

Abstract

A new Pd-catalyzed reaction for the stereoselective synthesis of cyclopentane-fused benzocyclobutenes is described. These transformations likely proceed via carbamate-directed carbopalladation followed by intramolecular C–H activation of an alkylpalladium intermediate. The mechanistic relationship between these transformations and Pd-catalyzed reactions of γ-(n-Boc-amino)alkenes with aryl bromides that afford pyrrolidines is discussed. Differences in reactivity between Pd-amino and Pd-amido complexes appear to play a key role in the outcome of these transformations.
During the course of studies on Pd-catalyzed carboamination reactions of N-protected γ-aminoalkenes1 we observed that the use of the weak base Cs2CO3 in transformations of terminal alkene substrates (e.g., 1) provided 2-benzylpyrrolidine derivatives (e.g., 2) in yields that were comparable to those obtained with the stronger base NaOtBu (eq 1).2 However, when the Cs2CO3 conditions were employed with cyclopentene-derived substrate 3, a surprising result was obtained. As shown below, the Pd-catalyzed reaction of 3 with 4-bromobiphenyl (4) in the presence of Cs2CO3 did not provide the expected product 5, but instead generated benzocyclobutene derivative 6 in 72% yield and >20:1 dr (eq 3). This result is in marked contrast with the reaction of 3 with 4 in the presence of NaOtBu,1b which affords the expected heterocycle 5 in 51% yield (eq 2).3
(1).

(2), (3).

The generation of benzocyclobutene 6 is both synthetically and mechanistically interesting.4 Benzocyclobutenes are widely employed as precursors to ortho-quinodimethides, which are known to undergo facile [4+2] cycloaddition reactions,5 and can also be employed in polymerizations.6 Moreover, the surprising effect of base on the reactivity of 3 raises questions about the mechanistic relationship between the reactions shown in eqs 1–3. In this Letter we describe our preliminary studies on the preparation of benzocyclobutenes via coupling of 3 with aryl bromides, and present a mechanistic hypothesis that accounts for the observed effect of base on these transformations.
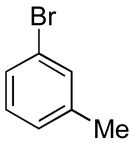
To explore the scope of the Pd-catalyzed benzocyclobutene forming process, we examined reactions of 3 with various p-, m- and o-substituted aryl bromides. As shown in Table 1, reactions of p-substituted starting materials afforded products substituted exclusively at the 2-position of the aromatic ring (entries 1–4). Similarly high regioselectivity was obtained in reactions of o-substituted aryl bromides, which afforded products bearing substituents at the 5-position (entries 5–6). The regioselectivities observed in reactions of m-substituted aryl bromides were dependent on the nature of the substituent. Although the reaction of 3 with m-bromotoluene proceeded with good regioselectivity (entry 7), the coupling of 3 with m-bromoanisole afforded a 2:1 mixture of regioisomers (entry 8).
Table 1.
Synthesis of Benzocyclobutenes from 3a
 | |||
|---|---|---|---|
| entry | ArBr | product | yield (%)b,c |
| 1 |

|

|
R = CH2OAc: 74 |
| 2 | R = Ph: 72 | ||
| 3 | R = Cl: 70 | ||
| 4 | R = OMe: 58 | ||
| 5 |

|

|
R = Me: 85 |
| 6 | R = Ph: 80 | ||
| 7 |
|

|
68 |
| 8 |

|

|
66
(2:1) |
Conditions: 1.0 equiv 3, 1.2. equiv ArBr, 2.3 equiv Cs2CO3, 4 mol % Pd(OAc)2, 8 mol % Dpe-Phos, dioxane (0.25 M), 100 °C.
Yields refer to average isolated yields obtained in two or more experiments.
All products were obtained with >20:1 dr and >20:1 regioselectivity unless otherwise noted.
The major side products formed in reactions between 3 and either electron-rich or –neutral aryl bromides were arylated cyclopentanes (7), although in many reactions trace amounts of bicyclic products 8–10 were detected by 1H NMR analysis of the crude reaction mixtures (Fig. 1).7
Figure 1.

Side products
Although the benzocyclobutene-forming reactions proceeded smoothly with many aryl bromides, the Pd-catalyzed coupling of 3 with the electron-poor substrate 3-bromobenzotrifluoride afforded a 26:39:23 mixture of 11:8:9 (eq 4);8 upon purification 11 was obtained in 24% yield. In addition, the Pd-catalyzed reaction of 3 with 1-bromo-2-methylnaphthalene proceeded slowly and in low conversion (ca. 40%) to afford 5-aryl azabicyclo[3.3.0]octane 12 in 26% isolated yield (eq 5).9
(4).

(5).

The dependence of base on the outcome of Pd-catalyzed reactions of 3 with aryl bromides (eqs 2–3) likely results from differences in reactivity of palladium amino complexes vs. palladium amido complexes.10 As shown in Scheme 1, oxidative addition of the aryl bromide to Pd(0) generates 13, which can bind the carbamate to provide 14. In the presence of a base, amino complex 14 can potentially be converted to amido complex 18. However, with the weak base Cs2CO3 this process should be relatively slow due to the low solubility of Cs2CO3,10 and the equilibrium between 14 and 18 may favor 14. In contrast, when the relatively strong, soluble base NaOtBu is employed, the conversion of 14 to 18 is relatively fast, and the equilibrium favors amido complex 18.10,11
Scheme 1.

The benzocyclobutene products formed when Cs2CO3 is used as base are likely generated via directed carbopalladation12,13 of amino complex 14 to provide the sterically hindered alkylpalladium intermediate 15, which lacks β-hydrogen atoms syn to the metal. Intramolecular aryl C–H bond activation4,14 of 15 provides 16 and an equivalent of HBr, which is neutralized by Cs2CO3 Complex 16 is then converted to benzocyclobutene 17 via C–C bond-forming reductive elimination.4,15 The arylated cyclopentane side product 7 may result from competing protonation of the Pd–C bond(s) of 15 or 16. The conversion of the electron-poor m-bromobenzotrifluoride to a mixture of 11, 8, and 9 is likely due to enhancement of the N–H proton acidity of 14 when the complex bears an electron-withdrawing aryl substituent, which would shift the 14/18 equilibrium toward 18.
Under conditions that facilitate rapid and/or thermodynamically favorable formation of amido complex 18, the reactions likely proceed via syn-amidopalladation as described previously to generate 19,1 which is converted to 8 via C–C bond-forming reductive elimination. Alternatively, 19 can also be transformed to 9 or 10 via β-hydride elimination/reinsertion processes.1a,c
The differences in reactivity observed between substrates bearing terminal alkenes (e.g., 1) and cycloalkene substrate 3 may be due either to the influence of alkene size on the position of the 18/19 equilibrium, the influence of substrate sterics on the rate of C–C bond-forming reductive elimination from 19, or the influence of alkene substitution on the relative rates of alkene insertion into the Pd–C bond of 14 vs. the Pd–N bond of 18. In addition, the fact that Pdcatalyzed reactions of 3 with aryl halides lacking o-hydrogen atoms are converted to azabicyclooctanes (e.g., 12) in the presence of Cs2CO3 suggests that the carbopalladation of 14 is either reversible,16 or very slow with bulky aryl groups. Our current data do not allow us to differentiate between these possibilities.
In conclusion, we have developed a new transformation for the conversion of 3 to cyclopentane-fused benzocyclobutenes via an unprecedented sequence of heteroatom directed carbopalladation followed by intramolecular aryl C–H bond activation. These are the first examples of reactions that exploit directed carbopalladation for the generation and functionalization of alkylpalladium intermediates that lack syn-β-hydrogen atoms. Importantly, the results described above illustrate that differences in reactivity between Pd-amino and Pd-amido complexes can be exploited to allow the construction of strikingly different products from common starting materials. Further studies on the scope of this method and applications of these concepts are currently underway.
Supplementary Material
Experimental procedures and characterization data for new compounds and x-ray crystal structure of 6 (34 pages). This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
The authors thank the NIH-NIGMS (GM–071650) for financial support of this work. Additional support was provided by the Camille and Henry Dreyfus Foundation, Research Corporation, Eli Lilly, Amgen, and 3M.
References
- 1.(a) Ney JE, Wolfe JP. Angew Chem, Int Ed. 2004;43:3605. doi: 10.1002/anie.200460060. [DOI] [PubMed] [Google Scholar]; (b) Bertrand MB, Wolfe JP. Tetrahedron. 2005;61:6447. [Google Scholar]; (c) Ney JE, Wolfe JP. J Am Chem Soc. 2005;127:8644. doi: 10.1021/ja0430346. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Bertrand MB, Wolfe JP. Org Lett. 2006;8:2353. doi: 10.1021/ol0606435. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wolfe JP. Eur J Org Chem. 2007:571. and references cited therein. [PMC free article] [PubMed] [Google Scholar]
- 2.Bertrand MB, Leathen ML, Wolfe JP. Org Lett. 2007;9:457. doi: 10.1021/ol062808f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dpe-Phos = bis(2-diphenylphosphinophenyl)ether.
- 4.For Pd-catalyzed reactions of aryl bromides with norbornene and related bicyclo[2.2.n]alkenes that afford benzocyclobutene products, see: Catellani M, Chiusoli GP, Ricotti S. J Organomet Chem. 1985;296:C11.Catellani M, Chiusoli GP, Ricotti S, Sabini F. Gazz Chim Ital. 1985;115:685.Catellani M, Ferioli L. Synthesis. 1996:769.
- 5.(a) Sadana AK, Saini RK, Billups WE. Chem Rev. 2003;103:1539. doi: 10.1021/cr010022j. [DOI] [PubMed] [Google Scholar]; (b) Mehta G, Kotha S. Tetrahedron. 2001;57:625. [Google Scholar]
- 6.Farona MF. Prog Polym Sci. 1996;21:505. [Google Scholar]
- 7.These structures were assigned by comparison of NMR spectra to those obtained for isolated samples of 5, 12, and a previously described N-aryl analog of 10. See reference 1c.
- 8.The remainder of the mixture consisted of 5% N-arylated substrate and 8% of unidentified products.
- 9.A similar result was obtained with 2-bromo-m-xylene, which afforded a 5-aryl azabicyclo[3.3.0]octane in 30% yield (ca. 40% conversion). See the Supporting Information for complete details.
- 10.Amino complex refers to coordination of the neutral carbamate, whereas amido complex refers to coordination of the deprotonated, anionic carbamate. For discussion of kinetic and thermodynamic effects in amido complex formation, see: Meyers C, Maes BUW, Loones KTJ, Bal G, Lemiere GLF, Dommisse RA. J Org Chem. 2004;69:6010. doi: 10.1021/jo049774e.Driver MS, Hartwig JF. Organometallics. 1997;16:5706.
- 11.When NaOtBu is employed as base the amido complex could also be generated via reaction of 13 with deprotonated carbamate, or through reaction of the carbamate with a LnPd(Ar)(OtBu) complex. See: Shekhar S, Hartwig JF. Organometallics. 2007;26:340. doi: 10.1021/om0607548.
- 12.For early examples, see: Kasahara A, Izumi T, Takeda T, Imamura H. B Chem Soc Jpn. 1974;47:183.Andersson C–M, Larsson J, Hallberg A. J Org Chem. 1990;55:5757.
- 13.For a recent review see: Oestreich M. Eur J Org Chem. 2005:783.
- 14.For recent reviews, see: Campeau L–C, Fagnou K. Chem Commun. 2006:1253. doi: 10.1039/b515481m.Catellani M. Synlett. 2003:298.
- 15.For tandem intermolecular carbopalladation/C–H functionalization processes involving alkenylpalladium intermediates, see: Pinto A, Neuville L, Retailleau P, Zhu J. Org Lett. 2006;8:4927. doi: 10.1021/ol062022h.Furuta T, Asakawa T, Iinuma M, Fujii S, Tanaka K, Kan T. Chem Commun. 2006:3648. doi: 10.1039/b607684j.
- 16.a) Campora J, Gutierrez-Puebla E, Lopez JA, Monge A, Palma P, del Rio D, Carmona E. Angew Chem, Int Ed. 2001;40:3641. doi: 10.1002/1521-3773(20011001)40:19<3641::aid-anie3641>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]; b) Catellani M, Frignani F, Rangoni A. Angew Chem, Int Ed. 1997;36:119. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and characterization data for new compounds and x-ray crystal structure of 6 (34 pages). This material is available free of charge via the Internet at http://pubs.acs.org.