

Abstract
In the preceding accompanying paper (1), the 1.5 Å resolution crystal structure of GFP variant S65T/H148D is presented and the possible consequences of an unusual short hydrogen bond (≤2.4 Å) between the carboxyl oxygen of Asp148 and phenol oxygen of the chromophore are discussed. In this work, we report the femtosecond time-resolved emission of this variant at pH 5.6 by ultrafast fluorescence upconversion spectroscopy. Following excitation at 400 nm, green fluorescence is observed at 510 nm with a rise on a timescale that is faster than the 170 femtosecond instrument response. Time-resolved emission spectra at 140 K also exhibit the immediate appearance of green fluorescence, and this extremely fast process is hardly affected by deuteration of exchangeable protons. These results appear to be dramatically different from those of wild-type GFP, in which the green fluorescence at 508 nm is produced on the picosecond timescale as a result of excited-state proton transfer from the state that is excited at 400 nm. The unique features observed in S65T/H148D and apparent ultrafast excited-state proton transfer are discussed in light of evidence for multiple states underlying the band at around 415 nm, as suggested by steady-state fluorescence spectra. The behavior of these different states may explain the novel photophysical properties observed for this GFP variant, including the ultrafast green fluorescence and the absence of completely matched decay in blue fluorescence. We speculate that two different orientations of the Asp introduced at position 148, not distinguishable by chromatography, mass spectrometry or x-ray crystallography, give rise to the two functionally distinct populations.
At room temperature wild-type green fluorescent protein (GFP) exhibits two absorption bands with maxima at 396 and 476 nm. Previous studies have established that these bands can be attributed to the neutral and anionic states of the chromophore, denoted A and B, respectively (2–4). Excitation of either form leads to green fluorescence at 508 nm with an essentially equivalent, high quantum yield. Scheme 1 represents the key photophysical features of wild-type GFP at room temperature (2). In this model, direct excitation of the anionic form B leads to green fluorescence. Upon excitation of the neutral form A, proton transfer occurs in the excited state and A* rapidly converts to I*, an anionic form that subsequently emits green fluorescence.
Scheme 1.
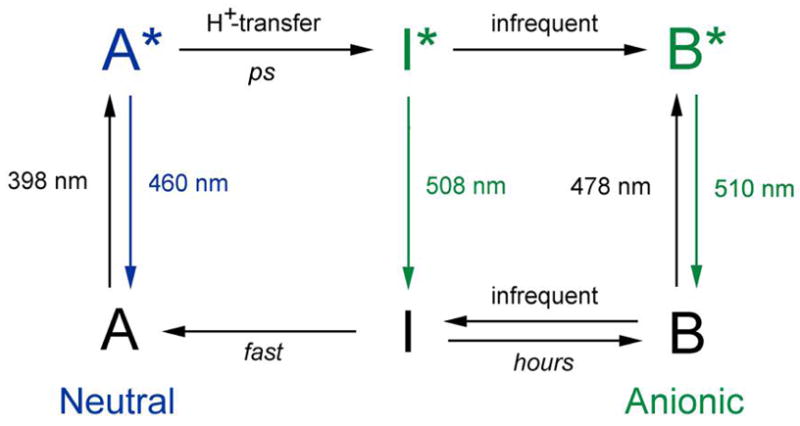
Spectroscopic and structural studies have suggested some differences between I and B (5–9). The blue emission from A* at 460 nm is observed only on the picosecond timescale and its decay kinetics match the rise kinetics of the green emission from I* at 508 nm. Both the A* decay and I* rise are slowed by replacing exchangeable protons with deuterons, which led to the suggestion that an excited-state proton transfer (ESPT) reaction connects these states (2, 10). Time-resolved vibrational studies provided further evidence for the ESPT pathway (11, 12). Recent studies on a class of dual-emission GFP (deGFP) variants suggest that the overall scheme still holds, although the rate of ESPT is decreased compared to the wild type and thus significant blue emission from A* can be observed even at steady state making this a useful class of GFPs for probing local pH in cells (13, 14). A high-resolution crystal structure of one deGFP indicates that the pH-dependent ESPT rate is a result of different hydrogen bond networks around the chromophore than in wild-type GFP (15). Although the working model in Scheme 1 provides a general framework for understanding the photochemistry and photophysics of wild-type GFP and deGFPs, a number of details and its general applicability remain open questions.
In the preceding accompanying paper, we reported the high resolution crystal structures of GFP S65T/H148D and several closely related mutants (1). The 1.5 Å resolution structure of S65T/H148D at pH 5.6 reveals an unusual short hydrogen bond (≤ 2.4 Å) between the phenol oxygen of the chromophore and the carboxyl oxygen of Asp148. In this paper, we investigate the femtosecond time-resolved emission of GFP S65T/H148D by fluorescence upconversion spectroscopy. At pH 5.6, we observe an instrument-response-limited rise in green fluorescence following excitation of the absorption band around 415 nm, nominally like band A in wild-type GFP, much faster than what was observed for the wild type or deGFPs. Absorption and steady-state fluorescence spectra exhibit a number of unusual features suggesting multiple states underneath the broad A band in S65T/H148D. We attempt to relate this ground-state landscape to the other novel photophysical properties observed for this GFP variant.
MATERIALS AND METHODS
Sample Preparation
GFP S65T/H148D was prepared as described previously (16). The samples were exchanged into 100 mM NaCl, 50 mM MES buffer solutions at pH 5.6 and pD 5.6, by concentrating the protein followed by resuspension in the desired buffer (repeated three times) in an Amicon Ultra-4 centrifugal filter unit with 10,000 MWCO (Millipore, Billerica, MA). The deuterated buffer solution was prepared as described previously (14) and corrected for the isotope effect on the pH meter (17). For temperature-dependent fluorescence and absorption studies, a 60% (v/v) glycerol (or glycerol-(OD)3) solution was prepared in buffer containing the protein at pH 5.6 (or pD 5.6). For the low-high-low pH cycling experiment, 100 mM NaCl, 50 mM Bis-Tris-Propane buffer solutions of pH 9.2 and pD 9.2 were prepared. The buffer exchange was performed in the following order: first pH 5.6, then pH 9.2, followed by pH 5.6; the same sequence was followed in deuterated buffers.
Variable Temperature Apparatus
A miniature Joule-Thomson refrigerator (MMR, Mountain View, CA) was used to vary the temperature between 298 and 140 K. To alleviate the concentration requirement to reach a sufficient optical density, we replaced the 75-μm path length cell that was previously used (14) with a 1-mm path length quartz cuvette (Starna Cells, Atascadero, CA) cut from 45 to 13 mm long to reduce the sample volume and heat loss. This cuvette contained ~ 80 μL sample and was clamped onto the cold finger in the refrigerator to improve the efficiency of heat transfer. The temperature in the sample was measured with a temperature-sensitive Pt resistor immersed in the sample inside the cuvette and was found to be somewhat higher than that on the cold finger (by at most ~ 20 K at 140 K). All the temperatures subsequently referred to are those measured by the sensor on the cold finger. Experiments below 140 K were not performed as the sample in glycerol/buffer in the 1-mm path length cuvette cracks at a temperature lower than 140 K.
Absorption and Steady-state Fluorescence Measurements
Absorption spectra were recorded on a PerkinElmer Lambda 12 UV/Vis spectrophotometer (PerkinElmer, Wellesley, MA). Fluorescence emission and excitation spectra were recorded with front-face geometry on a Spex FluoroLog fluorimeter with a Spex 1620 dual-grating emission monochromator (Spex, Metuchen, NJ) as described previously (14). For room-temperature studies, a standard 1-mm path length quartz cuvette containing the protein in buffer was used. Following excitation at 400 nm, fluorescence emission was collected from 420 to 650 nm and corrected as described (14). In addition, fluorescence excitation was detected from 320 to 490 nm at 510 nm emission. For temperature-dependent studies, the variable temperature apparatus with a shortened 1-mm path length cuvette containing the protein in glycerol/buffer was used. Absorption and fluorescence emission with 400 nm excitation were measured in ~ 20 K intervals after cooling at a rate less than 0.1 K/s and a 5-min equilibration time at the desire temperature (14). At 296, 210 and 140 K, fluorescence emission with 370 and 430 nm excitation and fluorescence excitation detected at 460, 510 and 550 nm were also measured using the same setup.
Fluorescence Upconversion Spectroscopy
The time-resolved fluorescence of GFP S65T/H148D at pH 5.6 and pD 5.6 in buffer at room temperature and in glycerol/buffer at 140 K was measured using the fluorescence upconversion setup described previously (14). Samples were excited at 400 nm by the second harmonic of a mode-locked Ti: Sapphire laser (Spectra Physics, Mountain View, CA), pumped by an Ar-ion laser. The cross-correlation of the gate beam and scattered excitation light was used as the instrument response function (IRF) with a full-width at half maximum (FWHM) of typically 170 fs. 10 mW of 400 nm light at 82 MHz, polarized at the magic angle with respect to the gate beam, was used to excite the sample in a 1-mm path length quartz cuvette. For room-temperature studies, the sample was stirred rapidly with a small magnetic stir bar. For 140 K studies, the sample was translated with the refrigerator by 16 μm after each time point in kinetics scans or wavelength point in spectral scans to avoid photobleaching. At both temperatures, the kinetics were measured at 475 and 510 nm within a time window of 1.3 ns. The data at 510 nm were fit to the convolution of the IRF at 400 nm with a model function composed of a sum of exponentials with two femtosecond components of negative amplitudes, a baseline and an offset of time zero. In addition to the kinetics measurements at discrete wavelengths, time-resolved emission spectra were obtained as described previously (14) at t = −0.3, −0.1, 0, 0.1, 0.3 and 0.5 ps at room temperature and t = −0.3, 0, 0.3, 10, 100 and 1000 ps at 140 K.
Selective Photobleaching
A frequency-doubled, diode-pumped solid-state CW laser at 457 nm (Melles Griot, Carlsbad, CA) with an average output power of ~ 200 mW was used to irradiate the sample while it was continuously stirred in a 1-mm path length quartz cuvette at room temperature. After photobleaching, the whole sample was transferred from the cuvette into a 1.5-mL microcentrifuge tube and centrifuged to remove the precipitate. The supernatant was then recovered by decanting. 50 mW of 450 nm light at 82 MHz, generated from the second harmonic of the Ti: Sapphire laser running at 900 nm was also used for photobleaching.
Stark Spectroscopy
Absorption and Stark spectra of GFP S65T/H148D at pH 5.6, pD 5.6, and pH 9.2 were measured and analyzed as described previously (18). For comparison, absorption and Stark spectra were also measured for three dual-emission GFPs, i.e. deGFP1, deGFP3, and deGFP4 at low pH, where band A dominates the absorption profile at 77 K.
RESULTS
Absorption and Steady-state Fluorescence at Room Temperature
The room temperature absorption and fluorescence spectra of GFP S65T/H148D at pH 5.6 are show in Figure 1. Elsliger et al. previously reported that the absorption profile displays two pH-dependent peaks at 415 and 487 nm, with a chromophore pKa of 7.8 (16). At pH 5.6, the higher energy band at 415 nm dominates the spectrum, whereas at pH 9.2, the equilibrium shifts and the absorption is dominated by the lower energy band at 487 nm (see Figure 1 in the succeeding paper (19)). As for wild type GFP (2) and dual-emission GFPs (13–15), these two electronic absorption bands of S65T/H148D are denoted A and B, respectively, noting that their identification with different protonation states is only a working model. In this paper, we focus mostly on the low -pH A absorption band; the characterization of the high-pH absorption band, B, is described in the accompanying papers (1, 19). Interestingly, band A in S65T/H148D is red-shifted by ~ 20 nm from that of the wild type and deGFPs, where band A lies at 395–400 nm (20, 21). Replacing exchangeable protons with deuterons at the same pH results in a small blue shift of band A to 411 nm (Figure 1A), while no such effect upon deuteration is observed in either the wild type (2) or deGFPs (13, 14). To demonstrate that the protons affecting band A are exchangeable, a sample was subjected to a low-high-low pH or pD cycling treatment. This process caused no change to the absorption spectrum after a cycle was finished (data not shown), so the exchange is fully reversible.
Figure 1.
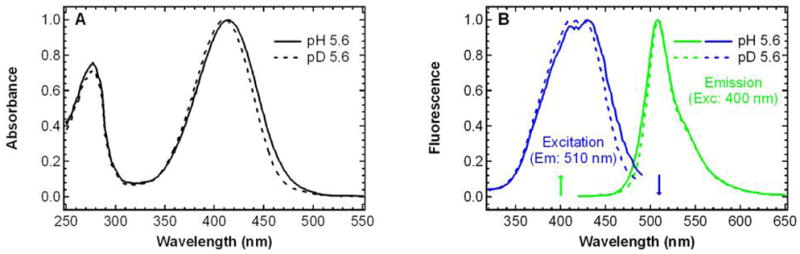
Steady-state absorption (A) and fluorescence (B) spectra of S65T/H148D GFP at pH 5.6 (solid) and pD 5.6 (dashed) at room temperature. In panel B, fluorescence emission spectra (λexc = 400 nm) and fluorescence excitation spectra (λem = 510 nm) are shown in green and blue, respectively. All spectra are normalized to the peak value of the spectrum.
Following excitation of band B at pH 9.2, the steady-state emission spectrum exhibits one peak at 510 nm, consistent with other members in the GFP family (20, 21). At pH 5.6, similar features are observed upon excitation of band A, and fluorescence between 420 and 470 nm that might be associated with emission from A* is barely detectable (Figure 1B). This bright green-emitting feature upon illumination at 400 nm is purely a result of exciting band A, as is manifest from the absorption profile seen in Figure 1A, in which only band A is seen while band B is negligible. Deuteration of exchangeable protons at pH 5.6 does not lead to a noticeable shift of the emission peak. However, a measurable isotope effect is observed in the fluorescence excitation profile as shown in Figure 1B. With the emission monitored at 510 or 550 nm, the excitation spectrum at pH 5.6 is red-shifted relative to that at pD 5.6 as indicated by Figure 1B and this shift is consistent with the trend observed in the absorption spectrum (Figure 1A). We stress that the fluorescence excitation maximum at pH 5.6 is at 420 nm, red-shifted from the absorption maximum (415 nm). The observation that the fluorescence quantum yield is greater by exciting on the red side of the absorption maximum is even more dramatic in a sample of S65T/H148D mixed with glycerol for temperature-dependent studies, where the excitation maximum for 510 nm emission is shifted to 435 nm (Figure 4E and F).
Figure 4.
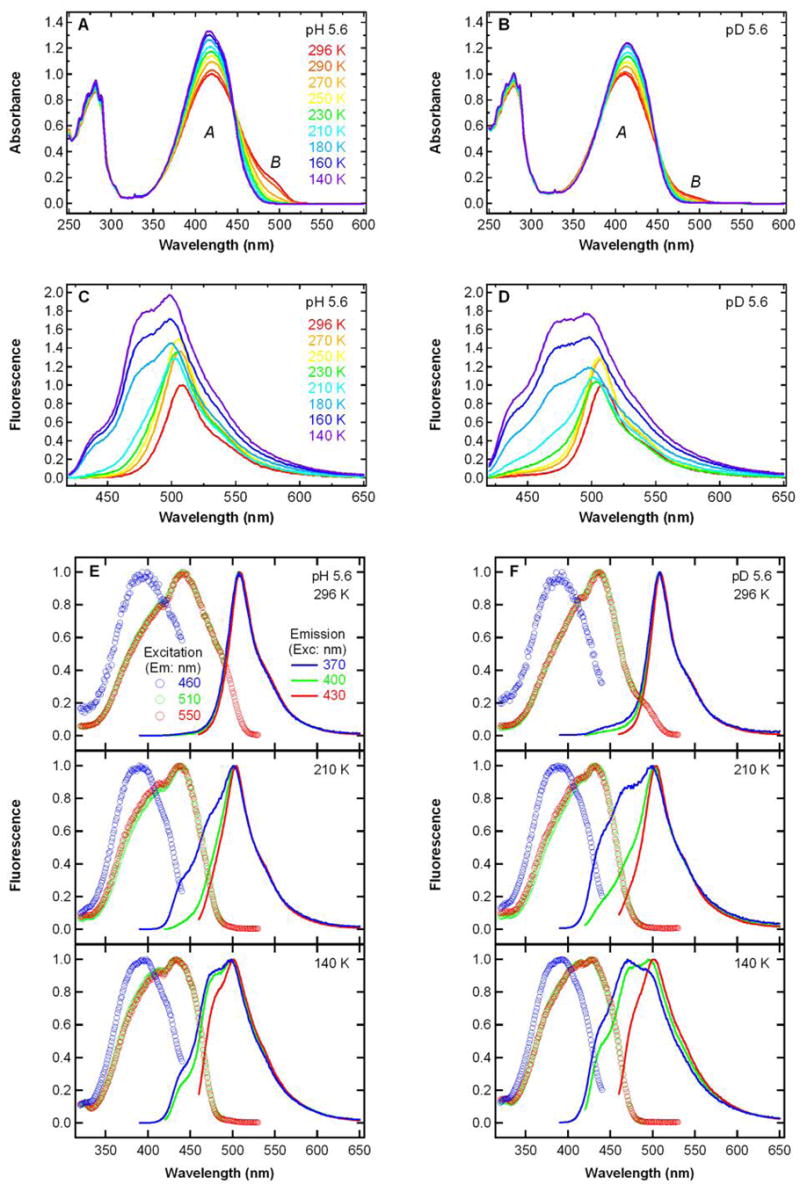
Steady-state absorption (A, B) and fluorescence emission (C, D) spectra (λexc = 400 nm) of S65T/H148D GFP from 296 K (red) to 140 K (violet) in ~ 20 K intervals at pH 5.6 (A, C) and pD 5.6 (B, D). All the spectra in panels A–D are normalized to the peak value of the spectrum at 296 K. Steady-state fluorescence excitation (circles) and emission (lines) spectra at 296, 210 and 140 K at pH 5.6 (E) and pD 5.6 (F). Excitation spectra with emission at 460, 510 and 550 nm are shown as the blue, green and red circles, respectively. Emission spectra with excitation at 370, 400 and 430 nm are shown as the blue, green and red lines, respectively. All the spectra in panels E and F are normalized to the peak value of the spectrum.
Time-resolved Fluorescence at Room Temperature
To understand how excitation of band A of S65T/H148D can produce green fluorescence and determine whether excited-state proton transfer is involved, we measured the time-resolved fluorescence following excitation at 400 nm at pH 5.6. Direct excitation of band A leads to a nearly instantaneous (within the instrument response) emission at 510 nm as can be seen in Figure 2A. The rising edge at 510 nm was fit to the convolution of the IRF measured at 400 nm with a sum of two exponentials; the major component, with a fit lifetime of 62 fs, comprises 91% of the total amplitude. By contrast, the rise of green emission following excitation anywhere in the 400 nm A band occurs on a much slower picosecond timescale in wild type (2) and deGFPs (13, 14) and mirrors the decay of A*. Emission from the higher energy state of S65T/H148D monitored at 475 nm exhibits a rapid decay as shown in Figure 2B. However, the kinetics of this decay in blue emission does not completely match the ultrafast rise in green emission from the lower energy state and this mismatch will be discussed later. Deuteration hardly affects the initial rise of the 510 nm emission as seen in Figure 2A, whereas in the wild type, deuteration leads to a dramatic slowing effect for both the rise of green fluorescence and decay of blue fluorescence (2).
Figure 2.
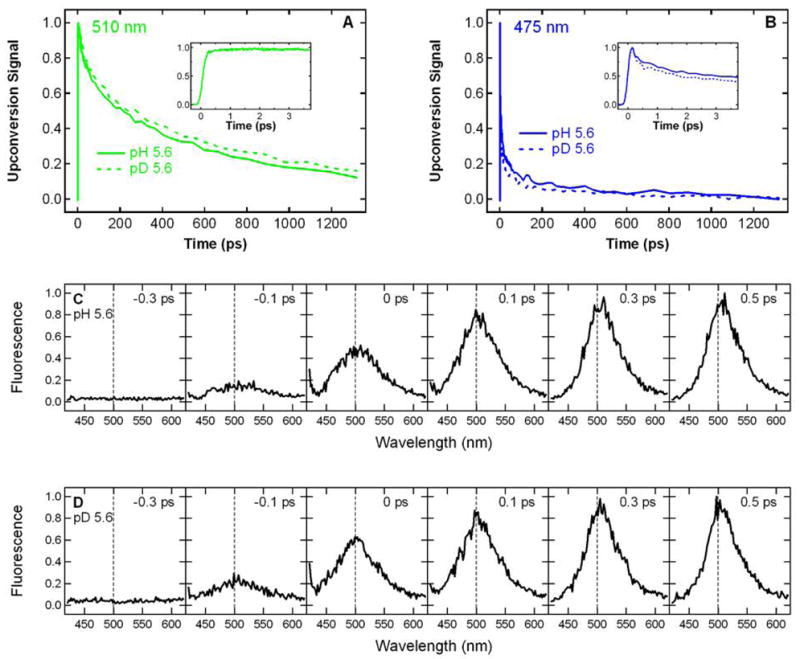
Room temperature time-resolved fluorescence of S65T/H148D GFP excited at 400 nm. The kinetics measured at 510 nm (A) and 475 nm (B) at pH 5.6 (solid) and pD 5.6 (dashed) are normalized to the peak intensity. The insets to (A) and (B) zoom in on the first few ps of the kinetics. The kinetics at pD 5.6 is also plotted in the inset to panel A and is essentially identical to what is observed at pH 5.6. Time-resolved emission spectra at t = − 0.3, − 0.1, 0, 0.1, 0.3 and 0.5 ps at pH 5.6 (C) and pD 5.6 (D) are normalized to the peak intensity in the spectrum at t = 0.5 ps. The dashed vertical line at 500 nm is shown in panels C and D to guide the eye.
In addition to the kinetics probed at two discrete wavelengths, time-resolved emission spectra following excitation at 400 nm were measured at pH 5.6 (Figure 2C). At negative time delays, the gate pulse arrives prior to the excitation pulse; thus, only the baseline is seen. With the relative time delay approaching zero, a wide emission band at ~ 500 nm appears and rapidly rises to its maximum intensity within hundreds of fs. At positive time delays, the initial band narrows to a band at ~ 510 nm, which is similar to the one observed at steady state (Figure 1B). The spectral evolution is barely altered by deuteration of exchangeable protons as seen in Figure 2D. These time-resolved spectra clearly demonstrate that the initial ultrafast rise in green fluorescence measured at 510 nm is primarily from a species whose emission is centered at 510 nm, rather than the red tail of a higher energy band at ~ 465 nm as was observed for one deGFP (deGFP2) whose spectral evolution has been studied in detail at low pH (14). Therefore, the kinetics detected at 510 nm can be used to describe the behavior of the green-emitting state.
Time-resolved Fluorescence at 140 K
To determine if the ultrafast rise in green emission can be slowed down by cooling, we measured the time-resolved fluorescence of S65T/H148D following excitation at 400 nm at 140 K. At this low temperature, the fluorescence monitored at 475 nm decays with a lifetime much longer than that at room temperature as seen by comparing Figure 3B and 2B. Nevertheless, the emission at 510 nm still appears within the instrument response as shown in Figure 3A. Exchanging protons with deuterons has no measurable effect on the initial rise in fluorescence at 510 nm (Figure 3A). Note that the 510 nm fluorescence decay at 140 K also exhibits a lifetime that is considerably longer than that at room temperature, that is, cooling slows down the nonradiative decay.
Figure 3.
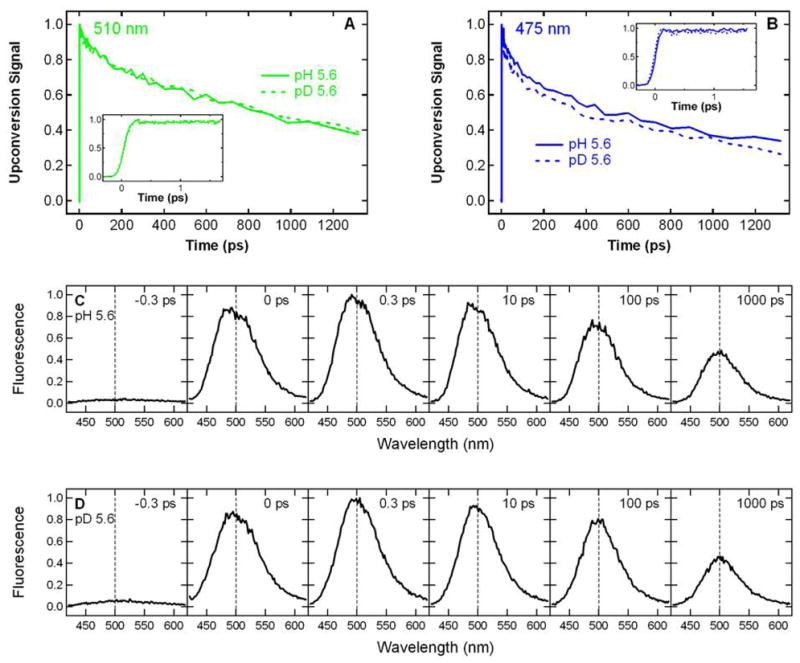
Time-resolved fluorescence of S65T/H148D GFP excited at 400 nm at 140 K. The kinetics measured at 510 nm (A) and 475 nm (B) at pH 5.6 (solid) and pD 5.6 (dashed) are normalized to the peak intensity. The insets to (A) and (B) zoom in on the first few ps of the kinetics. Time-resolved emission spectra at t = − 0.3, 0, 0.3, 10, 100 and 1000 ps at pH 5.6 (C) and pD 5.6 (D) are normalized to the peak intensity in the spectrum at t = 0.3 ps. The dashed vertical line at 500 nm is shown in panels C and D to guide the eye. Note that these spectra are taken at time points different from those at room temperature shown in Figure 2C and D.
Because the kinetics recorded at discrete wavelengths can be complicated due to overlapping bands, particularly at low temperatures for S65T/H148D (refer to Figure 4C), we also measured the time-resolved emission spectra at 140 K as shown in Figure 3C. In the negative time spectra, only the baseline is seen. The emission profile at zero time delay exhibits one broad band at ~ 500 nm, which quickly rises to its maximum amplitude within the instrument response. This band subsequently decays with a lifetime substantially longer than that at room temperature. Interestingly, the broadness of this band remains throughout the observation time window (1 ns), and the emission on the blue side of the spectrum is much more enhanced than that at room temperature (c.f. Figure 2C). We note that as a result of band overlap, the kinetics of this blue emission would significantly affect the single-wavelength kinetics if measured on the lower energy side. Within the signal-to-noise, deuteration hardly affects the spectral evolution as indicated by Figure 3D. These data obtained at 140K suggest that the ultrafast rise in the emission at 510 nm is from a broad feature peaked at ~ 500 nm, instead of completely from the red tail of a higher energy band at ~ 465 nm as was observed for one deGFP that has been studied in detail at low pH (14).
Temperature Dependence of Absorption and Steady-state Fluorescence
The temperature-dependent absorption and fluorescence spectra of S65T/H148D in glycerol/buffer solution at pH 5.6 are shown in Figure 4. Upon lowering the temperature, the peak absorbance of band A increases by up to 30%, while that of the small, lower energy band B decreases. It is noted that the peak position of band A remains almost constant (~ 415 nm) as the temperature is decreased, despite a modest change in the lineshape on the lower energy side of the peak. As can be seen in Figure 4A, the percentage of band B at 140 K is close to zero; thus, this observation excludes the possibility that the ultrafast rise in fluorescence following 400 nm excitation is due to direct excitation of the blue edge of band B and subsequent emission from B*. Deuteration causes little change to how the absorption lineshape evolves as a function of temperature (Figure 4B).
Following excitation at 400 nm, the steady-state fluorescence spectrum exhibits a dramatic change as the temperature is lowered. At pH 5.6, the peak at 510 nm increases in intensity from room temperature to 250 K and then exhibits a small decrease in intensity between 250 and 210 K. In this temperature range, the emission on the blue side between 420 and 470 nm becomes detectable (Figure 4C). Below 180 K, this blue fluorescence feature increases significantly as the temperature is decreased even further and some vibronic structure is visible. A deuteration effect is manifest mostly as a greater enhancement on the blue side of the emission spectrum at temperatures below 180 K (Figure 4D). The existence of a substantial blue feature is not consistent with an ultrafast, sub-ps ESPT mechanism alone, in which rapid deprotonation of the neutral chromophore in the excited state leads to quenching of the blue fluorescence and formation of a green-emitting, anionic state, as reflected in the working model in Scheme 1.
In addition to the emission spectra, fluorescence excitation spectra detected at 460, 510 and 550 nm emission were also recorded at 296, 210 and 140 K. Interestingly, the fluorescence excitation profile depends strongly on the emission energy monitored as seen in Figure 4E. The normalized excitation spectra with emission detected at 510 and 550 nm are superimposable and exhibit one peak at ~ 435 nm, whereas the one with emission at 460 nm shows another peak at ~ 400 nm across the whole temperature range examined. By contrast, only a peak at 415 nm is seen in the absorption spectrum (Figure 4A). We then recorded the fluorescence emission spectra with excitation on both the blue (λexc = 370 nm) and red sides (λexc = 430 nm) of the absorption peak. As can be seen in Figure 4E, a strong dependence on excitation energy is evident at low temperatures. At both 210 and 140 K, excitation on the higher energy side at 370 nm gives much more enhanced blue emission between 420 and 470 nm than when exciting on the lower energy side at 430 nm, while the emission lineshape above 500 nm hardly changes with excitation energy. Upon deuterium exchange, similar features are observed in the excitation and emission profile (Figure 4F). Note that the blue emission upon 370 nm excitation is even more enhanced at pD 5.6 than at pH 5.6, and at pD 5.6 the intensity of blue emission is even greater than that of the green emission above 500 nm at 140 K.
Selective Photobleaching
The temperature -dependent fluorescence excitation and emission spectra suggest the possibility of multiple states underlying band A in S65T/H148D at pH 5.6. To explore this further, we attempted to selectively destroy a component by exposure of the protein at pH 5.6 to 200 mW, 457 nm continuous wave laser light. As seen in Figure 5A, the overall absorbance of band A slowly diminishes, but as this occurs the peak shifts to the blue side. The increasing baseline offset seen in Figure 5A is caused by the scattering of precipitated protein as the photobleaching proceeds. Interestingly, the difference between the absorption spectrum after a certain amount of photobleaching time and the one at time zero exhibits a negative feature at ~ 435 nm as shown in Figure 5A. This differential loss feature, denoted Aβ, grows in the difference absorption spectra with the time of irradiation. Note that the observation of a blue-shifted absorption peak is not an artifact caused by the increasing baseline, since the supernatant of the sample obtained after centrifugation to remove the precipitated protein confirms this blue shift. After 10 hours of photobleaching, the supernatant shows an absorption peak at ~ 400 nm, the position for the neutral chromophore in the wild type (3) and deGFPs (15). More extended photobleaching of the supernatant continues to reduce the absorbance of this peak, but yields no further blue shift (data not shown). It is noted that a small fraction of the protein is converted to the anionic B form upon 457 nm irradiation, as indicated by the bump at ~ 490 nm seen in the fluorescence excitation spectrum (Figure 5B). Concomitant with the bleaching of the Aβpopulation and blue shift of the absorption band are changes in the fluorescence emission and excitation spectra of the supernatant as shown in Figure 5B. After photobleaching with 457 nm laser light, the fluorescence emission spectra when exciting at 400 nm exhibit enhanced blue emission between 420 and 470 nm relative to the green emission above 500 nm, and the ratio of blue/green fluorescence increases with the time of irradiation. Even though the remaining population absorbs around 400 nm, deuterium exchange of the supernatant did not significantly affect the excited-state dynamics, suggesting that the remaining protein does not exhibit ESPT as in conventional GFP’s. In the excitation spectra, the ratio of the higher energy side (400 nm) to lower energy side (435 nm) also increases with the time of irradiation, consistent with the selective bleaching of Aβ (Figure 5B). Purging the sample with Ar, deuterating exchangeable protons at the same pH or photobleaching with 50 mW, 450 nm pulsed laser light leads to at most minor changes in the evolution of the absorption profile upon photobleaching.
Figure 5.
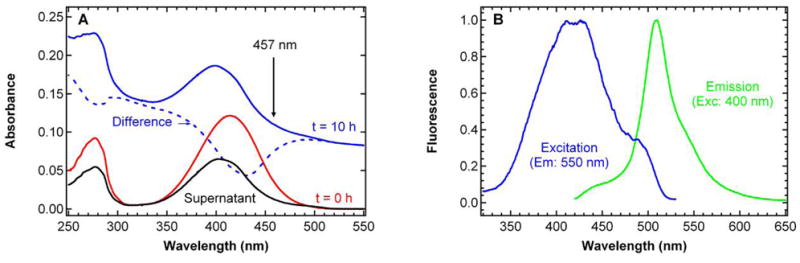
Steady-state absorption (A) and fluorescence (B) spectra of S65T/H148D GFP at pH 5.6 after 10 hours of photobleaching with 200 mW 457 nm CW laser at room temperature. In panel A, absorption spectra of the sample before and after photobleaching are shown as the solid red and blue lines, respectively. The difference spectrum is shown as the dashed blue line. The spectrum of the supernatant after centrifugation is shown as the solid black line. In panel B, the fluorescence emission spectrum (λexc = 400 nm) and the fluorescence excitation spectrum (λem = 550 nm) are shown for the photobleached sample in green and blue, respectively. Both spectra in panel B are normalized to the peak value of the spectrum.
Electronic Stark Effect Spectroscopy
The 77 K absorption and Stark spectra of S65T/H148D and deGFP3 in glycerol/buffer solutions at pH 5.6 and pD 5.6 are shown in Figure 6. As has been demonstrated in previous studies, Stark spectroscopy provides useful information on the electronic structure of the chromophore in wild type GFP, S65T GFP, BFP and DsRed (2, 22, 23). At pH 9.2, S65T/H148D exhibits absorption and Stark spectra (data not shown) with characteristics typical of an anionic chromophore in GFP. In particular, the Stark spectrum of S65T/H148D resembles the second derivative of the absorption and is very similar to the Stark lineshape of S65T, in which the anionic B form dominates the ground-state equilibrium even at neutral pH (22). Therefore, the high pH form of S65T/H148D can be considered a canonical B form, with the change in dipole moment, |Δμ| = 7.4 D/f, comparable to the value for S65T [7.0 ‘D/f, where f is the local field correction factor typically between 1 and 1.3 (22)].
Figure 6.
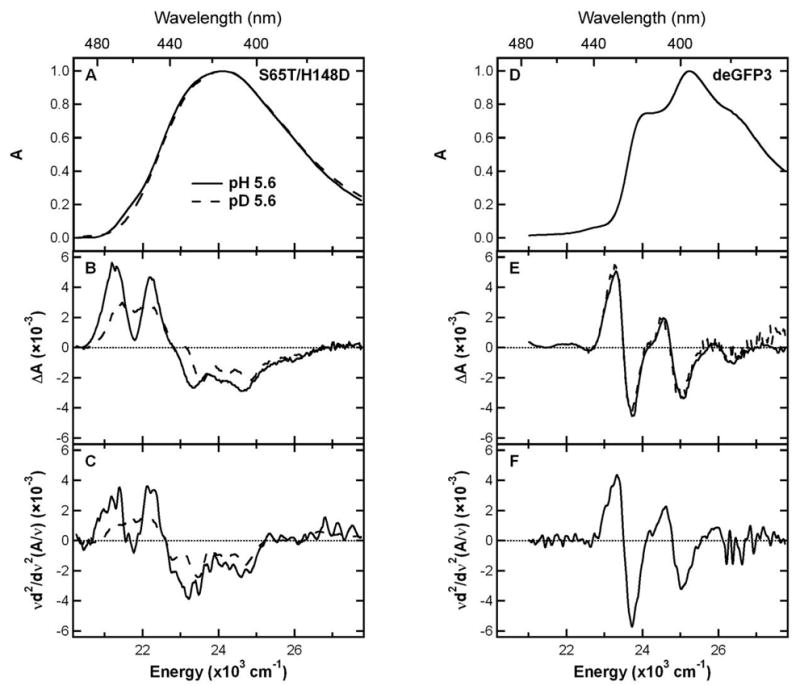
77 K Absorption, Stark spectra of S65T/H148D and deGFP3. Absorption spectra of S65T/H148D at pH 5.6 (solid) and pD 5.6 (dashed) in panel A and deGFP3 at pH 5.6 in panel D are normalized to the peak value of the spectrum. Stark spectra for S65T/H148D in panel B and deGFP3 in panel E are scaled to unity peak absorption and an electric field of 1.0 MV/cm. The 2nd-derivative component of absorption for S65T/H148D in panel C and deGFP3 in panel F are shown for comparison.
At pH 5.6, on the other hand, S65T/H148D exhibits features in the absorption and Stark spectra distinct from other GFPs studied thus far (Figure 6). In earlier work, blue fluorescent protein (BFP, Y66H/Y145F) was used as a model for the neutral A form as it was the only GFP variant available at the time with mostly A-state character (22). The deGFPs at low pH are a much better model as the A form dominates the ground-state equilibrium (14). Using deGFP3 as an example (other deGFPs are similar, not shown), the absorption spectrum exhibits some resolved vibronic structure (Figure 6D), and the Stark spectrum (Figure 6E) closely matches the second derivative of the absorption (Figure 6F) with |Δμ| = 7.0 D/f, interestingly similar to the value reported for the anionic B form (22). It is noteworthy that this value of |Δμ| for the A state is much larger than previously estimated from BFP (22), which may reflect the difference in chemical structure between the GFP and BFP chromophores.
In contrast to deGFP3, the absorption spectrum of S65T/H148D at pH 5.6 is fairly broad and nearly featureless, without any well-resolved vibronic progression (Figure 6A). Replacing exchangeable protons with deuterons at the same pH results in a noticeable difference on the long wavelength shoulder of the absorption band. The Stark spectra of S65T/H148D shown in Figure 6B are also clearly different from those of deGFPs. Furthermore, deuteration leads to a significant change in the Stark lineshape as shown in Figure 6B, while no such effect upon deuteration is observed for deGFP3 (Figure 6E). As seen in Figure 6C, the 2nd derivative of the absorption spectra for both the pH 5.6 and pD 5.6 samples of S65T/H148D do not compare as well to the Stark spectra (Figure 6B) as is the case for deGFP3 (Figures 6E and F). That is, although there are similar features seen in the Stark and the 2nd derivative of absorption, a good fit to the Stark would require a significant contribution from the 1st derivative of absorption, much larger than has been documented for a GFP chromophore. Furthermore, this sum-of-derivatives analysis yields an unusually large value of 13.5 D/f for |Δμ|, nearly twice of that for a typical GFP chromophore, which is physically unreasonable. Thus, while an improved fit to the Stark can be obtained by including contributions from other derivatives, the fluorescence excitation and photobleaching experiments reported above suggest that there may be more than one species under band A of S65T/H148D. Since Stark spectra are derivative spectra, they are dominated by the component with a relatively narrow feature, while for a broad and featureless component the derivatives are relatively small. Therefore, if the Stark contains such a relatively sharp feature, e.g. from an underlying component in absorption, a spurious value will be obtained for |Δμ| by fitting the entire lineshape with one set of electro-optic parameters. In addition, the deuterium effect on the Stark lineshape of S65T/H148D suggests deuteration may affect the small absorption component with a relatively sharp Stark feature. Thus, the Stark spectra reinforce the suggestion that band A is more complex.
DISCUSSION
The data presented here on GFP S65T/H148D reveal several interesting and unique features compared with conventional GFPs. In the following, w e consider the origin of the initial ultrafast rise in green fluorescence and how this unusual phenomenon may relate to the short hydrogen bond (≤2.4 Å) between the phenol oxygen of the chromophore and the carboxyl oxygen of Asp148 described in the accompanying paper (1). We have presented evidence that two populations, Aα and Aβ, are under band A in S65T/H148D in glycerol/buffer solution. Aαabsorbs in a wavelength region close to that of a conventional A form in GFP, but its excited-state dynamics are quite different. The absorption of Aβ is red-shifted relative to that of a conventional neutral chromophore. Once excited, Aβ quickly gives rise to a green-emitting state by a process that is not significantly influenced by either exchanging protons with deuterons or lowering the temperature. One way to rationalize this result is that the short hydrogen bond leads to a more delocalized proton, that is, there is only one potential well involved. Thus, after excitation only a small proton displacement is required to generate a green-emitting state, and this process in the excited state of Aβ is barely dependent upon the hydrogen isotope or temperature on the ultrafast timescale that we can detect.
Previous spectroscopic and structural studies on wild-type GFP suggest that excitation of the neutral chromophore at 400 nm leads to excited-state proton transfer from the chromophore to a proton acceptor on the tens of ps timescale, possibly through a hydrogen-bonding network, and this excited-state interconversion process can be slowed down significantly by deuterating exchangeable protons or cooling (2, 6–8, 10–12, 24). Recent studies on dual-emission GFPs are consistent with this overall scheme, though the rates of proton transfer and non-radiative processes and the identity of the proton acceptor and transfer pathway(s) may be different (13–15). Although it is reasonable to relate the bright green fluorescence detected upon excitation of the chromophore in S65T/H148D to ESPT, several aspects of the dynamics are very different from the earlier systems. The ultrafast rise in the amplitude of green emission at room temperature in buffer solution (Figure 2A and C) could be the result of an ultrafast (much faster than the 170 fs instrument response) ESPT and the beginning of the decay in blue emission, i.e. the first 200 fs, occurs within the instrument response and thus appears to be consistent with this ultrafast process. However, the rest of the decay in blue fluorescence occurs on a much slower ps timescale and does not fit well into the ultrafast ESPT model. This suggests that whatever process is responsible for the ultrafast rise in green fluorescence does not quench all the blue fluorescence.
Besides the unusual instantaneous appearance of green fluorescence following excitation at 400 nm, S65T/H148D also exhibits a number of unusual features in its steady-state fluorescence and absorption spectra. In glycerol/buffer solution at pH 5.6 with the fluorescence monitored on the green side (i.e. at 510 or 550 nm) of the emission peak, the excitation profile displays one peak at ~ 435 nm, while the excitation spectrum measured with the emission monitored on the blue side (i.e. at 460 nm) shows another peak at ~ 400 nm (Figure 4E). By comparison, dual-emission GFPs at low pH, where the neutral state of the chromophore dominates, exhibit only a single peak at ~ 400 nm, regardless of the emission energy monitored (15). The feature at 435 nm observed in the excitation profile of S65T/H148D in glycerol/buffer solution at pH 5.6 is hence indicative of a new population underneath band A in the absorption spectrum. The absorption of the two populations suggested by the excitation spectrum, Aα (λmax ≈ 435 nm) and Aβ (λmax ≈400 nm), overlaps, therefore the peak position appears red -shifted from that of a conventional neutral form, 400 nm, to 415 nm. The slightly smaller red shift to 411 nm observed in deuterated glycerol/buffer solution could be caused by a small change in the ratio of Aα and Aβ populations, though there is little independent information on this.
Upon excitation into band A of S65T/H148D in glycerol/buffer solution with 400 nm light, both Aα and Aβ are directly excited, the latter because of the broadness of the Aβ band. Fluorescence excitation spectra shown in Figure 4E clearly demonstrate that when Aβis excited at room temperature, blue emission at 460 nm is detected. We did not find direct evidence, such as a deuterium isotope effect, which would suggest that the excitation of Aα also produces green emission through ESPT. On the other hand, when Aβ is excited, mostly green emission is observed. In addition, we consider both the initial ultrafast decay in blue fluorescence and the corresponding ultrafast rise in green fluorescence to originate from a rapid process in Aβ, while the much slower phase of ps decay in blue emission to be associated with a slower process in Aα. In this model, excitation of Aβ quickly gives rise to a green-emitting state and this process occurs within the 170 fs instrument response. That is, both the decay in blue emission and rise in green emission are faster than what the instrument can resolve and thus appear to be instantaneous. In addition, this rapid process is hardly affected by deuteration of exchangeable protons as shown in Figure 2, suggesting that it is either not strongly coupled to proton motion or too strongly coupled and thus fast to allow the current instrument to detect any difference caused by deuteration given the time resolution achievable. We speculate the latter process, i.e. an ESPT that is faster than the 170 fs IRF is more likely, although direct observation of this process and its connection to proton motion would require better time resolution. Moreover, this ultrafast process appears to be hardly influenced by temperature since at least some green fluorescence still exhibits a nearly instantaneous rise (Figure 3A and C) even with cooling to 140 K. Interestingly, this may be reminiscent of the photophysical behavior of an anionic GFP chromophore, where instantaneous green fluorescence can be readily seen following direct excitation of the chromophore already deprotonated in the ground state (2). On the other hand, the decay of Aα* occurs on a much slower ps timescale as suggested by the residual blue fluorescence after the initial instrument-response-limited decay. There is no direct evidence suggesting that Aα* could subsequently produce a green-emitting state through ESPT. As the temperature is lowered, the blue emission is more enhanced corresponding to a slowed nonradiative decay in Aα* (Figure 4E). This is also reflected by a longer lifetime of blue fluorescence at lower temperatures seen in time-resolved experiments (Figure 3B).
The apparent similarity in photophysics between the Aβ population of S65T/H148D, and an anionic GFP chromophore, gives credence to the view that Aβ shares some characteristics in charge distribution with the anionic B or I form. With the 1.5 Å resolution crystal structure of S65T/H148D at pH 5.6 presented in the preceding accompanying paper (1), we can speculate on the conformational landscape of the chromophore in the ground state. S65T/H148D exhibits an overall β-barrel topology, as is typical for the GFP family (20, 21); however, an unusual short hydrogen bond (bond length ≤ 2.4 Å) is observed between the carboxyl oxygen of Asp148 and phenol oxygen of the chromophore. It is probable that the pKa of Asp148 in S65T/H148D is different from that of a free aspartic acid in buffer due to the strong interaction with the neighboring chromophore. That is, there exists some ambiguity in determining the protonation states of Asp 148 and the chromophore. As a result, an otherwise neutral chromophore may possess a proton displaced away from its ordinary position by Asp 148, and this argument, if true, is consistent with the 435 nm excitation maximum of Aβ, much red-shifted from a conventional A form of GFP. We are led to propose an alternative working model in which S65T/H148D in glycerol/buffer solution at pH 5.6 exists as two populations in the ground state, Aα and Aβ, each with a distinct location of the proton and this exact position determines the excitation maximum and excited-state dynamics of the corresponding species. There is no evidence for two chemically distinct populations by chromatography or mass spectrometry run on multiple batches of protein prepared in different labs. Likewise the x-ray structure shows no evidence for two populations and the resolution of the structure is excellent. Nonetheless, even at 1.5 Å resolution it is possible that the two rotomer forms of Asp 148 shown below and tentatively assigned to Aαand Aβ are present and not distinguishable:
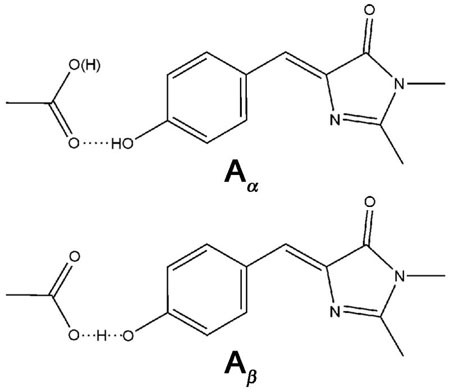
The differences in these populations involve the position of hydrogen(s) which are not seen in the structure. In the first population Aα, the chromophore is protonated at the phenol end and acts as a hydrogen donor to the Asp 148 acceptor. In the second population Aβ, the chromophore is at least partially deprotonated at the phenol end. Excitation of Aβrapidly leads to the formation of a green-emitting state and the complete bond-breaking is greatly facilitated by the already displaced proton; thus, like an anionic form, ultrafast green fluorescence appears. The crystal structure in the preceding accompanying paper (1) allows for the calculation of the bond lengths between the carbon and the two oxygen atoms of the carboxyl in Asp 148. For the proximal oxygen which is involved in the hydrogen bond with the chromophore, the C-O bond length is 1.29 Å. For the distal oxygen which is not involved in that hydrogen bond, the C-O bond length is only 1.22 Å. Previously Derissen et al. (25) observed the bond lengths of 1.202 and 1.306 Å for C=O and C-O(H), respectively for L-asp in crystals. Thus, the bond lengths in S65T/H148D suggest that the proximal C-O in Asp 148 carries, at least in part, single-bond nature and this is consistent with the argument about the protonation state of Asp 148, which is not completely anionic. Note that at the present resolution of x-ray diffraction studies of S65T/H148D, 1.5 Å, there exist uncertainties in the absolute values of bond length. Therefore, those bond lengths of C-O must be interpreted with great caution. Future structural studies of S65T/H148D with higher resolution may be able to address this issue more definitively.
In addition to the bond lengths, another piece of evidence was found from the titration of the absorption spectrum of S65T/H148D in a region close to pH 5.6, the value used for the most part of this study. With the pH increased from a value 0.6 pH unit below to above 5.6, in other words from 5.0 to 6.2, the absorption spectrum exhibits a small but detectable shift in maximum position that is on the order of 1.3 nm (Figure 7). This shift suggests that a modest change in the bulk pH can influence the effective ratio of the two populations described above, likely by affecting the protonation state of the distal oxygen of Asp148; thus, it is consistent with the scenario proposed above. Note that in this titration experiment, only the region close to pH 5.6 was examined. A broader region might give a larger shift in absorption maximum; however, a pH lower than 5.0 tends to precipitate the protein, while a pH higher than 6.2 starts to induce the formation of the anionic form, whose absorption is lower in energy at 487 nm as presented in the preceding accompanying paper (1). These would complicate the appearance of the absorption spectrum; therefore, we only considered the region from pH 5.0 to 6.2.
Figure 7.
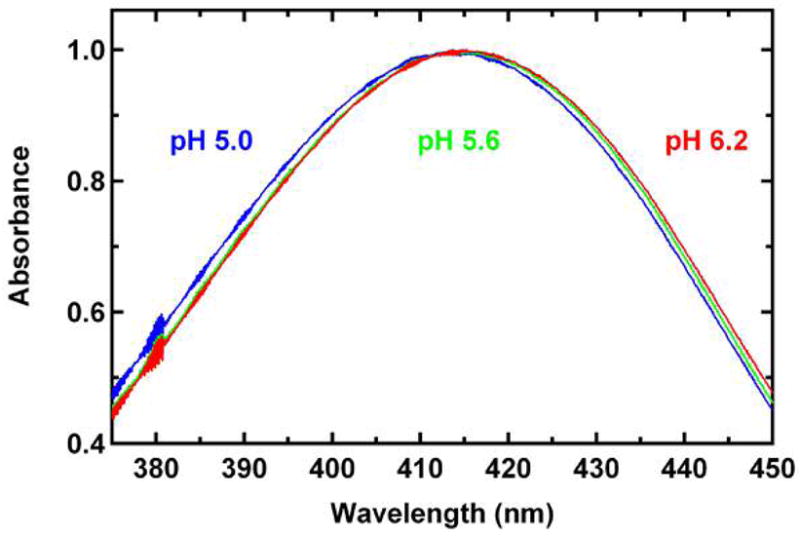
Steady-state absorption spectra of S65T/H148D GFP at pH 5.0 (blue), 5.6 (green), and 6.2 (red) at room temperature. All spectra are normalized to the peak value. Only the region from 375 to 450 nm is shown to make the difference in absorption maximum more visible. The wavelength interval is 0.1 nm. All the spectrum baselines are flat (550 to 700 nm, not shown) and within the noise of the instrument.
So far, we have discussed the possibility of multiple states underlying band A which fits with the rapid rise in green emission and the absence of completely matched decay in blue emission in S65T/H148D in glycerol/buffer solution. However, we realize that it may not be straightforward to apply the preceding model directly in the interpretation of the data obtained in buffer solution at pH 5.6 without glycerol. For example, the excitation maximum with green emission in buffer is not shifted by the same amount as in glycerol/buffer solution (Figure 1B, 4E and F). Nonetheless, the red side of the absorption maximum (415 nm) still exhibits higher excitation efficiency than the blue side does. In other words, the partitioning of the ground state into two populations may not appear to be as obvious as in glycerol/buffer solution, but there is still evidence for it. In fact, by selectively photobleaching a population of S65T/H148D at pH 5.6 with a 457 nm laser, a band centered at 435 nm is observed as a negative feature that grows in the difference absorption spectra with photobleaching time. This preferential photobleaching effect provides support to the existence of population Aβ Other evidence emerges from the. photophysical characteristics of GFP S65T/H148D in buffer, including the ultrafast rise in green fluorescence as well as the absorption maximum, which are very similar to those in glycerol/buffer solution.
The ultrafast green fluorescence and short hydrogen bond observed in GFP S65T/H148D are unusual. We propose the apparent ultrafast ESPT is a result of small proton displacement strongly facilitated by the short hydrogen bond between the phenol oxygen of chromophore and carboxyl oxygen of Asp 148. There is good precedent for ultrafast intramolecular EPST processes in simpler molecules. For example this was observed in a photoacid, 1,8-dihydroxyanthraquinone (chrysazin) in hexane at room temperature (26), and in a “tight” complex of acetate and a photoacid, 8-hydroxy-1,3,6-trisulfonate-pyrene (HPTS) (27). In order to further evaluate the nature of the proton involved in the short hydrogen bond in GFP S65T/H148D, one needs to acquire spectroscopic with better time and structural data at higher resolution or a neutron scattering structure to precisely locate the key proton(s).
Acknowledgments
We thank Tim McAnaney and Prof. Chris Chidsey for helpful discussions.
Abbreviations
- GFP
green fluorescent protein
- deGFP
dual-emission green fluorescent protein
- ESPT
excited-state proton transfer
- IRF
instrument response function
Footnotes
This work was supported in part by grants from the NIH (GM27738 to S.G.B. and R01 GM42618 to S.J.R.). The fluorescence upconversion facilities are supported by the Medical Free Electron Laser Program of the Air Force Office of Scientific Research (Grant No. F49620-00-1-0349). X.S. was supported by a William R. and Sara Hart Kimball Stanford Graduate Fellowship and P.A. by the Fonds de recherche sur la nature et les technologies and the National Science and Engineering Research Council of Canada.
References
- 1.Shu X, Kallio K, Shi X, Abbyad P, Kanchanawong P, Childs W, McAnaney TB, Boxer SG, Remington SJ. [Google Scholar]
- 2.Chattoraj M, King BA, Bublitz GU, Boxer SG. Ultra-fast excited state dynamics in green fluorescent protein: multiple states and proton transfer. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:8362–8367. doi: 10.1073/pnas.93.16.8362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heim R, Prasher DC, Tsien RY. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:12501–12504. doi: 10.1073/pnas.91.26.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cubitt AB, Heim R, Adams SR, Boyd AE, Gross LA, Tsien RY. Understanding, improving and using green fluorescent proteins. Trends in biochemical sciences. 1995;20:448–455. doi: 10.1016/s0968-0004(00)89099-4. [DOI] [PubMed] [Google Scholar]
- 5.Creemers TM, Lock AJ, Subramaniam V, Jovin TM, Volker S. Three photoconvertible forms of green fluorescent protein identified by spectral hole-burning. Nature structural biology. 1999;6:557–560. doi: 10.1038/9335. [DOI] [PubMed] [Google Scholar]
- 6.Brejc K, Sixma TK, Kitts PA, Kain SR, Tsien RY, Ormo M, Remington SJ. Structural basis for dual excitation and photoisomerization of the Aequorea victoria green fluorescent protein. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:2306–2311. doi: 10.1073/pnas.94.6.2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palm GJ, Zdanov A, Gaitanaris GA, Stauber R, Pavlakis GN, Wlodawer A. The structural basis for spectral variations in green fluorescent protein. Nature structural biology. 1997;4:361–365. doi: 10.1038/nsb0597-361. [DOI] [PubMed] [Google Scholar]
- 8.Kennis JT, Larsen DS, van Stokkum IH, Vengris M, van Thor JJ, van Grondelle R. Uncovering the hidden ground state of green fluorescent protein. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:17988–17993. doi: 10.1073/pnas.0404262102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jung G, Wiehler J, Zumbusch A. The photophysics of green fluorescent protein: influence of the key amino acids at positions 65, 203, and 222. Biophys J. 2005;88:1932–1947. doi: 10.1529/biophysj.104.044412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lossau H, Kummer A, Heinecke R, PollingerDammer F, Kompa C, Bieser G, Jonsson T, Silva CM, Yang MM, Youvan DC, MichelBeyerle ME. Time-resolved spectroscopy of wild-type and mutant Green Fluorescent Proteins reveals excited state deprotonation consistent with fluorophore-protein interactions. Chemical Physics. 1996;213:1–16. [Google Scholar]
- 11.Stoner-Ma D, Jaye AA, Matousek P, Towrie M, Meech SR, Tonge PJ. Observation of excited-state proton transfer in green fluorescent protein using ultrafast vibrational spectroscopy. Journal of the American Chemical Society. 2005;127:2864–2865. doi: 10.1021/ja042466d. [DOI] [PubMed] [Google Scholar]
- 12.van Thor JJ, Georgiev GY, Towrie M, Sage JT. Ultrafast and low barrier motions in the photoreactions of the green fluorescent protein. The Journal of biological chemistry. 2005;280:33652–33659. doi: 10.1074/jbc.M505473200. [DOI] [PubMed] [Google Scholar]
- 13.McAnaney TB, Park ES, Hanson GT, Remington SJ, Boxer SG. Green fluorescent protein variants as ratiometric dual emission pH sensors. 2. Excited-state dynamics. Biochemistry. 2002;41:15489–15494. doi: 10.1021/bi026610o. [DOI] [PubMed] [Google Scholar]
- 14.McAnaney TB, Shi X, Abbyad P, Jung H, Remington SJ, Boxer SG. Green fluorescent protein variants as ratiometric dual emission pH sensors. 3. Temperature dependence of proton transfer. Biochemistry. 2005;44:8701–8711. doi: 10.1021/bi050132a. [DOI] [PubMed] [Google Scholar]
- 15.Hanson GT, McAnaney TB, Park ES, Rendell ME, Yarbrough DK, Chu S, Xi L, Boxer SG, Montrose MH, Remington SJ. Green fluorescent protein variants as ratiometric dual emission pH sensors. 1. Structural characterization and preliminary application. Biochemistry. 2002;41:15477–15488. doi: 10.1021/bi026609p. [DOI] [PubMed] [Google Scholar]
- 16.Elsliger MA, Wachter RM, Hanson GT, Kallio K, Remington SJ. Structural and spectral response of green fluorescent protein variants to changes in pH. Biochemistry. 1999;38:5296–5301. doi: 10.1021/bi9902182. [DOI] [PubMed] [Google Scholar]
- 17.Glasoe PK, Long FA. Use of Glass Electrodes to Measure Acidities in Deuterium Oxide. Journal of Physical Chemistry. 1960;64:188–190. [Google Scholar]
- 18.Bublitz GU, Boxer SG. Stark spectroscopy: Applications in chemistry, biology, and materials science. Annual Review of Physical Chemistry. 1997;48:213–242. doi: 10.1146/annurev.physchem.48.1.213. [DOI] [PubMed] [Google Scholar]
- 19.Leiderman P, Genosar L, Huppert D, Kallio K, Remington SJ, Solntsev KM, Tolbert LM. doi: 10.1021/bi7009053. [DOI] [PubMed] [Google Scholar]
- 20.Tsien RY. The green fluorescent protein. Annual Review of Biochemistry. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 21.Zimmer M. Green fluorescent protein (GFP): Applications, structure, and related photophysical behavior. Chemical Reviews. 2002;102:759–781. doi: 10.1021/cr010142r. [DOI] [PubMed] [Google Scholar]
- 22.Bublitz G, King BA, Boxer SG. Electronic structure of the chromophore in green fluorescent protein (GFP) Journal of the American Chemical Society. 1998;120:9370–9371. [Google Scholar]
- 23.Lounis B, Deich J, Rosell FI, Boxer SG, Moerner WE. Photophysics of DsRed, a red fluorescent protein, from the ensemble to the single-molecule level. Journal of Physical Chemistry B. 2001;105:5048–5054. [Google Scholar]
- 24.Leiderman P, Huppert D, Agmon N. Transition in the temperature-dependence of GFP fluorescence: From proton wires to proton exit. Biophysical Journal. 2006;90:1009–1018. doi: 10.1529/biophysj.105.069393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Derissen JL, Endeman HJ, Peerdema Af. Crystal and Molecular Structure of L-Aspartic Acid. Acta Crystall B-Stru B. 1968;24:1349. [Google Scholar]
- 26.Arzhantsev SY, Takeuchi S, Tahara T. Ultrafast excited-state proton transfer dynamics of 1,8-dihydroxyanthraquinone (chrysazin) studied by femtosecond time-resolved fluorescence spectroscopy. Chemical Physics Letters. 2000;330:83–90. [Google Scholar]
- 27.Rini M, Magnes BZ, Pines E, Nibbering ETJ. Real-time observation of bimodal proton transfer in acid-base pairs in water. Science. 2003;301:349–352. doi: 10.1126/science.1085762. [DOI] [PubMed] [Google Scholar]