

Abstract
 The electrophilic cyclization of substituted propargylic aryl ethers by I2, ICl and PhSeBr produces 3,4-disubstituted 2H-benzopyrans in excellent yields. This methodology results in vinylic halides or selenides under mild reaction conditions, and tolerates a variety of functional groups, including methoxy, alcohol, aldehyde and nitro groups.
The electrophilic cyclization of substituted propargylic aryl ethers by I2, ICl and PhSeBr produces 3,4-disubstituted 2H-benzopyrans in excellent yields. This methodology results in vinylic halides or selenides under mild reaction conditions, and tolerates a variety of functional groups, including methoxy, alcohol, aldehyde and nitro groups.
Introduction
2H-1-Benzopyrans, commonly known as 2H-benzopyrans or 2H-chromenes, are key structural units of a variety of biologically important compounds, many of which are pharmaceutically significant. The 2H-benzopyran Daurichromenic acid is known to exhibit anti-HIV properties,1 while Coutareagenin possesses antidiabetic activity.2 Derivatives of 3,4-diphenylchromans are known to have estrogenic activity.3 Numerous derivatives of 2H-benzopyrans are useful for treatment of proliferative skin disorder and microbial infections4 and show potent antifungal activity.5 Derivatives of 2H-benzopyrans, like 2,4-diphenyl-2H-benzopyran and 2,2,4-triphenyl-2H-benzopyran, have been studied for their photochromic behavior.6 Due to their biological and pharmaceutical importance, the isolation and synthesis of 2H-benzopyrans has received considerable attention in the literature.
Substituted 2H-benzopyrans have been synthesized in our laboratories by the Pd(II)-catalyzed cyclization of allylic aryl ethers,7 while the palladium-catalyzed cross-coupling of 4-trifluoromethanesulfonyloxy-2H-benzopyrans with arylboronic acids has also been reported in the literature.8 Syntheses of 2H-benzopyrans are also known using platinum and gold catalysis,9 Hg(II)-mediated cyclizations,10 Grignard reagents,11 and microwave irradiation.12
A wide range of carbocycles and heterocycles have been constructed using the electrophilic cyclization of disubstituted alkynes13 and transition metal-catalyzed cyclizations.14,15 Many researchers, including those from our group, have utilized these cyclizations for the synthesis of benzofurans,16 furans,17 benzo[b]thiophenes,18 thiophenes,19 naphthols,20 indoles,21 quinolines,22 isoquinolines,23 isocoumarins,24 isochromenes25 and polycyclic aromatics.26 Some methods are not compatible with functionality, while some require the use of costly metals as catalysts. Recently Barluenga and co-workers reported the synthesis of 2H-benzopyrans by the cyclization of aryl propargylic ethers using costly IPy2BF4 and HBF4.27 They also reported two examples of the iodocyclization of propargylic ethers using I2 in water. We report herein many examples and a general synthesis of 2H-benzopyrans in good yields via electrophilic cyclization using the simple, inexpensive electrophiles I2, ICl, and PhSeBr in nitromethane.
Results and Discussion
Our early studies mainly focused on the iodocyclization of substituted propargylic aryl ethers to give 3,4-disubstituted 2H-benzopyrans in excellent yields. Phenyl 3-phenyl-2-propynyl ether (1) was used as a model system for optimization of the reaction conditions using I2 or ICl (eq 1).
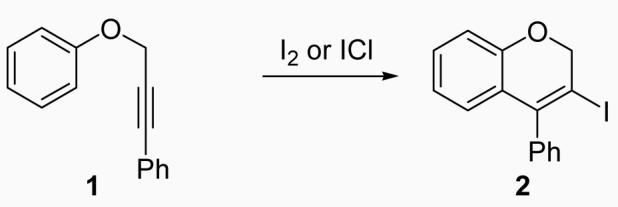
Early in this work, conditions similar to our previous iodocyclization reactions were used. For instance, the reaction was run with 0.25 mmol of 1, 2 equiv of NaHCO3 as the base, and 3 equiv of I2 in 5 ml of CH3CN at 25 °C to obtain a 61% isolated yield of the desired 3-iodo-4-phenyl-2H-benzopyran (2)27 (Table 1, entry 1). Solvents, like CH2Cl2, CH3OH and DMF, resulted in lower yields (entries 2-4), but CH3NO2 gave a yield of 77% (entry 5). No reaction was observed at 0 °C after 4 h (entry 6), while the reaction was messy and produced only a 42% yield at 40 °C (entry 7). Reducing the amount of I2 to 2 equiv decreased the yield significantly to 53% (entry 8), while an increase in the number of equivalents of I2 caused a slight decrease in the yield to 69% (entry 9). Reducing the reaction time to 12 h lowered the yield to 62% (entry 10), while there was no increase in yield when the reaction time was doubled to 48 h (entry 11). The presence of the base proved to be important for the reaction as the yield was reduced to 58% without the base and only 70% with one equiv of the base (entries 12 and 13). Several additional bases were examined in this reaction, but NaHCO3 was found to give the best yield (entries 14-16).
Table 1.
Optimization of the Cyclization of Phenyl 3-Phenyl-2-propynyl Ether using I2 as the Electrophile (eq 1)a
| entry | solvent | I2 (equiv) | base (equiv) | time (h) | % isolated yield |
|---|---|---|---|---|---|
| 1 | CH3CN | 3 | NaHCO3 (2) | 24 | 61 |
| 2 | CH2Cl2 | 3 | NaHCO3 (2) | 24 | 49 |
| 3 | CH3OH | 3 | NaHCO3 (2) | 24 | 41 |
| 4 | DMF | 3 | NaHCO3 (2) | 24 | 52 |
| 5 | CH3NO2 | 3 | NaHCO3 (2) | 24 | 77 |
| 6 | CH3NO2 | 3 | NaHCO3 (2) | 4 | 0b |
| 7 | CH3NO2 | 3 | NaHCO3 (2) | 24 | 42c |
| 8 | CH3NO2 | 2 | NaHCO3 (2) | 24 | 53 |
| 9 | CH3NO2 | 4 | NaHCO3 (2) | 24 | 69 |
| 10 | CH3NO2 | 3 | NaHCO3 (2) | 12 | 62 |
| 11 | CH3NO2 | 3 | NaHCO3 (2) | 48 | 76 |
| 12 | CH3NO2 | 3 | - | 24 | 58 |
| 13 | CH3NO2 | 3 | NaHCO3 (1) | 24 | 70 |
| 14 | CH3NO2 | 3 | NaOMe (2) | 24 | 69 |
| 15 | CH3NO2 | 3 | Na2CO3 (2) | 24 | 75 |
| 16 | CH3NO2 | 3 | KOH (2) | 24 | - |
Representative procedure: phenyl 3-phenyl-2-propynyl ether (0.25 mmol), I2, a base and the solvent (5 mL) were placed in a 4 dram vial and stirred at 25 °C for the indicated time.
The reaction was run at 0 °C.
The reaction was run at 40 °C.
In our previous cyclizations, ICl has proven to be a better electrophile for some substrates. Therefore, with a view to obtaining better yields, the cyclization of phenyl 3-phenyl-2-propynyl ether (1) was optimized using ICl as the electrophile, starting with our previously developed conditions. Because of the importance of the solvent in such reactions, the reaction was carried out using CH2Cl2, diethyl ether, THF, hexane, CH3OH and CH3NO2 at low temperatures (Table 2, entries 1-6). The solvent CH3NO2 gave the best yield of 69%, but the temperature had to be raised to −25 °C. The reaction was run at −25 °C with the second best solvent for the reaction, namely CH2Cl2, to get a slightly lower yield of 65% (entry 7). In all of these reactions, NaHCO3 was used as a base. The yield increased to 96% when the reaction was carried out without NaHCO3 (entry 8). Increasing the temperature to 0 °C gave undetermined side products with the desired compound being formed in less than a 5% yield (entry 9). Decreasing or increasing the amount of ICl gave slightly lower yields (entries 10 and 11). Thus, our optimized conditions using ICl are 0.25 mmol of 1, 1.5 equiv of ICl and 5 ml of CH3NO2 at −25 °C.
Table 2.
Optimization of the Cyclization of Phenyl 3-Phenyl-2-propynyl Ether using ICl as the Electrophile (eq 1)a
| entry | solvent | ICl (equiv) | temperature °C | time (h) | % yield |
|---|---|---|---|---|---|
| 1 | CH2Cl2 | 1.5 | −78 | 2 | 63b |
| 2 | Ether | 1.5 | −78 | 2 | 51b |
| 3 | THF | 1.5 | −78 | 2 | 58b |
| 4 | Hexane | 1.5 | −78 | 2 | 49b |
| 5 | CH3OH | 1.5 | −78 | 2 | -b |
| 6 | CH3NO2 | 1.5 | −25 | 2 | 69b |
| 7 | CH2Cl2 | 1.5 | −25 | 2 | 65b |
| 8 | CH3NO2 | 1.5 | −25 | 0.5 | 96 |
| 9 | CH3NO2 | 1.5 | 0 | 0.5 | <5 |
| 10 | CH3NO2 | 1.2 | −25 | 2 | 71 |
| 11 | CH3NO2 | 2.0 | −25 | 0.5 | 85 |
Representative procedure: phenyl 3-phenyl-2-propynyl ether (0.25 mmol), ICl and the solvent (5 mL) were placed in a 4 dram vial and stirred at the indicated temperature for the indicated time.
NaHCO3 (0.5 mmol) was added.
After obtaining our best conditions using either I2 or ICl, we decided to study the scope of this reaction on various substrates. Ethers 1, 4, 5 and 6 were obtained by standard Sonogashira chemistry28 using commercially available starting materials (Scheme 1). Ethers 9 and 10 were obtained by a two step approach, the first step being the synthesis of the substituted aryl propargylic ethers 7 and 8, followed by Sonogashira chemistry (Scheme 2). Ethers 11, 12 and 13 were synthesized by a Mitsunobu reaction (Scheme 3), while ethers 14,29 15,30 16,30 17,27 18,31 19,31 20,31 2132 and 2233 (refer to Table 3) were previously reported in the literature.
Scheme 1.

Scheme 2.

Scheme 3.

Table 3.
Synthesis of 3,4-Disubstituted 2H-Benzopyrans by Electrophilic Cyclization of Propargylic Aryl Ethers.a
| entry | ether | condition | product(s) | % isolated yield |
||
|---|---|---|---|---|---|---|
| 1 |  |
1 | A |  |
2 | 77 |
| 2 | 1 | B | 2 | 96 | ||
| 3 | 1 | C |  |
23 | 95 | |
| 4 | 4 | A | 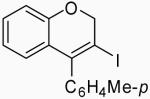 |
24 | 88 | |
| 5 | 4 | B | 24 | 76 | ||
| 6 | 5 | A | 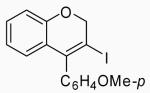 |
25 | 89 | |
| 7 | 5 | B | 25 | 92 | ||
| 8 | 14 | A | 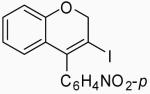 |
26 | 59 | |
| 9 | 14 | B | 26 | 53 | ||
| 10 |  |
6 | A | 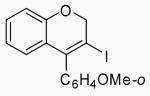 |
27 | 85 |
| 11 | 6 | B | 27 | 88 | ||
| 12 | 15 | A | 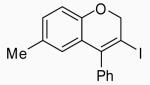 |
28 | 79 | |
| 13 | 15 | B | 28 | 74 | ||
| 14 | 16 | A | 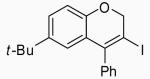 |
29 | 82 | |
| 15 | 16 | B | 29 | 77 | ||
| 16 | 17 | A | 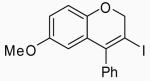 |
30 | 92 | |
| 17 | 17 | B | 30 | 61 | ||
| 18 | 18 | A | 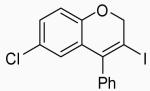 |
31 | 52 | |
| 19 | 18 | B | 31 | 75 | ||
| 20 | 9 | A | 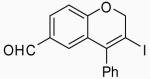 |
32 | 76b [2:1] |
|
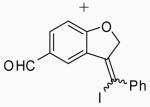 |
33 | |||||
| 21 | 9 | B | 32 + 33 | 78b [1:11] |
||
| 22 | 9 | D | 32 + 33 | 77b [6:1] |
||
| 23 | 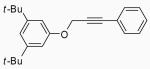 |
10 | A | 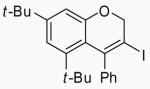 |
34 | 93 |
| 24 | 10 | B | 34 | 77 | ||
| 25 | 11 | A | 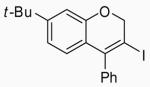 |
35 | 72 | |
| 26 | 11 | B | 35 | 79 | ||
| 27 | 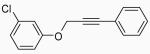 |
19 | A | 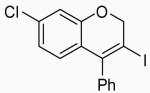 |
36 | 21 |
 |
37 | 28 | ||||
| 28 | 19 | B |
36 + |
27 | ||
| 37 | 42 | |||||
| 29 | 20 | B | 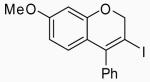 |
38 | 82b [2:3] |
|
 |
39 | |||||
| 30 | 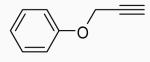 |
3 | A | - | ||
| 31 | 3 | B | - | |||
| 32 | 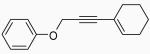 |
12 | A | 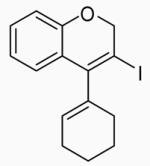 |
40 | 74 |
| 33 | 12 | B | 40 | 72 | ||
| 34 | 21 | A | - | |||
| 35 | 21 | B | - | |||
| 36 | 21 | C | 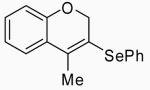 |
41 | 79 | |
| 37 | 22 | A | 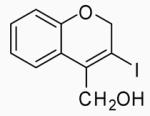 |
42 | 79 | |
| 38 | 22 | B | 42 | 72 | ||
| 39 | 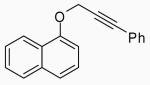 |
13 | A | 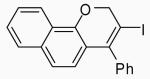 |
43 | 61 |
| 40 | 13 | B | 43 | 50 |
Representative procedure for conditions A: ether (0.25 mmol), NaHCO3 (0.50 mmol), I2 (0.75 mmol) and CH3NO2 (5 mL) were placed in a 4 dram vial and stirred at 25 °C for 24 h. Conditions B: ether (0.25 mmol), ICl (0.375 mmol) and CH3NO2 (5 mL) were placed in a 4 dram vial and stirred at −25 °C for 30 min. Conditions C: ether (0.25 mmol), PhSeBr (1.2 equiv) and CH2Cl2 (5 mL) were placed in a 4 dram vial and stirred at 25 °C for the indicated time. Conditions D: CH2Cl2 was used as the solvent for conditions B at −78 °C.
The ratios were determined by 1H NMR spectroscopy.
Cyclizations were then carried out using our optimized conditions for I2 (conditions A) or ICl (conditions B). 3,4-Disubstituted 2H-benzopyrans were obtained in good yields using I2 or ICl when the substituent on the propargylic alkyne was either a simple phenyl or an alkenyl group (Table 3, entries 1, 2, 32 and 33). An alkyl substituent on the alkyne terminus did not give the desired product with I2 or ICl as the electrophile, but worked with PhSeBr (entries 34-36). However, an hydroxymethyl-substituted alkyne gave good yields with I2, as well as ICl (entries 37 and 38). Simple phenyl propargyl ether (3) failed to give any of the desired product with I2 or ICl (entries 30 and 31).
The introduction of substituents on the aryl groups has a considerable effect on the yield of the reaction. Substituents were first introduced onto the aromatic ring attached to the alkyne. Electron-donating groups, like Me and MeO in the para or ortho positions, gave good yields (entries 4-7, 10 and 11), while an electron-withdrawing group, like a NO2 group, gave relatively poor yields of 59% with I2 and 53% with ICl (entries 8 and 9). Introducing substituents onto the aromatic ring attached to the oxygen moiety also has a pronounced effect. Electron-donating groups, like Me, t-Bu and MeO, on the phenyl ring para to the oxygen gave better yields with I2 as the electrophile (entries 12, 14 and 16) than those obtained using ICl (entries 13, 15 and 17). Compound 30 was thus obtained in improved yields without the use of ion exchange resins as additives, as was reported by Barluenga.27 The sterically-hindered ether 10 also gave good results (entries 23 and 24).
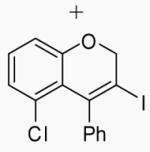
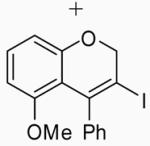
Placing an electron-withdrawing chlorine in the position para to the oxygen gave somewhat lower yields (entries 18 and 19) of the desired isomer. An aldehyde group in the position para to the oxygen gave a mixture of two inseparable regioisomers, the ratio of which depended on the electrophile used and the reaction temperature (entries 20 and 21). The benzopyran isomer was favored when the reaction was carried out at −78 °C with CH2Cl2 as the solvent and ICl as the electrophile (entry 22).
In order to study the regiochemistry of cyclization, the reaction was carried out with substituents meta to the oxygen moiety. A sterically bulky t-Bu group in the meta position gave selectively one product 35 (entries 25 and 26). A less bulky Cl in the meta position gave two regioisomers 36 and 37 (entries 27 and 28), while a MeO group gave a similar mixture of two inseparable regioisomers 38 and 39 (entry 29). α-Naphthyl propargylic ether 13 also gave the desired compound 43 in modest yields (entries 39 and 40).
Phenyl 3-phenyl-2-propynyl ether (3) gave a 95% yield of the cyclized product 23 when PhSeBr was used as the electrophile (entry 3). Surprisingly, ether 21, which failed to give the desired product with I2 or ICl, gave a 79% yield of compound 41 with PhSeBr as the electrophile (entry 36).
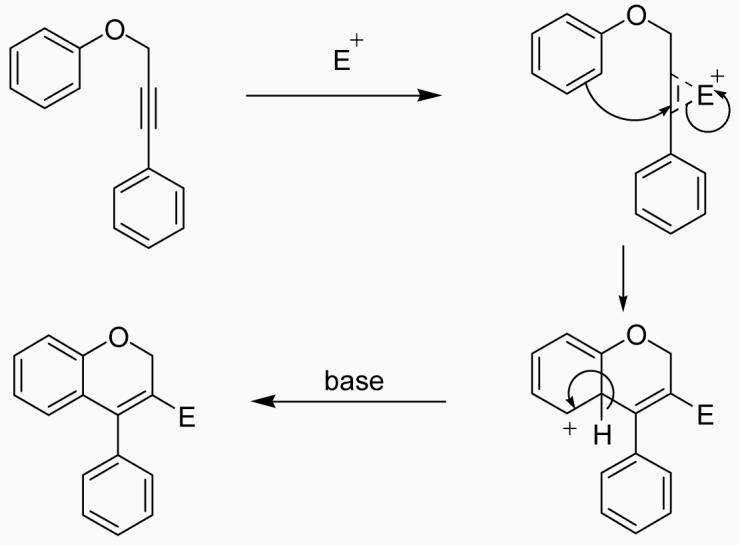
We believe that the mechanism of these cyclizations involves initial formation of an iodonium or selenonium intermediate by attack of the electrophile on the triple bond, followed by electrophilic attack on the electron cloud of the aromatic ring. Loss of a proton gives the 2H-benzopyran (Scheme 4).
Scheme 4.
During our cyclization studies, ether 9 gave a mixture of two isomers, one being the expected benzopyran product and the other a possible five-membered ring dihydrofuran product. This encouraged us to confirm the structure of our cyclized product 2 using X-ray crystallography (see the Supporting Information), which indeed proved to be a 2H-benzopyran.
The iodo-(2H)-benzopyrans obtained by iodocyclization appear highly promising as intermediates for the preparation of more highly substituted benzopyrans. Indeed 3-iodobenzopyrans, like the ones prepared here, have recently been further elaborated by palladium-catalysed cross-coupling reactions.27 To further prove the utility of our methodology, we have carried out the palladium/copper-catalyzed reaction of our product 2 with 5-ethynyl-2-fluorotoluene to obtain 44 in an 87 % yield. Palladium catalysed CO insertion in our product 42 gave compound 45 in an overall 72 % yield (Scheme 5).
Scheme 5.

Conclusions
3,4-Disubstituted 2H-benzopyrans have been obtained from starting materials that are easy to synthesize. The reaction conditions are mild and the products are easy to isolate in good yields. The iodine moiety in the products provides a useful handle for further functionalization of the resulting heterocycles. A polycyclic Sonogashira product 44 has been obtained in a good yield. Our methodology tolerates functional groups, including alcohol, aldehyde, methoxy and nitro groups. In addition to I2 and ICl, PhSeBr has also been used as the electrophile. The structure of 3-iodo-4-phenyl-2H-benzopyran (2) has been confirmed by X-ray crystallography.
Experimental Section
General procedure for the palladium/copper-catalyzed reaction of phenyl propargyl ether with aryl halides
To a solution of 2.5 mmol of the aryl halide in Et3N (15 ml) was added PdCl2(PPh3)2 (2 mol %), which was then stirred for 5 min. CuI (1.5 mol %) was then added and the flask was sealed and flushed with Ar. The reaction was stirred for 20 min. A solution of 3.0 mmol of phenyl propargyl ether in 2 mL of Et3N was then added dropwise and the reaction mixture was allowed to stir at room temperature for the desired time. After the reaction was over, the resulting solution was diluted with H2O (10 ml) and extracted with diethyl ether (3 × 15 mL). The combined ether fractions were dried over anhydrous Na2SO4 and concentrated under vacuum to yield the crude product. The crude product was purified by flash chromatography on silica gel using ethyl acetate/hexanes as the eluent.
Phenyl 3-p-tolylprop-2-yn-1-yl ether (4)
This compound was obtained as a white solid: mp 71-72 °C; 1H NMR (300 MHz, CDCl3) δ 2.30 (s, 3H), 4.86 (s, 2H), 6.94-7.08 (m, 5H), 7.18-7.33 (m, 4H); 13C NMR (75 MHz, CDCl3) δ 21.7, 56.9, 83.5, 87.5, 115.2, 119.5, 121.6, 129.3, 129.7, 132.0, 139.0, 158.1; IR (neat, cm−1) 3032, 2914, 1598, 1490, 1214, 1029; HRMS m/z 222.10477 (calcd C16H14O, 222.10447).
General procedure for the palladium/copper-catalyzed reaction of terminal alkynes with iodobenzene
To a solution of 4.5 mmol of iodobenzene in Et3N (15 ml), was added PdCl2(PPh3)2 (2 mol %), and CuI (1.5 mol %), and the mixture was stirred for 30 min under Ar. A solution of 3.0 mmol of the terminal alkyne in 2 mL of Et3N was then added dropwise and the reaction mixture was allowed to stir at room temperature for the desired time. After the reaction was over, the resulting solution was diluted with H2O (10 ml) and extracted with diethyl ether (3 × 15 mL). The combined ether fractions were dried over anhydrous Na2SO4 and concentrated under vacuum to yield the crude product. The crude product was purified by flash chromatography on silica gel using ethyl acetate/hexanes as the eluent.
4-(3-Phenylprop-2-yn-1-yloxy)benzaldehyde (9)
This compound was obtained as a brown solid: mp 86-87 °C; 1H NMR (300 MHz, CDCl3) δ 5.00 (s, 2H), 7.14 (d, J = 8.8 Hz, 2H), 7.25-7.32 (m, 3H), 7.41-7.44 (m, 2H), 7.87 (d, J = 8.8 Hz, 2H), 9.90 (s, 1H); 13C NMR (75 MHz, CDCl3) δ 57.0, 82.9, 88.1, 115.4, 122.0, 128.5, 129.1, 130.6, 132.0, 132.1, 162.8, 191.0; IR (neat, cm−1) 3078, 2827, 1690, 1598, 1250, 1009; HRMS m/z 236.08409 (calcd C16H12O2, 236.08373).
General procedure for the triphenylphosphine/diethyl azodicarboxylate-promoted formation of the substituted phenyl propargylic ethers
To a solution of 1.31 g of PPh3 (5.0 mmol) in dry benzene (15 ml) was added the substituted propargylic alcohol (5.0 mmol) and the substituted phenol (5.0 mmol) under an inert atmosphere with stirring. Diethyl azodicarboxylate (0.87 g, 5.0 mmol) was then added slowly and the reaction mixture was stirred at r.t. for 18 to 36 h. After the reaction was complete, the solvent was evaporated under reduced pressure and the crude product was purified by flash chromatography on silica gel using hexanes/ethyl acetate as the eluent.
3-tert-Butylphenyl 3-phenylprop-2-yn-1-y1 ether (11)
This compound was obtained as a yellow oil: 1H NMR (300 MHz, CDCl3) δ 1.30 (s, 9H), 4.86 (s, 2H), 6.83 (dd, J = 8.0, 2.3 Hz, 1H), 6.96-7.02 (m, 1H), 7.08 (t, J = 1.9 Hz, 1H), 7.19-7.25 (m, 4H), 7.39-7.42 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 31.4, 34.8, 56.6, 84.3, 87.2, 111.2, 113.1, 118.6, 122.4, 128.4, 128.7, 129.1, 131.9, 153.0, 157.7; IR (neat, cm−1) 3067, 2955, 2868, 1588, 1485, 1270, 1029; HRMS m/z 264.15187 (calcd C19H20O, 264.15142).
General procedure for iodocyclization
To a solution of 0.25 mmol of the ether and 3 mL of CH3NO2, 2.0 equiv of NaHCO3 and 3.0 equiv of I2 dissolved in 2 mL of CH3NO2 was added gradually. The reaction mixture was allowed to stir at room temperature for the desired time. Alternatively, to a solution of 0.25 mmol of the ether and 3 mL of CH3NO2 at −25 to −30 °C, 1.5 equiv of ICl dissolved in 2 mL of CH3NO2 was added gradually. The reaction mixture was allowed to stir at −25 to −30 °C for the desired time. The excess I2 or ICl was removed by washing with satd aq Na2S2O3. The mixture was then extracted by diethyl ether (3 × 10 mL). The combined ether layers were dried over anhydrous Na2SO4 and concentrated under vacuum to yield the crude product, which was purified by flash chromatography on silica gel using hexanes/ethyl acetate as the eluent.
3-Iodo-4-phenyl-2H-benzopyran (2)
This compound was obtained as a pale yellow solid: mp 99-100 °C; 1H NMR (300 MHz, CDCl3) δ 5.06 (s, 2H), 6.61 (dd, J = 7.7, 1.6 Hz, 1H), 6.76 (dt, J = 7.7, 1.1 Hz, 1H), 6.85 (dd, J = 8.0, 1.0 Hz, 1H), 7.14 (dd, J = 7.9, 1.6 Hz, 1H), 7.18-7.22 (m, 2H), 7.39-7.46 (m, 3H); 13C NMR (75 MHz, CDCl3) δ 75.1, 91.2, 116.1, 121.7, 124.2, 126.5, 128.3, 128.7, 129.5, 129.7, 140.0, 142.0, 153.3; IR (neat, cm−1) 3062, 2904, 1475, 1219, 1029, 994; HRMS m/z 333.98600 (calcd C15H11IO, 333.98547).
General procedure for the PhSeBr cyclizations
To a solution of 0.25 mmol of the substituted phenyl propargylic ether and CH2Cl2 (3 mL), 0.375 mmol of PhSeBr dissolved in 2 mL of CH2Cl2 was added dropwise. The mixture was allowed to stir at room temperature for the desired time. The reaction mixture was washed with 20 mL of water and extracted with diethyl ether (3 × 10 mL). The combined ether layers were dried over anhydrous Na2SO4 and concentrated under vacuum to yield the crude product, which was further purified by flash chromatography on silica gel using hexanes/ethyl acetate as the eluent.
4-Phenyl-3-phenylselenyl-2H-benzopyran (23)
This compound was obtained as a brown oil: 1H NMR (300 MHz, CDCl3) δ 4.84 (s, 2H), 6.84-6.87 (m, 2H), 6.94 (t, J = 7.4 Hz, 1H), 7.12-7.25 (m, 5H), 7.27-7.34 (m, 4H), 7.40-7.44 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 29.9, 71.1, 115.3, 121.4, 124.0, 128.2, 128.4, 128.8, 129.1, 129.2, 129.2, 129.3, 129.6, 134.6, 141.0, 158.4; IR (neat, cm−1) 3052, 2919, 1582, 1480, 1224, 1024; HRMS m/z 364.03707 (calcd C21H16OSe, 364.03664).
3-(4-Fluoro-3-methylphenylethynyl)-4-phenyl-2H-benzopyran (44)
This compound was prepared by the following procedure. To a solution of 0.17 g of 3-iodo-4-phenyl-2H-benzopyran (2) (0.5 mmol) in Et3N (5 ml), was added PdCl2(PPh3)2 (2 mol %) and CuI (1.5 mol %), and the mixture was stirred for 30 min under Ar. 0.6 Mmol of 5-ethynyl-2-fluorotoluene dissolved in 1 mL of Et3N was then added dropwise and the reaction mixture was allowed to stir at room temperature for 24 h. The reaction was monitored by TLC and an additional 0.4 mmol of the 5-ethynyl-2-fluorotoluene dissolved in 1 mL of Et3N was added slowly under an inert atmosphere and the reaction mixture was further allowed to stir at room temperature for another 24 h. After the reaction was over, the resulting solution was diluted with H2O (5 ml) and extracted with diethyl ether (3 × 10 mL). The combined ether fractions were dried over anhydrous Na2SO4 and concentrated under vacuum to yield the crude product, which was purified by flash chromatography on silica gel using hexanes/ethyl acetate as the eluent to obtain the desired compound 44 in an 87% yield as a pale yellow solid: mp 79-80 °C; 1H NMR (300 MHz, CDCl3) δ 2.18 (d, J = 1.6 Hz, 3H), 4.88 (s, 2H), 6.81-6.93 (m, 4H), 6.97-7.05 (m, 2H), 7.10-7.21 (m, 1H), 7.39-7.45 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 14.6 (d, J = 3.4 Hz), 68.0, 86.7 (d, J = 1.9 Hz), 95.1 (d, J = 0.8 Hz), 112.4, 115.3 (d, J = 9.4 Hz), 116.4, 118.9 (d, J = 3.8 Hz), 121.8, 124.3, 125.3 (d, J = 18.2 Hz), 126.8, 128.2 (d, J = 10.2 Hz), 129.8, 130.2, 130.8 (d, J = 8.4 Hz), 134.7 (d, J = 5.7 Hz), 136.6, 140.8, 154.6, 160.1, 162.6; IR (neat, cm−1) 3047, 2919, 2192, 1480, 1224, 1106; HRMS m/z 340.12694 (calcd C24H17FO, 340.12634).
1,4-Dihydro-2,5-dioxacyclopenta[a]naphthalen-3-one (45)
This compound was prepared by the following procedure. To a solution of 0.14 g of 4-hydroxymethyl-3-iodo-2H-benzopyran (42) (0.5 mmol) in DMF (5 ml) was added PdCl2(PPh3)2 (5 mol %) and K2CO3 (2 equiv), and the mixture was stirred for 6 h under an atmosphere of CO at 60 °C. The reaction was monitored by TLC and, after completion of the reaction, the resulting solution was cooled to room temperature, diluted with ether (15 ml), and washed with brine (15 ml). The aqueous layer was extracted with diethyl ether (3 × 15 mL). The combined ether fractions were dried over anhydrous Na2SO4 and concentrated under vacuum to yield the crude product, which was purified by flash chromatography on silica gel using hexanes/ethyl acetate as the eluent to obtain the desired compound 45 in an 72% yield as a brown solid: mp 151-152 °C; 1H NMR (400 MHz, CDCl3) 5.13-5.15 (m, 4H), 6.92-6.99 (m, 2H), 7.07 (dd, J = 7.5, 1.5 Hz, 1H), 7.34 (dt, J = 8.2, 0.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 63.3, 68.8, 116.4, 117.2, 118.3, 122.0, 124.2, 133.8, 154.1, 154.8, 170.7; IR (neat, cm−1) 2361, 1744, 1666, 1449, 1336, 1181, 1052; HRMS m/z 188.04776 (calcd C11H8O3, 188.04743).
Supplementary Material
Acknowledgements
We thank the National Institute of General Medical Sciences (GM070620) and the National Institutes of Health Kansas University Chemical Methodologies and Library Development Center of Excellence (P50 GM069663) for support of this research, Johnson Matthey, Inc., and Kawaken Fine Chemicals Co., Ltd., for donations of palladium catalysts and Dr. Arkady Ellern and the Molecular Structure Laboratory of Iowa State University for the single crystal X-ray results.
Footnotes
Supporting Information Available: General experimental procedures and spectral data for all previously unreported starting materials and products. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Zhendong J, Ying K. PCT Int. Appl. 2004;25 [Google Scholar]; (b) Iwata N, Wang N, Yao X, Kitanaka S. J. Nat. Prod. 2004;67:1106. doi: 10.1021/np0303916. [DOI] [PubMed] [Google Scholar]; (c) Hu H, Harrison TJ, Wilson PD. J. Org. Chem. 2004;69:3782. doi: 10.1021/jo049703f. [DOI] [PubMed] [Google Scholar]
- 2.Korec R, Sensch KH, Zoukas T. Arzneim. Forsch. 2000;50:122. [PubMed] [Google Scholar]
- 3.(a) Carney RWJ, Bencze WL, Wojtkunski J, Renzi AA, Dorfman L, De Stevens G. J. Med. Chem. 1966;9:516. doi: 10.1021/jm00322a017. [DOI] [PubMed] [Google Scholar]; (b) Gauthier S, Caron B, Cloutier J, Dory YL, Favre A, Larouche D, Mailhot J, Ouellet C, Schwerdtfeger A, Leblanc G, Martel C, Simard J, Merand Y, Belanger A, Labrie C, Labrie F. J. Med. Chem. 1967;40:2117. doi: 10.1021/jm970095o. [DOI] [PubMed] [Google Scholar]
- 4.Bonadies F, Di Fabio R, Bonini C. J. Org. Chem. 1984;49:1647. [Google Scholar]
- 5.Morandim AA, Bergamo DCB, Kato MJ, Cavalheiro AJ, Bolzani VS, Furlan M. Phytochem. Anal. 2005;16:282. doi: 10.1002/pca.843. [DOI] [PubMed] [Google Scholar]
- 6.(a) Martins CI, Coelho PJ, Carvalho LM, Oliveira-Campos AMF, Samat A, Guglielmetti R. Helv. Chim. Acta. 2003;86:570. [Google Scholar]; (b) Kodama Y, Nakabayashi T, Segawa K, Hattori E, Sakuragi M, Nishi N, Sakuragi H. J. Phys. Chem. A. 2000;104:11478. [Google Scholar]; (c) Padwa A, Andrew A, Lee GA, Owens W. J. Org. Chem. 1975;40:1142. [Google Scholar]
- 7.Larock RC, Wei L, Hightower TR. Synlett. 1998:522. [Google Scholar]
- 8.Eguchi T, Hoshino Y, Ayame A. Bull. Chem. Soc. Jpn. 2002;75:581. [Google Scholar]
- 9.(a) Pastine SJ, Youn SW, Sames D. Tetrahedron. 2003;59:8859. [Google Scholar]; (b) Nevado C, Echavarren AM. Chem. Eur. J. 2005;11:3155. doi: 10.1002/chem.200401069. [DOI] [PubMed] [Google Scholar]
- 10.Balasubramanian T, Balasubramanian KK. Tetrahedron Lett. 1991;32:6641. [Google Scholar]
- 11.Kirkiacharian BS, Danan A, Koutsourakis PG. Synthesis. 1991;10:879. [Google Scholar]
- 12.Dintzner MR, Lyons TW, Akroush MH, Wucka P, Rzepka AT. Synlett. 2005:1046. [Google Scholar]
- 13.(a) Knight DW, Redfern AL, Gilmore J. J. Chem. Soc., Perkin Trans. 1. 2002:622. [Google Scholar]; (b) Bellina F, Biagetti M, Carpita A, Rossi R. Tetrahedron Lett. 2001;42:2859. [Google Scholar]; (c) Bellina F, Biagetti M, Carpita A, Rossi R. Tetrahedron. 2001;57:2857. [Google Scholar]; (d) Djuardi E, McNelis E. Tetrahedron Lett. 1999;40:7193. [Google Scholar]; (e) Marshall JA, Yanik MM. J. Org. Chem. 1999;64:3798. [Google Scholar]; (f) Knight DW, Redfern AL, Gilmore J. Chem. Commun. 1998:2207. [Google Scholar]; (g) Redfern AL, Gilmore J. Synlett. 1998:731. [Google Scholar]; (h) Ren XF, Konaklieva MI, Shi H, Dicy H, Lim DV, Gonzales J, Turos E. J. Org. Chem. 1998;63:8898. [Google Scholar]; (i) Bew SP, Knight DW. Chem. Commun. 1996:1007. [Google Scholar]; (j) El-Taeb GMM, Evans AB, Jones S, Knight DW. Tetrahedron Lett. 2001;42:5945. [Google Scholar]
- 14.For a review, see: Larock RC. Top. Organomet. Chem. 2005;14:147.
- 15.(a) Roesch KR, Larock RC. Org. Lett. 1999;4:553. [Google Scholar]; (b) Nan Y, Miao H, Yang Z. Org. Lett. 2000;2:297. doi: 10.1021/ol991327b. [DOI] [PubMed] [Google Scholar]; (c) Nevado C, Echavarren AM. Synthesis. 2005:167. [Google Scholar]; (d) Larock RC, Yum EK. J. Am. Chem. Soc. 1991;113:6689. [Google Scholar]; (e) Larock RC, Yum EK, Refvik MD. J. Org. Chem. 1998;63:7652. [Google Scholar]; (f) Roesch KR, Larock RC. J. Org. Chem. 1998;63:5306. [Google Scholar]; (g) Cacchi S, Fabrizi G, Moro L. Tetrahedron Lett. 1998;39:5101. [Google Scholar]; (h) Cacchi S, Fabrizi G, Moro L. Synlett. 1998:741. [Google Scholar]; (i) Arcadi A, Cacchi S, Rosario MD, Fabrizi G, Marinelli F. J. Org. Chem. 1996;61:9280. [Google Scholar]; (j) Cacchi S, Carnicelli V, Marinelli F. J. Organomet. Chem. 1994;475:289. [Google Scholar]; (k) Arcadi A, Cacchi S, Marinelli F. Tetrahedron Lett. 1992;33:3915. [Google Scholar]; (l) Cacchi S. J. Organomet. Chem. 1999;576:42. [Google Scholar]
- 16.(a) Arcadi A, Cacchi S, Fabrizi G, Marinelli F, Moro L. Synlett. 1999:1432. [Google Scholar]; (b) Yue D, Yao T, Larock RC. J. Org. Chem. 2005;70:10292. doi: 10.1021/jo051299c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Larock RC, Stinn DE. Tetrahedron Lett. 1988;29:4687. [Google Scholar]
- 17.(a) Sniady A, Wheeler KA, Dembinski R. Org. Lett. 2005;7:1769. doi: 10.1021/ol050372i. [DOI] [PubMed] [Google Scholar]; (b) Yao T, Zhang X, Larock RC. J. Am. Chem. Soc. 2004;126:11164. doi: 10.1021/ja0466964. [DOI] [PubMed] [Google Scholar]; (c) Larock RC, Liu CL. J. Org. Chem. 1983;48:2151. [Google Scholar]
- 18.(a) Hessian KO, Flynn BL. Org. Lett. 2003;5:4377. doi: 10.1021/ol035663a. [DOI] [PubMed] [Google Scholar]; (b) Yue D, Larock RC. J. Org. Chem. 2002;67:1905. doi: 10.1021/jo011016q. [DOI] [PubMed] [Google Scholar]; (c) Larock RC, Yue D. Tetrahedron Lett. 2001;42:6011. [Google Scholar]
- 19.Flynn BL, Flynn GP, Hamel E, Jung MK. Bioorg. Med. Chem. Lett. 2001;11:2341. doi: 10.1016/s0960-894x(01)00436-x. [DOI] [PubMed] [Google Scholar]
- 20.Zhang X, Sarkar S, Larock RC. J. Org. Chem. 2006;71:236. doi: 10.1021/jo051948k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Yue D, Larock RC. Org. Lett. 2004;6:1037. doi: 10.1021/ol0498996. [DOI] [PubMed] [Google Scholar]; (b) Yao T, Yue D, Larock RC. J. Comb. Chem. 2005;7:809. doi: 10.1021/cc050062r. [DOI] [PubMed] [Google Scholar]; (c) Amjad M, Knight DW. Tetrahedron Lett. 2004;45:539. [Google Scholar]; (d) Barluenga J, Trincado M, Rubio E, Gonzalez JM. Angew. Chem., Int. Ed. 2003;42:2406. doi: 10.1002/anie.200351303. [DOI] [PubMed] [Google Scholar]
- 22.Zhang X, Campo MA, Yao T, Larock RC. Org. Lett. 2005;7:763. doi: 10.1021/ol0476218. [DOI] [PubMed] [Google Scholar]
- 23.Huang Q, Hunter JA, Larock RC. J. Org. Chem. 2002;67:3437. doi: 10.1021/jo020020e. [DOI] [PubMed] [Google Scholar]
- 24.Yao T, Larock RC. J. Org. Chem. 2003;68:5936. doi: 10.1021/jo034308v. [DOI] [PubMed] [Google Scholar]
- 25.Yue D, Della Cá N, Larock RC. Org. Lett. 2004;6:1581. doi: 10.1021/ol049690s. [DOI] [PubMed] [Google Scholar]
- 26.(a) Yao T, Campo MA, Larock RC. J. Org. Chem. 2005;70:3511. doi: 10.1021/jo050104y. [DOI] [PubMed] [Google Scholar]; (b) Yao T, Campo MA, Larock RC. Org. Lett. 2004;6:2677. doi: 10.1021/ol049161o. [DOI] [PubMed] [Google Scholar]
- 27.Barluenga J, Trincado M, Marco-Arias M, Ballesteros A, Rubio E, Gonzalez JM. Chem. Commun. 2005:2008. doi: 10.1039/b500303b. [DOI] [PubMed] [Google Scholar]
- 28.Sonogashira K. In: Metal-Catalyzed Cross-Coupling Reactions. Diederich F, Stang PJ, editors. Wiley-VCH: Weinheim; Germany: 1998. pp. 203–229. Chapter 5. [Google Scholar]; (b) Sonogashira K, Tohda Y, Hagihara N. Tetrahedron Lett. 1975:4467. [Google Scholar]
- 29.Berg TC, Bakken V, Gundersen L, Petersen D. Tetrahedron. 2006;62:6121. [Google Scholar]
- 30.Muldakhmetov ZM, Agelmenev ME, Sovetov ES. Zh. Fiz. Khim. 1999;73:2085. [Google Scholar]
- 31.Issei W, Junya I. Chem. Pharm. Bull. 1963;11:1042. [Google Scholar]
- 32.Pastine SJ, Youn SW, Sames D. Org. Lett. 2003;5:1055. doi: 10.1021/ol034177k. [DOI] [PubMed] [Google Scholar]
- 33.Tolstikov GA, Tolstikov AG, Miftakhov MS, Spirikhin LV. Zh. Org. Khim. 1986;22:1620. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.