

Abstract
Reactivity-based selection strategies have been used to enrich combinatorial libraries for encoded biocatalysts having revised substrate specificity or altered catalytic activity. This approach can also assist in artificial evolution of enzyme catalysis from protein templates without bias for predefined catalytic sites. The prevalence of covalent intermediates in enzymatic mechanisms suggests the universal utility of the covalent complex as the basis for selection. Covalent selection by phosphonate ester exchange was applied to a phage display library of antibody variable fragments (scFv) in order to sample the scope and mechanism of chemical reactivity in a naive molecular library. Selected scFv segregated into structurally related covalent and non-covalent binders. Clones that reacted covalently utilized tyrosine residues exclusively as the nucleophile. Two motifs were identified by structural analysis, recruiting distinct Tyr residues of the light chain. Most clones employed Tyr32 in CDR-L1, whereas a unique clone (A.17) reacted at Tyr36 in FR-L2. Enhanced phosphonylation kinetics and modest amidase activity of A.17 suggested a primitive catalytic site. Covalent selection may thus provide access to protein molecules that approximate an early apparatus for covalent catalysis.
INTRODUCTION
Mechanism-based selection of molecular display libraries represents a powerful approach to study the relationship between structure and function and to alter the properties of enzymes. Active site-directed ligands, including tight binding inhibitors, transition state analogs and suicide substrates, have been employed for affinity or covalent capture of functional enzyme molecules1,2. In particular, covalent selection enables efficient partitioning of molecules based on their characteristic enzymatic reactivity. This strategy is ideally suited to isolating enzymes that utilize active site residues in covalent catalysis. Thus, chemical modification at the nucleophilic serine residue can be exploited to select functional variants of serine hydrolases. Phosphonylating agents can recruit an enzyme’s catalytic power for enhanced reactivity at the hydroxyl group of the active site serine3. Phosphonofluoridates or, more conveniently, phosphonate esters provide a prototype for selective covalent capture reagents. Recent demonstrations include the directed evolution of lipase4 and subtilisin5 activity from enzyme phage display libraries. Selection reagents identified new substrate specificities by discriminating reactivity at the conserved serine within an active site diversified at proximal residues. This approach could similarly be applied to differentiate sites without a predefined catalytic apparatus in order to probe nucleophile repertoires useful in recreating enzyme-like reactivity. The discovery of new protein configurations bearing nucleophilic residues within a microenvironment that is suitably organized for acyl transfer chemistry could have implications for the biogenesis of covalent catalysis.
The antibody combining site has served to test various schemes for eliciting elementary enzyme-like function. Phage display antibodies are amenable to classical selection for catalytic activity by transition state analog binding6,7 as well as functional selection by chemical capture using suicide substrate reactivity8-10 or disulfide exchange11. Nucleophile-directed reactive selection was formally demonstrated by enrichment of an esterolytic antibody clone through its acyl intermediate12. Antibodies that employ unique nucleophilic residues for covalent catalysis have also been elicited by reactive immunization13. However, non-covalent binding to transition state analogs may also invoke covalent mechanisms in abzymes induced by immunization14-17. Conventional in vivo affinity maturation could thus take various paths toward specific combining sites in which chemically reactive residues are acquired en route18. By contrast, in vitro reactive selection as defined here demands a nucleophilic residue in each selection event. Preservation of an essential structural feature for evolution of covalent catalytic mechanisms is therefore enforced from the outset. Here we report the selection of new chemically reactive antibody fragments from an unbiased human Fv phage display library. We aimed to explore the scope of obtainable nucleophile diversity and to initiate a comparative structural and functional analysis of the reactive sites in order to identify possible pathways to covalent catalysis.
MATERIALS AND METHODS
Reagents and phage library
Reactive phosphonate esters 1, 2 and 5 were previously reported19. Diphenylphosphonates 3 and 4 were synthesized as described elsewhere20. Benzenesulfonylfluoride (AEBSF) was purchased from Sigma Aldrich (St. Louis, MO). Tripeptide methylcoumarinamides Pro-Phe-Arg-MCA, Boc-Gly-Gly-Arg-MCA, Phe-MCA and Boc-Ala-Ala-Phe-MCA were obtained from Peptides International (Louisville, KY). The Griffin.1 phage display library of semisynthetic scFv constructed from human germline V genes was obtained through the MRC, Center for Protein Engineering, Cambridge, UK. This library was derived by recloning VH and VL segments from human synthetic Fab lox library vectors into the phagemid vector pHEN221. Methods and procedures for phage production, manipulation and expression of scFv have been reported elsewhere22.
Reactive selection of scFv-phage
A phage stock was prepared from the library in TG-1 cells by superinfection with M13-K07 helper phage. An aliquot of the phage was used to infect TG-1 cells, which were then serially diluted and plated on LB/amp agar to determine the phage titer. Plastic 96-well microtiter plates (Nunc Maxisorb) were coated with 150 μl of 10 μg/ml streptavidin (Amersham) in PBS overnight at 4°C and blocked with 3% BSA in PBS (BPBS) 2 hours at room temperature. Phage particles (3x1011 cfu/ml) were pre-adsorbed on a plate coated with streptavidin and blocked with BSA and then reacted with 1 (1 - 40 μM final concentration) in PBS at 37°C for 1 h. After the reaction the phage were precipitated twice with 1/5 volume PEG/NaCl. Phage pellets were resuspended in BPBS and about 6x109 phage particles were applied to wells of a plate coated with streptavidin and blocked with BPBS (experimental wells) or with 1 mM biotin in BPBS (control wells). After 1 h at room temperature all wells were washed 10 times with PBS containing 0.1% Tween 20, 3 times with PBS, 2 times with 100 mM glycine-HCl, pH 2.7 and finally with PBS. Bound phage was eluted with using a 1mg/ml trypsin solution in PBS, incubating for 20 min room temperature. The titers of eluted phage were determined after each round of selection and used to calculate the ratio of phage recovery from experimental and control selections. The frequency of clones containing the scFv gene insert was determined by PCR screening using the primer pair CAGGAAACAGCTATGAC and GAATTTTCTGTATGAGG. Following three rounds of selection, the HB2151 non-suppressor E coli strain was infected with an aliquot of eluted phage and bacteria were plated on LB agar containing 2% glucose and 100 μg/mL ampicillin and grown overnight at 30°C. Individual colonies were checked by PCR for the presence of the expected VL-VH insert. Colonies containing a copy of the full-length insert were picked and used to express scFv by induction of a culture in log phase growth by addition of 1 mM isopropyl β-D-galactopyranoside.
Expression and purification of scFv
Five ml of 2xYT medium (containing 100 μg/mL ampicillin and 2.0% glucose) was inoculated with a single bacterial colony and grown overnight with shaking at 37°C. The overnight culture was used to inoculate 500 ml of 2xYT medium (containing 100 μg/mL ampicillin and 0.1% glucose) and the culture incubated at 37°C (two 2 L flasks, 200 rpm) for 2h. Then expression was induced by addition of IPTG to a final concentration of 0.2 mM. Incubation was continued for 12 h at 22°C, 200 rpm. The expressed scFv protein was purified by a three-step procedure. Cells were harvested and the periplasmic fraction was adsorbed on immobilized nickel-charged affinity resin (Ni-NTA, EMD Biosciences, La Jolla, CA). The resin was washed and the bound protein eluted with 0.15 M imidazole in phosphate buffer according to the manufacture’s protocol. Activity could be confirmed at this stage by reacting with 20 μM 1, followed by western blot analysis using streptavidin-HRP and ECL substrate (Amersham Biosciences). In large-scale preparations and amidase assay the protein recovered by metal ion chromatography was dialyzed against 10 mM Tris, pH 8 and further purified on a mono Q 5/5 column (Amersham Biosciences) using a gradient 0 - 0.25 M NaCl in 10 mM Tris, pH 8.0 in 30 min. The peak eluting at 90 - 130 mM NaCl was collected (0.5 ml/fraction) and activity was analyzed by fluorescence assay.
Reaction kinetics and ELISA
Reactions at concentrations of 2 ranging from 10 - 100 μM and 3-10 μM scFv were carried out in 0.1 M phosphate buffer, pH 7.8 at 22°C. Reaction rates were determined from absorbance changes at 400 nm due to 4-nitrophenol formation and rate constants determined using the buffer-specific extinction coefficient. Active antibody concentrations were extrapolated from the 400 nm Amax in the presence of excess of 2. Modification rate constants k1 were estimated by Kitz-Wilson analysis 23,24. The substrate concentrations were determined from the total p-nitrophenol released after addition of NaOH to a final concentration of 1M. A second order rate constant 2.87 × 10-4 M-1 min-1 for reference reaction of 2 with phenol was determined from initial rates in identical buffer conditions. For amidase activity peptide-MCA substrates μM were added to 10 mM Tris, pH 8.0 containing purified scFv to obtain final concentrations of 20 - 300 μM amide and 50 - 100 μg/ml of protein in the absence or presence of 2 (20 μM). Fluorescence changes (Ex 380 nm, Em 460 nm) were recorded on a Cary Eclipse spectrofluorimeter equipped with thermostated cell changer (Varian, Inc). Kinetic data were analyzed using Prizm 4 software (GraphPad Inc.). Immunoadsorption was performed by absorbing a solution of scFv A.17 or mutant A.17Y36F at 155 μg/ml with 1 ml of agarose conjugated anti-c-myc Ab (Sigma-Aldrich, St. Louis, MO) or a control agarose-polyclonal Ab. After 1 h at room temperature the unbound protein was recovered by spin filtration and used for fluorescence assay with F-MCA substrate. Non-covalent binding was determined by ELISA. Ni-NTA purified scFv (0.1 mg/ml) were reacted with 100 μM of 1 and adsorbed on plates coated with anti-c-Myc mAb (Sigma-Aldrich). Plates were washed with PBS-tween and bound scFv was detected with streptavidin-HPR and tetramethylbenzidine-peroxide substrate (Pierce, Rockford, IL)
DNA sequence analysis
Phagemid DNA was isolated from 5 mL overnight cultures of phosphonate-reactive clones by alkaline lysis followed by RNAase A digestion and ethanol precipitation. The nucleotide sequences of the VH and VL genes of selected clones were determined by automated sequencing on an ABI sequencer using the primers GCCACCTCCGCCTGAACC CTATGCGGCCCCATTCA. The sequences were compared with germ-line V gene sequences deposited in the online Vbase directory (http://vbase.mrc-cpe.cam.ac.uk).
Analysis of modifications by SELDI mass spectrometry
Generally, the scFv were labeled with 1 or 2 at final concentrations of 20-100 μM for 60 min at 37°C. After labeling, proteins were precipitated with 4 volumes of acetone. The protein pellets were resuspended in 6 M urea, 50 mM Tris-HCl pH 7.5, 5mM DTT and incubated 1 hour at 65°C. Reduced cysteine residues were alkylated with 10 mM of iodacetamide, 30 min at 37°C in the dark. Labeled and control proteins were run on 12% SDS-PAGE. The gel slice containing the bands corresponding to scFv was excised and treated with sequencing grade trypsin according to the Ciphergen ProteinChip® applications guide. Mass spectrometric analysis of tryptic fragments was performed on a Ciphergen SELDI, using alpha-cyano-4-hydroxy cinnamic acid (CHCA) as a matrix.
Analysis of peptides by MALDI-TOF MS and MS/MS
Mass spectra were recorded on an Ultraflex II MALDI ToF-ToF mass spectrometer (Bruker Daltonik, Germany) equipped with Nd laser. The MH+ molecular ions were measured in reflector mode with accuracy of mass peak measurement of 0.02%. Aliquots (1 μL) of the sample were mixed on a steel target with an equal volume of 2,5-dihydroxybenzoic acid solution (10 mg/mL in 30% acetonitrile/0.5% trifluoroacetic acid) (Aldrich Chemical Co.), and the droplet was left to dry at room temperature. Each spectrum was obtained as the sum of a minimum 500 laser shots. Fragment ion spectra were generated by laser-induced dissociation slightly accelerated by low-energy collision-induced dissociation using helium as a collision gas. Correspondence of the experimentally determined masses to the peptide and to MS/MS peptide fragments was manually interpreted with the help of GPMAW 4.04 software (Lighthouse data, Denmark) using 0.02-0.05% precision as a criterion.
Site-directed mutagenesis
A.5
Mutagenesis was performed by PCR. A 127 bp fragment containing the Tyr32L/Phe mutation was amplified using pHEN2A.5 as template by sequential PCR with primers a/b followed by a/c, with sequences
(a)CTGGCGGTAGTGCACTTCAGTCT,
(b)GCTGCTGATACCAGTCGACAAAATTACTTCCGATG and
(c)CGTTTCCGGAAGCTGCTGATACCAGTCGAC.
The product was digested with ApaL I and Kpn2 I and ligated with the phagemid pHEN2A.5 digested with ApaL I and Xma I. A PCR product containing TyrL36/Phe mutant was obtained using primers a/d and pHEN2A.5 as a template, where primer d is CGTTTCCGGAAGCTGCTGGAACCAGTCTACG. This product was digested with ApaL I and Kpn2 I and ligated with the phagemid pHEN2A.5 digested with ApaL I and Xma I.
A.17
Mutations at Tyr36 and Tyr32 were created by overlap extension with two DNA fragments, obtained by PCR using pHEN2A.17 as template and primers a/e and f/i for the pair encoding Tyr36Phe mutation, a/g and h/i for the pair encoding Tyr36Ser mutation and a/i and j/h for the pair encoding Tyr32Phe mutation, where additional primer sequences were:
(e)GGAGCTGCTGGAACCAGGATACA; (f) TGTATCCTGGTTCCAGCAGCTCC;
(g) GGAGCTGCTGGGACCAGGATACA; (h)TGTATCCTGGTCCCAGCAGCTCC;
(i) GATGTGCGGCCGCACCTAGGA; (j) GGAGCTGCTGGAACCAGGATACA;
(k) TGTATCCTGGTTCCAGCAGCTCC. Overlap extension amplification for each mutation was completed with primer pair a/h and the product was used to replace the insert between ApaL I and Avr II sites in the phagemid pHEN2A.17.
The double mutation at tyrosine 32L and 36L (A.17Y32/36F) was created similarly using pHEN2A.17Y36F as template and primers a/l and m/i, where additional primer sequences were:
(l) GGGAATAATTTTGTATCCTGGTTCCAGC and
(m) TTGTATCCTGGTTCCAGCAGCTGCCAGG
Molecular Modeling
Models were constructed with the Swiss PDB Viewer 3.7 program and Swiss Model server25. VL and VH chains were modeled separately using input coordinates a known homologous Ab (1F3R-B for VH and 2B0S-L for VL in the Protein Data Bank). The ternary structure of the VH/VL model was refined using Insight II software (Accelrys) for energy minimization calculations.
RESULTS
Selection and screening of scFv-phage
The classical covalent reactivity of phosphonate esters for active site serine nucleophiles3,20 was used to probe a phage library constructed of artificially diversified human Ig V-gene segments expressing scFv fragments.
Employing a moderately reactive phosphonate diester 1 (Chart 1) for solution phase labeling, absorption of phage onto avidin-coated wells, and an enzymatic elution procedure26 it was possible to efficiently enrich the library for covalent binders. After three rounds of selection and screening for full-length scFv inserts, 47 clones were identified which all bound 1 by ELISA. Nine clones randomly picked from the original library did not bind. In order to distinguish clones that bound the ligand covalently, scFv treated with 1 were resolved by SDS-PAGE, electroblotted and stained with streptavidin-peroxidase to reveal a product at 28 kDa. Eight unique covalent binders and 6 non-covalent binders were chosen for further analysis.
Chart 1.

Reactive phosphonates used for selection and assay of scFv.
Clonal analysis
Alignment of polypeptide sequences of selected clones showed greater homogeneity among covalent binding clones than in the simple binders (Figure 1). The latter were diversified in all VH CDRs, whereas the former differed mainly in CDR-H3. Genetic lineages and CDR3 polypeptide sequences of the selected and 9 non-selected clones were compared in order to further distinguish structures associated with binding and reactivity (Table 1). The Vλ1 family is common to all selected clones with the exception of S.9, which is derived from Vλ3. VH4 is preferred among the reactive clones. Only clone A.21 uses the VH1 germline. The Vλ1 DPL-3 segment is represented in 7/8 covalent binding clones and 4/6 non-covalent binding clones. While clone A.17 appears unique among covalent binders with Vλ1 DPL5 and VH4 DP-67 segments, there is strong homology between DPL5 and DPL3. Interestingly, the VH4 DP-67 segment is not found paired with a DPL3 in non-covalent binders. Also striking is the presence of highly charged VH CDR3 sequences in non-covalent binders possessing VH4 DP-67. Covalent binder A.21 is highly homologous with non-covalent binders S.1, S.7 and S.14, having the same Vλ/VH germline and DPL-3 segment, except in CDR-H3 where it deviates significantly. The non-selected clones were diversified in both VH and VL families as well as in their CDR3 sequences.
Figure 1.
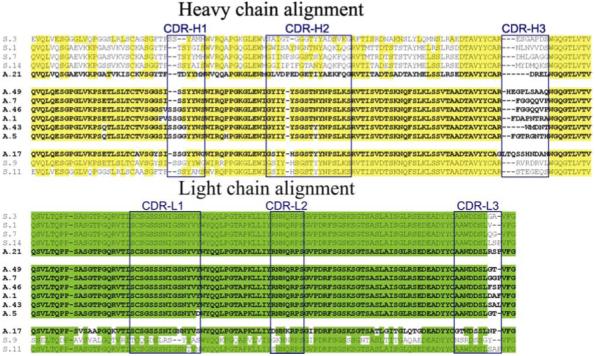
Alignment of covalent and non-covalent binding clones. The conserved amino acid residues of the heavy chain are highlighted in yellow and those of light chain in green. CDR regions are enclosed in boxes. Sequences of covalent binding clones (designated as A.nn), are shown in bold font.
Table 1.
CDR3 sequences and germline V gene segments of selected and non-selected scFv clones
| Clone | Light chain | Heavy chain | No. of copies | |||||
|---|---|---|---|---|---|---|---|---|
| Family | Segment | CDR3 * | Family | Segment | CDR3 | |||
| Non selected clones | ||||||||
| N4 | κ | A23* | MQATQFPRP | VH6 | DP-74 | FNTPTFDY | 1 | |
| N6 | λ | Vλ7 | DPL19 | LLSYSGANV | VH7 | DP-21 | SAMVNPV | 1 |
| N7 | λ | Vλ4 | DPL24 | GESHTIDGQV | VH4 | DP-66 | TSMHFRRWR | 1 |
| N8 | λ | Vλ1 | DPL3 | AAWDDSLTPT | VH2 | DP-27 | REGRVTDY | 1 |
| N9 | λ | Vλ3 | DPL16 | NSRDSSGNH | VH1 | DP-10 | SMNPTFDY | 1 |
| N17 | λ | Vλ9 | DPL22 | GADHGSGSSF | VH4 | DP-68 | VLFVTFDY | 1 |
| N43 | κ | A19* | MQALQALC | VH6 | DP-74 | TLGDPFDY | 1 | |
| N48 | λ | Vλ4 | DPL24 | GESHTIGGQVS | VH4 | DP-71 | CPRPTH | 1 |
| N55 | λ | Vλ1 | DPL2 | AAWDDSLTCC | VH1 | DP-14 | NVRNMWMW | 1 |
| Binding clones | ||||||||
| S.1 | λ | Vλ1 | DPL-3 | AAWDDSLV | VH1 | DP-14 | NLNVVDS | 1 |
| S.3 | λ | Vλ1 | DPL-3 | AAWDDSLGA | VH3 | DP-45 | ESGAPDS | 1 |
| S.7 | λ | Vλ1 | DPL-3 | AAWDDSLQG | VH1 | DP-7 | DHLGAGG | 1 |
| S.9 | λ | Vλ3 | DPL-16 | NSRDSSGY | VH4 | DP-67 | RVRDRVL | 2 |
| S.11 | λ | Vλ1 | DPL-2 | AAWDDSLSAP | VH3/VH4 | DP-47/DP-67 | STEGEQS | 1 |
| S.14 | λ | Vλ1 | DPL-3 | AAWDDSLLSP | VH1 | DP-10 | MYDMQKS | 1 |
| Reactive clones | ||||||||
| A.1 | λ | Vλ1 | DPL-3 | AAWDDSLDAF | VH4 | DP-66 | FDAPNTRA | 1 |
| A.46 | λ | Vλ1 | DPL-3 | AAWDDSLFSP | VH4 | DP-66 | FGGQQVP | 1 |
| A.5 | λ | Vλ1 | DPL-3 | AAWDDSLGT | VH4 | DP-65 | FGTRGNTH | 1 |
| A.43 | λ | Vλ1 | DPL-3 | AAWDDSLSAL | VH4 | DP-65 | WMDNT | 1 |
| A.7 | λ | Vλ1 | DPL-3 | AAWDDSLGGP | VH4 | DP-71 | FGGQQVP | 3 |
| A.49 | λ | Vλ1 | DPL-3 | AAWDDSLGT | VH4 | DP-71 | HEGPLSAAQ | 1 |
| A.21 | λ | Vλ1 | DPL-3 | AAWDDSLRSP | VH1 | DP-3 | DREL | 1 |
| A.17 | λ | Vλ1 | DPL-5 | GTWDSSLNP | VH4 | DP-67 | LTQSSHNDAN | 2 |
Underlined residues in CDR-L3 are germline encoded (codons were not varied during library construction).
Seven of the covalent binders were kinetically characterized in the reaction with phosphonate 2 (supporting information, Figure S1A). Six of these had similar kinetic reactivity with k1 of 0.012 - 0.035 min-1 while one (A.17) was an order of magnitude more reactive (k1 = 0.32 min-1). Dissociation constants varied from 35 to 213 μM (Table 2). Titration of the reactive scFv using excess 2 suggested that 90 -100% of the total protein was labeled. Stoichiometry consistent with a single site of modification was conclusively demonstrated in each case by mass spectrometry (supporting information, Figure S1B).
Table 2.
Kinetic properties of the covalent binding clones. Catalytic parameters were determined from the labeling assay with 2 at 22°C using the Kitz-Wilson kinetic scheme24
| Clone | k1, min-1 | Kd, μM | k1/Kd (M-1min-1) |
|---|---|---|---|
| A.17 | 0.32 ± 0.005 | 151 ± 21 | 2119 |
| A.5 | 0.035 ± 0.002 | 35 ± 23 | 1000 |
| A.21 | 0.032 ± 0.005 | 71 ± 18 | 451 |
| A.43 | 0.017 ± 0.004 | 81 ± 43 | 210 |
| A.46 | 0.021 ± 0.004 | 89 ± 49 | 236 |
| A.49 | 0.012 ± 0.004 | 56 ± 14 | 214 |
| A.7 | 0.037 ± 0.003 | 213 ± 86 | 174 |
Identification of the active site nucleophiles
SELDI MS of tryptic fragments of clone A.5 before and after the reaction with 1 identified one modified peptide (m/z = 3056), corresponding to the 29-mer fragment derived from CDR-L1 and parts of the flanking framework regions (VL-Val19-Lys45). The site of reaction in this peptide was determined by de novo sequencing by MALDI-MS/MS. From the observable peaks in the MS fragmentation pattern it was possible to deduce that the modification was borne at Tyr32. The same modification site was shown for clones A.7, A.21 and A.43. Labeling at the equivalent tryptic peptide (m/z 3058) was found for the reaction product of A.17 (supporting information, Figure S2). However, a different fragmentation pattern was obtained in MS/MS analysis of this peptide, suggesting modification had occurred at framework Tyr36 (supporting information, Figure S3). Additional analysis of the peptide by chymotrypsinolysis and direct MS was performed for confirmation (supporting information, Figure S4). Two principal products were observed, concordant with the unmodified N-terminal fragment Val19-Trp35 and the phosphonate-modified C-terminal fragment Tyr36-Lys45. Both Tyr32 and Tyr36 are encoded in the λ VL germline repertoire27. The position of the two nucleophiles and context within diversified Fv elements is illustrated in alignment of A.17 and A.5 polypeptides (Figure 2).
Figure 2.

Structural alignment of A.5 and A.17 VH and VL sequences. The CDR regions are indicated in colored highlight. The tryptic peptides spanning the residues modified by 1 are enclosed in the blue box. Reactive tyrosine residues are shown in red font. Residues of VH and VL and CDRs are identified according to the Kabat numbering system.
Site-directed mutagenesis studies were performed to corroborate the results implicating different nucleophilic Tyr residues in the two types of reactive clones. The mutant A.5Y32F failed to react with 1, whereas mutant A.5Y36F had reactivity like the wild type as determined by streptavidin blotting. Conversely, mutant scFv A.17Y32F had the wild type reactivity while A.17Y36F, A.17Y36S and A.17Y32/36F mutants were completely non-reactive. Comparable amounts of mutant scFv proteins were available for reaction as detected by c-Myc tag staining (Figure 3A). The functionally impaired mutants were also shown to have no capacity to bind the phosphonate by ELISA (Figure 3B).
Figure 3.
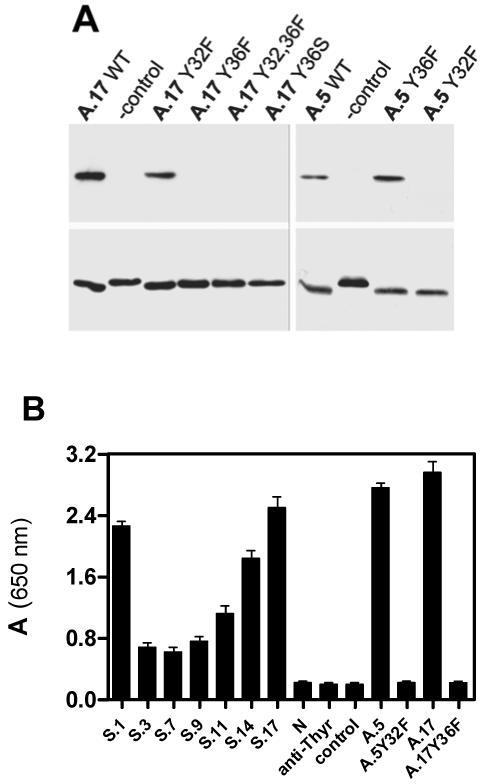
(A) Covalent reactivity of A.17 and A.5 and their mutants. Concentrations of scFvs were normalized as confirmed by comparable staining in anti-c-Myc blot (lower panel). (B) Binding efficiency of clones selected after the third round, including reactive clones A.5, A.17 and their non-reactive mutants A.5Y32F and A.17Y36F. Non-selected clone (N) and anti-thyroglobulin scFv (anti-Thyr) were included as negative controls. Binding was determined as described in Methods.
Phosphonylation and amidase activities
At high concentration diphenylphosphonates 3 and 4 and the serine protease inhibitor AEBSF could block labeling of wild type A.17 by 1 (Figure 4). However, covalent adduct formation with these ligands was not detected by mass analysis. Activated phosphonates 5 and 6 (sarin) also did not react, suggesting the A.17 phosphonylation site is more selective than common serine hydrolases.
Figure 4.
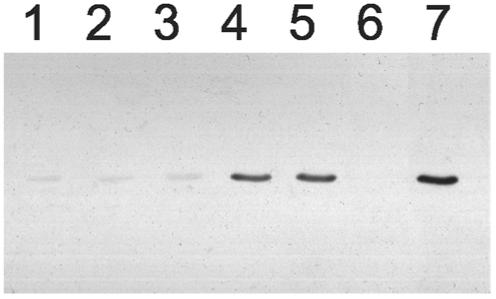
Reaction of A.17 with alternative substrates. A.17 was incubated for 1 hour at 37°C with 5 mM each of AEBSF (lane 1), 3 (lane 2), or 4 (lane 3), or 100 μM of 5 (lane 4), 6 (lane 5), or 2 (lane 6), or PBS buffer alone (lane 7). All samples were then incubated for 1 hour at 37°C with 100 μM of 1 and analyzed by western blot as in Figure 3A.
The two covalent binders, A.17 and A.5, were assessed for amidase activity using fluorogenic peptide methylcoumarinamides (MCA). After FPLC purification on mono Q sepharose neither protein reacted with typical trypsin substrates Pro-Phe-Arg-MCA or Boc-Gly-Gly-Arg-MCA. On the other hand hydrolysis of two hydrophobic substrates, Phe-MCA and Boc-Ala-Ala-Phe-MCA, was modestly accelerated in the presence of 50 μg/ml of A.17. To rule out possible artifacts or proteolytic contamination, wild type A.17 and the A.17Y36F mutant were similarly fractionated by ion exchange FPLC and their hydrolytic activities compared. For A.17 a peak of activity eluted at the scFv protein peak using two different salt gradients. In contrast no evidence of similar activity was obtained in the fractions spanning the scFv peak for mutant A.17Y36F (supporting information, Figure S5). Activity was specifically depleted by immunoadsorption with anti-c-Myc agarose, but not by a control agarose matrix. The reaction of purified A.17 with Phe-MCA was characterized by saturation kinetics with constants kcat of approximately 1.5 ± 0.8 × 10-3 min-1, and Km of 75 ± 15 μM. The activity was completely inhibited by preincubation with 20 μM of 2.
Molecular modeling of the active sites
Relatively few primary structure differences distinguished the covalent from non-covalent binding clones. The most remarkable differences were concentrated in CDR-H3 where the former are characterized by the presence of polar, neutral sequences in contrast to hydrophobic or highly charged sequences in the latter. Molecular modeling of the reaction center suggested the juxtaposition of CDR-L1 and FR-L2 with residues of the heavy chain CDR-H3 in both A.5 and A.17 (supporting information, Figure S6). Remarkably, modifiable Tyr36 is effectively buried in a deep cavity at the VH-VL interface and its side chain is oriented toward residues of CDR-H3, while the more common nucleophile, Tyr32 in CDR-L1, is exposed to the medium in a shallow site at the apex of the molecule.
DISCUSSION
Approaches for directed evolution of enzymes typically depart from the existing enzymes to modify or redirect activity for different purposes. This mimics natural adaptation of enzymes to new functions by gene duplication, diversification and selection. However, the origins of enzyme activity can presumably be traced to the acquisition of reactive residues or cofactors that participate chemically in the catalytic reactions. Directed evolution might similarly apply to primitive proteins to select chemical reactivity that is adaptable to enzyme mechanisms. Antibody combining sites are well suited to both natural and artificial diversification, providing for vast permutations of protein functional group presentation. In this study reactive selection was applied to identify scFv molecules that express covalent reactivity, a chemical trait that is virtually universal to catalysis. Chemical properties and structural insights derived from the reactive molecules could therefore suggest basic features for the design of natural or artificial enzymes.
Highly reactive phosphorylating/phosphonylating agents, including DFP and sarin, efficiently inhibit a wide range of serine hydrolases by modification at the active site serine residue. On the other hand, synthetic arylphosphonate esters have moderated reactivity, allowing greater inhibitor specificity based on substrate features incorporated into the molecules3,20. We used a p-nitrophenylphosphonate ester with intermediate reactivity to allow capture of nucleophilic residues having a wide range of reactivities in chemical selection. Reagents based on this structure had excellent attributes for in vitro applications, including good enzyme selectivity, low non-specific protein labeling and adequate stability to hydrolysis19. The selection procedure included reaction of phage particles with soluble biotinylated phosphonate 1, followed by affinity capture of biotinylated phage. Stringent washing conditions coupled with trypsin-mediated elution presumably accounted for efficient partitioning of non-covalent and covalently bound phage. Reduction of the reagent concentration in the final round of selection afforded another degree of control for kinetic differentiation of reactive scFv phage.
The identification of several clones with similar reaction kinetics suggested the possibility of a common structural basis for their reactivity. In fact, a common VL and similar VH4 germline genes were used in all these clones. The ten-fold more reactive clone A.17 used a unique VL germline, but included a similar VH4 gene. Interestingly, the non-reactive clones also presented one of the two canonical genes used in the reactive clones, but paired with a different VL or VH gene. Enhanced nucleophilicity of Tyr36 is consistent with a buried, solvent-shielded position in the combining site cleft of A.17, as suggested by modeling. The models also provide a rationale for the influence of CDR-H3 sequences in the reactivities of A.5 and A.17. A channel for ligand access to VL Tyr36 in A.17 is not obvious from the model. It is possible that access is gated by conformational mobility of a 7-residue hydrophilic loop in CDR-H3 (Table 1). By contrast, covalent binders reacting at Tyr32 have shorter CDR-H3 (7/7 clones), frequently including Pro (4/7 clones), which favors more rigid conformations.
The reference reaction of phosphonate 2 with phenol (k2 = 2.87 × 10-4 M-1 min-1) suggested a rate enhancement by A.17 of 7.3 × 106 or an effective molarity (EM = k1/k2) of 1.1 × 103 M, consistent with a proximity effect in nucleophilic catalysis28. Furthermore, mutants A.17Y36F and A.5Y32F did not bind 1, as detectable for selected non-covalent binders. The conservative Tyr32/36Phe replacements were unlikely to have a large effect on scFv structure or binding affinity. The specificity of these scFv for phenolic phosphonates and failure to react with the more activated phosphonofluoridate 6 suggested a significant influence of non-covalent binding on the reactivity of the Tyr nucleophile. The kinetic nature of reactive selection favors scFv clones that can utilize specific non-covalent interactions to accelerate the covalent reaction. The poor binding of phosphonate 1 by the mutants supports this interpretation and suggests that non-covalent binding is expressed in the transition state, as expected for an enzyme-like site.
Covalent catalysis is a long-standing goal in approaches to generate artificial enzymes by chemical design29, directed molecular evolution1,2 or immunization strategies30. Enzymes utilizing nucleophilic tyrosine are rare in nature. Our results most likely reflect properties of the selection reagent, as the nucleophilicity and pKa of tyrosine hydroxyl balances the p-nitrophenol leaving group of ester 1. In principle, the selection reagent could be tuned to demand more reactive nucleophiles, including serine or threonine residues.
Modest amide hydrolysis by A.17 demonstrates the potential for the selected nucleophile to engage in covalent catalysis and encourages further efforts to improve catalytic activity. No clones were identified using serine nucleophiles, as suggested in reports of scFv with peptidase activity recovered from an autoimmune repertoire31. Recent studies in experimental autoimmune encephalomyelitis suggest that such antibodies may have unique origins in pathogenic autoimmunity32. In earlier studies catalytic antibodies were generated with higher frequency in autoimmune prone animals, suggesting that reactive clones could be excluded from a normal repertoire by immune tolerance33. Immunization strategies culminating in reactive immunization proved the potential of affinity maturation to drive in vivo clonal selection to obtain antibodies with impressive catalytic activity13. Selection for covalent binding in vitro bypasses early sampling of non-covalent clones and could therefore take a direct path to sites having appropriate catalytic machinery. Restricted diversification of reactive clones coupled with variation of selection reagents and conditions in further rounds of selection could lead to rapid ascent in kinetic efficiencies or substrate specificities. The recognition of a CDR-H3 motif for reactivity could also be useful in creating biased scFv libraries. These studies demonstrate a structural basis for nascent protein nucleophiles and define possible entry points for in vitro evolution of new artificial enzymes.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the US National Institutes of Health grants CA90564 (A.T.) and BTEP No. 37 (A.G.G. and H.C.M.), the University of California, Davis - School of Medicine, Children’s Miracle Network (A.T.), the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases (H.C.M.), the Russian Foundation of Basic Research Grant 06-04-49727-a (N.A.P.), and the National Technical Base Ministry of Industry (Russian Federation) Grant 77 (A.G.G.).
REFERENCES
- (1).Fernandez-Gacio A, Uguen M, Fastrez J. Trends in Biotechnology. 2003;21:408–414. doi: 10.1016/S0167-7799(03)00194-X. [DOI] [PubMed] [Google Scholar]
- (2).Soumillion P, Fastrez J. Curr Opin Biotechnol. 2001;12:387–394. doi: 10.1016/s0958-1669(00)00232-9. [DOI] [PubMed] [Google Scholar]
- (3).Powers JC, Asgian JL, Ekici OD, James KE. Chem Rev. 2002;102:4639–4750. doi: 10.1021/cr010182v. [DOI] [PubMed] [Google Scholar]
- (4).Danielsen S, Eklund M, Deussen HJ, Graslund T, Nygren PA, Borchert TV. Gene. 2001;272:267–274. doi: 10.1016/s0378-1119(01)00527-3. [DOI] [PubMed] [Google Scholar]
- (5).Legendre D, Laraki N, Graslund T, Bjornvad ME, Bouchet M, Nygren PA, Borchert TV, Fastrez J. J Mol Biol. 2000;296:87–102. doi: 10.1006/jmbi.1999.3437. [DOI] [PubMed] [Google Scholar]
- (6).Baca M, Scanlan TS, Stephenson RC, Wells JA. Proc Natl Acad Sci U S A. 1997;94:10063–10068. doi: 10.1073/pnas.94.19.10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Fujii I, Fukuyama S, Iwabuchi Y, Tanimura R. Nat Biotechnol. 1998;16:463–467. doi: 10.1038/nbt0598-463. [DOI] [PubMed] [Google Scholar]
- (8).Janda KD, Lo LC, Lo CH, Sim MM, Wang R, Wong CH, Lerner RA. Science. 1997;275:945–948. doi: 10.1126/science.275.5302.945. [DOI] [PubMed] [Google Scholar]
- (9).Betley JR, Cesaro-Tadic S, Mekhalfia A, Rickard JH, Denham H, Partridge LJ, Pluckthun A, Blackburn GM. Angew Chem Int Ed Engl. 2002;41:775–777. doi: 10.1002/1521-3773(20020301)41:5<775::aid-anie775>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- (10).Cesaro-Tadic S, Lagos D, Honegger A, Rickard JH, Partridge LJ, Blackburn GM, Pluckthun A. Nat Biotechnol. 2003;21:679–685. doi: 10.1038/nbt828. [DOI] [PubMed] [Google Scholar]
- (11).Janda KD, Lo CH, Li T, Barbas CF, 3rd, Wirsching P, Lerner RA. Proc Natl Acad Sci U S A. 1994;91:2532–2536. doi: 10.1073/pnas.91.7.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Gao C, Lin CH, Lo CH, Mao S, Wirsching P, Lerner RA, Janda KD. Proc Natl Acad Sci U S A. 1997;94:11777–11782. doi: 10.1073/pnas.94.22.11777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wagner J, Lerner RA, Barbas CF., 3rd Science. 1995;270:1797–1800. doi: 10.1126/science.270.5243.1797. [DOI] [PubMed] [Google Scholar]
- (14).Thayer MM, Olender EH, Arvai AS, Koike CK, Canestrelli IL, Stewart JD, Benkovic SJ, Getzoff ED, Roberts VA. J Mol Biol. 1999;291:329–345. doi: 10.1006/jmbi.1999.2960. [DOI] [PubMed] [Google Scholar]
- (15).Zhou GW, Guo J, Huang W, Fletterick RJ, Scanlan TS. Science. 1994;265:1059–1064. doi: 10.1126/science.8066444. [DOI] [PubMed] [Google Scholar]
- (16).Wirsching P, Ashley JA, Benkovic SJ, Janda KD, Lerner RA. Science. 1991;252:680–685. doi: 10.1126/science.2024120. [DOI] [PubMed] [Google Scholar]
- (17).Tramontano A, Janda KD, Lerner RA. Proc Natl Acad Sci U S A. 1986;83:6736–6740. doi: 10.1073/pnas.83.18.6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Karlstrom A, Zhong G, Rader C, Larsen NA, Heine A, Fuller R, List B, Tanaka F, Wilson IA, Barbas CF, 3rd, Lerner RA. Proc Natl Acad Sci U S A. 2000;97:3878–3883. doi: 10.1073/pnas.97.8.3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Tramontano A, Ivanov B, Gololobov G, Paul S. Appl Biochem Biotechnol. 2000;83:233–242. doi: 10.1385/abab:83:1-3:233. [DOI] [PubMed] [Google Scholar]
- (20).Oleksyszyn J, Powers JC. Methods Enzymol. 1994;244:423–441. doi: 10.1016/0076-6879(94)44032-8. [DOI] [PubMed] [Google Scholar]
- (21).Griffiths AD, Williams SC, Hartley O, Tomlinson IM, Waterhouse P, Crosby WL, Kontermann RE, Jones PT, Low NM, Allison TJ, et al. Embo J. 1994;13:3245–3260. doi: 10.1002/j.1460-2075.1994.tb06626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Marks JD, Bradbury A. Methods Mol Biol. 2004;248:161–176. doi: 10.1385/1-59259-666-5:161. [DOI] [PubMed] [Google Scholar]
- (23).Hixson CS, Krebs EG. J Biol Chem. 1979;254:7509–7514. [PubMed] [Google Scholar]
- (24).Kitz R, Wilson IB. J Biol Chem. 1962;237:3245–3249. [PubMed] [Google Scholar]
- (25).Schwede T, Kopp J, Guex N, Peitsch MC. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Goletz S, Christensen PA, Kristensen P, Blohm D, Tomlinson I, Winter G, Karsten U. J Mol Biol. 2002;315:1087–1097. doi: 10.1006/jmbi.2001.5314. [DOI] [PubMed] [Google Scholar]
- (27).Kabat EA, National Institutes of Health (U.S.) Columbia University . Sequences of proteins of immunological interest. 5th ed. U.S. Dept. of Health and Human Services Public Health Service National Institutes of Health; Bethesda, MD: 1991. [Google Scholar]
- (28).Fersht A. 2nd ed. W.H. Freeman; New York: 1985. Enzyme structure and mechanism. [Google Scholar]
- (29).Breslow R. Artificial enzymes. Wiley-VCH; Weinheim: 2005. [Google Scholar]
- (30).Lerner RA, Benkovic SJ, Schultz PG. Science. 1991;252:659–667. doi: 10.1126/science.2024118. [DOI] [PubMed] [Google Scholar]
- (31).Paul S, Tramontano A, Gololobov G, Zhou YX, Taguchi H, Karle S, Nishiyama Y, Planque S, George S. J Biol Chem. 2001;276:28314–28320. doi: 10.1074/jbc.M102530200. [DOI] [PubMed] [Google Scholar]
- (32).Ponomarenko NA, Durova OM, Vorobiev II, Belogurov AA, Jr., Kurkova IN, Petrenko AG, Telegin GB, Suchkov SV, Kiselev SL, Lagarkova MA, Govorun VM, Serebryakova MV, Avalle B, Tornatore P, Karavanov A, Morse HC, III, Thomas D, Friboulet A, Gabibov AG. PNAS. 2006;103:281–286. doi: 10.1073/pnas.0509849103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Tawfik DS, Chap R, Green BS, Sela M, Eshhar Z. Proc Natl Acad Sci U S A. 1995;92:2145–2149. doi: 10.1073/pnas.92.6.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.