

Abstract
This study was aimed at evaluating the potent and specific aldose reductase inhibitor fidarestat, on diabetes-associated cataract formation, and retinal oxidative-nitrosative stress, glial activation, and apoptosis. Control and streptozotocin-diabetic rats were treated with or without fidarestat (16 mg kg-1d-1) for 10 weeks after an initial 2-week period without treatment. Lens changes were evaluated by indirect ophthalmoscopy and portable slit lamp. Nitrotyrosine, poly(ADP-ribose), and glial fibrillary acidic protein expression were assessed by immunohistochemistry. The rate of apoptosis was quantified in flat-mounted retinas by TUNEL assay with immunoperoxidase staining. To dissect the effects of high glucose exposure in retinal microvascular cells, primary bovine retinal pericytes and endothelial cells were cultured in 5 or 30 mM glucose, with or without fidarestat (10 μM) for 3-14 days. Apoptosis was assessed by TUNEL assay, nitrotyrosine and poly(ADP-ribose) by immunocytochemistry, and Bax and Bcl-2 expression by Western blot analyses. Fidarestat treatment prevented diabetic cataract formation and counteracted retinal nitrosative stress, and poly(ADP-ribose) polymerase activation, as well as glial activation. The number of TUNEL-positive nuclei (mean ± SEM) was increased approximately 4-fold in diabetic rats vs. controls (207±33 vs. 49±4, p<0.01), and this increase was partially prevented by fidarestat (106±34, p<0.05 vs. untreated diabetic group). The apoptotic cell number increased with the prolongation of exposure of both pericytes and endothelial cells to high glucose levels. Fidarestat counteracted nitrotyrosine and poly(ADP-ribose) accumulation and apoptosis in both cell types. Antiapoptotic effect of fidarestat in high glucose-exposed retinal pericytes was not associated with the inhibition of Bax or increase in Bcl-2 expression. In conclusion, the findings, i) support an important role for aldose reductase in diabetes-associated cataract formation, and retinal oxidative-nitrosative stress, glial activation, and apoptosis, and ii) provide a rationale for the development of aldose reductase inhibitors, and, in particular, fidarestat, for the prevention and treatment of diabetic ocular complications.
Keywords: aldose reductase, apoptosis, diabetic cataract, fidarestat, glial activation, nitrosative stress, poly(ADP-ribose) polymerase
Introduction
Multiple mechanisms including increased sorbitol pathway activity (1,2), non-enzymatic glycation and glycoxidation (3,4), enhanced oxidative-nitrosative stress (5,6), protein kinase C (7,8), poly(ADP-ribose) polymerase (9,10) and lipoxygenase (11,12) activation, and others have been implicated in the pathogenesis of chronic diabetic complications. The relations between individual mechanisms are not completely understood and require specific studies. On the one hand, numerous reports indicate that increased activity of the first enzyme of the sorbitol pathway, aldose reductase (AR), contributes to diabetes-associated enhanced oxidative-nitrosative stress [reviewed in (13)], and, in particular, lipid peroxidation (2,13), increased production of superoxide and peroxynitrite (13-16), and depletion of the important antioxidants, reduced glutathione, ascorbate and taurine (2,13,17-20). On the other hand, findings from several groups suggest that AR metabolises highly reactive lipid peroxidation-derived aldehydes and thus acts as an ‘antioxidant enzyme’ (21,22). Thus, in the opinion of these authors (23), increased AR activity plays a protective, rather than a pathogenetic, role in diabetic complications, which explains the failure of a number of AR inhibitors in clinical trials.
The interaction between increased AR activity and oxidative-nitrosative stress in diabetic ocular complications has been inadequately explored. Furthermore, a large proportion of the studies with AR inhibitors has been performed in galactose, rather than diabetic, models of cataract (24,25) and retinopathy (26,27). The present study evaluated the effect of fidarestat on diabetic cataract formation and early retinal changes in streptozotocin-diabetic rats. We were particularly interested in the relations between increased AR activity and oxidative-nitrosative stress, and its direct consequence, premature apoptosis, in the whole diabetic retina and high glucose-exposed retinal microvascular cells.
Methods
Reagents
Unless otherwise stated, all chemicals were of reagent-grade quality, and were purchased from Sigma Chemical Co., St. Louis, MO. Rabbit polyclonal anti-nitrotyrosine (NT) antibody was purchased from Upstate, Lake Placid, NY, and mouse monoclonal anti-poly(ADP-ribose) from Trevigen, Inc., Gaithersburg, MD. Secondary Alexa Fluor 488 goat anti-rabbit and Alexa Fluor 488 goat anti-mouse antibodies, and Prolong Gold Antifade Reagent with 4′,6-diamidino-2-phenylindole (DAPI) were purchased from Invitrogen, Eugene, OR. Biotinylated anti-rabbit and anti-mouse antibody, Avidin/Biotin Blocking Kit, Vectastain Elite ABC Kit (Standard), and DAB Substrate Kit were obtained from Vector Laboratories, Burlingame, CA. Mouse monoclonal anti-glial fibrillary acidic protein (GFAP) antibody, ApopTag® Plus Fluorescein In Situ Apoptosis Detection Kit, and ApopTag® Peroxidase In Situ Apoptosis Detection Kit were purchased from Chemicon International, Inc., Temecula, CA. Micromount mounting medium was purchased from Surgipath Medical Ind., Richmond, IL. Other reagents for immunohistochemistry were purchased from Dako Laboratories, Inc., Santa Barbara, CA.
Animals
The experiments were performed in accordance with regulations specified by the National Institutes of Health ‘Principles of Laboratory Animal Care’ (1985 revised version) and Pennington Biomedical Research Center Protocol for Animal Studies. Male Wistar rats (Charles River, Wilmington, MA), body weight 250-300 g, were fed a standard rat chow (PMI Nutrition Int., Brentwood, MO) and had access to water ad libitum. Streptozotocin (STZ)-diabetes was induced as described (2,15). Blood samples for glucose measurements were taken from the tail vein ∼48 h after the STZ injection and the day before the animals were sacrificed. Rats with blood glucose ≥13.8 mM were considered diabetic. The experimental groups comprised control and diabetic rats treated with or without the AR inhibitor fidarestat (Sanwa Kagaku Kenkyusho Co., Ltd., Nagoya, Japan) (16 mg kg-1d-1, in the drinking water), for 10 weeks after an initial 2-week period without treatment). This dose was selected from our previous studies in which it essentially inhibited retinal sorbitol pathway activity and oxidative-nitrosative stress in male Wistar rats with short-term STZ-induced diabetes (15,28). At the end of the 12-week study, lens changes were evaluated by indirect ophthalmoscopy and portable slit lamp (Kowa Co., Tokyo, Japan). Evaluations were preceded by mydriasis with topical 1% tropicamide hydrochloride. Lens clarity was subjectively classified into the following stages: i) clear lenses, ii) equatorial valuoles, iii) cortical opacities, and iv) mature cataract.
Anesthesia, euthanasia and tissue sampling
The animals were sedated by CO2, and immediately sacrificed by cervical dislocation. Both eyes were rapidly enucleated. One eye from each rat was fixed in normal buffered 4% formalin for further assessment of NT, poly(ADP-ribose), and GFAP immuno-reactivities by conventional immunohistochemistry as we described previously (15). NT, poly(ADP-ribose) and GFAP accumulations reflect severity of nitrosative stress, and extent of PARP activation and retinal glial activation, respectively. The other eye was fixed in 4% paraformaldehyde in PBS for preparation of flat-mounted retinas and quantitation of apoptosis. A number of the rats from each experimental group were used for the assessment of retinal glucose and sorbitol pathway intermediate concentrations. Several retinal sections from control and diabetic rats were used to obtain representative images of apoptotic nuclei using the ApopTag Plus Fluorescein In Situ Apoptosis Detection Kit.
Specific methods employed in animal studies
Retinal glucose and sorbitol pathway intermediate concentrations
Retinal glucose, sorbitol and fructose concentrations were assessed by spectrofluorometric enzymatic methods with hexokinase/glucose 6-phosphate dehydrogenase, sorbitol dehydrogenase, and fructose dehydrogenase as we have previously described in detail (28-30).
Immunohistochemical studies
All flat-mounted retinas were processed by a single investigator and evaluated in a blinded manner. The rate of apoptosis was quantified by TUNEL assay with immunoperoxidase staining, using the ApopTag Peroxidase In Situ Apoptosis Detection Kit. The samples were processed as described previously (31,32) with a minor modification. Endogenous peroxidase quenching was skipped, as this has been noted to cause DNA breaks and lower TdT-enzyme activity. Retinas were stained with 3-amino-9-ethylcarbazole substrate (Sigma, St. Louis, MO) according to the manufacturer’s instructions. The total number of TUNEL-positive cells per retina was counted under a light microscope. NT, poly(ADP-ribose), and GFAP immuno-reactivities in retinal sections were assessed by conventional immunohistochemistry. In brief, the sections were deparaffinized in xylene, hydrated in decreasing concentrations of ethanol and washed in water. Non-specific binding was blocked by 10% goat serum containing 1% BSA in TBS for 1 h, and the Avidin/Biotin Blocking kit, according to the manufacturer’s instructions. Then, rabbit polyclonal anti-NT, mouse monoclonal anti-poly(ADP-ribose), or mouse monoclonal anti-GFAP antibodies were used in the working dilutions 1:200, 1:100 and 1:500, respectively. Primary antibodies were omitted in negative controls. Secondary biotinylated anti-rabbit and anti-mouse antibodies were applied in a working dilution 1:200. Sections were counter-stained with Gill’s hematoxylin, dehydrated and mounted in Micromount mounting medium. At least ten fields of each section were examined to select one representative image. Low power observations of retinal sections stained for NT, poly(ADP-ribose), and GFAP were made using a Zeiss Axioskop microscope. Color images were captured with a Zeiss Axiocam HRc CCD camera at 1300×1030 resolution. Low power images were generated with a 40× acroplan objective using the automatic capturing feature of the Zeiss Axiovision software (Ver. 3.1.2.1). The intensity of color was graded from 1 to 4 (1, no staining; 2, faint; 3, moderate; 4, intense), and the immunohistochemistry score was expressed as mean ± SEM for each experimental group. Low power observations of retinal sections stained for TUNEL-positive cells (ApopTag Plus Fluorescein In Situ Apoptosis Detection Kit) were made using a Zeiss Axioplan 2 imaging microscope. Fluorescent images were captured with a Photometric CoolSNAP™ HQ CCD camera at 1392×1040 resolution. Low power images were generated with a 40× acroplan objective using the RS Image™ 1.9.2 software.
Cell culture studies
Bovine retinal pericyte and endothelial cell cultures were established from fresh cow eyes as described previously (33,34). Passages 4-6 were used for all experiments. Purity of cultures was confirmed by characteristic pericyte and endothelial cell morphology and by the use of specific pericyte (α smooth muscle actin) and endothelial cell (von Willebrand factor) markers. On average, in pericyte experiments, 98.8±1.4% of the isolated cells were identified as pericytes. In endothelial cell experiments, 99.5±1.1% of the isolated cells were identified as endothelial cells.
To dissect the effects of high glucose exposure and AR inhibition, pericytes and endothelial cells were cultured (at least three 6-well plates per condition) for 3-14 days in the DMEM-medium containing 20% serum, 100 U/ml penicillin, 100 mg/ml streptomycin, and, for endothelial cells only, 50 μg/ml of endothelial growth supplement. The cells were placed on round glass cover slips and coated with gelatin or fibronectin (for pericytes and endothelial cells, respectively). At 80% confluency, pericyte and endothelial cell cultures were exposed to 5.5 or 30 mM glucose for 3-14 days. A number of 14-day cultures in 30 mM glucose were exposed to 10 μmol/l fidarestat treatment. At 3, 7 and 14 days, the rate of apoptosis was quantified with the ApopTag Plus Fluorescein In Situ Apoptosis Detection Kit. For fluorescent immunohistochemistry, cover slips with pericyte or endothelial cells were washed in PBS and fixed in 4% paraformaldehyde for 10 min. The fixed cells were washed in PBS and preincubated with 0.2% Triton X-100 in PBS for 15 min. Cover slips were blocked with 1% BSA containing 10% goat serum for 1 h. Then the cells were treated with either mouse monoclonal anti-poly(ADP-ribose) antibody (1:100 dilution) or rabbit polyclonal anti-NT antibody (1:200 dilution). Secondary Alexa Fluor 488 goat anti-mouse antibody or Alexa Fluor 488 goat anti-rabbit antibody were applied in working dilutions 1:200. Primary antibody was omitted in negative controls. Cover slips were mounted in Prolong Gold Antifade Reagent and placed on a slide. Images of immunostained cells were captured with a Photometric CoolSNAP HQ CCD camera at 1392×1040 resolutions. Fluorescence was quantified using the ImageJ 1.32 software (National Institutes of Health, Bethesda, MD). Seven to ten representative images with ∼30 cells each were quantified per experimental condition, and the average was calculated.
Statistical analysis
The results are expressed as mean ± SEM. Data were subjected to equality of variance F-test, and then to log transformation, if necessary, before one-way analysis of variance. Where overall significance (p<0.05) was attained, individual between-group comparisons were made using the Student-Newman-Keuls multiple range test. Significance was defined at p≤0.05. When between-group variance differences could not be normalized by log transformation (datasets for body weights and plasma glucose), the data were analyzed by the nonparametric Kruskal-Wallis one-way analysis of variance, followed by the Bonferroni/Dunn test for multiple comparisons.
Results
The initial (prior to STZ administration) body weights were similar in control and diabetic rats treated with or without fidarestat The final body weights were similarly reduced in untreated and fidarestat-treated diabetic rats compared with the control group (Table I). Initial blood glucose concentrations were 4.5- and 4.6-fold higher in untreated and fidarestat-treated diabetic rats than in non-diabetic controls. In a similar fashion, final blood glucose concentrations were 4.7-fold higher in untreated and fidarestat-treated diabetic rats than in non-diabetic controls. Aldose reductase (AR) inhibition did not affect either weight gain or blood glucose concentrations in non-diabetic rats.
Table I.
Initial and final body weights and blood glucose concentrations in control and diabetic rats, with and without fidarestat treatment
| Body weight (g) |
Blood glucose (mmol/l) |
|||
|---|---|---|---|---|
| Initial | Final | Initial | Final | |
| Control | 291±2.3 | 565±22 | 5.7±0.16 | 5.5±0.4 |
| Control + fidarestat | 295±9.0 | 570±42 | 5.5±0.13 | 5.3±0.3 |
| Diabetic | 288±3.8 | 353±13a | 25.4±1.23a | 26.1±1.3a |
| Diabetic + fidarestat | 293±8.0 | 362±21a | 26.3±0.93a | 25.7±1.2a |
Data are means ± SEM, n=6-10 per group.
p<0.01 vs. controls.
At the end of the study, 25% of untreated diabetic rat eyes displayed the vacuolar stage of cataract, 44% had cortical opacities, and 25% mature cataracts (Table II). Fidarestat treatment completely prevented the development of cataractous changes in diabetic rats.
Table II.
Retinal glucose and sorbitol pathway intermediate concentrations (nmol mg protein-1) in control rats and diabetic rats, with and without fidarestat treatment
| Glucose | Sorbitol | Fructose | |
|---|---|---|---|
| Control | 10.9±0.4 | 1.90±0.11 | 1.20±0.10 |
| Diabetic | 89.2±6.5a | 10.60±0.75a | 4.80±1.23a |
| Diabetic + fidarestat | 87.8±6.1a | 1.84±0.35b | 1.99±0.40b |
Data are means ± SEM, n = 6-10 per group.
p<0.01 vs. controls
p<0.01 vs. untreated diabetic group.
Retinal glucose, sorbitol, and fructose concentrations were 8.2-, 5.6-, and 3.9-fold higher in untreated diabetic rats than in the control group (Table III). Diabetes-induced increase in retinal glucose concentrations was not affected by fidarestat treatment, whereas elevation in retinal sorbitol concentration was completely prevented. Retinal fructose concentration remained slightly elevated in fidarestat-treated diabetic rats, but the difference with the non-diabetic control group did not achieve statistical significance.
Table III.
Rates of different stages of cataractogenesis in control and diabetic rats, with and without fidarestat treatment
| Clear lenses | Vacuoles | Opacities | Mature cataract | |
|---|---|---|---|---|
| Control (12) | 12 | 0 | 0 | 0 |
| Control + fidarestat (12) | 12 | 0 | 0 | 0 |
| Diabetic (16) | 1 | 4 | 7 | 4 |
| Diabetic + fidarestat (20) | 20 | 0 | 0 | 0 |
Number of eyes in each group is indicated in parentheses.
Retinal nitrotyrosine immunoreactivity was increased in diabetic rats compared with controls, and this increase was essentially prevented by fidarestat treatment (Fig. 1). AR inhibition did not affect nitrotyrosine immunoreactivity in non-diabetic rats. In a similar fashion, retinal poly(ADP-ribose) immunoreactivity was increased in diabetic rats compared with controls, and this increase was essentially prevented by fidarestat treatment (Fig. 2). AR inhibition did not affect poly(ADP-ribose) immunoreactivity in non-diabetic rats.
Figure 1.

(A) Representative microphotographs of nitrotyrosine immunostaining in the retinas of control and diabetic rats, with and without fidarestat treatment. Magnification ×40. (B) Immunohistochemistry scores of nitrotyrosine immunostaining in experimental groups. Mean ± SEM, n=6-10 per group. **p<0.01 vs. controls; ##p<0.01 vs. untreated diabetic group.
Figure 2.

(A) Representative microphotographs of poly(ADP-ribose) immunostaining in the retinas of control and diabetic rats, with and without fidarestat treatment. Magnification ×40. (B) Immunohistochemistry scores of poly(ADP-ribose) immunostaining in experimental groups. Mean ± SEM, n=6-10 per group. **p<0.01 vs. controls; ##p<0.01 vs. untreated diabetic group.
Retinal GFAP immunoreactivity was increased in diabetic rats compared with controls, and this increase was essentially prevented by fidarestat treatment (Fig. 3). AR inhibition did not affect GFAP immunoreactivity in non-diabetic rats.
Figure 3.
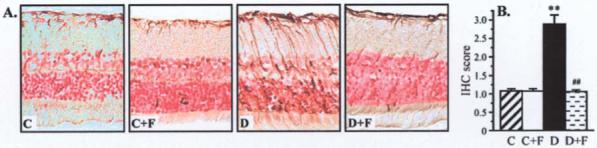
Representative microphotographs of glial fibrillary acidic protein immunostaining in the retinas of control and diabetic rats, with and without fidarestat treatment. Magnification ×40. (B) Immunohistochemistry scores of poly(ADP-ribose) immunostaining in experimental groups. Mean ± SEM, n=6-10 per group. **p<0.01 vs. controls; ##p<0.01 vs. untreated diabetic group.
Representative images of apoptotic cells (TUNEL fluorescence) in the retinal sections of control and diabetic rats are shown in Fig. 4A. The number of TUNEL-positive nuclei in flat-mounted retinas was increased ∼4-fold in diabetic rats compared with the control group, and this increase was partially corrected by fidarestat treatment (Fig. 4B and C).
Figure 4.

(A) Representative microphotographs of apoptotic cells (TUNEL fluorescence, shown by arrows) in the retinal sections of control and diabetic rats. Magnification ×40. (B) Representative microphotographs of apoptotic cells (TUNEL immunostaining) in the flat-mounted retinas of control and diabetic rats, with and without fidarestat treatment. Magnification ×20. (C) Apoptotic cell numbers per retina in control and diabetic rats, with and without fidarestat treatment. Mean ± SEM, n= 6-10 per group. **p<0.01 vs. controls; ##p<0.01 vs. untreated diabetic group.
Nitrotyrosine fluorescence was increased in retinal pericytes (Fig. 5A) and endothelial cells (Fig. 5B) cultured in 30 mM glucose, compared with those cultured in 5 mM glucose, and this increase was essentially prevented by fidarestat.
Figure 5.
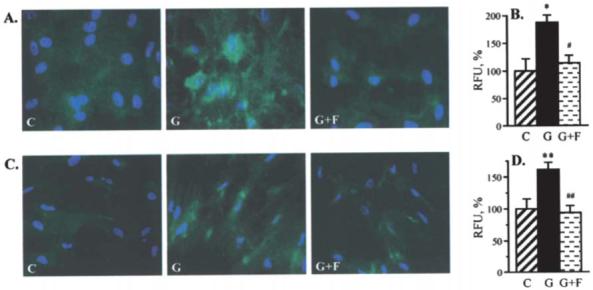
Representative microphotographs of nitrotyrosine fluorescence (green) in bovine retinal pericytes (A) and endothelial cells (C) cultured in 5 or 30 mM glucose with or without fidarestat. Magnification ×100. Blue fluorescence corresponds to 4′,6-diamidino-2-phenylindole-stained nuclei. Nitrotyrosine fluorescence (relative fluorescence units per cell) in bovine retinal pericytes (B) and endothelial cells (D) cultured in 5 or 30 mM glucose with or without fidarestat. Fluorescence per cell in pericytes or endothelial cells cultured in 5.5 mM glucose is taken as 100%. C, 5 mM glucose; G, 30 mM glucose; G+F, 30 mM glucose plus 10 μM fidarestat. Mean ± SEM, n=7-10 per group. *p<0.05 and **p<0.01 vs. cells cultured in 5 mM glucose; #p<0.05 and ##p<0.01 vs. cells cultured in 30 mM glucose without fidarestat.
The percentage of poly(ADP-ribose)+ nuclei was increased in retinal pericytes (Fig. 6A) and endothelial cells (Fig. 6B) cultured in 30 mM glucose compared with those cultured in 5 mM glucose. An AR-inhibitor treatment reduced, but did not completely normalize, the percentage of poly(ADP-ribose)+ nuclei in both retinal microvascular cell types.
Figure 6.

Representative microphotographs of poly(ADP-ribose) fluorescence (green) in bovine retinal pericytes (A) and endothelial cells (C) cultured in 5 or 30 mM glucose with or without fidarestat. Magnification ×100. Blue fluorescence corresponds to 4′,6-diamidino-2-phenylindole -stained nuclei. (Right) Percentage of poly(ADP-ribose)+ nuclei in bovine retinal pericytes (B) and endothelial cells (D) cultured in 5 or 30 mM glucose with or without fidarestat. C, 5 mM glucose; G, 30 mM glucose; G+F, 30 mM glucose plus 10 μM fidarestat. Mean ± SEM, n=7-10 per group. *p<0.05 and **p<0.01 vs. cells cultured in 5 mM glucose; #p<0.05 vs. cells cultured in 30 mM glucose without fidarestat.
The number of aptoptotic cells increased with the prolongation of exposure of both retinal pericytes (Fig. 7A) and endothelial cells (Fig. 7B) to high glucose levels. AR inhibition counteracted high glucose-induced apoptosis in both cell types, with more clearly manifest effect in retinal pericytes. Antiapoptotic effect of fidarestat in high glucose-exposed retinal pericytes was not associated with the inhibition of Bax or increase in Bcl-2 expression (Fig. 8).
Figure 7.

(A and C) Representative microphotographs of apoptotic cells (TUNEL fluorescence, green) in bovine retinal pericyte (A) and endothelial cell (C) cultures. Magnification ×100. C, 5 mM glucose, 3 days; G3, 30 mM glucose, 3 days; G7, 30 mM glucose, 7 days; G14, 30 mM glucose, 14 days; G14+F, 30 mM glucose plus 1 μM fidarestat, 14 days. Blue fluorescence corresponds to 4′,6-diamidino-2-phenylindole-stained nuclei. (B and D) Percentage of TUNEL-positive cells in bovine retinal pericyte (B) and endothelial cell (D) cultures. C, 5 mM glucose; G, 30 mM glucose; G+F, 30 mM glucose plus 10 μM fidarestat. Mean ± SEM, n=7-10 per group. *p<0.05 and **p<0.01 vs. cells cultured in 5 mM glucose; #p<0.05 vs. cells cultured in 30 mM glucose without fidarestat.
Figure 8.

(A) Representative Western blot of Bax and Bcl-2 expressions in bovine retinal pericyte cultures. Equal protein loading was confirmed with ß-actin antibody. Bax (B) and Bcl-2 (C) expressions in bovine retinal pericyte cultures. Bax and Bcl-2 expressions in bovine retinal pericytes cultured in 5 mM glucose are taken as 100%. C, 5 mM glucose; G, 30 mM glucose; G+F, 30 mM glucose plus 1 μM fidarestat. Mean ± SEM, n=3 per group.
Discussion
Numerous findings implicate increased AR activity in diabetes-associated pathophysiological and pathological changes in the cornea, iris, ciliary body, choroidal vasculature and optic nerve, as well as in cataracts and retinopathy [reviewed in (35)]. The role of AR in diabetic cataracto-genesis has been confirmed in numerous studies in young diabetic and galactose-fed rats (24,26,35). Both models display lens polyol accumulation, swelling, membrane permeation, vacuole and cleft formation, disturbance to the intracellular environment, protein aggregation and modification, light scatter, and, finally, rapidly developing mature cataracts. In contrast, only one group (36) which employed visual, rather than instrumental, assessment of cataract formation, reported a beneficial effect of an aldose reductase inhibitor (ARI) treatment on slow-developing diabetic cataracts in mature STZ-diabetic rats treated with suboptimal doses of insulin. These rats do not display weight loss, are free from acute diabetic complications such as ketoacidosis, and represent a more clinically relevant model for studying diabetic cataractogenesis than galactose-fed or even young weanling diabetic rats, because cataract typically develops in human diabetic subjects of mature age, treated with insulin or oral hypoglycemic agents. Using this animal model and indirect ophthalmoscopy and portable slit lamp examination, we demonstrated that the ARI fidarestat, completely blocked slow-developing diabetic cataract formation. The important role of AR in diabetic cataractogenesis is in line with studies in other diabetic animal models including, i) the South American rodent, Octodon degus, which has a very high AR expression in the lens, and rapidly develops cataracts after spontaneously becoming diabetic in a laboratory environment (37,38), and ii) transgenic mice selectively expressing human AR in the lens, which, in contrast to wild-type diabetic mice that do not accumulate sorbitol in the lens and do not develop cataracts, display clearly manifest lens sorbitol accumulation and opacification (39).
Our findings also demonstrated that AR inhibition counteracts numerous changes characteristic of early diabetic retinopathy. In particular, fidarestat treatment counteracted retinal glial activation manifest in GFAP accumulation. The latter is consistent with previous studies with other ARIs, sorbinil (40) and ARI-809 (36). It has been hypothesized that retinal neurodegenerative changes including increased glial cell reactivity and microglial activation, together with altered glutamate metabolism and premature apoptosis, are a critical component of diabetic retinopathy (41). Notably, however, a recent study in the STZ-diabetic mouse model showed that the diabetes-induced degeneration of retinal capillaries can develop independent of neuronal loss or chronic GFAP upregulation in glial cells (42).
Evidence for the important role of the potent oxidant peroxynitrite, a product of superoxide anion radicals and nitric oxide (6,43), in diabetic complications including endothelial dysfunction (44), peripheral (45-48) and autonomic (49) neuropathy, and retinopathy (50) is emerging. Peroxynitrite causes multiple cytotoxic effects including lipid peroxidation, protein nitration and nitrosylation, DNA breakage and base modification, impairment of cell signaling, depolarization of mitochondrial membrane, PARP and metalloproteinase activation, and, in extreme cases, necrosis and premature apoptosis (6,43). The present findings revealed accumulation of NT, a product of peroxynitrite-induced injury, in the retinas of rats with a 12-week duration of diabetes as well as in high glucose-exposed retinal pericytes and endothelial cells. Nitrosative stress in both diabetic retina and retinal vascular cells was prevented by fidarestat. The latter is consistent with the alleviation of peroxynitrite-induced injury by AR inhibition in aorta, peripheral nerve, vasa nervorum, renal cortex, and retina of STZ-diabetic rats with a shorter duration of diabetes and high glucose-exposed human Schwann and bovine endothelial cells in previous studies by our group (15,16,51).
Peroxynitrite-induced injury is closely linked to another phenomenon recently implicated in the development of diabetic complication, i.e. PARP activation. PARP activation results in NAD+ depletion and energy failure, changes in transcriptional regulation and gene expression, the accumulation of glutamate and cytosolic Ca++, and mitogen-activated protein kinase activation (9,10,52). Evidence for the important role of PARP activation in diabetic endothelial dysfunction, peripheral and autonomic neuropathy, cardiomyopathy, retinopathy and nephropathy is emerging (9,10,52-58). The present study reveals clearly manifest PARP activation in the retina, and, in particular, retinal inner neuronal and ganglion cell layers, of diabetic rats as well as in high glucose-exposed cultured retinal pericytes and endothelial cells. AR inhibition blunted PARP activation in both diabetic rat retina and high glucose-exposed retinal microvascular cells. Notably, however, in contrast to the high glucose-induced nitrotyrosine accumulation that was completely prevented by fidarestat treatment in both pericytes and endothelial cells, the percentage of poly(ADP-ribose)+ nuclei remained slightly elevated, compared to the corresponding microvascular cells cultured in normoglycemic conditions. Recently, certain factors, other than oxidative stress and resultant DNA single-strand breakage have been implicated in PARP activation under pathological conditions associated with oxidative stress. In particular, it has been shown that PARP can be activated by phosphorylation performed by phosphorylated ERK (59). Specific studies are needed to assess the contributions of oxidative stress vs. other factors to PARP activation in tissue sites for diabetic complications including the retina and lens.
The increased rate of retinal neural cell apoptosis has been identified at the very early stage of diabetes (31). Furthermore, the frequency of early apoptosis in retinal capillary cells has been found to predict the development of the histologic lesions of retinopathy in diabetes and galactosemia (60). In the present study, the number of apoptotic cells was increased 4-fold in flat-mounted retinas of mature diabetic rats compared with non-diabetic controls. It progressively increased with the prolongation of exposure of cultured bovine retinal pericytes and endothelial cells to high glucose levels. Our findings are consistent with other studies suggesting that AR inhibition suppresses early apoptosis in the neural retina of diabetic rats (36,40) as well as high glucose-exposed cultured retinal pericytes (61). Furthermore, they support and complement our previous findings of the inhibitory effect of the ARI zopolrestat on caspase-3 activation, an indirect measure of cell death, in high glucose-exposed cultured retinal endothelial cells (62). Notably, the effect of fidarestat on retinal pericyte apoptosis is not associated with changes in the proapoptotic protein Bax or antiapoptotic protein Bcl-2, expression.
In conclusion, fidarestat treatment counteracted cataract formation, retinal oxidative-nitrosative stress, glial activation, and apoptosis in mature streptozotocin-diabetic rats. The results, consistent with previous reports on the alleviation of diabetic complications with fidarestat treatment (63-66), support a pathogenetic, rather than protective, role for increased aldose reductase activity in diabetic cataractogenesis and early retinopathy, and provide a rationale for the development of aldose reductase inhibitors, and, in particular, fidarestat, for their prevention and treatment.
Acknowledgements
This study was supported by the Juvenile Diabetes Research Foundation International Grant 1-2005-223 and the National Institutes of Health Grant DK 071566-01 (both to IGO), and the Intramural Research Program of the National Institutes of Health / National Institute of Alcohol Abuse and Alcoholism (to PP).
References
- 1.Oates PJ, Mylari BL. Aldose reductase inhibitors: therapeutic implications for diabetic complications. Expert Opin Investig Drugs. 1999;8:2095–2119. doi: 10.1517/13543784.8.12.2095. [DOI] [PubMed] [Google Scholar]
- 2.Obrosova IG, Van Huysen C, Fathallah L, Cao XC, Greene DA, Stevens MJ. An aldose reductase inhibitor reverses early diabetes-induced changes in peripheral nerve function, metabolism, and antioxidative defense. FASEB J. 2002;16:123–125. doi: 10.1096/fj.01-0603fje. [DOI] [PubMed] [Google Scholar]
- 3.Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation. 2006;114:597–605. doi: 10.1161/CIRCULATIONAHA.106.621854. [DOI] [PubMed] [Google Scholar]
- 4.Yan SF, Barile GR, D’Agati V, Du Yan S, Ramasamy R, Schmidt AM. The biology of RAGE and its ligands: uncovering mechanisms at the heart of diabetes and its complications. Curr Diab Rep. 2007;7:146–153. doi: 10.1007/s11892-007-0024-4. [DOI] [PubMed] [Google Scholar]
- 5.Pennathur S, Heinecke JW. Oxidative stress and endothelial dysfunction in vascular disease. Curr Diab Rep. 2007;7:257–264. doi: 10.1007/s11892-007-0041-3. [DOI] [PubMed] [Google Scholar]
- 6.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes. 1998;47:859–866. doi: 10.2337/diabetes.47.6.859. [DOI] [PubMed] [Google Scholar]
- 8.Das Evcimen N, King GL. The role of protein kinase C activation and the vascular complications of diabetes. Pharmacol Res. 2007;55:498–510. doi: 10.1016/j.phrs.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 9.Jagtap P, Szabó C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov. 2005;4:421–440. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]
- 10.Obrosova IG, Julius UA. Role for poly(ADP-ribose) polymerase activation in diabetic nephropathy, neuropathy and retinopathy. Curr Vasc Pharmacol. 2005;3:267–283. doi: 10.2174/1570161054368634. [DOI] [PubMed] [Google Scholar]
- 11.Natarajan R, Nadler JL. Lipid inflammatory mediators in diabetic vascular disease. Arterioscler Thromb Vasc Biol. 2004;24:1542–1548. doi: 10.1161/01.ATV.0000133606.69732.4c. [DOI] [PubMed] [Google Scholar]
- 12.Hao CM, Breyer MD. Physiologic and pathophysiologic roles of lipid mediators in the kidney. Kidney Int. 2007;71:1105–1115. doi: 10.1038/sj.ki.5002192. [DOI] [PubMed] [Google Scholar]
- 13.Obrosova IG. Increased sorbitol pathway activity generates oxidative stress in tissue sites for diabetic complications. Antioxid Redox Signal. 2005;7:1543–1552. doi: 10.1089/ars.2005.7.1543. [DOI] [PubMed] [Google Scholar]
- 14.Gupta S, Chough E, Daley J, Oates P, Tornheim K, Ruderman NB, Keaney JF., Jr Hyperglycemia increases endothelial superoxide that impairs smooth muscle cell Na+-K+-ATPase activity. Am J Physiol Cell Physiol. 2002;282:C560–C566. doi: 10.1152/ajpcell.00343.2001. [DOI] [PubMed] [Google Scholar]
- 15.Obrosova IG, Pacher P, Szabó C, Zsengeller Z, Hirooka H, Stevens MJ, Yorek MA. Aldose reductase inhibition counteracts oxidative-nitrosative stress and poly(ADP-ribose) polymerase activation in tissue sites for diabetes complications. Diabetes. 2005;54:234–242. doi: 10.2337/diabetes.54.1.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.El-Remessy AB, Abou-Mohamed G, Caldwell RW, Caldwell RB. High glucose-induced tyrosine nitration in endothelial cells: role of eNOS uncoupling and aldose reductase activation. Invest Ophthalmol Vis Sci. 2003;44:3135–3143. doi: 10.1167/iovs.02-1022. [DOI] [PubMed] [Google Scholar]
- 17.Lou MF, Dickerson JE, Jr, Garadi R, York BM., Jr Glutathione depletion in the lens of galactosemic and diabetic rats. Exp Eye Res. 1988;46:517–530. doi: 10.1016/s0014-4835(88)80009-5. [DOI] [PubMed] [Google Scholar]
- 18.Obrosova IG, Fathallah L. Evaluation of an aldose reductase inhibitor on lens metabolism, ATPases and antioxidative defense in streptozotocin-diabetic rats: an intervention study. Diabetologia. 2000;43:1048–1055. doi: 10.1007/s001250051488. [DOI] [PubMed] [Google Scholar]
- 19.Stevens MJ, Hosaka Y, Masterson JA, Jones SM, Thomas TP, Larkin DD. Downregulation of the human taurine transporter by glucose in cultured retinal pigment epithelial cells. Am J Physiol. 1999;277:E760–E771. doi: 10.1152/ajpendo.1999.277.4.E760. [DOI] [PubMed] [Google Scholar]
- 20.Obrosova IG. How does glucose generate oxidative stress in peripheral nerve? Int Rev Neurobiol. 2002;50:3–35. doi: 10.1016/s0074-7742(02)50071-4. [DOI] [PubMed] [Google Scholar]
- 21.Srivastava S, Chandra A, Wang LF, Seifert WE, Jr, DaGue BB, Ansari NH, Srivastava SK, Bhatnagar A. Metabolism of the lipid peroxidation product, 4-hydroxy-trans-2-nonenal, in isolated perfused rat heart. J Biol Chem. 1998;273:10893–10900. doi: 10.1074/jbc.273.18.10893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pladzyk A, Ramana KV, Ansari NH, Srivastava SK. Aldose reductase prevents aldehyde toxicity in cultured human lens epithelial cells. Exp Eye Res. 2006;83:408–416. doi: 10.1016/j.exer.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 23.Srivastava SK, Ramana KV, Bhatnagar A. Role of aldose reductase and oxidative damage in diabetes and the consequent potential for therapeutic options. Endocr Rev. 2005;26:380–392. doi: 10.1210/er.2004-0028. [DOI] [PubMed] [Google Scholar]
- 24.Morikubo S, Takamura Y, Kubo E, Tsuzuki S, Akagi Y. The inhibitory effect of aldose reductase inhibitor for proliferation in the lens epithelial cells of rat sugar cataract. Nippon Ganka Gakkai Zasshi. 2003;107:565–570. [PubMed] [Google Scholar]
- 25.Kador PF, Betts D, Wyman M, Blessing K, Randazzo J. Effects of topical administration of an aldose reductase inhibitor on cataract formation in dogs fed a diet high in galactose. Am J Vet Res. 2006;67:1783–1787. doi: 10.2460/ajvr.67.10.1783. [DOI] [PubMed] [Google Scholar]
- 26.Robison WG, Jr, Jacot JL, Glover JP, Basso MD, Hohman TC. Diabetic-like retinopathy: early and late intervention therapies in galactose-fed rats. Invest Ophthalmol Vis Sci. 1998;39:1933–1941. [PubMed] [Google Scholar]
- 27.Cusick M, Chew EY, Ferris F, III, Cox TA, Chan CC, Kador PF. Effects of aldose reductase inhibitors and galactose withdrawal on fluorescein angiographic lesions in galactose-fed dogs. Arch Ophthalmol. 2003;121:1745–1751. doi: 10.1001/archopht.121.12.1745. [DOI] [PubMed] [Google Scholar]
- 28.Obrosova IG, Minchenko AG, Vasupuram R, White L, Abatan OI, Kumagai AK, Frank RN, Stevens MJ. Aldose reductase inhibitor fidarestat prevents retinal oxidative stress and vascular endothelial growth factor overexpression in streptozotocin-diabetic rats. Diabetes. 2003;52:864–871. doi: 10.2337/diabetes.52.3.864. [DOI] [PubMed] [Google Scholar]
- 29.Obrosova IG, Stevens MJ. Effect of dietary taurine supplementation on GSH and NAD(P)-redox status, lipid peroxidation, and energy metabolism in diabetic precataractous lens. Invest Ophthalmol Vis Sci. 1999;40:680–688. [PubMed] [Google Scholar]
- 30.Obrosova IG, Stevens MJ, Lang HJ. Diabetes-induced changes in retinal NAD-redox status: pharmacological modulation and implications for pathogenesis of diabetic retinopathy. Pharmacology. 2001;62:172–180. doi: 10.1159/000056091. [DOI] [PubMed] [Google Scholar]
- 31.Barber AJ, Lieth E, Khin SA, Antonetti DA, Buchanan AG, Gardner TW. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest. 1998;102:783–791. doi: 10.1172/JCI2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.El-Remessy AB, Al-Shabrawey M, Khalifa Y, Tsai NT, Caldwell RB, Liou GI. Neuroprotective and blood-retinal barrier-preserving effects of cannabidiol in experimental diabetes. Am J Pathol. 2006;168:235–244. doi: 10.2353/ajpath.2006.050500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Podestà F, Romeo G, Liu WH, Krajewski S, Reed JC, Gerhardinger C, Lorenzi M. Bax is increased in the retina of diabetic subjects and is associated with pericyte apoptosis in vivo and in vitro. Am J Pathol. 2000;156:1025–1032. doi: 10.1016/S0002-9440(10)64970-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sone H, Deo BK, Kumagai AK. Enhancement of glucose transport by vascular endothelial growth factor in retinal endothelial cells. Invest Ophthalmol Vis Sci. 2000;41:1876–1884. [PubMed] [Google Scholar]
- 35.Kador PF. Ocular pathology of diabetes mellitus. In: Tasman W, Jaeger EA, editors. Duane’s Ophthalmology. Vol. 3. Wolters Kluwer/Lippicott Williams and Wilkins; Philadelphia: 2007. pp. pp1–84. [Google Scholar]
- 36.Sun W, Oates PJ, Coutcher JB, Gerhardinger C, Lorenzi M. A selective aldose reductase inhibitor of a new structural class prevents or reverses early retinal abnormalities in experimental diabetic retinopathy. Diabetes. 2006;55:2757–2762. doi: 10.2337/db06-0138. [DOI] [PubMed] [Google Scholar]
- 37.Datiles MB, III, Fukui H. Cataract prevention in diabetic Octodon degus with Pfizer’s sorbinil. Curr Eye Res. 1989;8:233–237. doi: 10.3109/02713688908997564. [DOI] [PubMed] [Google Scholar]
- 38.Brown C, Donnelly TM. Cataracts and reduced fertility in degus (Octodon degus). Contracts secondary to spontaneous diabetes mellitus. Lab Anim. 2001;30:25–26. [PubMed] [Google Scholar]
- 39.Lee AY, Chung SK, Chung SS. Demonstration that polyol accumulation is responsible for diabetic cataract by the use of transgenic mice expressing the aldose reductase gene in the lens. Proc Natl Acad Sci USA. 1995;92:2780–2784. doi: 10.1073/pnas.92.7.2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dagher Z, Park YS, Asnaghi V, Hoehn T, Gerhardinger C, Lorenzi M. Studies of rat and human retinas predict a role for the polyol pathway in human diabetic retinopathy. Diabetes. 2004;53:2404–2411. doi: 10.2337/diabetes.53.9.2404. [DOI] [PubMed] [Google Scholar]
- 41.Barber AJ. A new view of diabetic retinopathy: a neurodegenerative disease of the eye. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:283–290. doi: 10.1016/S0278-5846(03)00023-X. [DOI] [PubMed] [Google Scholar]
- 42.Feit-Leichman RA, Kinouchi R, Takeda M, Fan Z, Mohr S, Kern TS, Chen DF. Vascular damage in a mouse model of diabetic retinopathy: relation to neuronal and glial changes. Invest Ophthalmol Vis Sci. 2005;46:4281–4287. doi: 10.1167/iovs.04-1361. [DOI] [PubMed] [Google Scholar]
- 43.Szabó C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 44.Szabó C, Mabley JG, Moeller SM, Shimanovich R, Pacher P, Virag L, Soriano FG, Van Duzer JH, Williams W, Salzman AL, Groves JT. Part I: pathogenetic role of peroxynitrite in the development of diabetes and diabetic vascular complications: studies with FP15, a novel potent peroxynitrite decomposition catalyst. Mol Med. 2002;8:571–580. [PMC free article] [PubMed] [Google Scholar]
- 45.Obrosova IG, Mabley JG, Zsengellér Z, Charniauskaya T, Abatan OI, Groves JT, Szabó C. Role for nitrosative stress in diabetic neuropathy: evidence from studies with a peroxynitrite decomposition catalyst. FASEB J. 2005;19:401–403. doi: 10.1096/fj.04-1913fje. [DOI] [PubMed] [Google Scholar]
- 46.Vareniuk I, Pavlov IA, Drel VR, Lyzogubov VV, Ilnytska O, Bell SR, Tibrewala J, Groves JT, Obrosova IG. Nitrosative stress and peripheral diabetic neuropathy in leptin-deficient (ob/ob) mice. Exp Neurol. 2007;205:425–436. doi: 10.1016/j.expneurol.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 47.Obrosova IG, Drel VR, Oltman CL, Mashtalir N, Tibrewala J, Groves JT, Yorek MA. Role of nitrosative stress in early neuropathy and vascular dysfunction in streptozotocin-diabetic rats. Am J Physiol Endocrinol Metab. 2007;293:E1645–E1655. doi: 10.1152/ajpendo.00479.2007. [DOI] [PubMed] [Google Scholar]
- 48.Drel VR, Pacher P, Vareniuk I, Pavlov IA, Ilnytska O, Lyzogubov VV, Bell SR, Groves JT, Obrosova IG. Evaluation of the peroxynitrite decomposition catalyst Fe(III) tetra-mesitylporphyrin octasulfonate on peripheral neuropathy in a mouse model of type 1 diabetes. Int J Mol Med. 2007;20:783–792. [PMC free article] [PubMed] [Google Scholar]
- 49.Nangle MR, Cotter MA, Cameron NE. Effects of the peroxynitrite decomposition catalyst, FeTMPyP, on function of corpus cavernosum from diabetic mice. Eur J Pharmacol. 2004;502:143–148. doi: 10.1016/j.ejphar.2004.08.033. [DOI] [PubMed] [Google Scholar]
- 50.Zheng L, Du Y, Miller C, Gubitosi-Klug RA, Kern TS, Ball S, Berkowitz BA. Critical role of inducible nitric oxide synthase in degeneration of retinal capillaries in mice with streptozotocin-induced diabetes. Diabetologia. 2007;50:1987–1996. doi: 10.1007/s00125-007-0734-9. [DOI] [PubMed] [Google Scholar]
- 51.Drel VR, Pacher P, Stevens MJ, Obrosova IG. Aldose reductase inhibition counteracts nitrosative stress and poly(ADP-ribose) polymerase activation in diabetic rat kidney and high-glucose-exposed human mesangial cells. Free Radic Biol Med. 2006;40:1454–1465. doi: 10.1016/j.freeradbiomed.2005.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garcia Soriano F, Virág L, Jagtap P, Szabó E, Mabley JG, Liaudet L, Marton A, Hoyt DG, Murthy KG, Salzman AL, Southan GJ, Szabó C. Diabetic endothelial dysfunction: the role of poly(ADP-ribose) polymerase activation. Nat Med. 2001;7:108–113. doi: 10.1038/83241. [DOI] [PubMed] [Google Scholar]
- 53.Pacher P, Liaudet L, Soriano FG, Mabley JG, Szabó E, Szabó C. The role of poly(ADP-ribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes. 2002;51:514–521. doi: 10.2337/diabetes.51.2.514. [DOI] [PubMed] [Google Scholar]
- 54.Obrosova IG, Li F, Abatan OI, Forsell MA, Komjáti K, Pacher P, Szabó C, Stevens MJ. Role of poly(ADP-ribose) polymerase activation in diabetic neuropathy. Diabetes. 2004;53:711–720. doi: 10.2337/diabetes.53.3.711. [DOI] [PubMed] [Google Scholar]
- 55.Zheng L, Szabó C, Kern TS. Poly(ADP-ribose) polymerase is involved in the development of diabetic retinopathy via regulation of nuclear factor-kappaB. Diabetes. 2004;53:2960–2967. doi: 10.2337/diabetes.53.11.2960. [DOI] [PubMed] [Google Scholar]
- 56.Ilnytska O, Lyzogubov VV, Stevens MJ, Drel VR, Mashtalir N, Pacher P, Yorek MA, Obrosova IG. Poly(ADP-ribose) polymerase inhibition alleviates experimental diabetic sensory neuropathy. Diabetes. 2006;55:1686–1694. doi: 10.2337/db06-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gibson TM, Cotter MA, Cameron NE. Effects of poly(ADP-ribose) polymerase inhibition on dysfunction of non-adrenergic non-cholinergic neurotransmission in gastric fundus in diabetic rats. Nitric Oxide. 2006;15:344–350. doi: 10.1016/j.niox.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 58.Szabó C, Biser A, Benko R, Böttinger E, Suszták K. Poly(ADP-ribose) polymerase inhibitors ameliorate nephropathy of type 2 diabetic Leprdb/db mice. Diabetes. 2006;55:3004–3012. doi: 10.2337/db06-0147. [DOI] [PubMed] [Google Scholar]
- 59.Kauppinen TM, Chan WY, Suh SW, Wiggins AK, Huang EJ, Swanson RA. Direct phosphorylation and regulation of poly(ADP-ribose) polymerase-1 by extracellular signal-regulated kinases 1/2. Proc Natl Acad Sci USA. 2006;103:7136–7141. doi: 10.1073/pnas.0508606103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kern TS, Tang J, Mizutani M, Kowluru RA, Nagaraj RH, Romeo G, Podesta F, Lorenzi M. Response of capillary cell death to aminoguanidine predicts the development of retinopathy: comparison of diabetes and galactosemia. Invest Ophthalmol Vis Sci. 2000;41:3972–3978. [PubMed] [Google Scholar]
- 61.Miwa K, Nakamura J, Hamada Y, Naruse K, Nakashima E, Kato K, Kasuya Y, Yasuda Y, Kamiya H, Hotta N. The role of polyol pathway in glucose-induced apoptosis of cultured retinal pericytes. Diabetes Res Clin Pract. 2003;60:1–9. doi: 10.1016/s0168-8227(02)00248-6. [DOI] [PubMed] [Google Scholar]
- 62.El-Remessy AB, Bartoli M, Platt DH, Fulton D, Caldwell RB. Oxidative stress inactivates VEGF survival signaling in retinal endothelial cells via PI 3-kinase tyrosine nitration. J Cell Sci. 2005;118:243–252. doi: 10.1242/jcs.01612. [DOI] [PubMed] [Google Scholar]
- 63.Kato N, Mizuno K, Makino M, Suzuki T, Yagihashi S. Effects of 15-month aldose reductase inhibition with fidarestat on the experimental diabetic neuropathy in rats. Diabetes Res Clin Pract. 2000;50:77–85. doi: 10.1016/s0168-8227(00)00164-9. [DOI] [PubMed] [Google Scholar]
- 64.Hotta N, Toyota T, Matsuoka K, Shigeta Y, Kikkawa R, Kaneko T, Takahashi A, Sugimura K, Koike Y, Ishii J, Sakamoto N. SNK-860 Diabetic Neuropathy Study Group: Clinical efficacy of fidarestat, a novel aldose reductase inhibitor, for diabetic peripheral neuropathy: a 52-week multicenter placebo-controlled double-blind parallel group study. Diabetes Care. 2001;24:1776–1782. doi: 10.2337/diacare.24.10.1776. [DOI] [PubMed] [Google Scholar]
- 65.Kato N, Yashima S, Suzuki T, Nakayama Y, Jomori T. Long-term treatment with fidarestat suppresses the development of diabetic retinopathy in STZ-induced diabetic rats. J Diabetes Complications. 2003;17:374–379. doi: 10.1016/s1056-8727(02)00193-9. [DOI] [PubMed] [Google Scholar]
- 66.Hotta N, Yasuda K, Sumita Y, Sano T, Kakuta H, Nagashima M, Hayashi Y, Yamamoto M, Wakao T, Okuyama M, Kobayashi M, Mori K. Effects of a novel aldose reductase inhibitor, fidarestat (SNK-860), on vibration perception threshold and subjective symptoms in patients with diabetic polyneuropathy: An open-label pilot study. Clin Drug Investig. 2004;24:671–680. doi: 10.2165/00044011-200424110-00006. [DOI] [PubMed] [Google Scholar]