

Abstract
The absolute configuration of gymnocin-B has been determined to be (S)-10 and (S)-37. Three challenges towards the configurational assignment of this largest of the polyether marine toxin include (i) introduction of p-(meso-triphenylporphyrin)-cinnamate group (TPPcinnamate) on sterically hindered 10-, 37-hydroxyls under mild conditions, (ii) conformational analysis in the presence of TPPcinnamates at C-10 and C-37 positions on the flexible seven-membered rings embodied in a large polyether ladder-like scaffold structure, and (iii) determination of the chirality at C-10 and C-37 on the basis of porphyrin/porphyrin circular dichroism exciton coupled interaction over a large distance. The experimentally obtained positive exciton couplet by CD and FDCD, of the bis-TPPcin derivative of gymnocin-B, is in good agreement with that of theoretically calculated CD of the MMFF optimized structures, by employing DeVoe's coupled oscillator approach, thus establishing the full absolute configuration of gymnocin-B.
Introduction
Gymnocins are a series of cytotoxic marine polycyclic ethers, produced by the red tide dinoflagellate Karenia (formerly Gymnodinium) mikimotoi. The dinoflagellate Karenia mikimotoi is one of the most notorious red tide species that cause devastating damages to aquaculture and marine ecosystems worldwide and yet the mechanism of the toxic effect remains unknown.1,2 Recently, gymnocin-A, which exhibits potent cytotoxicity against mouse lymphoide P388 cells, has been isolated from K. mikimotoi (Chart 1).3 The structure of gymnocin-A, characterized by 14 contiguous saturated ether rings and a 2-methyl-2-butenal side-chain at 5-position of A ring, was elucidated by 2D-NMR analysis and collision-induced MS/MS experiments.3 Furthermore, the absolute configuration of gymnocin-A was also determined by applying the modified Mosher method at the 50-OH group. Very recently, an elegant total synthesis of this complex molecule has been achieved by Sasaki and co-workers.4
Chart 1.

Structures of gymnocin-A and gymnocin-B.
In our continuous effort to isolate and characterize new gymnocin congeners, we recently isolated a new toxin, gymnocin-B from the dinoflagellate K. mikimotoi collected at Kushimoto, Wakayama in Japan, in addition to other congeners including gymnocin-A (Chart 1).5 Based on extensive NMR and MS analysis, the structure and the relative configuration of gymnocin-B, shown in Chart 1, were recently elucidated;5 Similar to gymnocin-A, this new toxin contains 15 contiguous polyether skeleton and a 2-methyl-2-butenal side-chain at the C-5 position. The number of contiguous ether rings is the largest among the polycyclic ethers hitherto known. However, the absolute configuration of gymnocin-B remains unsolved, since the three sterically hindered pseudo-axial hydroxyls, namely, 10-, 37- and 54-hydroxyls hampered straightforward application of the Mosher method which relies on esterification with MTPA reagents.
The CD exciton chirality method based on the coupled oscillator theory has been widely used for determining the absolute configurations of various organic compounds.6,7 The presence of two hydroxyl groups at C-10 and C-37 in gymnocin-B, separated by 20 Å, leads to the possibility of applying the exciton chirality method by introducing potent CD reporter groups at these hydroxyl moieties. However, this method faces the following three challenges. Firstly, since the amount of gymnocin-B available from the natural source is very limited, an efficient microscale protocol is required to derivatize the sterically hindered 10- and 37-hydroxyls with strong UV/Vis absorbing CD reporter group, which are commonly bulky π-π electron systems. Secondly, a precise conformational analysis of this large polyether structure including the critical 10-OH, 37-OH is required because of the five flexible seven-membered rings. Thirdly, difficulties are expected in measurement of exciton couplet over a large interchromophoric distance of ca. 30 Å. In this paper, we describe our strategy for overcoming these difficulties and successful assignment of absolute configuration of gymnocin-B.
Results and Discussion
1. Model study with sterically hindered secondary 17-OH of 3-oxo-5α-androstan-17-ol (1) and bis-steroid (5) (Scheme 2)
Scheme 2.

Introduction of TPP and TBP at 17-OH of 5α-androstan-3-one derivatives.
The extremely small amount of gymnocin-B available was the first challenge for determination of its absolute stereochemistry. Therefore, we carried out several model studies aimed at selecting the appropriate CD reporter groups capable of long range exciton coupling8,9 and determining optimal reaction conditions for gymnocin-B derivatization. We have already shown that p-(meso-triphenylporphyrin)-carboxylic acid (TPP-CO2H) introduced at the hydroxyl or amino groups via acylates, represents one of the most potent CD reporter groups, especially suited for the observation of long range exciton coupling.7,10 Based on this precedence, we investigated a new tetraphenylporphyrin chromophore, p-(meso-triphenylporphyrin)-cinnamate (TPPcin, 419 nm, ε 387,000 in CH2Cl2, and 415 nm, ε 362,000 in MeOH),11 which was introduced at the secondary 17-OH of 3-oxo-5α-androstan-17-ol 1 and bis-steroid 5.12 Since the secondary 17-OH group present in templates 1 and 5 is sterically hindered due to the neighboring C-18 methyl group and C-13 quaternary carbon, introduction of porphyrin chromophores in 1 and 5 is challenging. Furthermore, since the bis-steroid 5 has quite a rigid steroid skeleton with two remote pseudo-equatorial hydroxyls at the 17 and 17' positions separated by ca. 19 Å, a distance similar to that between gymnocin-B 10- and 37-hydroxyls (ca. 20 Å) (Fig. 1a and 4a), this template represents a particularly important model not only to test the simultaneous introduction of the two TPPcin at the hindered hydroxyls, but also to evaluate the efficiency of long range chromophoric interaction of new TPPcin chromophore, and conformational preference of the cinnamate moiety, i.e., s-cis or s-trans.
Fig. 1.
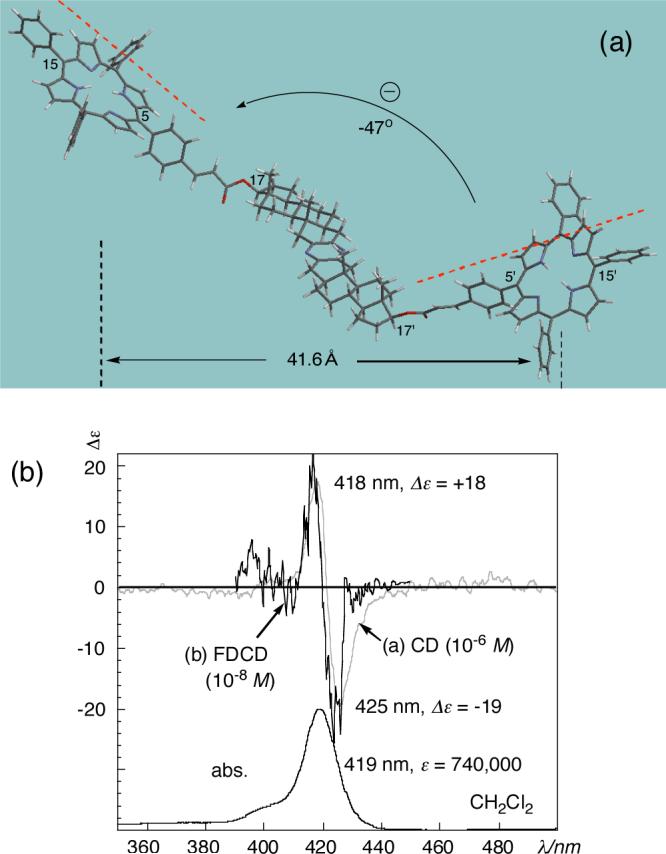
(a) The most stable conformation of bis-TPPcin 7 calculated by Monte Carlo/MMFF94s with Spartan 02. (b) CD and FDCD spectra of 7 in CH2Cl2. Gray line: CD at 1.1 × 10−6 M. Black line: FDCD at 1.1 × 10−8 M, light-cul filter, 520 nm, HT voltage, 586 V using Jasco J-465 devices with 4.0 mm balancing masks.
Fig. 3.
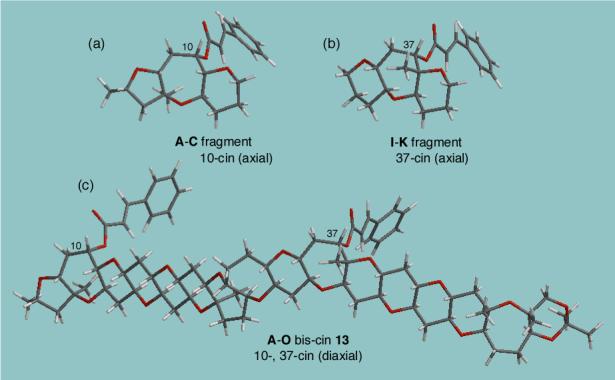
The most stable conformations of bis-cinnamate 13 and mono-cinnamate truncated models: (a) A-C fragment: 10-cin (axial). (b) I-K fragment: 37-cin (axial). (c) A-O fragment 13: 10-, 37-bis-cin (diaxial). All conformation was optimized by Monte Carlo/MMFF94s with Spartan 02. Similarly, the fragments H-K, G-K, and F-K show the lowest energy conformations with 37-cin axial.
1−1. Derivatization
An efficient method for introducing a CD reporter group at sterically hindered –OH groups under mild conditions, especially bulky porphyrinoids for the purpose of observing long-range chromophoric interactions, would greatly expand the versatility of the exciton coupled CD method for stereochemical analysis of large natural products.13-16 So far, stereochemical analysis of sterically hindered hydroxyls has been achieved by introducing (i) benzyl chromophores and/or (ii) benzoate chromophores generated directly by oxidation of benzyl ether derivatives.17-21 However, the benzyl/benzoate approach is not suitable for the analysis of gymnocin-B for several reasons; the simple benzyl/benzoate chromophores cannot provide exciton coupling over a long range due to their weak UV absorbance. Furthermore, the benzylation conditions, namely, the reaction with p-substituted benzyl chloride in the tetramethylurea/DMF/THF solvent system using sodium hydride as a strong base, is not suited for minuscule amounts of natural products, in particular, for complex molecules exemplified by gymnocins. In addition, there is the conformational ambiguity arising from the rotation of benzyloxy moiety. The harsh oxidation condition (RuCl3/NaIO4) of the benzyl ether to benzoate derivatives is also not applicable to substrates containing sensitive functional groups, such as polyether moieties and/or the oxidation-sensitive porphyrin derivatives. For such reasons, we investigated a new approach to introduce the porphyrin-cinnamate chromophore at the sterically hindered secondary –OH groups.
Recently, we reported that the olefin cross metathesis reaction22,23 can be applied successfully for introducing styrene chromophores as CD reporter groups, and based on this, a chemical/chiroptical method for configurational assignment of the olefin containing natural and synthetic compounds was developed.24,25 Usage of the recently developed Grubbs' second generation ruthenium catalyst26-28 leads to the replacement of a variety of C=C double bonds with the styrene group in high yield under mild conditions. Furthermore, we investigated the application of this transition metal catalyzed reaction towards the derivatization of sterically hindered hydroxyls. This new protocol involves the combined method of acylation of such hydroxyls with small and reactive acryloyl chloride reagent (toxicity data shown in Experimental Section)29-33 followed by cross metathesis with vinyl-porphyrins (Scheme 1). Thus, a variety of the cinnamate-type porphyrin chromophores, such as tetraphylporphyrin or tetrabenzoporphyrin, shown in Scheme 2, can be introduced at hindered hydroxyls under mild conditions.
Scheme 1.

Introduction of chromophores at sterically hindered hydroxyl.
The acylation of model substrate 1 with tetraphenylporphyrin carboxylic acid (TPP-CO2H) under EDC coupling conditions requires more than 10 equivalents of the carboxylic acid reagent and a long reaction time (24 hours in dichloromethane under reflux condition). Furthermore, the EDC coupling between 1 and the sterically congested tetrabenzoporphyrin-carboxylic acid (TBP-CO2H) due to phenyl groups at β, β'-positions of porphyrin34 failed to give the corresponding ester derivatives, while the reactions at elevated temperature lead to decomposition of the sensitive benzoporphyrin chromophore.
According to the procedure reported here, the 17-OH of steroid 1 was first acylated with acryloyl chloride in the presence of diisopropylethylamine in THF to give 2 in almost quantitative yield. Since the reaction was also achieved with a weaker base, such as triethylamine and DMAP, the acryroylation can also be performed with base-sensitive substrates. The vinyl group in 2 was then subjected to cross metathesis with 6 equivalents of vinyl-tetraphenylporphyrin (vinyl-TPP) 3a in the presence of Grubbs' second generation ruthenium catalyst to provide 4a in 72 % yield (see detailed condition in Experimental Section). As reported by Grubbs and others,22-25 the (E)-stereoisomer is the sole product under conditions of excess styrene derivatives and presence of the second generation catalyst which shifts the metathesis equilibrium toward the thermodynamically more stable trans-C=C double bonds. The stereoselective construction of the (E)-double bond by reaction with excess vinyl-TPP 3a makes this approach attractive since the geometry of the cinnamate double bond may occasionally affect the sign of CD exciton chirality. Noteworthy, the vinyl-tetrabenzoporphyrin (vinyl-TBP) 3b could also be introduced by cross metathesis under equilibrium conditions to provide 4b as a single (E)-stereoisomer in 65 % yield; the intense deep colored porphyrin derivatives are also readily detectable by thin layer chromatography (TLC). Thus, the entire procedure including reaction monitoring and product isolation can be performed at the microgram-scale down to 100 μg.
The simultaneous derivatization of the two secondary 17-OHs in bis-steroid 5 was next attempted. Our attempts for the simultaneous bis-acylation of 5 at the two secondary 17-hydroxyls by TPP-CO2H was not successful; the product obtained under several different acylation conditions, e.g., with EDC and DMAP in dichloromethane, surprisingly gave only the mono-acylate.
However, two TPPcin chromophores could be efficiently introduced at both hindered hydroxyls by a new approach; bis-steroid 5 was first reacted with the reactive acryloly chloride, 77 % yield, and the two vinyl groups introduced in 6 were then functionalized by “ double cross metathesis” with 12 equivalents of vinyl-TPP 3a. This method yielded the bis-porphyrin derivative 7 with (E)-stereochemistry at both locations in ca. 50 % yield from 5.
1−2. Long range chromophoric interaction of bis-TPPcin derivative (7)
The lowest energy conformation of bis-TPPcinnamate 7 obtained by a MMFF94s/Monte Carlo conformational analysis is shown in Figure 1a. For both TPPcin groups introduced at 17 and 17' hydroxyls, the cinnamate moiety possesses an s-cis conformation, in agreement with the preferred conformation of regular methyl cinnamates; the s-cis conformation is of 0.97 kcal/mol lower energy than the corresponding s-trans isomer, as evaluated by DFT/BLYP/6−31G* level calculations. In the case of bis-TPPcin 7with known absolute configuration, the most stable conformation as calculated by MC/MMFF94s with Spartan 02 led to a negative twist of −47 ° between the porphyrin effective transition moments (see later) along the 5−15 and 5'−15' axes. Note that the CD spectrum of 7 (Figure 1b) shows a clear negative exciton couplet with an amplitude of −37, reflecting the negative twist orientation between the remote stereogenic centers at 17 and 17' positions over a distance of 42Å. Furthermore, this relatively weak long range exciton coupling was measured by FDCD at a two-fold higher sensitivity, namely, 10−8 M, due to the favorable fluorescent properties of TPPcinnamate (fluorescent quantum yield Φf = 0.13).11 Thus, the highly sensitive FDCD analysis should prove very useful for detection of weak coupling resulting from long range interactions and/or limited sample amount.
The successful “ TPPcinnamate” microscale approach performed for the conformationally rigid dimeric steroid 5 provides an intriguing protocol for the stereochemical analysis of many large natural products carrying a variety of functional groups, e.g., sterically hindered hydroxyl and amino groups as exemplified by gymnocin-B.
2. Derivatization of gymnocin-B
Based on these successful model studies described above, we performed derivatization of gymnocin-B 8 using the 300 μg amount of available samples (Scheme 3). In order to differentiate the 10- and 37-hydroxyls, out of the four hydroxyls in 8, the 1,3-diol moiety in the O ring was first protected as the acetonide. After aqueous work up with saturated NaHCO3 solution and quick filtration through silica gel, the product was immediately reacted with acryloyl chloride in the presence of triethylamine and DMAP in CH2Cl2 at 60 °C for 6 hours. After direct purification of the reaction mixture by silica gel chromatography, the resulting bis-acryloyl derivative 10 was subjected to cross metathesis with excess of vinyl-TPP 3a (10 equivalents) under equilibrium conditions to afford the red-colored bis-TPPcin derivative 11, ca. 10 μg, as estimated from its UV-Vis spectrum. Although the presence of two chromophores is supported by observed exciton coupled CD, despite repeated attempts, the molecular ion peak of 11 could not be observed. The highest peak we found was at 1846 resulting from a loss of one TPPcinnamic acid group (see Experimental Section).
Scheme 3.

Derivatization of gymnocin-B.
3. Conformational analysis
The gymnocin-B bis-TPPcinnamate 11 is an unique and large molecule (MW = 2530) consisting of 15 polyether ring systems, five of them being conformationally flexible 7-membered rings (B, G, H, J, and O rings). An extensive conformational analysis was necessary in order to predict the dihedral angles between the transition dipoles of two porphyrins present in such an unusual system as 11 (Chart 2). Especially, critical was the precise conformational analysis around the 10-TPPcinnamate and 37-TPPcinnamate groups in rings B and J, respectively, since the pseudo-axial and/or pseudo-equatorial orientations of the hydroxyl groups could sensitively affect the absolute sense of twist between the porphyrins (vide infra). The conformational analysis started from the NOE analysis of gymnocin-B 8 and its tetrabenzoate derivative 12. Subsequently, the molecular modeling study was applied to bis-cinnamate 13 and bis-TPPcinnamate 14, (simplified model of 11), by Molecular Mechanics (MM) using the Merck Molecular Force Field (MMFF94) developed by Halgren35 in its “ static” version MMFFs, which is the default in Macromodel 7.1.36
Chart 2.
Gymnocin-B derivatives for conformational analysis.
3−1. NOE investigation of native gymnocin-B (8) and tetrabenzoate derivative (12) (Figure 2)
Figure 2.

NOE correlations at B-C and J-K rings of gymnocin-B.
A small coupling constant (1 Hz) between 10-H and 11-H and an NOE between 10-H/11-H in gymnocin-B 8 indicated that the 10-H/11-H dihedral angle was close to 90 degrees (Figure 2a).3,5 Therefore, in the oxepane ring B, this leads to 10-H pseudo equatorial and 10-OH pseudo axial orientation, similarly to gymnocin-A. The pseudo axial disposition for 10-OH is corroborated by NOEs, 8-H/9β-H, 8-H/11-H, 9α-H /10-H, and 9β-H /10-H.
On the other hand, a 37-OH/39-H NOE indicates a similar orientation for 37-OH and 39-H in ring J (Figure 2b). NOE correlation was observed between 37-H/60-Me and 36β-H/60Me, but not between 35-H/37-H. Thus, similar to 10-OH, the NOE data support a pseudo axial orientation for 37-OH. The NOE analysis of tetrabenzoate derivative 12 also led to a diaxial conformation of 10-OBz and 37-OBz.37
3−2. Molecular modeling
The complexity of such a large polyether structure on one hand, and the difficulties in parameterization of such a large molecule by MMFF94s on the other, necessitated the performance of a conformational analysis of several simpler truncated models of gymnocin-B that carry the simpler regular cinnamate moiety at 10-OH and 37-OH, instead of the bulkier TPPcinnamates (see structure 13 in Chart 2). The molecular modeling results for the truncated models were then compared with that of the gymnocin biscinnamate 13 and with NOE analysis of gymnocin-B 8, and its tetrabenzoate 12. Finally, the molecular modeling of the real system 14, i.e., bis-TPPcin of 8, was undertaken in order to evaluate the projection angle between the 5−15 direction of the two porphyrins, which according to theoretical studies38 is considered as the direction of porphyrin Soret band effective transition moment.
The conformational analysis was performed by molecular mechanics calculations using the MMFF94s force field with Monte Carlo conformational search using a Spartan ‘02 package (see the Computational Section for details). The main degrees of conformational freedom for both truncated models and gymnocin-B bis-cinnamate derivatives are (i) the C10-O and/or C37-O rotamerisms, (ii) the s-cis/s-trans C=C-C=O bond isomerism of the cinnamate moiety, and (iii) seven membered-ring conformations, i.e., chair- or boat-like conformation of B, G, H, J, and O rings. In order to simplify the calculations, the unsaturated aldehyde side chain at C-5 of A-ring was replaced by the methyl group (structures 13 and 14 in Chart 2). The (10S, 37S) absolute configurations of gymnocin-B shown in Chart 2 were chosen for all calculations.
3−2−1. Truncated models of 13; mono-cinnamate models
While only the A-C ring system of 13 was considered as the truncated model for obtaining the 10-cinnamate conformation, more extensive molecular modeling was required for obtaining geometrical information of the 37-cinnamate on J-ring. For this purpose, four truncated models I-K, H-K, G-K, and F-K fragments were studied, expecting that an enlargement of the ring system from the I-K fragment to the F-K fragment may provide more precise data for the seven membered J ring and the 37-cinnamate, located at the right and suspected more flexible side of gymnocin-B (Chart 2).
The conformational analysis of the A-C truncated system revealed four conformations within 2 kcal/mol from the most stable conformation. In the lowest energy conformation the 10-cinnamate is pseudo-axial with an s-cis orientation between the carbonyl and C=C double bond as shown in Figure 3a. The slight difference between four minima arises from the s-cis and s-trans rotamerism of the cinnamate, but importantly, only the chair-like conformation for B ring and the axial conformation for 10-cinnamate were found.
Similarly, the MMFF94s calculation of I-K fragment showed an axially oriented 37-cinnamate at the J-ring being the most stable conformation (Figure 3b). Again, only chair-like conformations of seven membered J-ring and pseudo-axial 37-cinnamate were found for all other three conformations obtained within a 2 kcal/mol energy window.
Furthermore, the continuous conformational analysis of the H-K, G-K, and F-K truncated models (structures not shown) with an enlarged polyether ring system similarly exhibited preference for the pseudo-axial 37-cinnamate conformations among the lowest energy conformations obtained within 2 kcal/mol. These results indicate that the presence of F, G, and H ring systems attached to the I-K fragment have nil, or only negligible, effects on the axial conformation of 37-cinnamate. Thus, the molecular modeling of these truncated models showed a strong preference for the diaxial conformation of both 10- and 37-cinnamates.
3−2−2. Bis-cinnamate 13
The lowest energy conformation resulting from conformational analysis of the whole structure of 13 with A-O ring system (Chart 2) is shown in Figure 3c. In this structure, both 10- and 37-cinnamates also adopt pseudo-axial orientation, consistent with the stable conformations obtained for the truncated mono-cinnamate models. Furthermore, the ten lowest conformations within a 2 kcal/mol energy window, all show the 10-cinnamate with a pseudo-axial orientation. The 37-cinnamate also shows a preference for pseudo-axial orientation, except in one case where the orientation is equatorial at 1.7 kcal/mol higher energy level than the most stable conformation (Boltzman-weighted population of about 5 %). As described previously, the NOE study of gymnocin-B 8 and the tetrabenzoate derivative 12 (Figure 2) has predicted the pseudo-diaxial conformation for both the 10- and 37-benzoates. This is in good agreement with the MMFFs molecular modeling results of the bis-cinnamates 13 conformation, thus validating the MMFFs conformational analysis of a large polyether gymnocin-B.
3−2−3. Bis-TPPcinnamate 14
Finally, an MMFF94s conformational analysis was performed on gymnocin-B bis-TPPcinnamate 14 (Chart 2). Due to the complexity of structure 14 containing a large gymnocin polyether skeleton with two large TPPcinnamate moieties, it was safer to take into consideration the conformations obtained within a rather large energy window. Therefore we considered a 7 kcal/mol energy window as well as the distributions of structures characterized by the sign and the dihedral angle between the transition dipole moments of the two porphyrins. A total 16 main conformations were found for bis-TPP cinnamate 14 within 7 kcal/mol (Table 1). The most stable conformation with s-cis orientation of both 10- and 37-TPPcin is shown in Figure 4a. Interestingly, while the 10-TPPcin is found at the axial position similar to previous cases of simple cinnamates, the 37-TPPcin clearly adopts a pseudo-equatorial orientation (Figure 4a). This lowest energy conformation with 10-axial and 37-equatorial TPPcin results in a clockwise twist between the two porphyrin effective transitions defined by the 5−15 and 5'−15' directions (dihedral angle of +29°), hence leading to positive exciton chirality. Similarly, the survey of the other lowest structures obtained within 7 kcal/mol (Table 1) gave rise to a preferred equatorial 37-TPPcin orientation, except for conformations 3 and 6, while 10-TPPcin always remains axial in all calculated conformations. This reveals a preferred clockwise twist between the porphyrin effective transitions in 15 of a total of 16 conformations. From two conformations 3 and 6 with axial 37-TPPcin, only 3 shows a negative interporphyrin twist (dihedral angles of −9°, Figure 4b) and is of 1.72 kcal/mol higher energy, i.e., 5 % of the Boltzmann-weighted population, therefore expecting to provide a negligible contribution to CD. The second conformation found with axial 37-TPPcin, e.g., 6, however interestingly exhibits a positive interporphyrin helicity, presumably as a result of slight distortion of J-ring from its usual chair-like conformation. This conformational analysis therefore clearly predicts a positive CD exciton chirality in the porphyrin region for the chosen absolute configurations of 10S, 37S. A more quantitative evaluation of exciton coupled CD based on the Boltzmann-weighted DeVoe calculation will be described in the next section.
Table 1.
Conformational analysis of gymnocin-B-bisTPPcinnamate 14.a
| conformation | ΔG (Kcal/mol)b | orientation of 10-TPPcin | orientation of 37-TPPcin | τ12 (deg)c and predicted interporphyrin twist |
|---|---|---|---|---|
| 1 | 0 | ax | eq | 29 (+) |
| 2 | 1.35 | ax | eq | 33 (+) |
| 3 | 1.72 | ax | ax | −9 (−) |
| 4 | 2.21 | ax | eq | 35 (+) |
| 5 | 2.59 | ax | eq | 76 (+) |
| 6 | 3.34 | ax | ax | 12 (+) |
| 7 | 3.96 | ax | eq | 35 (+) |
| 8 | 4.10 | ax | eq | 81 (+) |
| 9 | 4.12 | ax | eq | 109 (+) |
| 10 | 4.12 | ax | eq | 34 (+) |
| 11 | 4.25 | ax | eq | 31 (+) |
| 12 | 4.82 | ax | eq | 95 (+) |
| 13 | 4.98 | ax | eq | 33 (+) |
| 14 | 5.93 | ax | eq | 48 (+) |
| 15 | 6.24 | ax | eq | 66 (+) |
| 16 | 6.42 | ax | eq | 16 (+) |
Conformational analysis was performed by molecular mechanics calculations using the MMFF94s force field with Monte Carlo conformational search by Spartan02 package.
Relative free energy: Boltzman-weighted population at 298 K consists of 1 (86 %), 2 (9 %), and 3 (5 %) conformation, respectively.
Projection angle between 5−15 directions of porphyrins (see Figure 4).
Fig. 4.
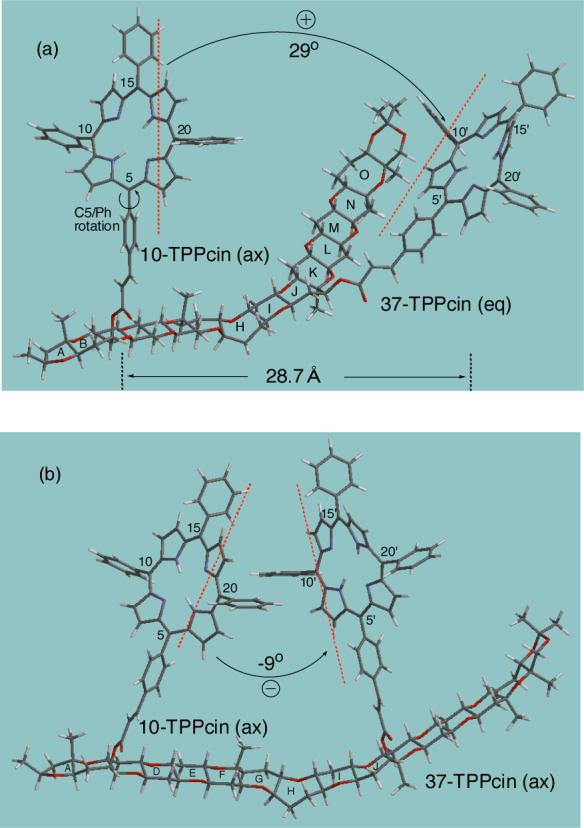
Two low energy conformations of bis-TPPcinnamate 14 obtained by Monte Carlo/MMFF94s with Spartan 02. (a) The lowest energy conformation of 14 with 10-axial, 37-equatorial. (b) conformation with 10-, 37-diaxial of 1.72 kcal/mol higher energy.
It is not surprising that the introduction of the tetraphenylporphyrins often results in an unusual conformational preference due to their steric bulkiness, aromaticity, and hydrophobicity. For example, it is known that the TPP-diester derivative of trans-1,2-cyclohexanol exhibits an unusual 1,2-diaxial conformation due to the severe porphyrin/porphyrin steric interaction of the 1,2-diequatorial conformation.10 Yet, the equatorial preference of 37-TPPcinnamate conformation in 14, in contrast to the conformational preference of the regular axial cinnamate, was an interesting finding. An additional molecular modeling of the A-K fragment of 14 without the L-O ring system was conducted. Similar to the case of 14, equatorial 37-TPPcin was found as the lowest energy conformation, but interestingly, also axial 37-TPPcin was found only at a 0.19 kcal/mol higher energy level. Furthermore, out of eighteen energy minima obtained within 7 kcal/mol, eleven conformations were found with axial 37-TPPcin orientation (see Supporting Information). The comparison of these MMFFs molecular modeling results of 14 and its truncated A-K fragment reflects some effects played by the L-O polyether tail, the presence of which most likely changes the K-ring strain. This in turn affects axial/equatorial position of 37-TPPcin and leads to some distortion of J-ring. (cf. Ref. 39)
4. CD spectroscopic analysis of bis-TPP derivative of gymnocin-B
The CD spectrum of bis-TPPcinnamate 11 in MeOH at 3.0 × 10−6 M is shown in Figure 5 (solid line). A clear almost conservative CD exciton couplet was observed with extrema at 419 nm (Δε +11) and 414 nm (Δε −15). The positive exciton chirality led to the assignment of gymnocin-B with (S)-10 and (S)-37 as depicted in this paper (see absolute structure in Figure 4a).
Fig. 5.
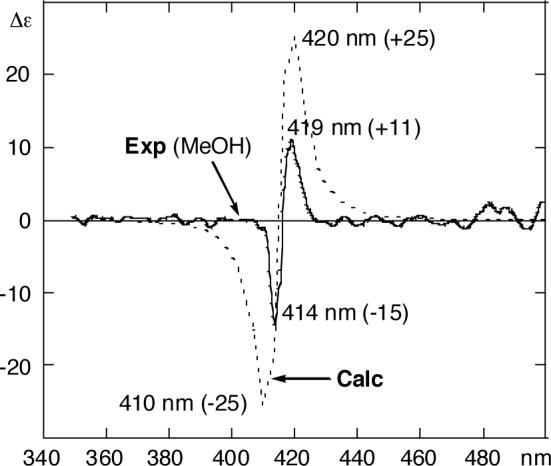
Experimental and calculated CD spectra of compound 11. Solid line: experimental, c = 3.0 × 10−6 M in MeOH. Dotted line: Boltzman-weighted (at 298 K) average CD calculated with DeVoe's method and porphyrin hybrid approach (D = 77 D2 for 5−15 dipoles and 39 D2 for 10−20 dipoles, νmax = 24.1 kK, Lorentzian band with Δν1/2 = 0.75 kK width) on lowest energy MC/MMFF94s structures within 2 kcal/mol (see Table 1).
In order to investigate the nature of the exciton coupling operating at this large interchromophoric distance of ca. 30 Å (Figure 4a), quantitative CD calculations were also performed by the DeVoe method.40-43 A hybrid approach was used to describe the Soret transition,10,38 where the 5−15 component was given full dipolar strength (77 D2, extrapolated from the absorption spectrum of mono-TPPcin 4a, 415 nm, ε 362,000 in MeOH), while the 10−20 component was given half dipolar strength (39 D2). We have previously found that this approach is well justified when a largely unrestricted libration around the phenylporphyrin bonds exists.38 The MC/MMFF94s conformational search revealed three conformations within 2 kcal/mol energy window (Table 1). The corresponding Boltzmann-weighted calculated CD spectrum (Figure 5, dotted line), was found in good agreement with the observed CD spectrum (solid line). To summarize, both the experimental and calculated CD data lead to the assignment of absolute configuration of gymnocin-B as depicted in Chart 1.
Conclusion
In conclusion, we have determined the absolute configuration of gymnocin-B, a cytotoxic marine natural product containing the largest 15-polyether skeleton isolated so far. The three challenges encountered during the course of configurational assignments of this intriguing natural product are the following: (i) introduction of bulky porphyrinoid reporter groups at the sterically hindered axial 10- and 37-hydroxyls, (ii) conformational flexibility arising from the presence of five seven membered rings, and especially the investigation of critical 10- and 37-axial and/or equatorial -OH orientation in the B and J rings, and (iii) measurement of clear bisignate CD arising from exciton coupled interaction between very remote chromopores (distance over ca. 30 Å) attached to a flexible polyether scaffold. We have developed a microscale method for introducing a bulky TPPcin chromophore into sterically hindered hydroxyls by acryloylation/cross metathesis. This resulted in a clear positive CD exciton coupling over a large distance. Extensive conformational analysis of gymnocin-B and its truncated models by MMFF94s/Monte Carlo calculation resulted in 10-axial and 37-equatorial TPPcin conformation in 11, namely, prediction of a positive interporphyrin twist for arbitrary chosen 10(S), 37(S)-configuration. Finally, the Boltzman-weighted calculated CD by DeVoe's coupled oscillator method was in full agreement with the experimental CD of 11, those establishing the absolute configuration of gymnocin-B as 10(S), 37(S), and hence, the absolute configurations of remaining 31 stereogenic centers based on the relative configuration of gymnocin-B previously determined by NMR.5
Experimental Section
Acryloylation of sterically hindered hidroxyls: acryloylated derivative of mono-steroid (2)
To a solution of 5α-androstan-17-β-ol-3-one 1 (50 mg, 0.172 mmol) and diisopropylethylamine (60.0 μL, 0.344 mmol) in dichloromethane (1.0 mL) was slowly added acryloyl chloride* (28.0 μL, 0.344 mmol) at 0 °C. After the mixture was warmed to room temperature and stirred for an additional 1 h, H2O and saturated aqueous NH4Cl solution was added, and the resulting mixture was extracted with ethyl acetate. The organic layers were combined, washed with brine, dried over Na2SO4, filtered and concentrated in vacuo to give the crude products. Column chromatography on silica gel (gradually 9 % to 25 % ethyl acetate in hexane) gave 2 (60 mg, 100 %) as a white solid: 1H NMR (400 MHz, CDCl3) δ 0.73−1.80 (m, 16H), 0.84 (s, 3H), 1.02 (s, 3H), 1.99−2.04 (m, 1H), 2.07−2.12 (m, 1H), 2.16−2.43 (m, 4H), 4.68 (dd, 1H, J = 8.8, 8.8 Hz), 5.81 (dd, 1H, J = 10.4, 1.3 Hz), 6.12 (dd, 1H, J = 17.3, 10.4 Hz), 6.38 (dd, 1H, J = 17.4, 1.3 Hz); HRFABMS calcd for C22H33O3 [M+H]+ 345.2429, found 345.2435. *Toxicity data of acryloyl chloride; IHL-MUS LC50 (inhalation-mouse lethal concentration 50 percent kill) = 92 mg/m3/2h.
Cross Metathesis with Vinyl-TPP (3a): TPPcinnamate derivative of mono-steroid (4a)
To a solution of 2 (3.0 mg, 8.71 μmol) in dichloromethane (2.0 mL) were added Grubbs' second generation ruthenium catalyst (1.7 mg, 1.99 μmol) and vinyl-TPP 3a (33.5 mg, 52.3 μmol) at room temperature, and the mixture was stirred at 80 °C for 3 h. The reaction mixture was concentrated in vacuo to give the crude product which was purified by column chromatography on silica gel (ethyl acetate in hexane, gradually from 9 % to 20 %) to afford 4a (6.0 mg, 72 %) as a deep red colored solid. The reaction was also performed using 100 μg of 2: 1H NMR (300 MHz, CDCl3) δ 0.97 (s, 3H), 1.05 (s, 3H), 0.82−2.45 (m, 22H), 4.85 (dd, 1H, J = 7.9, 7.9 Hz), 6.75 (d, 1H, J = 16.0 Hz), 7.72−7.79 (m, 9H), 7.93 (d, 2H, J = 8.1 Hz), 8.00 (d, 1H, J = 16.1 Hz), 8.20−8.26 (m, 8H), 8.83−8.87 (m, 8H); HRFABMS calcd for C66H61O3N4 [M+H]+ 957.4743, found 957.4766.
Cross Metathesis with Vinyl-TBP (3b): TBPcinnamate derivative of mono-steroid (4b)
To a solution of 2 (693 μg, 2.01 μmol) in dichloromethane (2.0 mL) was added Grubbs' second generation ruthenium catalyst (513 μg, 0.604 μmol) and vinyl-TBP 3b (3.70 mg, 6.04 μmol) at room temperature, and the mixture was stirred at 80 °C for 5 h. The reaction mixture was concentrated in vacuo to give the crude product, which was purified by column chromatography on silica gel (33 % ethyl acetate in hexane) to afford 4b (1.2 mg, 64 %) as a green colored solid. Due to benzoporphyrins aggregation in solution, only TLC and MS analysis were possible for identification of the product: Rf = 0.40 (50 % ethyl acetate in hexane); HRFABMS calcd for C64H56O3N4 [M]+ 928.4352, found 928.4384.
Bis-acryloylated Derivative of bis-steroid (6)
To a solution of bis-steroid 5 (10 mg, 17.5 μmol) and diisopropylethylamine (6.08 μL, 34.9 μmol) in dichloromethane (2.0 mL) was slowly added acryloyl chloride (3.12 μL, 38.4 μmol) at room temperature. After the mixture was stirred 3.5 h at this temperature, H2O and saturated aqueous NH4Cl solution was added, and the resulting mixture was extracted with chloroform. The organic layers were combined, washed with brine, dried over Na2SO4, filtered and concentrated in vacuo to give the crude products. Column chromatography on silica gel (gradually 17 % to 25 % ethyl acetate in hexane) gave bis-acryloylated derivative 6 (9.1 mg, 77 %) as a white solid: 1H NMR (300 MHz, CDCl3) δ 0.81 (s, 6H), 0.84 (s, 6H), 0.88−1.84 (m, 30H), 2.15−2.27 (m, 2H), 2.50−2.63 (m, 4H), 2.78 (dd, 2H, J = 17.8, 5.2 Hz), 2.91 (d, 2H, J = 16.7 Hz), 4.70 (dd, 2H, J = 8.0, 8.0 Hz), 5.81 (dd, 2H, J = 10.4, 1.6 Hz), 6.12 (dd, 2H, J = 17.3, 10.4 Hz), 6.39 (dd, 2H, J = 17.3, 1.6 Hz); HRFABMS calcd for C44H61O4N2 [M+H]+ 681.4632, found 681.4631.
Bis-TPPcinnamate derivative of bis-steroid (7)
To a solution of 6 (3.0 mg, 4.41 μmol) and vinyl-TPP 3a (33.9 mg, 52.9 μmol) in dichloromethane (2.0 mL) at 80 °C were gradually added a solution of excess amount of Grubbs' second generation ruthenium catalyst (4.49 mg, 5.29 μmol) in dichloromethane (5.0 mL) over 5h with vigorously stirring. The reaction mixture was concentrated in vacuo to give the crude product which was purified by column chromatography on silica gel twice (ethyl acetate in hexane, gradually from 25 % to 33 %) to afford 7 (5.0 mg, 60 %) as a deep red colored solid: 1H NMR (400 MHz, CDCl3) δ 0.82−2.30 (m, 44H), 2.51−2.59 (m, 4H), 2.77 (brs, 2H), 2.89−2.97 (m, 2H), 4.87 (dd, 2H, J = 8.3, 8.3 Hz), 6.75 (d, 2H, J = 16.0 Hz), 7.73−7.80 (m, 18H), 7.93 (d, 4H, J = 8.1 Hz), 8.01 (d, 2H, J = 16.3 Hz), 8.20−8.26 (m, 16H), 8.84−8.87 (m, 16H); FABMS calcd for C132H116O4N10 [M]+ 1906.40, found 1906.91.
Acetonide Derivative of gymnocin-B (9)
To a solution of gymnocin-B 8 (300 μg, 0.259 μmol) in acetone (1.0 mL) was added p-toluenesulfonic acid monohydrate (1.2 mg, 6.31 μmol) at room temperature. After the mixture was stirred at room temperature for 10 h, saturated aqueous NaHCO3 solution was added, and the resulting mixture was extracted with CHCl3. The organic layers were combined, washed with brine, dried over MgSO4, filtered on silica gel (9 % MeOH in CHCl3) and concentrated in vacuo to give a crude product as white solid, which was analyzed by MS, and immediately subjected for acryloylation without further purification: HRFABMS calcd for C65H97O20 [M+H]+ 1197.6573, found 1197.6584.
Bis-acryloylated derivative of gymnocin-B (10)
To a solution of the crude 9, obtained as described above, in dichloromethane (1.0 mL) were added acryloyl chloride (4.21 μL, 51.8 μmol), triethylamine (7.23 μL, 51.8 μmol), and dimethylaminopyridine (633 μg, 51.8 μmol) at room temperature, and the mixture was stirred at 60 °C for 6 h. The reaction mixture was concentrated in vacuo to give the crude product which was purified by preparative TLC on silica gel (50 % ethyl acetate in hexane), and then by column chromatography on silica gel (from 50 % ethyl acetate in hexane to 6 % MeOH in CHCl3) to provide 10 as a white solid: 1H NMR (400 MHz, CDCl3) Representative signals: δ 5.35 (brs, 1H), 5.44 (d, 1H, J = 10.6 Hz), 5.83 (d, 1H, J = 10.2 Hz), 5.85 (d, 1H, J = 10.4 Hz), 6.12 (dd, 1H, J = 17.4, 10.4 Hz), 6.13 (dd, 1H, J = 17.4, 10.6 Hz), 6.38 (d, 2H, J = 17.9 Hz), 6.49 (dd, 1H, J = 7.0, 7.0 Hz), 9.41 (s, 1H); FABMS calcd for C71H100O22 [M]+ 1306, found 1306.
Bis-TPPcinnamate derivative of gymnocin-B (11)
To a solution of 10 obtained above in dichloromethane (1.0 mL) was added Grubbs' second generation ruthenium catalyst (137 μg, 0.161 μmol) and vinyl-TPP 3a (491 μg, 0.766 μmol) at room temperature, and the mixture was stirred at 80 °C for 9 h. The reaction mixture was concentrated in vacuo to give the crude product which was purified twice by column chromatography on silica gel (from 50 % ethyl acetate in hexane to 6 % MeOH in CHCl3) to afford 11 (ca. 10 μg) as a deep red colored solid: Rf = 0.07 (50 % ethyl acetate in hexane); UV/vis (MeOH) 415 nm, ε = 720,000; CD (MeOH) 419 nm (Δε = +11), 414 nm (Δε = −15); Despite repeated attempts, it was not possible to observe the molecular ion peak of bis-TPPcinnamate derivative 11 (calculated MW: 2531), either by ESI or MALDI. Instead an intense peak corresponding to the loss of a TPPcinnamic acid fragment (McLafferty rearrangement) was observed. MALDI-TOFMS calcd for C112H124O20N4 [M-C47H32O2N4 (TPPcinnamic acid)]+ 1846, found 1846.
Computational methods
Molecular modeling was performed by Spartan '02 (Wavefunction, Inc., Irvine, CA), using default parameters and convergence criteria. The conformations were calculated with Merck force field (MMFF94s)36 using the Monte Carlo conformational search method with 1000 iteration steps. The coupled oscillator CD calculations were performed by DeVoe method using Fortran program developed by W. Hug.44 Spectral parameters for DeVoe calculation were extrapolated from the experimental UV/Vis spectrum of the mono-TPPcin chromophore 4a (D = 77 D2 for 5−15 dipoles and 39 D2 for 10−20 dipoles, νmax = 24.1 kK, Lorentzian band with Δν1/2 = 0.75 kK width). The conformations optimized by MMFF94s were used as the input geometries and the Boltzmann-weighted CD spectrum was calculated at 298K.
Supplementary Material
Acknowledgement
We are grateful for the discussions to Dr. Gennaro Pescitelli and Prof. Lorenzo DiBari, University of Pisa, Prof. Wolfgang Kreiser, University of Dortmund, and to Prof. Carlo Rosini and Dr. Egidio Giorgio, University of Potenza, who also made available to us the DeVoe program. We acknowledge the Jasco Corporation for generously providing us with a new FDCD J-465 device. This research is supported by the National Institutes of Health, NIH Grant GM 34509 (K.N. and N.B.). K.T. is grateful to the JSPS Postdoctoral Fellowships for Research Abroad.
Footnotes
Supporting Information Available. Copies of COSY and ROESY spectra of gymnocin-B, 1H NMR and MS spectra of the compounds 2, 4a, 4b, 6, 7, and 9-11, as well as some conformational data. This material is available free of charge via the internet at http://pubs.acs.org.
REFERENCES
- 1.Honjo T. Rev. Fish Sci. 1994;2:225–253. [Google Scholar]
- 2.Jenkinson IR, Arzul G. 1998. Xunta de Galicia and IOC of UNESCO: Flance.
- 3.Satake M, Shoji M, Oshima Y, Naoki H, Yasumoto T. Tetrahedron Lett. 2002;43:5829–5832. [Google Scholar]
- 4.a Tsukano C, Sasaki M. J. Am. Chem. Soc. 2003;125:14294–14295. doi: 10.1021/ja038547b. [DOI] [PubMed] [Google Scholar]; b Tsukano C, Ebine M, Sasaki M. J. Am. Chem. Soc. 2005;127:4326–4335. doi: 10.1021/ja042686r. [DOI] [PubMed] [Google Scholar]
- 5.Satake M, Tanaka Y, Ishikura Y, Oshima Y, Naoki H, Yasumoto T. submitted.
- 6.Harada N, Nakanishi K. Circular Dichroic Spectroscopy - Exciton Coupling in Organic Stereochemistry. University Science Books; Mill Valley, CA: 1983. [Google Scholar]
- 7.Berova N, Nakanishi K. In: Circular Dichroism: Principles and Applications. 2nd ed. Berova N, Nakanishi K, Woody RW, editors. Wiley-VCH; New York: 2000. pp. 337–382. [Google Scholar]
- 8.Chen S-ML, Harada N, Nakanishi K. J. Am. Chem. Soc. 1974;96:7352–7354. [Google Scholar]
- 9.Canceill J, Collet A, Jacques J. J. Chem. Soc., Perkin II. 1982:83. [Google Scholar]
- 10.Matile S, Berova N, Nakanishi K, Fleischhauer J, Woody RW. J. Am. Chem. Soc. 1996;118:5198–5206. [Google Scholar]
- 11.Tanaka K, Pescitelli G, Nakanishi K, Berova N. Monatsh. Chem. 2005;136:367–395. [Google Scholar]
- 12.Cerny I, Pouzar V, Budesinsky M, Drasar P. Collect. Czech. Chem. Commun. 2000;65:1597–1608. [Google Scholar]
- 13.Yasumoto T, Murata M. Chem. Rev. 1993;93:1897–1909. [Google Scholar]
- 14.Satake M, Murata M, Yasumoto T. J. Am. Chem. Soc. 1993;115:361–362. [Google Scholar]
- 15.Faulkner DJ. Nat. Prod. Rep. 1999;16:155–198. [Google Scholar]
- 16.Faulkner DJ. Nat. Prod. Rep. 2002;19:1–48. doi: 10.1039/b009029h. [DOI] [PubMed] [Google Scholar]
- 17.Wiesler WT, Berova N, Ojika M, Meyers HV, Chang M, Zhou P, Lo LC, Niwa M, Takeda R, Nakanishi K. Helv. Chim. Acta. 1990;73:509–51. [Google Scholar]
- 18.Takeda R, Zask A, Nakanishi K, Park MH. J. Am. Chem. Soc. 1987;109:914–915. [Google Scholar]
- 19.Takeda R, Park MH, Nakanishi K. Isr. J. Chem. 1989;29:239–242. [Google Scholar]
- 20.Nakanishi K, Kuroyanagi M, Nambu H, Oltz EM, Takeda R, Verdine GL, Zask A. Pure Appl. Chem. 1984;56:1031–1048. [Google Scholar]
- 21.Chang M, Meyers HV, Nakanishi K, Ojika M, Park JH, Park MH, Takeda R, Vazquez J, Wiesler WT. Pure Appl. Chem. 1989;61:1193–1200. [Google Scholar]
- 22.Connon SJ, Blechert S. Angew. Chem., Int. Ed. 2003;42:1900–1923. doi: 10.1002/anie.200200556. [DOI] [PubMed] [Google Scholar]
- 23.Chatterjee AK, Choi T-L, Sanders DP, Grubbs RH. J. Am. Chem. Soc. 2003;125:11360–11370. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]
- 24.Tanaka K, Nakanishi K, Berova N. J. Am. Chem. Soc. 2003;125:10802–10803. doi: 10.1021/ja036847n. [DOI] [PubMed] [Google Scholar]
- 25.Tanaka K, Pescitelli G, Di Bari L, Xiao TL, Nakanishi K, Armstrong DW, Berova N. Org. Biomol. Chem. 2004;2:48–58. doi: 10.1039/b312542d. [DOI] [PubMed] [Google Scholar]
- 26.Chatterjee AK, Toste FD, Choi T-L, Grubbs RH. Adv. Synth. Catal. 2002;344:634–637. [Google Scholar]
- 27.Scholl M, Ding S, Lee CW, Grubbs R,H. Org. Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 28.Scholl M, Trnka TM, Morgan JP, Grubbs RH. Tetrahedron Lett. 1999;40:2247–2250. [Google Scholar]
- 29.Olsson T, Stern K, Westman G, Sundell S. Tetrahedron. 1990;46:2473–2482. [Google Scholar]
- 30.Mizutani H, Watanabe M, Honda T. Tetrahedron. 2002;58:8929–8936. [Google Scholar]
- 31.Langer P, Albrecht U. Synlett. 2002:1841–1842. [Google Scholar]
- 32.Honda T, Namiki H, Kaneda K, Mizutani H. Org. Lett. 2004;6:87–89. doi: 10.1021/ol0361251. [DOI] [PubMed] [Google Scholar]
- 33.Wang C, Russell GA. J. Org. Chem. 1999;64:2066–2069. doi: 10.1021/jo982388a. [DOI] [PubMed] [Google Scholar]
- 34.Giraud-Roux M, Proni G, Nakanishi K, Berova N. Heterocycles. 2003;61:417–432. [Google Scholar]
- 35.a Halgren TA. J. Comput. Chem. 1996;17:490–519. [Google Scholar]; b Halgren TA. J. Comput. Chem. 1996;17:520–552. [Google Scholar]; c Halgren TA. J. Comput. Chem. 1996;17:553–586. [Google Scholar]; d Halgren TA, Nachbar RB. J. Comput. Chem. 1996;17:587–615. [Google Scholar]; e Halgren TA. J. Comput. Chem. 1996;17:616–641. [Google Scholar]
- 36.Mohamadi F, Richards NGJ, Guida WC, Liskamp R, Lipton M, Caufield C, Chang G, Hendrickson T, Still WC. J. Comput. Chem. 1990;11:440–467. [Google Scholar]
- 37.Satake M, Tanaka Y, Ishikura Y, Oshima Y. personal communication.
- 38.Pescitelli G, Gabriel S, Wang Y, Fleischhauer J, Woody RW, Berova N. J. Am. Chem. Soc. 2003;125:7613–7628. doi: 10.1021/ja030047v. [DOI] [PubMed] [Google Scholar]
- 39.Eliel EL, Allinger NL, Angyal SJ, Morrison GA. Conformational Analysis. John Wiley & Sons, Inc.; New York: 1965. pp. 189–210. [Google Scholar]
- 40.DeVoe H. J. Chem. Phys. 1964;41:393–400. [Google Scholar]
- 41.DeVoe H. J. Chem. Phys. 1965;43:3199–208. [Google Scholar]
- 42.Rosini C, Zandomeneghi M, Salvadori P. Tetrahedron: Asymm. 1993;4:545–54. [Google Scholar]
- 43.Superchi S, Giorgio E, Rosini C. Chirality. 2004;16:422–451. doi: 10.1002/chir.20056. [DOI] [PubMed] [Google Scholar]
- 44.Hug W, Ciardelli F, Tinoco I., Jr J. Am. Chem. Soc. 1974;96:3407–3410. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.