

Abstract
This study identifies ataxia-telangiectasia mutated (ATM) as a further component of the complex signalling network of radiation-induced DNA damage in non-targeted bystander cells downstream of ataxia-telangiectasia and Rad3-related (ATR) and provides a rationale for molecular targeted modulation of these effects.
In directly irradiated cells ATR, ATM and DNA-PK deficiency resulted in reduced cell survival as predicted by the known important role of these proteins in sensing DNA damage. A decrease in clonogenic survival was also observed in ATR/ATM/DNA-PK proficient, non-irradiated bystander cells, but this effect was completely abrogated in ATR and ATM but not DNA-PK deficient bystander cells. ATM activation in bystander cells was found to be dependent on ATR function. Furthermore, the induction and co-localisation of ATR, 53BP1, ATM-S1981P, p21 and BRCA1 foci in non-targeted cells was demonstrated, suggesting their involvement in bystander DNA-damage signalling and providing additional potential targets for its modulation. 53BP1 bystander foci were induced in an ATR dependent manner predominantly in S-phase cells, similar to γH2AX foci induction.
In conclusion these results provide a rationale for the differential modulation of targeted and non-targeted effects of radiation.
Keywords: ataxia-telangiectasia and Rad3-related (ATR), ataxia-telangiectasia mutated (ATM), 53BP1, DNA damage response, radiation-induced bystander signalling
Introduction
There is now an extensive body of evidence for the spatial and temporal transmission of adverse effects from irradiated cells to unirradiated ‘bystander’ cells. Such ionising radiation-induced non-targeted effects have been reported for a range of endpoints (1-6) including the induction of γH2AX foci (7-11) which serve as a marker for DNA double strand breaks (12). Links between bystander responses in non-targeted cells and DNA repair pathways have been proposed (reviewed in (13)).
Most recently, ataxia-telangiectasia and Rad3-related (ATR) has been identified as a central figure within the bystander signalling cascade leading to γH2AX foci formation, whereas ataxia-telangiectasia mutated (ATM) and DNA-dependent protein kinase (DNA-PK) function were not essential for the induction of bystander γH2AX foci. Bystander foci induction has been shown to be cell cycle dependent and was observed predominantly in S-phase cells (8). These observations support the hypothesis of an accumulation of stalled replication forks in bystander cells which induces γH2AX foci in an ATR-dependent manner. In support of this hypothesis, H2AX phosphorylation at sites of gemcitabine-induced stalled replication forks has recently been reported (14).
DNA- replication fork stalling can be caused by double or single stranded DNA breaks produced by reactive oxygen species (ROS), or by adverse secondary structures of the DNA. ATR is involved in the recognition of stalled replication forks, as failure to stabilise them results in their collapse and ultimately in genetic instability (reviewed in (15)).
ATR, ATM and DNA-PK are members of the phosphoinositol 3-kinase-like kinase (PIKK) family which act as sensors of DNA damage and translate the signal into responses of cell cycle arrest and DNA repair (16). ATR is associated with an activating subunit ATRIP which responds to single stranded DNA-RPA-complexes (17). ATR recruitment to DNA damage induced by ionising radiation (IR) depends on the ATM-MRN complex and results in Chk1 phosphorylation (18). In contrast, ATR is recruited to sites of stalled replication and UV damage independent of ATM, and the phosphorylation and activation of ATM in response to UV treatment or replication fork stalling is ATR dependent (19). There is also substantial overlap between ATR and ATM downstream signalling which merges into an extensive signalling network (20).
This study reports a radiation-induced decrease in clonogenic survival in ATR/ATM proficient bystander cells but a complete abrogation of this effect in ATR/ATM but not DNA-PK deficient cells. We conclude that both ATR and ATM participate in bystander signalling leading to decreased survival in non-targeted cells. In contrast, in directly targeted cells survival decreased upon ATR, ATM and DNA-PK inhibition suggesting the possibility of differential modulation of targeted and non-targeted effects through ATM and ATR inhibitors. Furthermore, we demonstrate the induction and co-localisation of ATR, 53BP1, ATM-S1981P, p21 (WAF1/CIP1) and BRCA1 foci in non-targeted cells. 53BP1 foci induction occurred, similar to γH2AX foci, in an ATR dependent manner in S-phase bystander cells, and ATM activation was also found to be dependent on ATR function.
Material and Methods
Cell culture
T98G glioma cells, GM05849 ATM-/- fibroblasts and M059J (DNA-PK mutated) glioma cells were cultured in RPMI 1640 medium (Cambrex, Verviers, Belgium) supplemented with 10 % FBS (PAA, Pasching, Austria), 2 mM L-glutamine, 100 units/ml penicillin and 100 μg/ml streptomycin (all Cambrex, Verviers, Belgium). T98G cells were obtained from the European Collection of Cell Cultures (ECACC), and GM05849 ATM-/- fibroblasts were obtained from Cancer Research UK. M059J glioma cells were a kind gift from A. Kiltie, Leeds Institute of Molecular Medicine, UK. The DNA-PK mutant cell line M059J which lacks the p350 DNA-PK subunit (21) has been used in previous studies on the impact of DNA-PK deficiency on a range of biological endpoints (22-25).
ATR mutated Seckel cells (F02/98 hTERT (26)) and matched ATR proficient fibroblasts (48BR hTERT), received as gift from P. Jeggo, University of Sussex, UK, were cultivated in MEM medium supplemented with 15 % FBS (PAA, Pasching, Austria), 2 mM L-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin (all Cambrex, Verviers, Belgium) and 0.4 μg / ml puromycin (Sigma, Poole, UK).
All cells were incubated at 37°C, 5 % CO2. For all experiments non-confluent cell cultures were used.
For medium transfer experiments cells were seeded on 22 × 22 mm coverslips placed in 6-well tissue culture dishes and were treated with filtered medium obtained from T98G cells, which had been irradiated with 2 Gy of X-rays (Pantak, 240KV) followed by 30 - 60 minutes of incubation. The recipient cells were incubated with the conditioned medium at 37°C, 5 % CO2.
Clonogenic cell survival
Cells were seeded in T25 tissue culture flasks containing filtered medium derived from irradiated (2 Gy, X-rays) or sham-irradiated T98G cells, and incubated for 10 – 14 days at 37°C, 5 % CO2. For experiments involving direct irradiation, individual doses were 0, 1, 2 and 5 Gy. Flasks were stained with crystal violet staining solution, and individual colonies were counted. Average survival fractions and standard error (SEM) of 2 - 4 independent experiments performed in triplicate were calculated.
The final concentration for the ATM inhibitor KU-55933 (2-Morpholin-4-yl-6-thianthren-1yl-pyran-4-one, Calbiochem / Merck, Nottingham, UK) and the DNA-PK inhibitor NU7026 (KuDos, Cambridge, UK) was 10 μM. Caffeine (Sigma, Poole, UK) was used as an ATR/ATM inhibitor at a concentration of 0.1, 0.5 and 1 mM. The inhibitors were added to the culture medium 15 min prior to direct irradiation, or were added into the conditioned medium prior to transfer to recipient cells for bystander experiments.
Immunocytochemistry and microscopy
For immunocytochemistry, cells were fixed for 15 min with 4 % paraformaldehyde, permeabilized with 0.5 % Triton-X 100 (both Sigma, Poole, UK) and blocked with 3 % FBS (PAA, Pasching, Austria) in PBS for 30 min at room temperature. Incubation with a primary antibody specific for H2AX p139S (Upstate, Chandlers Ford, UK), 53BP1 (Novus Biologicals, Littleton, CO, USA), BRCA1 (Oncogene, Cambridge, MA, USA), p21/Cip1/WAF1 (PharMingen / BD Bioscience, San Diego, CA, USA), ATM p1981S (Upstate, Chandlers Ford, UK) and ATR (N-19, Santa Cruz Biotechnology, Santa Cruz, CA, US) for 1 h at room temperature was followed by incubation with a matching Alexa Fluor 488 or 567 labelled secondary antibody (Molecular Probes, Leiden, Netherlands) and nuclear counterstaining with DAPI. Cells were grown on coverslips and the inverted coverslip was placed on a glass slide with VectaShield mounting medium for fluorescence microscopy (Vector Laboratories, Burlingame, CA) after staining was completed, and the edges were sealed with clear nail varnish. A fluorescence microscope was used for imaging and analysis (Zeiss, Welwyn Garden City, UK). For quantification of foci numbers, cells were microscopically analysed for individual foci. Co-staining for 53BP1 and γH2AX was analysed in one representative experiment scoring ≥ 50 cells per data point. For 53BP1 / bromodeoxyuridine (BrdU) double staining, ≥99 cells per slide were scored for 53BP1 foci numbers in BrdU+ and BrdU− cells in one representative experiment. For single analysis of 53BP1, γH2AX and ATM-S1981P foci induction, 2-5 independent experiments were evaluated; average induced foci and standard error (SEM) were calculated. Average foci per cell and distribution of foci numbers in the analysed cell population were calculated using Microsoft Excel. For the calculation of induced foci per cell background foci numbers were subtracted.
For the detection of BrdU incorporation, cells were incubated with medium containing 20 μM BrdU for 15 min prior to fixation. A BrdU specific primary antibody followed by a matching Alexa Fluor 488 labelled secondary antibody (both Molecular Probes, Leiden, Netherlands) was applied after treatment with 2M HCl for 15 min at room temperature and neutralisation with 0.1M sodium borate buffer. For co-localisation analysis, ImageJ software and Colocalisation Test plugin (T Collins & W Rasband, http://www.uhnresearch.ca/facilities/wcif/software/Plugins/colocalisation_test.html ) were used to calculate Pearson's correlation coefficients (27) and determine the significance of correlation coefficients for individual cells using Costes' spatial statistics approach (28). DAPI images were used as masks to restrict the analysis to the cell nucleus.
Statistical analysis
Average foci numbers per cell, average survival fractions and standard errors were calculated using Microsoft Excel. The significance of reported findings was tested with a t-test comparing treated samples with their sham treated controls. Significance levels of p<0.05 (*) and p<0.01 (**) were indicated in the diagrams.
Results
Reduced clonogenic survival in bystander cells
A robust bystander response has previously been demonstrated for T98G glioma cells for various endpoints including the induction of γH2AX foci and micronuclei (1, 8, 29). γH2AX bystander foci have previously also been demonstrated in 48BR hTERT cells, but did not occur in ATR mutated F02-98 hTERT cells (8). In this study, we confirmed significant γH2AX bystander foci induction (p<0.01, t-test) in DNA-PK mutated M059J cells and ATM mutated GM05849 fibroblasts after 0.5 h and 24 h incubation with conditioned medium derived from irradiated T98G cells (Figure 1A). Induced foci per cell describe the average increase in foci per cell after subtraction of average background foci which are 1.7 foci per cell for GM05849 and 3.9 foci per cell for M059J cells.
Figure 1.

(A) Bystander γH2AX foci induction in DNA-PK mutated M059J cells and ATM mutated GM05849 fibroblasts. Induced foci numbers per cell in bystander cultures were calculated as average foci number per cell minus average foci number in control cultures. (B) Clonogenic survival in bystander cells. DNA-PK mutated M059J cells, ATM mutated GM05849 fibroblasts, T98G glioma cells, 48BR hTERT fibroblasts and ATR mutated F02-98 hTERT fibroblasts were treated with medium derived from irradiated T98G cells. (C) Clonogenic survival in directly irradiated cells. DNA-PK mutated M059J cells, ATM mutated GM05849 fibroblasts, T98G glioma cells, 48BR hTERT fibroblasts and ATR mutated F02-98 hTERT fibroblasts were irradiated with 1, 2 or 5 Gy of x-rays. All error bars are standard errors (SEM). * p<0.05, ** p<0.01 (paired t-test)
We also investigated the effect of medium transferred from irradiated T98G cells to non-irradiated bystander cells on clonogenic survival in a colony formation assay. A reduction of clonogenic survival by 20% was demonstrated for T98G bystander cells (p<0.05, t-test), and a similar effect was seen in 48BR hTERT fibroblasts (p=0.059) and DNA-PK mutated M059J cells (p<0.05). In contrast, ATM mutated GM05849 fibroblasts as well as ATR mutated F02-98 hTERT cells did not show a bystander effect for clonogenic survival (Figure 1B).
As expected, following direct irradiation, M059J, F02-98 hTERT and GM05849 cells that bear mutations in DNA-PK, ATR or ATM respectively, were much more radiosensitive than T98G cells and 48BR hTERT fibroblasts that are DNA-PK, ATR and ATM proficient (Figure 1C).
Modulation of the bystander effect on clonogenic survival
The above results suggest a dependency of the bystander effect, measured as reduced clonogenic survival, on the presence of functional ATR and ATM. In order to test this hypothesis, T98G cells were treated with Caffeine at 0.1, 0.5 and 1 mM and either incubated with medium derived from irradiated cells (Figure 2A) or subjected to direct irradiation (Figure 2A & C). Caffeine has been reported to inhibit ATM and ATR causing G2 checkpoint abrogation (30) although there is evidence for other pathways being involved (31). For directly irradiated cells, a significant radiosensitisation was detected with 1mM Caffeine (p<0.05 at 5 Gy, t-test), whereas a smaller effect was seen at 0.1 and 0.5 mM. In contrast, an abrogation of the bystander effect on clonogenic survival was demonstrated at all concentrations tested.
Figure 2.

Modulation of clonogenic survival in directly irradiated T98G cells or T98G bystander cells treated with medium derived from irradiated cells. (A) Caffeine pre-treatment. (B) Pre-treatment with 10 μM of the ATM inhibitor (ATMi) KU-55933 or the DNA-PK inhibitor (DNA-PKi) NU7026. (C) Dose response curves for direct irradiation after pre-treatment with caffeine, ATMi and DNA-PKi. All error bars are standard errors (SEM). * p<0.05, ** p<0.01 (paired t-test)
In a further experiment, the DNA-PK inhibitor NU7026 and the ATM inhibitor KU-55933 were tested in T98G cells for a modulation of clonogenic survival in directly irradiated and bystander cells. For direct irradiation, both inhibitors caused significant radiosensitisation of T98G cells at 10 μM (p<0.05 for DNA-PKi and p<0.01 for ATMi at 5 Gy, t-test; Figure 2C). The ATM inhibitor KU-55933 abrogated the bystander effect, whereas in cells treated with the DNA-PK inhibitor NU7026 a significant bystander response was still observed (p<0.01, t-test; Figure 2B). These results suggest a key role for ATM in the bystander response but do not identify a role for DNA-PK thus confirming the results observed in the mutant cell lines.
Co-localisation of γH2AX foci with DNA repair associated proteins in bystander cells
The induction of γH2AX foci, a marker for DNA damage, has been shown previously in bystander cells (7-11),.and therefore, co-localisation of γH2AX bystander foci with other proteins associated with the recognition of DNA damage and repair was predicted. Accordingly, 48BR hTERT fibroblasts and T98G cells were analysed for the induction of γH2AX, 53BP1, ATR, ATMpS1981, p21/Cip1/WAF1 and BRCA1 foci (Figure 3) 30 min after the transfer of conditioned medium from irradiated cells. γH2AX bystander foci were found to co-localise with 53BP1 foci and with ATR foci, and co-localisation was also observed for 53BP1 with p21/Cip1/WAF1 foci, 53BP1 with BRCA1 foci and 53BP1 with ATMpS1981 foci in bystander cells. Pearson correlation coefficients were calculated for channel 1 vs channel 2 (left and centre column in Figure 3) and Costes' spatial statistics approach demonstrated significant co-localisation for all tested combinations, i.e. in each case Pearson coefficients were greater than any correlation coefficient calculated between channel 1 and 10 randomly scrambled channel 2s.
Figure 3.

Co-localisation of proteins involved in DNA-damage recognition and repair and in cell cycle control in 48BR hTERT or T98G bystander cells at 30 min after transfer of medium from irradiated T98G cells. R values are Pearson correlation coefficients.
ATR dependent 53BP1 foci induction in bystander cells
53BP1 is a central figure in the DNA damage response (32-36). The induction of 53BP1 foci has previously been investigated in a study of effects induced by targeted cytoplasmic irradiation (37).
Here, a similar induction of 53BP1 and γH2AX foci in bystander cells could be detected in T98G glioma cells after 30 min incubation with conditioned medium from irradiated cells (Figure 4A). The induction of 53BP1 foci depended on functional ATR within the recipient cells, as ATR mutated F02-98 hTERT fibroblasts did not show 53BP1 foci induction in bystander cells in contrast to 48BR hTERT ATR proficient fibroblasts (Figure 4B). Background 53BP1 foci numbers were cell line dependent and highly variable between different cell lines with an average of 1.8 foci in F02-98 hTERT, 5.2 foci in 48BR hTERT and 8.4 in T98G cells. In T98G cells, high 53BP1 foci numbers (≥10 foci per cell) were predominantly observed in BrdU positive bystander cells (Figure 4C) confirming a link between S-phase / DNA replication and the early bystander response. These results demonstrate an ATR dependent 53BP1 bystander foci induction in S-phase cells comparable to the previously reported induction of γH2AX foci.
Figure 4.

Induction of 53BP1 bystander foci at 30 min after transfer of medium from irradiated T98G cells. (A) 53BP1 vs γH2AX foci in T98G bystander cells. (B) 53BP1 foci in ATR proficient 48BR hTERT vs ATR mutated F02-98 hTERT cells. All error bars are standard errors (SEM). ** p<0.01 (paired t-test) (C) Distribution of 53BP1 foci in bystander and control cell cultures after BrdU pulse labelling.
ATR dependent ATM phosphorylation in bystander cells
To further investigate the mechanism of ATM activation in the bystander signalling cascade, ATR proficient 48BR hTERT cells and ATR mutated F02-98 hTERT cells were analysed for ATM phosphorylation (Serine 1981) in directly irradiated and bystander cells (Figure 5). For both 48BR hTERT and F02-98 hTERT cells, ATM phosphorylation was detected in directly irradiated cells (1 Gy, X-rays) as early as 30 min after irradiation and these phospho-ATM foci co-localised with 53BP1 foci. When medium was transferred from irradiated to non-irradiated cells, recipient bystander 48BR hTERT cells developed, on average, 4 additional phospho-ATM foci per cell co-localised with 53BP1 foci whereas recipient bystander F02-98 hTERT cells showed only 0.3 additional phospho-ATM foci per cell consistent with previously described residual ATR activity in F02-98 hTERT cells (19). These findings support the hypothesis of a dependency of ATM phosphorylation on ATR signalling in bystander cells similar to 53BP1 foci formation. Background ATMS1981P foci numbers were 0.7 in F02-98 hTERT and 4.0 in 48BR hTERT cells.
Figure 5.

Phosphorylation of ATM (Serine 1981) and co-localisation with 53BP1 foci in directly irradiated, bystander and control cells at 30 min after irradiation / medium transfer. (A) ATR proficient 48BR hTERT cells. (B) ATR mutated F02-98 hTERT cells. (C) Bars: Average induced phospho-ATM foci per cell; error bars: standard error (SEM). ** p<0.01 (paired t-test)
Discussion
This study contributes to our understanding of mechanisms of radiation-induced signalling in non-targeted bystander cells which differs in several key aspects from the response of targeted cells. We have previously reported the ATR dependent induction of γH2AX foci for a prolonged time in bystander cells (8), and in this study we report a similar response for 53BP1 foci induction. This suggests an induction of DNA damage presumably linked to stalled replication forks in S-phase cells and the consecutive activation of proteins involved in DNA damage recognition and repair as well as cell cycle checkpoint control involving ATM, BRCA-1 and p21/Cip1/WAF1.
In this study we have demonstrated a reduction in clonogenic bystander cell survival in T98G, 48BR hTERT and DNA-PK mutated M059J cells but not in ATM-deficient GM05849 and ATR mutated F02-98 hTERT cells. These findings support the hypothesis of a dependency on ATR and ATM, but not DNA-PK, for bystander signalling resulting in reduced clonogenic survival. Further confirmation was obtained by treatment of T98G bystander cells with the ATM/ATR inhibitor Caffeine (30) and the specific ATM inhibitor KU-55933 (38) which both abrogated the effect. In contrast, the specific DNA-PK inhibitor NU7026 (39) did not block the bystander effect on clonogenic survival in T98G cells. Following direct irradiation, clonogenic survival was reduced by each of the PIKK inhibitors Caffeine, KU-55933 and NU7026 as would have been predicted for these radiosensitizers.
Further potential downstream effects of this emerging bystander signalling network are cell cycle checkpoint and apoptosis induction, the latter having already been reported as part of radiation-induced bystander responses (40, 41). An updated model for a bystander signalling network is proposed, taking into account the novel findings derived from this study (Figure 6). The initial DNA damage in bystander cells is thought to be caused by radical oxygen species (ROS) which are amplified by the induction of Transforming Growth Factor beta 1 (TGFß1). Oxidative DNA damage interferes with the replication fork in S-phase cells and single stranded DNA is exposed at sites of stalled replication initially attracting RPA and ATR/ATRIP which initiate a DNA damage response and lead to the recruitment of DNA repair factors including γH2AX, 53BP1 and BRCA1 with the aim of replication fork stabilisation and DNA repair. ATR can further phosphorylate a number of target proteins such as ATM and Chk1, potentially promoting cell cycle arrest and apoptosis induction.
Figure 6.
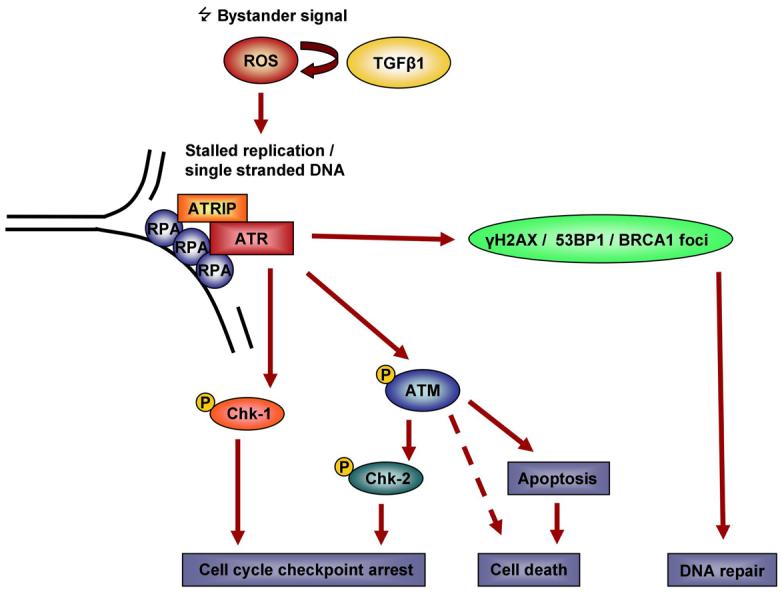
Model of ATR/ATM involvement in bystander signalling.
p53 status appears not to be critical for the bystander responses investigated in this study. T98G cells express mutant although probably partly functional p53 (42) whereas M059J, 48BR, F02-98 and GM05849 ATM-/- fibroblasts to our knowledge carry no p53 mutations. Previous bystander studies involved CHO cells (43) that carry a p53 mutation (44) and primary cells, e.g. primary human astrocytes (8) that most probably are not p53 mutated. More recently, p53 status was not found to play a role in the production or response to a bystander signal leading to mutation induction (45). Therefore, for certain endpoints the bystander DNA damage signalling presumably is independent of or can bypass p53 although it is a known ATM target. Nevertheless, it may be of interest to investigate a potential p53 involvement for selected bystander responses, e.g. bystander cell apoptosis.
From a therapeutic aspect, our study suggests ATR, ATM and their downstream effectors to be suitable therapeutic targets to differentially modulate targeted and non-targeted effects of radiation. Understanding the role of ATR and ATM pathways for radiation induced effects in non-targeted cells will complement the already established research into potential radiosensitising effects in directly targeted cells. Further research beyond this study will have to establish the fate of cells that are spared from obvious bystander responses due to impaired ATR or ATM function. In particular, long-term effects, such as increased mutation rates and genomic instability (46, 47), in these surviving cells have to be considered since drugs inhibiting ATR, ATM and Chk-1 are under development or already undergoing clinical trials. In combination with radiotherapy, the abrogation of bystander cell killing in ATR and ATM deficient cells could have unwanted late effects including the potential of secondary cancers arising from surviving bystander cells accumulating DNA damage.
Acknowledgements
This work was supported by grants from Cancer Research UK [CRUK] grant number C1513/A7047, the European NOTE project (FI6R 036465) and the US National Institutes of Health (5P01CA095227-02). We thank P. Jeggo, University of Sussex, UK for the provision of ATR mutated Seckel cells (F02/98 hTERT) and matched ATR proficient fibroblasts (48BR hTERT). The DNA-PK inhibitor NU7026 was a kind gift from Dr. Graeme Smith, KuDos, Cambridge, UK. Liz Ainsbury, Health Protection Agency Chilton, UK kindly provided support and advice with the statistical analysis.
Grant support:
Cancer Research UK, grant number C1513/A7047, the European NOTE project (FI6R 036465) and the US National Institutes of Health (5P01CA095227-02) to K.M. Prise
Cancer Research UK, grant number C14504/A6116, to K. Rothkamm
References
- 1.Shao C, Stewart V, Folkard M, Michael BD, Prise KM. Nitric oxide-mediated signaling in the bystander response of individually targeted glioma cells. Cancer Res. 2003;63:8437–42. [PubMed] [Google Scholar]
- 2.Shao C, Folkard M, Michael BD, Prise KM. Targeted cytoplasmic irradiation induces bystander responses. Proc Natl Acad Sci U S A. 2004;101:13495–500. doi: 10.1073/pnas.0404930101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mothersill C, Seymour CB. Radiation-induced bystander effects--implications for cancer. Nat Rev Cancer. 2004;4:158–64. doi: 10.1038/nrc1277. [DOI] [PubMed] [Google Scholar]
- 4.Belyakov OV, Mitchell SA, Parikh D, et al. Biological effects in unirradiated human tissue induced by radiation damage up to 1 mm away. Proc Natl Acad Sci U S A. 2005;102:14203–8. doi: 10.1073/pnas.0505020102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang L, Kim PM, Nickoloff JA, Morgan WF. Targeted and nontargeted effects of low-dose ionizing radiation on delayed genomic instability in human cells. Cancer Res. 2007;67:1099–104. doi: 10.1158/0008-5472.CAN-06-3697. [DOI] [PubMed] [Google Scholar]
- 6.Morgan WF, Sowa MB. Effects of ionizing radiation in nonirradiated cells. Proc Natl Acad Sci U S A. 2005;102:14127–8. doi: 10.1073/pnas.0507119102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sokolov MV, Smilenov LB, Hall EJ, et al. Ionizing radiation induces DNA double-strand breaks in bystander primary human fibroblasts. Oncogene. 2005;24:7257–65. doi: 10.1038/sj.onc.1208886. [DOI] [PubMed] [Google Scholar]
- 8.Burdak-Rothkamm S, Short SC, Folkard M, Rothkamm K, Prise KM. ATR-dependent radiation-induced gammaH2AX foci in bystander primary human astrocytes and glioma cells. Oncogene. 2007;26:993–1002. doi: 10.1038/sj.onc.1209863. [DOI] [PubMed] [Google Scholar]
- 9.Han W, Wu L, Chen S, et al. Constitutive nitric oxide acting as a possible intercellular signaling molecule in the initiation of radiation-induced DNA double strand breaks in non-irradiated bystander cells. Oncogene. 2007;26:2330–9. doi: 10.1038/sj.onc.1210024. [DOI] [PubMed] [Google Scholar]
- 10.Yang H, Asaad N, Held KD. Medium-mediated intercellular communication is involved in bystander responses of X-ray-irradiated normal human fibroblasts. Oncogene. 2005;24:2096–103. doi: 10.1038/sj.onc.1208439. [DOI] [PubMed] [Google Scholar]
- 11.Hu B, Wu L, Han W, et al. The time and spatial effects of bystander response in mammalian cells induced by low dose radiation. Carcinogenesis. 2006;27:245–51. doi: 10.1093/carcin/bgi224. [DOI] [PubMed] [Google Scholar]
- 12.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 13.Prise KM, Folkard M, Kuosaite V, et al. What role for DNA damage and repair in the bystander response? Mutat Res. 2006;597:1–4. doi: 10.1016/j.mrfmmm.2005.06.034. [DOI] [PubMed] [Google Scholar]
- 14.Ewald B, Sampath D, Plunkett W. H2AX phosphorylation marks gemcitabine-induced stalled replication forks and their collapse upon S-phase checkpoint abrogation. Mol Cancer Ther. 2007;6:1239–48. doi: 10.1158/1535-7163.MCT-06-0633. [DOI] [PubMed] [Google Scholar]
- 15.Andreassen PR, Ho GP, D'Andrea AD. DNA damage responses and their many interactions with the replication fork. Carcinogenesis. 2006;27:883–92. doi: 10.1093/carcin/bgi319. [DOI] [PubMed] [Google Scholar]
- 16.Rouse J, Jackson SP. Interfaces between the detection, signaling, and repair of DNA damage. Science. 2002;297:547–51. doi: 10.1126/science.1074740. [DOI] [PubMed] [Google Scholar]
- 17.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–8. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 18.Adams KE, Medhurst AL, Dart DA, Lakin ND. Recruitment of ATR to sites of ionising radiation-induced DNA damage requires ATM and components of the MRN protein complex. Oncogene. 2006;25:3894–904. doi: 10.1038/sj.onc.1209426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stiff T, Walker SA, Cerosaletti K, et al. ATR-dependent phosphorylation and activation of ATM in response to UV treatment or replication fork stalling. Embo J. 2006;25:5775–82. doi: 10.1038/sj.emboj.7601446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsuoka S, Ballif BA, Smogorzewska A, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–6. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 21.Lees-Miller SP, Godbout R, Chan DW, et al. Absence of p350 subunit of DNA-activated protein kinase from a radiosensitive human cell line. Science. 1995;267:1183–5. doi: 10.1126/science.7855602. [DOI] [PubMed] [Google Scholar]
- 22.Reitsema T, Klokov D, Banath JP, Olive PL. DNA-PK is responsible for enhanced phosphorylation of histone H2AX under hypertonic conditions. DNA Repair (Amst) 2005;4:1172–81. doi: 10.1016/j.dnarep.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 23.Karimi-Busheri F, Rasouli-Nia A, Allalunis-Turner J, Weinfeld M. Human polynucleotide kinase participates in repair of DNA double-strand breaks by nonhomologous end joining but not homologous recombination. Cancer Res. 2007;67:6619–25. doi: 10.1158/0008-5472.CAN-07-0480. [DOI] [PubMed] [Google Scholar]
- 24.Povirk LF, Zhou RZ, Ramsden DA, Lees-Miller SP, Valerie K. Phosphorylation in the serine/threonine 2609-2647 cluster promotes but is not essential for DNA-dependent protein kinase-mediated nonhomologous end joining in human whole-cell extracts. Nucleic Acids Res. 2007;35:3869–78. doi: 10.1093/nar/gkm339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daido S, Yamamoto A, Fujiwara K, et al. Inhibition of the DNA-dependent protein kinase catalytic subunit radiosensitizes malignant glioma cells by inducing autophagy. Cancer Res. 2005;65:4368–75. doi: 10.1158/0008-5472.CAN-04-4202. [DOI] [PubMed] [Google Scholar]
- 26.O'Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet. 2003;33:497–501. doi: 10.1038/ng1129. [DOI] [PubMed] [Google Scholar]
- 27.Manders EM, Stap J, Brakenhoff GJ, van Driel R, Aten JA. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J Cell Sci. 1992;103(Pt 3):857–62. doi: 10.1242/jcs.103.3.857. [DOI] [PubMed] [Google Scholar]
- 28.Costes SV, Daelemans D, Cho EH, et al. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J. 2004;86:3993–4003. doi: 10.1529/biophysj.103.038422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shao C, Folkard M, Michael BD, Prise KM. Bystander signaling between glioma cells and fibroblasts targeted with counted particles. Int J Cancer. 2005;116:45–51. doi: 10.1002/ijc.21003. [DOI] [PubMed] [Google Scholar]
- 30.Sarkaria JN, Busby EC, Tibbetts RS, et al. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999;59:4375–82. [PubMed] [Google Scholar]
- 31.Cortez D. Caffeine inhibits checkpoint responses without inhibiting the ataxia-telangiectasia-mutated (ATM) and ATM- and Rad3-related (ATR) protein kinases. J Biol Chem. 2003;278:37139–45. doi: 10.1074/jbc.M307088200. [DOI] [PubMed] [Google Scholar]
- 32.Adams MM, Carpenter PB. Tying the loose ends together in DNA double strand break repair with 53BP1. Cell Div. 2006;1:19. doi: 10.1186/1747-1028-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anderson L, Henderson C, Adachi Y. Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Mol Cell Biol. 2001;21:1719–29. doi: 10.1128/MCB.21.5.1719-1729.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bekker-Jensen S, Lukas C, Melander F, Bartek J, Lukas J. Dynamic assembly and sustained retention of 53BP1 at the sites of DNA damage are controlled by Mdc1/NFBD1. J Cell Biol. 2005;170:201–11. doi: 10.1083/jcb.200503043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mochan TA, Venere M, DiTullio RA, Jr., Halazonetis TD. 53BP1, an activator of ATM in response to DNA damage. DNA Repair (Amst) 2004;3:945–52. doi: 10.1016/j.dnarep.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 36.Ward IM, Minn K, Jorda KG, Chen J. Accumulation of checkpoint protein 53BP1 at DNA breaks involves its binding to phosphorylated histone H2AX. J Biol Chem. 2003;278:19579–82. doi: 10.1074/jbc.C300117200. [DOI] [PubMed] [Google Scholar]
- 37.Tartier L, Gilchrist S, Burdak-Rothkamm S, Folkard M, Prise KM. Cytoplasmic irradiation induces mitochondrial-dependent 53BP1 protein relocalization in irradiated and bystander cells. Cancer Res. 2007;67:5872–9. doi: 10.1158/0008-5472.CAN-07-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hickson I, Zhao Y, Richardson CJ, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 39.Veuger SJ, Curtin NJ, Richardson CJ, Smith GC, Durkacz BW. Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA-dependent protein kinase and poly(ADP-ribose) polymerase-1. Cancer Res. 2003;63:6008–15. [PubMed] [Google Scholar]
- 40.Belyakov OV, Folkard M, Mothersill C, Prise KM, Michael BD. Bystander-induced apoptosis and premature differentiation in primary urothelial explants after charged particle microbeam irradiation. Radiat Prot Dosimetry. 2002;99:249–51. doi: 10.1093/oxfordjournals.rpd.a006775. [DOI] [PubMed] [Google Scholar]
- 41.Lyng FM, Maguire P, Kilmurray N, et al. Apoptosis is initiated in human keratinocytes exposed to signalling factors from microbeam irradiated cells. Int J Radiat Biol. 2006;82:393–9. doi: 10.1080/09553000600803904. [DOI] [PubMed] [Google Scholar]
- 42.Enns L, Bogen KT, Wizniak J, Murtha AD, Weinfeld M. Low-dose radiation hypersensitivity is associated with p53-dependent apoptosis. Mol Cancer Res. 2004;2:557–66. [PubMed] [Google Scholar]
- 43.Nagasawa H, Little JB. Unexpected sensitivity to the induction of mutations by very low doses of alpha-particle radiation: evidence for a bystander effect. Radiat Res. 1999;152:552–7. [PubMed] [Google Scholar]
- 44.Hu T, Miller CM, Ridder GM, Aardema MJ. Characterization of p53 in Chinese hamster cell lines CHO-K1, CHO-WBL, and CHL: implications for genotoxicity testing. Mutat Res. 1999;426:51–62. doi: 10.1016/s0027-5107(99)00077-9. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, Zhou J, Held KD, et al. Deficiencies of Double-Strand Break Repair Factors and Effects on Mutagenesis in Directly gamma-Irradiated and Medium-Mediated Bystander Human Lymphoblastoid Cells. Radiat Res. 2008;169:197–206. doi: 10.1667/RR1189.1. [DOI] [PubMed] [Google Scholar]
- 46.Bowler DA, Moore SR, Macdonald DA, et al. Bystander-mediated genomic instability after high LET radiation in murine primary haemopoietic stem cells. Mutat Res. 2006;597:50–61. doi: 10.1016/j.mrfmmm.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 47.Lorimore SA, Coates PJ, Wright EG. Radiation-induced genomic instability and bystander effects: inter-related nontargeted effects of exposure to ionizing radiation. Oncogene. 2003;22:7058–69. doi: 10.1038/sj.onc.1207044. [DOI] [PubMed] [Google Scholar]