ischemia-Reperfusion.)
Myocardial ischemia decreased complex III activity in both SSM and IFM from aged hearts (Figure 2). Reperfusion did not result in a further decrease of complex III activity, providing additional evidence that damage during ischemia was dominant (Figure 2). This observation focused our attention on specific molecular targets of ischemia-induced mitochondrial damage in the aged heart.
Figure 2.
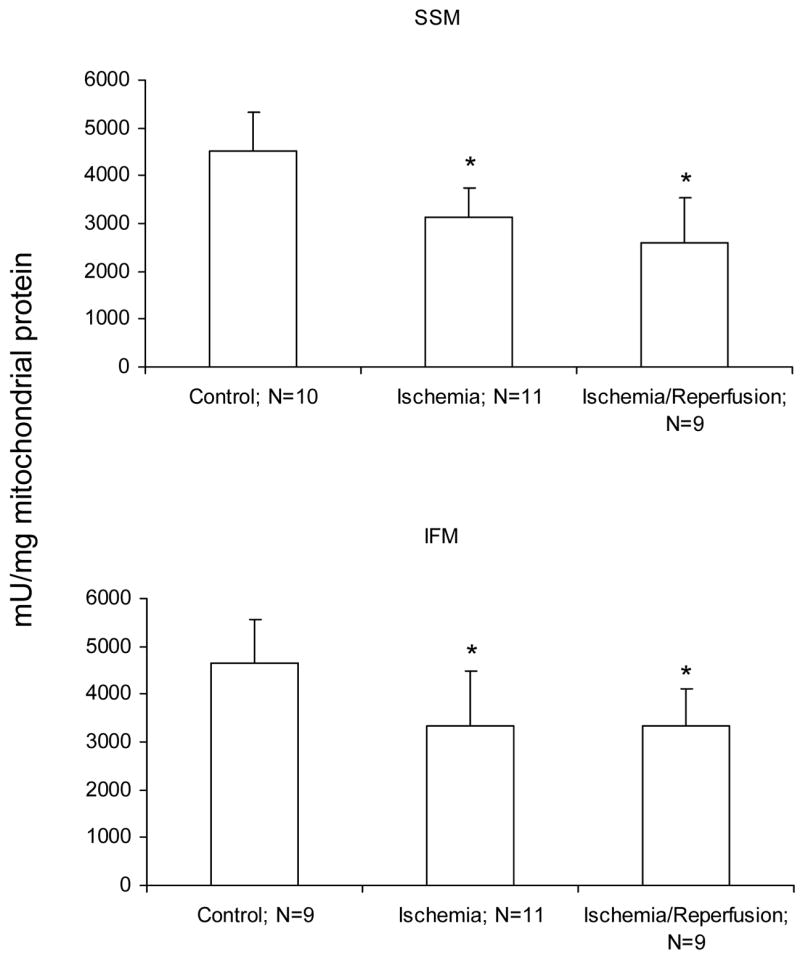
Ischemia markedly decreases complex III activity (decylubiquinone:cytochrome c reductase) in both subsarcolemmal (SSM) and interfibrillar (IFM) mitochondria obtained from the 24 month old Fischer 344 rat heart. Reperfusion does not result in additional decreases in complex III activity. The decrease in the rate of respiration are similar in the mitochondria isolated both following 25′ ischemia and following 25′ ischemia plus 30′ reperfusion. (Mean ± SD; * p<0.05; p=NS Ischemia vs. Ischemia-Reperfusion.)
Ischemia-leads to oxidative modification to cardiolipin in the aged, but not adult heart
We asked if ischemia or reperfusion altered the content or composition of cardiolipin in the adult or aged heart. Adult and aged hearts underwent 25 min. ischemia with or without 30 min. reperfusion. The content of cardiolipin measured by phosphate was unchanged in both adult and aged hearts following ischemia and reperfusion (Table 1). The average recovery of cardiolipin in both adult and aged hearts following ischemia and reperfusion was 97±4%. Analysis of cardiolipin molecular species was performed using reversed phase HPLC, followed by electrospray ionization mass spectrometry. As seen in Figure 3A the predominant cardiolipin species in IFM following 25 minutes of ischemia and 30 minutes of reperfusion in the adult 6 month heart has an m/z of 1448 (potassium salt m/z 1486) and represents tetra-linoleic-cardiolipin. In the IFM from the 24 month aged F344 rat heart after ischemia and reperfusion, the reversed phase HPLC of the cardiolipin fraction revealed a new molecular species of cardiolipin with a m/z 1496 (potassium salt m/z 1535) (Figure 3B) with a shortened retention time (data not shown). The mass of the new molecular species (1496) was increased by 48 compared to the tetra-linoleic cardiolipin species. The collision induced dissociation spectra in the MS2 and MS3 revealed that the +48 mass was present in a single acyl group of cardiolipin. This rapid modification of cardiolipin by +48 most likely represents the addition of three oxygen atoms and the earlier elution time is consistent with the increase in polarity with the addition of the oxygen atoms. The relative increase in the fractional content of the new oxidized species was estimated using the ratio of the integrated peak areas of the 1496 species to the 1448 species (Table 1). The content of the oxidized species increased at least three-fold in the aged heart compared to the adult heart (Table 1). These findings indicate markedly increased the selective oxidation of cardiolipin in the aged heart during ischemia or reperfusion. We next asked if these changes were observed during ischemia. Neither the content nor the composition of the molecular species of cardiolipin were different following 25 minutes of ischemia compared to that observed with ischemia/reperfusion (data not shown). Thus, substantial oxidative modification to cardiolipin occurs during ischemia in the aged heart.
Table 1.
Cardiolipin content and composition in the adult and aged heart following ischemia and reperfusion.
| SSM | IFM | |||
|---|---|---|---|---|
| Adult | Aged | Adult | Aged | |
| Cardiolipin (nmol/mg protein) | 49±4 | 50±6 | 53±8 | 42±8 |
| 1496/1448 (total ion current ratio) | 0.075±0.06 | 0.387±0.27* | 0.079±0.041 | 0.317±0.095* |
Mean±SD;
p<0.05 vs. adult; adult-6 month F344; aged 24 month F344
Figure 3.


Electrospray ionization mass spectrometry of cardiolipin molecular species separated by reverse phase HPLC (see Methods). (A) Cardiolipin from IFM from an adult rat following ischemia and reperfusion Fragmentation pattern of the 1486 amu (K+-adduct of the 1448-intact cardiolipin) major molecular species demonstrating the mass spectral “foot print”: 869-glycerophosphatidic acid fragment; 607-phosphatidic acid; 415-lysophosphatidic acid; 279-linoleic acid. (B) Cardiolipin from IFM from an aged rat following ischemia and reperfusion Fragmentation pattern of the new 1535 amu (K+-adduct of the 1496-intact cardiolipin) molecular species demonstrating the mass spectral “foot print”: ; 917-glycerophosphatidic acid fragment; 655-phosphatidic acid; 453-lysophosphatidic acid; 327-oxidized linoleic acid. The collision-induced dissociation demonstrates that the +48 addition tracks thorugh each fragment to the acyl-group.
DISCUSSION
The aged heart sustains increased injury during ischemia and reperfusion. Mitochondria are key contributors to myocardial injury during ischemia as well as during reperfusion. Ischemic damage is superimposed upon preexisting age-related defects in mitochondrial metabolism. Based upon the study of cardiolipin, oxidative damage to mitochondria occurs mainly during ischemia in the aged heart. Thus, oxidative processes are likely contributors to the mitochondrial damage that occurs during ischemia in the aged heart. Future work is needed in order to evaluate if the ischemic damage to mitochondria leads to myocyte injury and cell death during ischemia or during the early reperfusion period.
The evolution of oxidative damage during ischemia initially may appear to pose a conundrum. The generation of a detectable burst of ROS early in reperfusion is widely accepted [54–59]. It was originally thought that during cardiac ischemia the oxygen content rapidly decreases to complete anoxia. However, during the initial progression of myocardial ischemia, oxygen remains available [60]. During simulated ischemia in cardiomyocytes, under conditions of oxygen depletion, the ROS generation actually increases [61, 62]. In the setting of low flow ischemia, ROS production occurs continuously [58]. Even during stop flow ischemia in the isolated perfused heart, tissue oxygen remains detectable by sensitive electron paramagnetic resonance measurement at least during the initial ten minutes of ischemia [60]. Consistent with the presence of oxygen during the evolution of ischemic injury, ROS production monitored on line increases during global ischemia in the intact heart [56, 63]. Mitochondria are the major source for ROS production during ischemia [61, 62, 64]. Superoxide (•O2−) production during simulated ischemia in cardiac myocytes [61, 62] and during stop-flow ischemia in the isolated heart [63] is decreased by blockade of electron transfer into complex III. Complex III, a dominant site for the extra mitochondrial release of ROS [65, 66], is a likely site for the production of ROS that are detected within the cardiomyocyte during ischemia.
Cardiolipin, a phospholipid unique to mitochondria, is located in the inner mitochondrial membrane in close proximity to the sites of ROS production. Cardiolipin is highly enriched in oxidatively sensitive linoleic acid (C18:2) groups susceptible to lipid peroxide formation [67–70] and thus is a prime target of the ROS produced from the ETC during ischemia. In vitro, ROS production from the ETC, especially complex III [71], can deplete cardiolipin. In pathologic settings including oxidative stress, during ischemia, cytochrome c alters its interaction with cardiolipin resulting in a new tertiary structure of cytochrome c [72, 73], allowing ROS to react via the heme prosthetic group of cytochrome c to peroxidize cardiolipin [74]. The role of the cytochrome c-cardiolipin peroxidase in ischemia or reperfusion is unknown. Potential mechanisms of cardiolipin peroxidation during ischemia include heavy metal, most likely iron, catalyzed chain propagation reactions to generate oxidized linoleic acid residues in cardiolipin [67, 70], potential formation of the cytochrome c-cardiolipin peroxidase with subsequent cardiolipin peroxidation, or other as yet unknown mechanisms of mitochondrial-medicated peroxidation of linoleic acid. The formation of +48 amu compounds resulting from mitochondrial-mediated peroxidation of linoleic acid has been described [75–77]. The formation of a specific +48 amu product, rather than a mixture of products, suggests the presence of a specific peptide or enzymatic mechanism of peroxidation, rather than a stochastic chemical lipid peroxidation process. Oxidative damage to cardiolipin was increased in both SSM and IFM in the aged heart. Oxidative damage to cardiolipin, in turn, favors the delocalization and release of cytochrome c from the inner membrane, predisposing to its release from mitochondria and the activation of cell death programs [44]. The presence of oxidative damage in both populations of mitochondria suggests that age-induced defects localized to IFM may not be the sole mechanism of the enhanced oxidative damage observed during ischemia in the aged heart.
We hypothesized that the distal electron transport chain, including complex III (perhaps in concert with the cytochrome c-peroxidase), contributes to the ischemic damage to the ETC. To test this hypothesis, the ETC was reversibly blocked proximal to complex III using amobarbital (AMO). AMO, a short-acting barbiturate, specifically inhibits complex I [78] based on studies in isolated [90] and in situ cardiac mitochondria [63]. AMO treatment (2.5 mM) of the adult F344 rat heart immediately before 25 min. ischemia protects SSM and IFM against damage during ischemia, with preserved OXPHOS and markedly decreased cytochrome c loss [49]. Protection of OXPHOS during ischemia allows a critical test of the contribution of ischemic mitochondrial damage to myocardial injury during reperfusion. Protection of mitochondria led to improved contractile recovery, reduced cardiac enzyme release, and results in the formation of smaller infarcts in the adult F344 rat measured following 30 min. reperfusion [79]. The cardiac protection observed when the heart is reperfused in the setting of preserved mitochondrial function provides support for the working hypothesis that ischemic damage to mitochondria is a key mechanism of myocardial injury during reperfusion in the adult heart.
Reversible blockade of electron transport immediately before 25 min. ischemia in the aged heart attenuates ischemic damage to OXPHOS, reduces cytochrome c release, and decreases ischemic damage to complex III, even in IFM that contain the aging defect [80]. Ischemic mitochondrial damage, in turn, contributes to cardiac injury in the aged heart. Reperfusion of the aged heart in the absence of superimposed ischemic mitochondrial damage improves hemodynamic recovery and lessens cardiomyocyte death as assessed by the release of lactate dehydrogenase during reperfusion [80]. Integrated respiration and complex III that were protected from ischemic injury by blockade of electron transport did not sustain additional injury during reperfusion [80], again supporting the notion that mitochondrial damage occurs mainly during ischemia, in line with the findings of the current study in untreated hearts. Thus, the mitochondrial damage that occurs during ischemia contributes to injury in the aged heart during reperfusion.
In contrast to ischemic defects, the age-related defect in complex III independently contributes to cardiac injury during ischemia and reperfusion. We studied if an intervention that improved age-related defects in OXPHOS before ischemia would decrease myocardial injury during subsequent ischemia and reperfusion. Treatment of aged rats with acetylcarnitine (AcCN) increased the maximal rate of OXPHOS in IFM and restored complex III activity to rates observed in the adult heart [81]. Adult and aged F344 rat hearts underwent 25 min. ischemia and 30 min. reperfusion. In the aged heart, AcCN improved the recovery of left ventricular developed pressure and decreased the extent of myocardial cell death as measured by lactate dehydrogenase release during reperfusion [81]. In contrast, in the adult heart, contractile recovery and lactate dehydrogenase release during reperfusion was unchanged by treatment. These observations support the contribution of age-related defects to the increased damage observed following ischemia and reperfusion. Thus, pretreatment of aging defects rendered the aged hear t more resistant to ischemic injury [81, 82].
Restoration of OXPHOS and the complex III phenotype in the aged heart decreases cardiac injury during ischemia and reperfusion. Limitation of electron flow into complex III during ischemia blunts mitochondrial damage and decreases myocardial injury in the adult heart. Thus, modulation of the ETC can attenuate cardiac injury during ischemia and reperfusion. Although the concept that disrupting effective oxidative metabolism to protect the heart initially appears counterintuitive, blockade of electron transport decreases the production and release of ROS from mitochondria and preserves mitochondrial integrity, leading to the retention of cytochrome c. Modulation of mitochondrial metabolism by therapeutic intervention [81, 82] is a key potential mechanism of cardiac protection during ischemia and reperfusion for both the adult and aged heart. However, this conceptual approach, though intriguing, requires intervention before and during ischemia, which limits application in many settings of cardiac ischemia. The exciting option to utilize pretreatment of age-related defects in a preemptive fashion to decrease ischemia-reperfusion cardiac injury represents a second novel cardioprotective strategy for the aged heart. This approach treats aging defects to inoculate the high-risk aged heart against injury before ischemia occurs. Additional work is needed to evaluate if pretreatment to reverse age-related defects also impacts on ischemic mitochondrial damage. In a similar fashion, it will be important to determine if the improvement in age-related defects in complex III attenuates the age-enhanced production of oxidized cardiolipin during ischemia in the elderly heart. An improved understanding of the contributions of aging-defects in relationship to ischemic defects in OXPHOS is of major significance in reducing cardiac injury in the elderly heart.
Ischemia damages the ETC in the aged heart, setting the stage for cardiac injury during reperfusion. Reperfusion of the myocardium in the setting of preserved OXPHOS decreases ROS production and reduces infarct size, underscoring the role of ischemic ETC damage in the genesis of cardiac injury. The ability to prevent ischemic damage via reversible blockade of electron transport in the aged heart provides the first experimental approach to directly assess the responses of mitochondria that have been damaged by ischemia compared to mitochondria exposed to in situ ischemia but are devoid of injury. Future studies of the responses of the two mitochondrial phenotypes to the conditions of reperfusion should advance our understanding of the mechanisms whereby ischemic damage to the ETC is translated into mitochondrial-driven cardiomyocyte injury during early reperfusion in the aged heart. We hypothesize that the heart in human beings might be protected during reperfusion from the deleterious consequences of resumption of aerobic metabolism by ischemia-damaged mitochondria by modulating the respiratory function of the damaged mitochondria during reperfusion.
Acknowledgments
This work was supported by the National Institutes of Health (2PO1AG15885) and by the Office of Research and Development, Medical Research Service, Department of Veterans Affairs. We thank Dr. Bernard Tandler for editorial review and Mr. Paul Minkler for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lesnefsky EJ, Lundergan CF, Hodgson JM, Nair R, Reiner JS, Greenhouse SW, Califf RM, Ross AM. Increased left ventricular dysfunction in elderly patients despite successful thrombolysis: the GUSTO-I angiographic experience. J Am Coll Cardiol. 1996;28:331–337. doi: 10.1016/0735-1097(96)00148-9. [DOI] [PubMed] [Google Scholar]
- 2.Lesnefsky EJ, Gallo DS, Ye J, Whittingham TS, Lust WD. Aging increases ischemia-reperfusion injury in the isolated, buffer-perfused heart. J Lab Clin Med. 1994;124:843–851. [PubMed] [Google Scholar]
- 3.Liu L, Azhar G, Gao W, Zhang X, Wei JY. Bcl-2 and Bax expression in adult rat hearts after coronary occlusion: age-associated differences. Am J Physiol. 1998;275:R315–R322. doi: 10.1152/ajpregu.1998.275.1.R315. [DOI] [PubMed] [Google Scholar]
- 4.Lucas DT, Szweda LI. Cardiac reperfusion injury: aging, lipid peroxidation, and mitochondrial dysfunction. Proc Natl Acad Sci U S A. 1998;95:510–514. doi: 10.1073/pnas.95.2.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tani M, Suganuma Y, Hasegawa H, Shinmura K, Ebihara Y, Hayashi Y, Guo X, Takayama M. Decrease in ischemic tolerance with aging in isolated perfused Fischer 344 rat hearts: relation to increases in intracellular Na+ after ischemia. J Mol Cell Cardiol. 1997;29:3081–3089. doi: 10.1006/jmcc.1997.0533. [DOI] [PubMed] [Google Scholar]
- 6.Frolkis VV, Frolkis RA, Mkhitarian LS, Fraifeld VE. Age-dependent effects of ischemia and reperfusion on cardiac function and Ca2+ transport in myocardium. Gerontology. 1991;37:233–239. doi: 10.1159/000213266. [DOI] [PubMed] [Google Scholar]
- 7.Ataka K, Chen D, Levitsky S, Jimenez E, Feinberg H. Effect of aging on intracellular Ca2+, pHi, and contractility during ischemia and reperfusion. Circulation. 1992;86:II371–376. [PubMed] [Google Scholar]
- 8.Azhar G, Gao W, Liu L, Wei JY. Ischemia-reperfusion in the adult mouse heart influence of age. Exp Gerontol. 1999;34:699–714. doi: 10.1016/s0531-5565(99)00031-5. [DOI] [PubMed] [Google Scholar]
- 9.Fryer RM, Patel HH, Hsu AK, Gross GJ. Stress-activated protein kinase phosphorylation during cardioprotection in the ischemic myocardium. Am J Physiol Heart Circ Physiol. 2001;281:H1184–H1192. doi: 10.1152/ajpheart.2001.281.3.H1184. [DOI] [PubMed] [Google Scholar]
- 10.Murry CE, Richard VJ, Reimer KA, Jennings RB. Ischemic preconditioning slows energy metabolism and delays ultrastructural damage during a sustained ischemic episode. Circ Res. 1990;66:913–931. doi: 10.1161/01.res.66.4.913. [DOI] [PubMed] [Google Scholar]
- 11.Abete P, Testa G, Ferrara N, De Santis D, Capaccio P, Viati L, Calabrese C, Cacciatore F, Longobardi G, Condorelli M, Napoli C, Rengo F. Cardioprotective effect of ischemic preconditioning is preserved in food-restricted senescent rats. Am J Physiol Heart Circ Physiol. 2002;282:H1978–H1987. doi: 10.1152/ajpheart.00929.2001. [DOI] [PubMed] [Google Scholar]
- 12.Tani M, Honma Y, Hasegawa H, Tamaki K. Direct activation of mitochondrial K(ATP) channels mimics preconditioning but protein kinase C activation is less effective in middle-aged rat hearts. Cardiovasc Res. 2001;49:56–68. doi: 10.1016/s0008-6363(00)00240-6. [DOI] [PubMed] [Google Scholar]
- 13.Ishihara M, Sato H, Tateishi H, Kawagoe T, Shimatani Y, Ueda K, Noma K, Yumoto A, Nishioka K. Beneficial effect of prodromal angina pectoris is lost in elderly patients with acute myocardial infarction. Am Heart J. 2000;139:881–888. doi: 10.1016/s0002-8703(00)90021-8. [DOI] [PubMed] [Google Scholar]
- 14.Lee TM, Su SF, Chou TF, Lee YT, Tsai CH. Loss of preconditioning by attenuated activation of myocardial ATP-sensitive potassium channels in elderly patients undergoing coronary angioplasty. Circulation. 2002;105:334–340. doi: 10.1161/hc0302.102572. [DOI] [PubMed] [Google Scholar]
- 15.Suh JH, Heath SH, Hagen TM. Two subpopulations of mitochondria in the aging rat heart display heterogenous levels of oxidative stress. Free Radic Biol Med. 2003;35:1064–1072. doi: 10.1016/s0891-5849(03)00468-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Phaneuf S, Leeuwenburgh C. Cytochrome c release from mitochondria in the aging heart: a possible mechanism for apoptosis with age. Am J Physiol Regul Integr Comp Physiol. 2002;282:R423–R430. doi: 10.1152/ajpregu.00296.2001. [DOI] [PubMed] [Google Scholar]
- 17.Wei JY. Age and the cardiovascular system. New England Journal Of Medicine. 1992;327:1735–1739. doi: 10.1056/NEJM199212103272408. [DOI] [PubMed] [Google Scholar]
- 18.Palmer JW, Tandler B, Hoppel CL. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem. 1977;252:8731–8739. [PubMed] [Google Scholar]
- 19.Fannin SW, Lesnefsky EJ, Slabe TJ, Hassan MO, Hoppel CL. Aging selectively decreases oxidative capacity in rat heart interfibrillar mitochondria. Arch Biochem Biophys. 1999;372:399–407. doi: 10.1006/abbi.1999.1508. [DOI] [PubMed] [Google Scholar]
- 20.Paradies G, Ruggiero FM, Dinoi P, Petrosillo G, Quagliariello E. Decreased cytochrome oxidase activity and changes in phospholipids in heart mitochondria from hypothyroid rats. Arch Biochem Biophys. 1993;307:91–95. doi: 10.1006/abbi.1993.1565. [DOI] [PubMed] [Google Scholar]
- 21.Lesnefsky EJ, Hoppel CL. Oxidative phosphorylation and aging. Ageing Res Rev. 2006;5:402–433. doi: 10.1016/j.arr.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 22.Lesnefsky EJ, Gudz TI, Moghaddas S, Migita CT, Ikeda_Saito M, Turkaly PJ, Hoppel CL. Aging decreases electron transport complex III activity in heart interfibrillar mitochondria by alteration of the cytochrome c binding site. J Mol Cell Cardiol. 2001;33:37–47. doi: 10.1006/jmcc.2000.1273. [DOI] [PubMed] [Google Scholar]
- 23.Crofts AR, Barquera B, Gennis RB, Kuras R, Guergova_Kuras M, Berry EA. Mechanism of ubiquinol oxidation by the bc(1) complex: different domains of the quinol binding pocket and their role in the mechanism and binding of inhibitors. Biochemistry. 1999;38:15807–15826. doi: 10.1021/bi990962m. [DOI] [PubMed] [Google Scholar]
- 24.Crofts AR, Guergova_Kuras M, Huang L, Kuras R, Zhang Z, Berry EA. Mechanism of ubiquinol oxidation by the bc(1) complex: role of the iron sulfur protein and its mobility. Biochemistry. 1999;38:15791–15806. doi: 10.1021/bi990961u. [DOI] [PubMed] [Google Scholar]
- 25.Trumpower BL. The protonmotive Q cycle. Energy transduction by coupling of proton translocation to electron transfer by the cytochrome bc1 complex. J Biol Chem. 1990;265:11409–12. [PubMed] [Google Scholar]
- 26.Crofts AR, Shinkarev VP, Dikanov SA, Samoilova RI, Kolling D. Interactions of quinone with the iron-sulfur protein of the bc(1) complex: is the mechanism spring-loaded? Biochim Biophys Acta. 2002;1555:48–53. doi: 10.1016/s0005-2728(02)00253-0. [DOI] [PubMed] [Google Scholar]
- 27.Iwata S, Lee JW, Okada K, Lee JK, Iwata M, Rasmussen B, Link TA, Ramaswamy S, Jap BK. Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science. 1998;281:64–71. doi: 10.1126/science.281.5373.64. [DOI] [PubMed] [Google Scholar]
- 28.Moghaddas S, Hoppel CL, Lesnefsky EJ. Aging defect at the Qo site of complex III augments oxyradical production in rat heart interfibrillar mitochondria. Arch Biochem Biophys. 2003;414:59–66. doi: 10.1016/s0003-9861(03)00166-8. [DOI] [PubMed] [Google Scholar]
- 29.Lemasters JJ, Nieminen AL. Mitochondrial oxygen radical formation during reductive and oxidative stress to intact hepatocytes. Biosci Rep. 1997;17:281–291. doi: 10.1023/a:1027332611839. [DOI] [PubMed] [Google Scholar]
- 30.Duan J, Karmazyn M. Relationship between oxidative phosphorylation and adenine nucleotide translocase activity of two populations of cardiac mitochondria and mechanical recovery of ischemic hearts following reperfusion. Can J Physiol Pharmacol. 1989;67:704–709. doi: 10.1139/y89-114. [DOI] [PubMed] [Google Scholar]
- 31.Piper HM, Sezer O, Schleyer M, Schwartz P, Hutter JF, Spieckermann PG. Development of ischemia-induced damage in defined mitochondrial subpopulations. J Mol Cell Cardiol. 1985;17:885–896. doi: 10.1016/s0022-2828(85)80102-4. [DOI] [PubMed] [Google Scholar]
- 32.Kajiyama K, Pauly DF, Hughes H, Yoon SB, Entman ML, McMillin-Wood JB. Protection by verapamil of mitochondrial glutathione equilibrium and phospholipid changes during reperfusion of ischemic canine myocardium. Circ Res. 1987;61:301–310. doi: 10.1161/01.res.61.2.301. [DOI] [PubMed] [Google Scholar]
- 33.Rouslin W. Mitochondrial complexes I, II, III, IV, and V in myocardial ischemia and autolysis. Am J Physiol. 1983;244:H743–48. doi: 10.1152/ajpheart.1983.244.6.H743. [DOI] [PubMed] [Google Scholar]
- 34.Flameng W, Andres J, Ferdinande P, Mattheussen M, Van Belle H. Mitochondrial function in myocardial stunning. J Mol Cell Cardiol. 1991;23:1–11. doi: 10.1016/0022-2828(91)90034-j. [DOI] [PubMed] [Google Scholar]
- 35.Shin G, Sugiyama M, Shoji T, Kagiyama A, Sato H, Ogura R. Detection of mitochondrial membrane damages in myocardial ischemia with ESR spin labeling technique. J Mol Cell Cardiol. 1989;21:1029–1036. doi: 10.1016/0022-2828(89)90801-8. [DOI] [PubMed] [Google Scholar]
- 36.Ueta H, Ogura R, Sugiyama M, Kagiyama A, Shin G. O2−. spin trapping on cardiac submitochondrial particles isolated from ischemic and non-ischemic myocardium. J Mol Cell Cardiol. 1990;22:893–899. doi: 10.1016/0022-2828(90)90120-q. [DOI] [PubMed] [Google Scholar]
- 37.Lesnefsky EJ, Tandler B, Ye J, Slabe TJ, Turkaly J, Hoppel CL. Myocardial ischemia decreases oxidative phosphorylation through cytochrome oxidase in subsarcolemmal mitochondria. Am J Physiol. 1997;273:H1544–H1554. doi: 10.1152/ajpheart.1997.273.3.H1544. [DOI] [PubMed] [Google Scholar]
- 38.Lesnefsky EJ, Gudz TI, Migita CT, Ikeda-Saito M, Hassan MO, Turkaly PJ, Hoppel CL. Ischemic injury to mitochondrial electron transport in the aging heart: damage to the iron-sulfur protein subunit of electron transport complex III. Arch Biochem Biophys. 2001;385:117–128. doi: 10.1006/abbi.2000.2066. [DOI] [PubMed] [Google Scholar]
- 39.Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am J Physiol Cell Physiol. 2008;294:C460–C466. doi: 10.1152/ajpcell.00211.2007. [DOI] [PubMed] [Google Scholar]
- 40.Hoch FL. Cardiolipins and biomembrane function. Biochim Biophys Acta. 1992;1113:71–133. doi: 10.1016/0304-4157(92)90035-9. [DOI] [PubMed] [Google Scholar]
- 41.Rytomaa M, Kinnunen PK. Evidence for two distinct acidic phospholipid-binding sites in cytochrome c. J Biol Chem. 1994;269:1770–1774. [PubMed] [Google Scholar]
- 42.Salamon Z, Tollin G. Interaction of horse heart cytochrome c with lipid bilayer membranes: effects on redox potentials. J Bioenerg Biomembr. 1997;29:211–221. doi: 10.1023/a:1022401825287. [DOI] [PubMed] [Google Scholar]
- 43.Spooner PJ, Watts A. Cytochrome c interactions with cardiolipin in bilayers: a multinuclear magic-angle spinning NMR study. Biochemistry. 1992;31:10129–1038. doi: 10.1021/bi00156a037. [DOI] [PubMed] [Google Scholar]
- 44.Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci U S A. 2002;99:1259–1263. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shidoji Y, Hayashi K, Komura S, Ohishi N, Yagi K. Loss of molecular interaction between cytochrome c and cardiolipin due to lipid peroxidation. Biochem Biophys Res Commun. 1999;264:343–347. doi: 10.1006/bbrc.1999.1410. [DOI] [PubMed] [Google Scholar]
- 46.Borutaite V, Budriunaite A, Morkuniene R, Brown GC. Release of mitochondrial cytochrome c and activation of cytosolic caspases induced by myocardial ischaemia. Biochim Biophys Acta. 2001;1537:101–109. doi: 10.1016/s0925-4439(01)00062-x. [DOI] [PubMed] [Google Scholar]
- 47.Lesnefsky EJ, Hoppel CL. Ischemia-reperfusion injury in the aged heart: role of mitochondria. Arch Biochem Biophys. 2003;420:287–297. doi: 10.1016/j.abb.2003.09.046. [DOI] [PubMed] [Google Scholar]
- 48.Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL. Mitochondrial dysfunction in cardiac disease: ischemia-reperfusion, aging, and heart failure. J Mol Cell Cardiol. 2001;33:1065–1089. doi: 10.1006/jmcc.2001.1378. [DOI] [PubMed] [Google Scholar]
- 49.Chen Q, Hoppel CL, Lesnefsky EJ. Blockade of Electron Transport before Cardiac Ischemia with the Reversible Inhibitor Amobarbital Protects Rat Heart Mitochondria. J Pharmacol Exp Ther. 2006;316:200–207. doi: 10.1124/jpet.105.091702. [DOI] [PubMed] [Google Scholar]
- 50.Moghaddas S, Stoll MS, Minkler PE, Salomon RG, Hoppel CL, Lesnefsky EJ. Preservation of cardiolipin content during aging in rat heart interfibrillar mitochondria. J Gerontol A Biol Sci Med Sci. 2002;57:B22–B28. doi: 10.1093/gerona/57.1.b22. [DOI] [PubMed] [Google Scholar]
- 51.Krahenbuhl S, Chang M, Brass EP, Hoppel CL. Decreased activities of ubiquinol:ferricytochrome c oxidoreductase (complex III) and ferrocytochrome c:oxygen oxidoreductase (complex IV) in liver mitochondria from rats with hydroxycobalamin[c-lactam]-induced methylmalonic aciduria. J Biol Chem. 1991;266:20998–21003. [PubMed] [Google Scholar]
- 52.Lesnefsky EJ, Stoll MS, Minkler PE, Hoppel CL. Separation and quantitation of phospholipids and lysophospholipids by high-performance liquid chromatography. Anal Biochem. 2000;285:246–254. doi: 10.1006/abio.2000.4783. [DOI] [PubMed] [Google Scholar]
- 53.Ingalls ST, Kriaris MS, Xu Y, DeWulf DW, Tserng KY, Hoppel CL. Method for isolation of non-esterified fatty acids and several other classes of plasma lipids by column chromatography on silica gel. J Chromatogr. 1993;619:9–19. doi: 10.1016/0378-4347(93)80441-6. [DOI] [PubMed] [Google Scholar]
- 54.Ambrosio G, Zweier JL, Duilio C, Kuppusamy P, Santoro G, Elia PP, Tritto I, Cirillo P, Condorelli M, Chiariello M, et al. Evidence that mitochondrial respiration is a source of potentially toxic oxygen free radicals in intact rabbit hearts subjected to ischemia and reflow. J Biol Chem. 1993;268:18532–18541. [PubMed] [Google Scholar]
- 55.Bolli R, McCay PB. Use of spin traps in intact animals undergoing myocardial ischemia/reperfusion: a new approach to assessing the role of oxygen radicals in myocardial "stunning". Free Radic Res Commun. 1990;9:169–180. doi: 10.3109/10715769009145674. [DOI] [PubMed] [Google Scholar]
- 56.Kevin LG, Camara AK, Riess ML, Novalija E, Stowe DF. Ischemic preconditioning alters real-time measure of O2 radicals in intact hearts with ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2003;284:H566–H574. doi: 10.1152/ajpheart.00711.2002. [DOI] [PubMed] [Google Scholar]
- 57.Maupoil V, Rochette L, Tabard A, Clauser P, Harpey C. Evolution of free radical formation during low-flow ischemia and reperfusion in isolated rat heart. Cardiovasc Drugs Ther. 1990;4:791–795. doi: 10.1007/BF00051276. [DOI] [PubMed] [Google Scholar]
- 58.Merrill GF. Acetaminophen and low-flow myocardial ischemia: efficacy and antioxidant mechanisms. Am J Physiol Heart Circ Physiol. 2002;282:H1341–H1349. doi: 10.1152/ajpheart.00716.2001. [DOI] [PubMed] [Google Scholar]
- 59.Pietri S, Culcasi M, Cozzone PJ. Real-time continuous-flow spin trapping of hydroxyl free radical in the ischemic and post-ischemic myocardium. Eur J Biochem. 1989;186:163–173. doi: 10.1111/j.1432-1033.1989.tb15191.x. [DOI] [PubMed] [Google Scholar]
- 60.Ilangovan G, Liebgott T, Kutala VK, Petryakov S, Zweier JL, Kuppusamy P. EPR oximetry in the beating heart: myocardial oxygen consumption rate as an index of postischemic recovery. Magn Reson Med. 2004;51:835–8. 42. doi: 10.1002/mrm.20000. [DOI] [PubMed] [Google Scholar]
- 61.Becker LB, vanden Hoek TL, Shao ZH, Li CQ, Schumacker PT. Generation of superoxide in cardiomyocytes during ischemia before reperfusion. Am J Physiol. 1999;277:H2240–H2246. doi: 10.1152/ajpheart.1999.277.6.H2240. [DOI] [PubMed] [Google Scholar]
- 62.Vanden Hoek TL, Li C, Shao Z, Schumacker PT, Becker LB. Significant levels of oxidants are generated by isolated cardiomyocytes during ischemia prior to reperfusion. J Mol Cell Cardiol. 1997;29:2571–2583. doi: 10.1006/jmcc.1997.0497. [DOI] [PubMed] [Google Scholar]
- 63.Aldakkak M, Stowe DF, Chen Q, Lesnefsky EJ, Camara AK. Inhibited mitochondrial respiration by amobarbital during cardiac ischaemia improves redox state and reduces matrix Ca2+ overload and ROS release. Cardiovasc Res. 2008;77:406–415. doi: 10.1016/j.cardiores.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 64.Davies MJ. Direct detection of radical production in the ischaemic and reperfused myocardium: current status. Free Radic Res Commun. 1989;7:275–284. doi: 10.3109/10715768909087952. [DOI] [PubMed] [Google Scholar]
- 65.Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: Central role of complex III. J Biol Chem. 2003;278:36027–36031. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 66.Han D, Antunes F, Canali R, Rettori D, Cadenas E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J Biol Chem. 2003;278:5557–5563. doi: 10.1074/jbc.M210269200. [DOI] [PubMed] [Google Scholar]
- 67.Sevanian A, Wratten ML, McLeod LL, Kim E. Lipid peroxidation and phospholipase A2 activity in liposomes composed of unsaturated phospholipids: a structural basis for enzyme activation. Biochim Biophys Acta. 1988;961:316–327. doi: 10.1016/0005-2760(88)90079-3. [DOI] [PubMed] [Google Scholar]
- 68.Halliwell B, Gutteridge JM. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem J. 1984;219:1–14. doi: 10.1042/bj2190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sevanian A, Kim E. Phospholipase A2 dependent release of fatty acids from peroxidized membranes. J Free Radic Biol Med. 1985;1:263–271. doi: 10.1016/0748-5514(85)90130-8. [DOI] [PubMed] [Google Scholar]
- 70.O’Brien PJ. Intracellular mechanisms for the decomposition of a lipid peroxide. I. Decomposition of a lipid peroxide by metal ions, heme compounds, and nucleophiles. Can J Biochem. 1969;47:485–492. doi: 10.1139/o69-076. [DOI] [PubMed] [Google Scholar]
- 71.Paradies G, Petrosillo G, Pistolese M, Ruggiero FM. The effect of reactive oxygen species generated from the mitochondrial electron transport chain on the cytochrome c oxidase activity and on the cardiolipin content in bovine heart submitochondrial particles. FEBS Lett. 2000;466:323–326. doi: 10.1016/s0014-5793(00)01082-6. [DOI] [PubMed] [Google Scholar]
- 72.Belikova NA, Vladimirov YA, Osipov AN, Kapralov AA, Tyurin VA, Potapovich MV, Basova LV, Peterson J, Kurnikov IV, Kagan VE. Peroxidase activity and structural transitions of cytochrome c bound to cardiolipin-containing membranes. Biochemistry. 2006;45:4998–5009. doi: 10.1021/bi0525573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vladimirov YA, Proskurnina EV, Izmailov DY, Novikov AA, Brusnichkin AV, Osipov AN, Kagan VE. Mechanism of activation of cytochrome C peroxidase activity by cardiolipin. Biochemistry (Mosc) 2006;71:989–997. doi: 10.1134/s0006297906090070. [DOI] [PubMed] [Google Scholar]
- 74.Vladimirov YA, Proskurnina EV, Izmailov DY, Novikov AA, Brusnichkin AV, Osipov AN, Kagan VE. Cardiolipin activates cytochrome c peroxidase activity since it facilitates H(2)O(2) access to heme. Biochemistry (Mosc) 2006;71:998–1005. doi: 10.1134/s0006297906090082. [DOI] [PubMed] [Google Scholar]
- 75.Iwase H, Takatori T, Nagao M, Iwadate K, Nakajima M. Monoepoxide production from linoleic acid by cytochrome c in the presence of cardiolipin. Biochem Biophys Res Commun. 1996;222:83–89. doi: 10.1006/bbrc.1996.0701. [DOI] [PubMed] [Google Scholar]
- 76.Iwase H, Takatori T, Nagao M, Nijima H, Iwadate K, Matsuda Y, Kobayashi M. Formation of keto and hydroxy compounds of linoleic acid in submitochondrial particles of bovine heart. Free Radic Biol Med. 1998;24:1492–1503. doi: 10.1016/s0891-5849(98)00028-8. [DOI] [PubMed] [Google Scholar]
- 77.Imagawa T, Kasai S, Matsui K, Nakamura T. Methyl hydroperoxy-epoxy-octadecenoate as an autoxidation product of methyl linoleate: a new inhibitor-uncoupler of mitochondrial respiration. J Biochem. 1982;92:1109–1121. doi: 10.1093/oxfordjournals.jbchem.a134027. [DOI] [PubMed] [Google Scholar]
- 78.Degli Esposti M, Ghelli A, Crimi M, Estornell E, Fato R, Lenaz G. Complex I and complex III of mitochondria have common inhibitors acting as ubiquinone antagonists. Biochem Biophys Res Commun. 1993;190:1090–1096. doi: 10.1006/bbrc.1993.1161. [DOI] [PubMed] [Google Scholar]
- 79.Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Reversible blockade of electron transport during ischemia protects mitochondria and decreases myocardial injury following reperfusion. J Pharmacol Exp Ther. 2006;319:1405–1412. doi: 10.1124/jpet.106.110262. [DOI] [PubMed] [Google Scholar]
- 80.Tanaka-Esposito CC, Chen Q, Moghaddas S, Lesnefsky EJ. Reversible Blockade Of Electron Transport During Ischemia Protects Mitochondrial Function And Decreases Injury In The Aged Heart. Circulation. 2006;114:II-275. abstract 1436. [Google Scholar]
- 81.Lesnefsky EJ, He D, Moghaddas S, Hoppel CL. Reversal of mitochondrial defects before ischemia protects the aged heart. Faseb J. 2006;20:1543–1545. doi: 10.1096/fj.05-4535fje. [DOI] [PubMed] [Google Scholar]
- 82.Chen Q, Camara AK, Stowe DF, Hoppel CL, Lesnefsky EJ. Modulation of electron transport protects cardiac mitochondria and decreases myocardial injury during ischemia and reperfusion. Am J Physiol Cell Physiol. 2007;292:C137–C147. doi: 10.1152/ajpcell.00270.2006. [DOI] [PubMed] [Google Scholar]