


Abstract
Werner syndrome (WS) is a premature aging disorder caused by mutations in the WS gene (WRN). Although WRN has been suggested to play an important role in DNA metabolic pathways, such as recombination, replication and repair, its precise role still remains to be determined. WRN possesses ATPase, helicase and exonuclease activities. Previous studies have shown that the WRN exonuclease is inhibited in vitro by certain lesions induced by oxidative stress and positioned in the digested strand of the substrate. The presence of the 70/86 Ku heterodimer (Ku), participating in the repair of double-strand breaks (DSBs), alleviates WRN exonuclease blockage imposed by the oxidatively induced DNA lesions. The current study demonstrates that WRN exonuclease is inhibited by several additional oxidized bases, and that Ku stimulates the WRN exonuclease to bypass these lesions. Specific lesions present in the non-digested strand were shown also to inhibit the progression of the WRN exonuclease; however, Ku was not able to stimulate WRN exonuclease to bypass these lesions. Thus, this study considerably broadens the spectrum of lesions which block WRN exonuclease progression, shows a blocking effect of lesions in the non-digested strand, and supports a function for WRN and Ku in a DNA damage processing pathway.
INTRODUCTION
Accumulation of oxidatively induced DNA damage has been implicated in cancer, aging and neurodegenerative diseases (1). The ability to remove oxidatively damaged DNA is essential for maintaining proper cellular functions and genome integrity. Base excision repair (BER) is the predominant pathway responsible for the removal of DNA damage induced by oxidative stress, via the sequential actions of a DNA glycosylase, apurinic/apyrimidinic endonuclease (APE1), DNA polymerase and DNA ligase (2).
A large number of different lesions is produced in DNA by hydroxyl radicals generated by ionizing radiation and metal-ion-catalyzed Fenton reactions (3). Major products of purine oxidation are 7,8-dihydro-8-oxoguanine (8oxo-Gua), 7,8-dihydro-8-oxoadenine (8oxo-Ade) and 2-oxoadenine (2OH-Ade), as well as imidazole ring-opened Fapy lesions, 2,6-diamino-4-hydroxy-5-formamidopyrimidine (Fapy-Gua) and 4,6-diamino-5-formamidopyrimidine (Fapy-Ade). Depending on the reaction conditions, such as the presence or absence of oxygen and presence of reducing agents etc., the proportion between these oxidized purines may vary (4,5). 8oxo-Gua, 8oxo- and 2OH-Ade are characterized by strong miscoding potential and weak blocking of DNA synthesis (6–8). Fapy-Ade and Fapy-Gua moderately block selected DNA polymerases in vitro, and efficiently cause incorporation of non-cognate nucleotides (9–11). However, in E.coli, FapyGua was shown to be strongly mutagenic and was bypassed by replicative DNA polymerase with the efficiency similar to that of 8oxo-Gua (12,13). FapyGua was also mutagenic in simian kidney cells, whereas FapyAde was only weakly mutagenic in these cells (14). Elevated levels of all these lesions have been found in different types of human cancers (3,5,15,16).
The two most common oxidized cytosine derivatives are 5-hydroxycytosine (5OH-Cyt) and 5-hydroxyuracil (5OH-Ura). The levels of oxidized cytosine in DNA after γ-irradiation can be as high as those of 8oxo-Gua (17), and oxidized cytosines have been shown to be highly miscoding. They effectively induce GC→AT transitions, which are the most frequently occurring base substitution mutations in aerobic organisms (18). Increased levels of 5OH-Cyt and 5OH-Ura have been found in several types of cancers (3,5,19).
Another pyrimidine lesion, 5-hydroxymethyluracil (5OHMe-Ura), can be formed from reactive oxygen species (ROS)-damaged thymine, or from oxidized and deaminated 5-methylcytosine, which is often present in CpG islands in promoter regions of the genome (2). Formation of 5OHMe-Ura from 5-methylcytosine could be particularly important, as altered methylation may play a role in the development of cancer and aging (2). Interestingly, reduced levels of 5OHMe-Ura DNA glycosylase were found in cells from patients suffering from the premature aging syndrome Werner Syndrome (WS) (20).
WS is a human autosomal recessive disorder, which is characterized by the premature onset of phenotypical changes associated with normal aging, as well as an increased frequency of cancers (21). The gene responsible for WS encodes a 1432 aa protein, WRN, which belongs to the evolutionary conserved family of RecQ helicases (22). All RecQ helicases possess an ATP-dependent 3′ to 5′ helicase activity (23). In contrast to other human RecQ family members, WRN also possesses a 3′ to 5′ exonuclease activity (22), which shares homology with the human DnaQ family of replicative proofreading exonucleases (24) and RNase D from E. coli (25). Several nucleases have been demonstrated to participate in DNA repair. Although WRN interacts with proteins required for DNA repair, transcription, replication and telomere maintenance (21,23,26), the precise roles of the two enzymatic activities of WRN have still not been fully elucidated. WRN interacts with several oxidatively stress-related factors (27) and in contrast to normal human fibroblasts, WS fibroblasts continue to proliferate after extensive H2O2-induced DNA damage, whereby oxidation-induced lesions accumulate (28).
Cells derived from WS patients display premature replicative senescence, as do cells lacking any of the two subunits of the 70/86 Ku heterodimer (Ku) (29,30). Ku is a component of the DNA-PK complex required for the repair of DNA double-strand breaks (DSBs), via the non-homologous end joining (NHEJ) pathway. In addition to its role in DNA repair, Ku is also involved in transcription, chromatin silencing and telomere biology (31,32). Interestingly, Ku interacts with WRN and selectively stimulates its exonuclease activity on a substrate mimicking a DSB repair intermediate (33,34). Furthermore, interaction with Ku increases the processivity of the WRN exonuclease, allowing digestion of DNA structures, which the WRN exonuclease is not able to digest by itself (33,34). Moreover, studies from our group have shown that in the presence of Ku, WRN exonuclease is able to bypass certain oxidatively induced DNA lesions (35). This interaction has been suggested to play an important role in the processing of 3′ ends of duplex DNA during the repair of strand breaks (35). As both Ku and WRN localize to telomeres (36,37), the interaction between these two proteins may also be significant for telomere maintenance and processing.
It has previously been demonstrated that oxidative stress efficiently induces DNA damage at the telomere (38,39), and it is believed that this damage plays an important role in telomere shortening with age. Since our knowledge about the role of specific DNA lesions in this process is limited, we have here extended our previous studies with a selection of duplex DNA oligonucleotide substrates harbouring a variety of oxidatively induced DNA base lesions, in order to investigate the influence of these lesions on the progression of the WRN exonuclease.
Furthermore, we have made comparisons to human APE1, another prominent human 3′ to 5′ exonuclease, which is a very abundant protein in human cells. APE1 plays an important role in the repair of DNA base lesions via the BER pathway, and we aimed to examine, whether it may play a role as a sensor of oxidative damage proposed previously for WRN exonuclease (40). We found that only the exonuclease activity of WRN, and not of APE1, was significantly inhibited by 5OH-Ura and 5OH-Cyt lesions positioned in the digested strand of the DNA substrate. WRN exonuclease blockage at these lesions was alleviated by the addition of Ku, but the Ku stimulation of WRN exonuclease was not due to stabilization of WRN binding to DNA. Moreover, we observed that oxidatively induced DNA lesions positioned in the non-digested strand, including abasic sites (AP site) and 5OH-Ura, also affected the progression of the WRN exonuclease. In contrast, we did not observe such blockage in the case of APE1. Interestingly, we found that Ku was not able to stimulate WRN bypass of these base lesions in the non-digested strand. These findings provide new insights into the mechanisms of WRN exonuclease activity during DNA metabolic events related to DNA repair and telomere maintenance.
MATERIALS AND METHODS
Proteins
Recombinant His-tagged WRN and Ku protein were purified using an insect cell expression system, as described previously (37,41). Briefly, recombinant human WRN was over-expressed in insect cells and purified by chromatography, using DEAE-Sepharose, Q-Sepharose and Ni-NTA columns. The purity of WRN was checked by SDS-PAGE and Coomassie staining. Human wild-type Ku was purified using Ni-NTA, MonoQ HR5/5 chromatography and native DNA cellulose column, as previously described (41). Ku preparations were later tested for the presence of contaminating nucleic acids (41). Human recombinant APE1 protein was kindly provided by Dr David Wilson (National Institute on Aging, Baltimore) (42,43).
Oligonucleotides
Oligodeoxynucleotides with and without modified bases were purchased from DNA Technology, Aarhus, Denmark, except the oligonucleotide fragments containing the Fapy-G and Fapy-A lesions (G-16-mer and A-30-mer), which were kindly provided by Prof. Dr Marc M. Greenberg (Department of Chemistry, Johns Hopkins University, Baltimore, MD, USA). Oligonucleotide sequences are presented in Tables 1–3.
Table 1.
Oligonucleotides used to generate double stranded 3′-recessed DNA substrates
 |
Table 2.
Oligonucleotides used to generate double stranded forked DNA substrates
 |
Table 3.
Oligonucleotides used to generate double stranded forked DNA substrates containing Fapy lesions
 |
Labelling of oligonucleotides
Oligonucleotides (10 mM) were purified employing preparative gel electrophoresis and 20 pmol oligonucleotide was then radiolabelled in reaction buffer containing 1 × PNK reaction buffer (50 mM Tris-HCl (pH 7.6 at 25°C), 10 mM MgCl2, 5 mM DTT, 0.1 mM spermidine and 0.1 mM EDTA; Fermentas), 2 μl [γ-32P]-ATP (160 μCi/μl, Life Science, Denmark) and 2 μl PNK (10 U/μl, Fermentas). The reaction was incubated for 90 min at 37°C. Subsequently, the products were purified on a G25 Sephadex column (Molecular Imaging, Amersham). Ten picomoles of the radiolabelled strand was then duplexed with an equal amount of the complementary non-labelled strand, to a final concentration of 200 fmol/μl (200 μM). Radiolabelled oligonucleotides were stored at 4°C.
In some cases, radiolabelled duplexed oligonucleotides were further purified. Briefly, radiolabelled oligonucleotide (20 pmol) was incubated overnight with a 5-fold excess of the unlabelled strand. The duplexed oligonucleotides were separated from the single-stranded DNA by means of gel electrophoresis and cut out from the gel. The labelled duplex was eluted from the gel, ethanol precipitated, resuspended in a final volume of 20 μl H2O and stored at 4°C.
Labelling and purification of oligonucleotides containing Fapy-Gua and Fapy-Ade lesions
Oligodeoxynucleotides containing Fapy lesion (kindly provided by Dr M. Greenberg) were too short (FapyGua—16-mer, FapyAde—30-mer) for the construction of the WRN substrate. Therefore, these fragments were elongated by ligation with oligodeoxynucleotides of appropriate lengths (5′ end fragments: G—33-mer, A—24-mer), which were complementary to undamaged bottom strand (49-mer for oligo-containing Fapy-G or 54-mer for oligo-containing Fapy-A). The formed substrates were forked and had a similar duplex length to the substrates used in the previous assays. As a control, the same substrates were prepared, however without Fapy lesions (Table 3). Prior to ligation, 20 pmol of the G-16-mer or A-30-mer (undamaged or containing Fapy lesions, respectively), was phosphorylated in reaction buffer containing 1 × PNK buffer (Fermentas), 0,1 mM ATP and 2 μl PNK (10 U/μl, Fermentas). The reaction was incubated for 90 min at 37°C. In parallel, 30 pmol of the G-33-mer or A-24-mer oligos were radiolabelled for 90 min at 37°C, in reaction buffer containing 1 × PNK buffer (Fermentas), 2 μl [γ-32P]-ATP (160 μCi/μl, Life Science, Denmark) and 2 μl PNK (10 U/μl, Fermentas). After phosphorylation, DNA fragments were purified on a G25 Sephadex column (Molecular Imaging, Amersham). Fapy-containing substrates were constructed by mixing 20 pmol of the phosphorylated G-16-mer or A-24-mer, 30 pmol of the radiolabelled G-33-mer or A-30-mer with 60 pmol of the respective cold bottom strand oligo (G-49-mer or A-54-mer) in reaction buffer containing 1 × ligase buffer (New England Biolabs, UK). The oligos were annealed by heating 5 min at 80°C, and allowed to cool down to room temperature. After annealing, the top strand fragments were ligated by adding 500 U of the T4 DNA ligase (New England Biolabs, UK) and ATP to a final concentration of 1 mM, and incubating for 2 h at 16°C. Subsequently, 30 μl of FA buffer (80% formamide, 10 mM EDTA pH 8, 1 mg/ml xylene cyanol and 1 mg/ml bromophenol blue) was added to the oligo mixture, the mixture was denatured 5 min at 80°C, and the ligated top strand of the substrate was separated from the non-ligated top strand fragments by means of denaturing PAGE. The full-length oligonucleotide was cut out of the gel and eluted. After ethanol purification, the DNA was resuspended in 9–20 μl of TE buffer, depending on the yield of purified DNA, and stored at 4°C.
WRN exonuclease assays
For assays with forked substrates, radiolabelled DNA substrate (14 or 21 fmol for Fapy oligos) was diluted in 1 × exonuclease reaction buffer (40 mM Tris HCl pH 8; 4 mM MgCl2; 5 mM DTT and 1 μg/ml BSA). Reactions were initiated by the addition of WRN (0, 10, 20, 40, 60 fmol), incubated for 15 min at 37°C, and terminated with 10 μl FA buffer. Reactions were heat-denatured for 5 min at 90°C and reaction products were separated on a 14% acrylamide denaturing gel. The gels were dried and exposed to a phosphorimager screen. For reactions containing Ku, recombinant Ku (7.5 fmol) was added to the reaction prior to initiation of the reaction with WRN.
For assays with recessed substrates, the reaction buffer was supplemented with 2 mM ATP. Reactions were incubated with WRN (0–240 fmol), in the presence or absence of 128 fmol Ku, and incubated for 1 h at 37°C. Reactions were terminated by the addition of 10 μl FA buffer. Reaction products were separated and visualized as described above.
Quantification of WRN exonuclease blockage
For each concentration of WRN, the extent of bypass was quantified by measuring the intensity of all bands below the site of damage, relative to the total intensity of the lane. For comparison between experiments, bypass of the undamaged substrate was set to 1. Average values are based on a minimum of 3 experiments for each substrate.
APE1 exonuclease assay
Radiolabelled DNA substrate (13 fmol) was incubated in exonuclease reaction buffer supplemented with 100 fmol of appropriate unlabelled double-stranded DNA. Reactions were initiated by the addition of recombinant APE1 (0–50 fmol), incubated for 15 min at 37°C, and terminated with 10 μl FA buffer. Reaction products were separated and visualized as described above. For reactions containing Ku, the reaction buffer did not contain unlabelled double-stranded DNA and Ku (7.5–45 fmol) was added prior to APE1.
RESULTS
We have previously shown that certain modified nucleotides, as well as abasic sites and bulky adducts, such as cholesterol, located in the degraded strand of the DNA substrate, severely inhibit the progression of the WRN exonuclease (40). In addition, 8oxo-Ade and 8oxo-Gua, two of the most frequent free-radical-induced purine lesions, efficiently block WRN exonuclease on a 3′ recessed substrate (40). In this report, we investigated the influence of additional ROS-induced lesions on WRN exonuclease activity. First, we determined whether WRN exonuclease was affected by oxidation products of pyrimidines, such as 5OH-Cyt and 5OH-Ura. We introduced the lesions into the recessed strand of the previously used DNA substrate, as shown in Table 1, and measured the degradation of this strand by WRN.
In the presence of increasing amounts of WRN, the undamaged substrate was digested in proportion with the WRN concentration (Figure 1A). In contrast, progression of WRN exonuclease activity was inhibited on substrates containing 5OH-Ura and 5OH-Cyt. The blockage of WRN exonuclease by these lesions was not relieved by increasing the concentration of WRN, suggesting, that similar to the situation for 8oxo-Gua and 8oxo-Ade, the 5OH-Cyt and 5OH-Ura lesions impose a steric hindrance for WRN. For both lesions, the digestion was inhibited one nucleotide 3′ to the lesion (Figure 1A). This is in agreement with previous reports on the inhibition of WRN exonuclease by 8oxo-Gua and 8oxo-Ade (40).
Figure 1.


WRN exonuclease activity on substrates containing 5OH-Ura and 5OH-Cyt in the digested strand of the substrate. (A) WRN exonuclease blockage on recessed substrates. Recessed substrates (15 fmol) containing undamaged (left panel) or 5OH-Cyt (middle panel) and 5OH-Ura (right panel) in the digested strand were incubated in the absence or presence of WRN (0, 120 or 240 fmol) as indicated, for 1 h at 37°C. Reaction products were separated on a 14% polyacrylamide denaturing gel and visualized by phosphorimaging. (B–D) WRN exonuclease blockage on forked substrates. Unmodified or modified DNA substrates (B,C—14 fmol, D—21 fmol), containing lesions in the digested strand of the substrate were incubated with increasing amounts of WRN (10, 20, 40, 60 fmol) for 15 min at 37°C. Reaction products were separated and visualized as described in (A). Positions of the lesions are indicated by arrows. Und, undamaged; 8oxo-Ade, 8-oxoadenine; 8oxo-Gua, 8-oxoguanine; 5OH-Cyt, 5-hydroxycytosine; 5OH-Ura, 5-hydroxyuracil; 5OHMe-Ura, 5-hydroxymethyluracil; 2OH-Ade, 2-hydroxyadenine, Fapy-Gua, 2,6-diamino-4-hydroxy-5-formamidopyrimidine; Fapy-Ade, 4,6-diamino-5-formamidopyrimidine.
Next, we investigated whether blockage of WRN exonuclease activity by oxidatively induced DNA lesions was structure specific. To address this, we used a forked substrate containing a duplex fragment similar in length to the recessed substrates used above (Tables 2 and 3). As forked duplexes can be processed by both WRN exonuclease and helicase activities (44), we omitted ATP from the reactions to focus solely on the exonuclease products generated by WRN acting on the blunt end of this substrate.
Exonuclease assays with forked substrates containing 8oxo-Ade and 8oxo-Gua in the digested strand are shown in Figure 1B. In agreement with previous findings using recessed substrates (40), we observed a dramatic blockage of WRN exonuclease activity by 8oxo-Gua and 8oxo-Ade lesions in the forked duplexes. Almost no digested products were observed beyond the position of the lesion and the blockage occurred one nucleotide 3′ to the lesion (Figure 1B). The exonuclease activity of WRN on forked substrates containing 5OH-Ura, 5OH-Cyt, 5OHMe-Ura and 2OH-Ade is presented in Figure 1C. When compared to 8oxo-Gua and 8oxo-Ade, 5OH-Ura and 5OH-Cyt blocked WRN exonuclease activity less effectively, as some exonuclease products were observed beyond the position of the lesions in the substrate (Figure 1C). In contrast to the lesions tested thus far, 5OHMe-Ura and 2OH-Ade did not significantly block progression of the WRN exonuclease, even at the lowest WRN concentrations tested, but caused a slight pausing of WRN exonuclease (Figure 1C).
Next, we investigated the effects of Fapy-Gua or Fapy-Ade lesions on the progression of WRN exonuclease. Similar to the exonuclease assays with 8oxo-Gua and 8oxo-Ade, the Fapy-Gua lesion effectively blocked WRN exonuclease (Figure 1D), as only very few digested products were observed beyond the position of the lesion. The Fapy-Ade lesion also effectively blocked the progression of WRN exonuclease. As observed for other substrates, the blockage of WRN exonuclease occurred 3′ to the damaged nucleotide (Figure 1D).
The degree of WRN exonuclease blockage was quantified for each damaged substrate. Thus, the amount of digestion beyond the lesion was compared to the corresponding position in the undamaged substrate, at 40 fmol of WRN for substrates shown in Figure 1B and C, and at the identical molar ratio of enzyme to substrate for the Fapy-containing oligonucleotides (60 fmol WRN). The bypass in the undamaged substrate was set to 100%, and the relative blockage of the digestion for damaged oligonucleotides was calculated. The most potent inhibitors of WRN exonuclease progression were Fapy-Gua, Fapy-Ade, 8oxo-Gua and 8oxo-Ade where the WRN bypass was reduced by approximately 70% or more, when compared to the unmodified substrate (Table 4). 5OH-Ura and 5OH-Cyt blocked the WRN exonuclease to a lesser degree (40–50% reduction in WRN bypass), allowing a moderate degree of bypass in both substrates. The weakest blockage was observed with 5OHMe-Ura, which had almost no influence on the progression of the WRN exonuclease on the forked substrate.
Table 4.
Blockage of WRN exonuclease progression by oxidative lesions in the digested strand
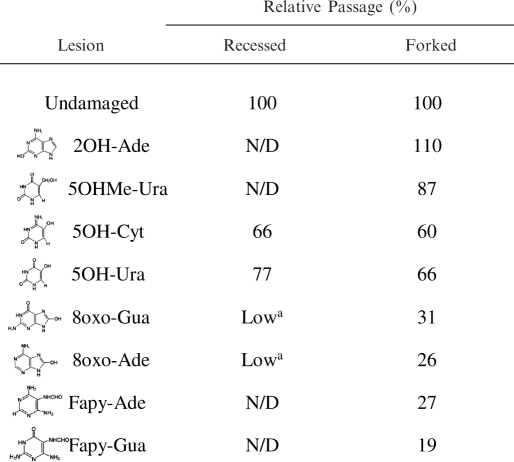 |
aBased on Machwe et al., Nucleic Acids Res., 2000.
N/D: not determined.
We have previously shown that Ku interacts with WRN and stimulates its exonuclease activity (34,45). Inhibition of the WRN exonuclease by 8oxo-Ade and 8oxo-Gua could be alleviated by the addition of Ku (35). Therefore, we next investigated whether Ku could also stimulate WRN digestion of substrates containing 5OH-Ura and 5OH-Cyt lesions.
As demonstrated above, WRN digestion of the undamaged substrate was proportional to the concentration of WRN, while in the presence of 5OH-Ura and 5OH-Cyt, WRN exonuclease progression was efficient up to, but not beyond, the modified nucleotides (Figure 2A). Ku efficiently stimulated the 3′ to 5′ exonuclease activity of WRN both on the undamaged and damaged recessed substrates. On substrates containing 5OH-Cyt and 5OH-Ura, the addition of Ku enhanced the exonuclease activity of WRN and allowed WRN exonuclease to digest beyond the position of the lesion (Figure 2A, middle and right panel).
Figure 2.

Ku stimulation of WRN exonuclease on substrates containing 5OH-Ura and 5OH-Cyt in the digested strand of the substrate. (A) Ku stimulation of WRN exonuclease past lesions positioned in recessed substrates. Undamaged recessed substrate (15 fmol) (left panel or recessed substrate containing 5OH-Cyt or 5OH-Ura in the digested strand (middle and right panel, respectively), were incubated with the indicated amounts of WRN protein, in the absence or presence of Ku (128 fmol) for 1 h at 37°C. (B) Ku stimulation of WRN exonuclease activity on forked substrates. Forked DNA substrates (10 fmol, undamaged or containing a single lesion in the digested strand) were incubated for 15 min at 37°C with WRN (20 fmol), in the absence or presence of Ku (7.5 fmol). Reactions were terminated and reaction products were processed as described for Figure 1. The positions of the lesions are indicated by the arrows. Und, undamaged substrate; 5OH-Cyt, 5-hydroxycytosine; 5OH-Ura, 5-hydroxyuracil, 8oxo-Gua, 8-oxoguanine.
Similar to the results with the recessed substrates, the addition of Ku resulted in significant stimulation of WRN exonuclease activity on forked substrates containing 5OH-Ura and 5OH-Cyt (Figure 2B). Again, we observed that WRN alone extensively digested the undamaged substrate, while on the damaged substrates the progression of WRN exonuclease was limited to the first few nucleotides prior to the DNA modification (5OH-Ura, 5OH-Cyt or 8oxo-Gua). The addition of Ku facilitated WRN digestion of the substrates containing 5OH-Cyt, 5OH-Ura and 8oxo-Gua, however pausing 3′ to the site of the lesion was still observed (Figure 2B). Thus, Ku also stimulated WRN exonuclease to bypass the oxidatively induced lesions 5OH-Ura and 5OH-Cyt in the context of forked duplex substrates. In summary, the processivity of WRN exonuclease activity is stimulated by Ku, whether or not a lesion is present. Ku alone did not exhibit any exonuclease activity on the substrate (Figure 2B).
Previous studies showed that Ku allows the digestion of certain DNA structures that cannot be digested by WRN alone (33,34). In addition, DNA-binding studies have shown that Ku efficiently recruits WRN to DNA (45,46), suggesting that the mechanism of Ku stimulation may involve stabilization of WRN binding to DNA. However, trapping assays with WRN in the presence or absence of Ku failed to confirm these suggestions (data not shown).
Blockage of WRN exonuclease at oxidatively induced lesions has previously been proposed to serve as a DNA damage sensor (40), and therefore, we next addressed whether this could be carried out by other human 3′ to 5′ exonucleases, such as APE1. APE1 is an abundant protein in human cells, and the major protein processing abasic sites (AP sites) during BER, which is the predominant pathway for removing oxidatively modified bases from DNA (47). Apart from its significant AP-endonuclease activity, APE1 also possesses a 3′ to 5′ exonuclease activity (47). In contrast to WRN, the APE1 exonuclease was not found to be blocked by 8oxo-Gua and 8oxo-Ade lesions in forked duplexes, but readily digested through the damaged nucleotides (Figure 3A). 5OH-Cyt and 5OH-Ura resulted in some pausing of APE1 exonuclease progression, and bands of higher intensities were visible at the positions of these lesions (Figure 3A). However, the intensity of the bands beyond the position of the lesion indicated that even if the APE1 exonuclease paused at these sites, it was still able to digest through these modifications. Similar to the results obtained with the WRN exonuclease, no inhibition of APE1 exonuclease activity was observed using substrates containing 5OHMe-Ura and 2OH-Ade (Figure 3A).
Figure 3.

APE1 exonuclease activity on substrates containing oxidative lesions in the digested strand of the substrate. (A) Forked substrates (14 fmol, undamaged or containing a single lesion in the digested strand) were incubated with APE1 (50 fmol) for 15 min at 37°C. (B) Forked substrates (5 fmol, undamaged or containing 5OH-Ura) were incubated for 15 min at 37°C with APE1 (140 fmol) and Ku (45 fmol) as indicated. Positions of the lesions are indicated by arrows. Reactions were terminated and reaction products were processed as described for Figure 1. Und, undamaged; 8oxo-Ade, 8-oxoadenine; 8oxo-Gua, 8-oxoguanine; 5OH-Cyt, 5-hydroxycytosine; 5OH-Ura, 5-hydroxyuracil; 5OHMe-Ura, 5-hydroxymethyluracil; 2OH-Ade, 2-hydroxyadenine.
We next investigated the effect of Ku on APE1 exonuclease activity. For both the undamaged substrate, and the substrate containing a 5OH-Ura lesion, no difference in the degree of digestion was observed between lanes containing APE1 only, or APE1 and Ku (Figure 3B). Thus, Ku did not stimulate APE1 exonuclease activity, as it did for WRN, demonstrating the specificity of the Ku stimulation of WRN exonuclease activity.
After investigating the effect of ROS-induced lesions located in the digested strand of the DNA substrate, we next asked whether WRN exonuclease activity was affected by the presence of lesions in the non-digested strand of the substrate. We used a forked duplex substrate with a single 8oxo-Gua, 5OH-Ura or abasic site (AP site) located in the non-digested strand (Table 2). The digestion pattern of WRN on a substrate containing 8oxo-Gua in the non-digested strand was similar to the digestion pattern observed with the undamaged substrate (Figure 4A). Quantification of the digestion (Table 5) confirmed that 8oxo-Gua in the non-digested strand did not have any influence on the progression of the WRN exonuclease. In contrast to 8oxo-Gua, WRN digestion of a substrate containing an AP site resulted in pausing 1 nucleotide before the DNA modification (Figure 4A, lesion indicated by an arrow), suggesting that an AP site in the non-digested strand of the substrate impaired the progression of the WRN exonuclease. The blockage was not robust, as WRN was able to digest beyond the position of the lesion (Figure 4A), without a significant decrease in the amount of total digested products (Table 5). Similar to results observed for the lesion positioned in the digested strand of the substrate (40), the position of the intense band was 3′ of the modification.
Figure 4.

WRN exonuclease activity in the presence of lesions positioned in the non-digested strand of the substrates. (A) Forked substrates (20 fmol, undamaged or containing a single lesion in the non-digested strand) were incubated for 15 min at 37°C with increasing amounts of WRN (10, 20, 40, 60 fmol). (B) Forked substrates (20 fmol, undamaged or containing a single lesion in the non-digested strand) were incubated for 15 min at 37°C without or with WRN (20 fmol) and with Ku (7.5 fmol) as indicated. Reactions were terminated and reaction products were processed as described for Figure 1. Positions of the lesions are indicated by arrows. Und, undamaged; 8oxo-Gua, 8-oxoguanine; AP site, abasic site; 5OH-Ura, 5-hydroxyuracil.
Table 5.
Blockage of WRN exonuclease progression by oxidative lesions in the non-digested strand
| Lesion | Relative passage (%) |
|---|---|
| Undamaged | 100 |
| 8oxo-Gua | 102 |
| AP site | 87 |
| 50H-Ura | 67 |
On substrates containing 5OH-Ura in the non-digested strand, the WRN exonuclease digestion pattern produced intense bands 3′ to the lesion, indicating some inhibition of the WRN exonuclease prior to the lesion (Figure 4A). Bypass of this lesion was decreased to a moderate degree (approximately 28% reduction in bypass), compared to the undamaged substrate (Table 5).
Thus, we show here for the first time, that certain base lesions such as an abasic site and 5OH-Ura, positioned in the non-digested strand of the DNA substrate, may retard WRN exonuclease activity. In contrast, other lesions, such as 8oxo-Gua, positioned in the non-digested strand, did not influence the progression of the WRN exonuclease.
Having established the influence of modified nucleotides in the non-digested strand on the exonuclease activity of WRN, we addressed whether Ku could stimulate WRN bypass of an AP-site or 5OH-Ura lesion located in the non-digested strand. For substrates containing an AP site and 5OH-Ura, the intensities of the bands surrounding each lesion were similar both in the absence and the presence of Ku (Figure 4B, note arrows), suggesting that in contrast to the results obtained with lesions positioned in the digested strand of the substrate, Ku does not stimulate WRN exonuclease activity (i.e. reduce stalling or pausing) on substrates containing lesions in the non-digested strand.
DISCUSSION
In this report, we investigated WRN exonuclease activity on two structure-specific substrates, each containing one of several different free-radical-induced lesions positioned in the digested strand. We found that Fapy-Gua, Fapy-Ade, 8oxo-Gua and 8oxo-Ade dramatically blocked WRN exonuclease activity, while 5OH-Ura and 5OH-Cyt inhibited WRN to a moderate degree, and 5OHMe-Ura and 2OH-Ade only had a minor effect. We included three different substrates to investigate whether sequence context or structure affected the degree of blockage by the oxidatively induced lesions, and we observed similar effects regardless of the sequence or the structure of the substrate (Table 4). As the recessed and forked substrates resembled intermediates of different pathways of DNA metabolism (40,44), our results suggest that the progression of the WRN exonuclease may be blocked by ROS-related DNA damage during processes such as DNA repair, replication, recombination and/or telomere maintenance.
In general, the greatest inhibition of WRN exonuclease progression was seen with lesions which were derivatives of purines, with a hydroxyl group close to the edge of the major groove of DNA (Fapy lesions and 8-oxopurines) (48). In contrast, pyrimidines with a C5 hydroxyl group, positioned more centrally in the major groove, like 5OH-Ura and 5OH-Cyt, had less effect on WRN exonuclease.
Our results suggest that the presence of a hydroxyl group precisely oriented in the major groove of the DNA helix is essential for the ability of lesions to block WRN, as lesions having the hydroxyl groups facing the minor groove of DNA (2OH-Ade, 5OHMe-Ura) (49,50) had very little effect on the progression of WRN exonuclease (Table 4). Under our experimental conditions, the hydroxyl groups at all oxidized positions of DNA lesions remained predominantly in the oxo form (51–55), suggesting that the difference in the blockage between purine and pyrimidine derivatives was not related to tautomerism of the additional hydroxyl groups.
In our study, Fapy lesions are not imposing more blockage on WRN exonuclease, than 8oxo-purines. This could appear surprising, considering the lack of the planar structure of imidazole ring in Fapy lesions and the reported moderate blockage of some DNA polymerases in vitro (9,11). However, it might be explained by the ability of Fapy lesions to adopt multiple rotameric/tautomeric forms, with an oxo group at the C8 position, and the preferential rotameric form of Fapy resembling a two-ring purine structure, with the stacking between the relevant adjacent base pairs similar to that of 8-oxopurines situated in the same sequence context (51,52). Due to the presence of a carbonyl group at C8, and flexibility of the imidazole fragment, properties of Fapy lesions could resemble 8oxo-Gua, and thus block WRN exonuclease similar to 8oxo-Gua. Comparable miscoding properties of Fapy-Gua and 8oxo-Gua (14), and similar recognition by 8oxo-Gua glycosylases (48,56) further support this suggestion.
Interestingly, 8oxo-Gua and 8oxo-Ade, which are some of the most efficient inhibitors of WRN exonuclease progression, have been observed to accumulate with age in normal individuals (1,15), and in cells from prematurely aged WS patients (57), indicating that WRN may play a role in the detection and/or removal of these lesions. Furthermore, the WRN exonuclease was also blocked by 5OH-Ura and 5OH-Cyt, which are among the most frequent derivatives of cytosine, and induce C to T transitions with very high frequencies (18).
The distinct inhibition of WRN exonuclease progression by base lesions containing a hydroxyl group suggests that the inhibition may be due to spatial constraints in the active site of the WRN exonuclease, which might then impair the catalysis process. An alternative mechanism by which modified nucleotides could inhibit the WRN exonuclease, may be a previously suggested destabilization of the double-stranded DNA in the proximity of the lesion (35). As WRN exonuclease is not able to digest single-stranded DNA (58,59), such changes induced by DNA modifications would greatly influence the progression of WRN. However, since abasic sites and bulky adducts have a stronger effect on the stability of the DNA helix compared to minor base modifications such as 8oxo-Gua and 5OH-Ura (47,60), this putative mechanism of inhibition seems unlikely. It remains unclear, how a non-distorting DNA lesion, such as 5-OHU in the non-digested strand, exerts more of an effect than a missing base.
In contrast to the results obtained with the WRN exonuclease, APE1 exonuclease activity was only moderately influenced by the presence of oxidatively-induced lesions in the digested strand of the substrate. The APE1 exonuclease only paused slightly at 5OH-Ura and 5OH-Cyt lesions, and was not affected by 8oxo-Gua or 8oxo-Ade, which efficiently blocked the WRN exonuclease. The differential effect of lesions on APE1 and WRN exonuclease most likely reflects differences in the structure of the active sites of the proteins and their modes of DNA binding (25,61). Our results suggest that recognition of damaged DNA is likely to be a unique feature of the WRN exonuclease and not a general characteristic of all human 3′ to 5′ exonucleases, which is in agreement with the recent structural data (24), and the previously suggested role for WRN as a sensor of DNA damaged bases (40).
Many processes of DNA metabolism on one of the two strands of double-stranded DNA, involve recognition of the opposite strand for proper execution. One example is the single strand cleavage of a DNA substrate by topoisomerase I, where the cleavage on one strand is affected by the presence of abasic sites or G methylation on the opposite strand (62). Furthermore, the interaction of E.coli formamidopyrimidine-DNA glycosylase (Fpg) and human 8-oxoguanine-DNA glycosylase (hOgg1) with DNA containing a single 8oxo-Gua in one strand, involves contacting both strands of the DNA duplex (63). In addition, the yeast XPC orthologue, Rad4, recognizes the undamaged strand of the substrate during the repair of cyclobutane pyrimidine dimers (64). As the WRN exonuclease has been proposed to function in the recognition of damaged DNA bases (40), we investigated whether WRN progression was also influenced by lesions located in the non-digested strand of the substrate. Here, we show for the first time, that digestion by the WRN exonuclease is influenced by lesions located in the non-digested strand, and we demonstrate that 5OH-Ura and an AP site positioned in the non-digested strand decrease WRN degradation of the digested strand (Table 4). Our results suggest that WRN interacts with the non-digested strand when degrading the opposite strand, consistent with the ring model of WRN exonuclease proposed by Perry et al. (24), where both DNA strands appear to contact the exonuclease domain. Furthermore, the mouse WRN exonuclease appears to work as a multimer (65). As there is no information currently available regarding the structure of WRN exonuclease binding longer double-stranded DNA substrates, more studies are required to fully explain our results.
It was previously shown that Ku stimulates WRN on substrates containing the oxidatively induced base lesions 8oxo-Gua and 8oxo-Ade, but not cholesterol or abasic sites (35). We now present evidence that Ku promotes WRN bypass of other free-radical-induced lesions as well, including 5OH-Ura and 5OH-Cyt (Figure 2). Our results extend the spectrum of lesions on which Ku can stimulate WRN exonuclease and confirm that the stimulation by Ku is a phenomenon unique to WRN exonuclease, as no effects of Ku were observed on the exonuclease activity of APE1. In addition, we demonstrate that Ku is not able to stimulate the WRN exonuclease to bypass oxidation-induced lesions which are present in the non-digested strand. Results by Tainer and co-workers (24) suggest that Ku may provide a suitable DNA orientation for the WRN exonuclease, but this does not explain the difference in the effect on the digested versus non-digested strand.
The genomic instability and cellular senescence exhibited by cells deficient in WRN or Ku, as well as similarities in the phenotypes of WRN and Ku80 knockout mice (29,30), suggest that these proteins function in a common DNA metabolic pathway. Ku and WRN are likely to participate together in the repair of DSBs via the non-homologous end joining (NHEJ) pathway, and cooperatively process the 3′ ends at the sites of DNA breaks (35). This interaction could be even more important in the context of DSBs induced by ionizing radiation and radiomimetic drugs, which are known to generate a considerable amount of oxidatively induced base lesions in addition to DNA strand breaks (66).
Due to the observation that both proteins also localize to telomeres (36,37), the Ku–WRN interaction has been suggested to play an important role in telomere metabolism, possibly in opening the telomeric D-loop during processes of DNA replication and repair (37). It is also likely that both proteins are involved in the repair of DSBs in that region. The G-rich regions of telomeres are particularly susceptible to oxidative damage (38,67), and may contribute to accelerated telomere shortening. As short telomeres are related to aging, the ability of Ku to stimulate the WRN exonuclease to bypass oxidative stress-induced lesions is likely important during DNA repair events and further supports the essential role of WRN and Ku in the maintenance of genome stability.
ACKNOWLEDGEMENTS
We thank Susanne Trillingsgaard Venø for help with performing the assays with Fapy lesions. This work was supported in part by the National Institute of Health/National Institute on Aging Intramural Program. It was furthermore supported by the European Union Research Training Network programme (contract no. HPRN-CT-2002-00240), as well as the Danish Research Council (22-03-0253), the Danish Cancer Society (DP05024), The NovoNordisk Foundation, and The Danish Aging Research Center. Funding to pay the Open Access publication charges for this article was provided by Danish Cancer Society.
Conflict of interest statement. None declared.
REFERENCES
- 1.Ames BN. Endogenous DNA damage as related to cancer and aging. Mutat. Res. 1989;214:41–46. doi: 10.1016/0027-5107(89)90196-6. [DOI] [PubMed] [Google Scholar]
- 2.Kow YW. Repair of deaminated bases in DNA. Free Radic. Biol. Med. 2002;33:886–893. doi: 10.1016/s0891-5849(02)00902-4. [DOI] [PubMed] [Google Scholar]
- 3.Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 2003;17:1195–1214. doi: 10.1096/fj.02-0752rev. [DOI] [PubMed] [Google Scholar]
- 4.Spencer JP, Jenner A, Aruoma OI, Cross CE, Wu R, Halliwell B. Oxidative DNA damage in human respiratory tract epithelial cells. Time course in relation to DNA strand breakage. Biochem. Biophys. Res. Commun. 1996;224:17–22. doi: 10.1006/bbrc.1996.0977. [DOI] [PubMed] [Google Scholar]
- 5.Olinski R, Zastawny T, Budzbon J, Skokowski J, Zegarski W, Dizdaroglu M. DNA base modifications in chromatin of human cancerous tissues. FEBS Lett. 1992;309:193–198. doi: 10.1016/0014-5793(92)81093-2. [DOI] [PubMed] [Google Scholar]
- 6.Watanabe T, van Geldorp G, Najrana T, Yamamura E, Nunoshiba T, Yamamoto K. Miscoding and misincorporation of 8-oxo-guanine during leading and lagging strand synthesis in Escherichia coli. Mol. Gen. Genet. 2001;264:836–841. doi: 10.1007/s004380000373. [DOI] [PubMed] [Google Scholar]
- 7.Tudek B, Laval J, Boiteux S. SOS-independent mutagenesis in lacZ induced by methylene blue plus visible light. Mol. Gen. Genet. 1993;236:433–439. doi: 10.1007/BF00277144. [DOI] [PubMed] [Google Scholar]
- 8.Kamiya H. Mutagenic potentials of damaged nucleic acids produced by reactive oxygen/nitrogen species: approaches using synthetic oligonucleotides and nucleotides: survey and summary. Nucleic Acids Res. 2003;31:517–531. doi: 10.1093/nar/gkg137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Graziewicz MA, Zastawny TH, Olinski R, Speina E, Siedlecki J, Tudek B. Fapyadenine is a moderately efficient chain terminator for prokaryotic DNA polymerases. Free Rad. Biol. Med. 2000;28:75–83. doi: 10.1016/s0891-5849(99)00208-7. [DOI] [PubMed] [Google Scholar]
- 10.Wiederholt CJ, Greenberg MM. Fapy.dG instructs Klenow exo(-) to misincorporate deoxyadenosine. J. Am. Chem. Soc. 2002;124:7278–7279. doi: 10.1021/ja026522r. [DOI] [PubMed] [Google Scholar]
- 11.Weledji YN, Wiederholt CJ, Delaney MO, Greenberg MM. DNA polymerase bypass in vitro and in E. coli of a C-nucleotide analogue of Fapy-dG. Bioorg. Med. Chem. 2008;16:4029–4034. doi: 10.1016/j.bmc.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patro JN, Wiederholt CJ, Jiang YL, Delaney JC, Essigmann JM, Greenberg MM. Studies on the replication of the ring opened formamidopyrimidine, Fapy.dG in Escherichia coli. Biochemistry. 2007;46:10202–10212. doi: 10.1021/bi700628c. [DOI] [PubMed] [Google Scholar]
- 13.Graziewicz MA, Zastawny TH, Olinski R, Tudek B. SOS-dependent A→G transitions induced by hydroxyl radical generating system hypoxanthine/xanthine oxidase/Fe3+/EDTA are accompanied by the increase of Fapy-adenine content in M13 mp18 phage DNA. Mutat. Res. 1999;434:41–52. doi: 10.1016/s0921-8777(99)00012-9. [DOI] [PubMed] [Google Scholar]
- 14.Kalam MA, Haraguchi K, Chandani S, Loechler EL, Moriya M, Greenberg MM, Basu AK. Genetic effects of oxidative DNA damages: comparative mutagenesis of the imidazole ring-opened formamidopyrimidines (Fapy lesions) and 8-oxo-purines in simian kidney cells. Nucleic Acids Res. 2006;34:2305–2315. doi: 10.1093/nar/gkl099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang YJ, Ho YS, Lo MJ, Lin JK. Oxidative modification of DNA bases in rat liver and lung during chemical carcinogenesis and aging. Chem. Biol. Interact. 1995;94:135–145. doi: 10.1016/0009-2797(94)03327-5. [DOI] [PubMed] [Google Scholar]
- 16.Fraga CG, Shigenaga MK, Park JW, Degan P, Ames BN. Oxidative damage to DNA during aging: 8-hydroxy-2′-deoxyguanosine in rat organ DNA and urine. Proc. Natl Acad. Sci. USA. 1990;87:4533–4537. doi: 10.1073/pnas.87.12.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burcham PC. Internal hazards: baseline DNA damage by endogenous products of normal metabolism. Mutat. Res./Genetic Toxicol. Environ. Mutagen. 1999;443:11–36. doi: 10.1016/s1383-5742(99)00008-3. [DOI] [PubMed] [Google Scholar]
- 18.Kreutzer DA, Essigmann JM. Oxidized, deaminated cytosines are a source of C→T transitions in vivo. Proc. Natl Acad. Sci. USA. 1998;95:3578–3582. doi: 10.1073/pnas.95.7.3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wallace SS. Biological consequences of free radical-damaged DNA bases. Free Rad. Biol. Med. 2002;33:1–14. doi: 10.1016/s0891-5849(02)00827-4. [DOI] [PubMed] [Google Scholar]
- 20.Ganguly T, Duker NJ. Reduced 5-hydroxymethyluracil-DNA glycosylase activity in Werner's syndrome cells. Mutat. Res. 1992;275:87–96. doi: 10.1016/0921-8734(92)90012-e. [DOI] [PubMed] [Google Scholar]
- 21.Opresko PL, Cheng WH, von Kobbe C, Harrigan JA, Bohr VA. Werner syndrome and the function of the Werner protein; what they can teach us about the molecular aging process. Carcinogenesis. 2003;24:791–802. doi: 10.1093/carcin/bgg034. [DOI] [PubMed] [Google Scholar]
- 22.Huang S, Beresten S, Li B, Oshima J, Ellis NA, Campisi J. Characterization of the human and mouse WRN 3′→5′ exonuclease. Nucleic Acids Res. 2000;28:2396–2405. doi: 10.1093/nar/28.12.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hickson ID. RecQ helicases: caretakers of the genome. Nat. Rev. Cancer. 2003;3:169–178. doi: 10.1038/nrc1012. [DOI] [PubMed] [Google Scholar]
- 24.Perry JJ, Yannone SM, Holden LG, Hitomi C, Asaithamby A, Han S, Cooper PK, Chen DJ, Tainer JA. WRN exonuclease structure and molecular mechanism imply an editing role in DNA end processing. Nat. Struct. Mol. Biol. 2006;13:414–422. doi: 10.1038/nsmb1088. [DOI] [PubMed] [Google Scholar]
- 25.Zuo Y, Wang Y, Malhotra A. Crystal structure of Escherichia coli RNase D, an exoribonuclease involved in structured RNA processing. Structure. 2005;13:973–984. doi: 10.1016/j.str.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 26.Khakhar RR, Cobb JA, Bjergbaek L, Hickson ID, Gasser SM. RecQ helicases: multiple roles in genome maintenance. Trends Cell Biol. 2003;13:493–501. doi: 10.1016/s0962-8924(03)00171-5. [DOI] [PubMed] [Google Scholar]
- 27.Pagano G, Zatterale A, Degan P, d'Ischia M, Kelly FJ, Pallardo FV, Kodama S. Multiple involvement of oxidative stress in Werner syndrome phenotype. Biogerontology. 2005;6:233–243. doi: 10.1007/s10522-005-2624-1. [DOI] [PubMed] [Google Scholar]
- 28.Von Kobbe C, May A, Grandori C, Bohr VA. Werner syndrome cells escape hydrogen peroxide-induced cell proliferation arrest. FASEB J. 2004;18:1970–1972. doi: 10.1096/fj.04-1895fje. [DOI] [PubMed] [Google Scholar]
- 29.Gu Y, Jin S, Gao Y, Weaver DT, Alt FW. Ku70-deficient embryonic stem cells have increased ionizing radiosensitivity, defective DNA end-binding activity, and inability to support V(D)J recombination. Proc. Natl Acad. Sci. USA. 1997;94:8076–8081. doi: 10.1073/pnas.94.15.8076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vogel H, Lim DS, Karsenty G, Finegold M, Hasty P. Deletion of Ku86 causes early onset of senescence in mice. Proc. Natl Acad. Sci. USA. 1999;96:10770–10775. doi: 10.1073/pnas.96.19.10770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song K, Jung D, Jung Y, Lee S-G, Lee I. Interaction of human Ku70 with TRF2. FEBS Lett. 2000;481:81–85. doi: 10.1016/s0014-5793(00)01958-x. [DOI] [PubMed] [Google Scholar]
- 32.Koike M. Dimerization, translocation and localization of Ku70 and Ku80 proteins. J. Radiat. Res. 2002;43:223–236. doi: 10.1269/jrr.43.223. [DOI] [PubMed] [Google Scholar]
- 33.Li B, Comai L. Functional interaction between Ku and the werner syndrome protein in DNA end processing. J. Biol. Chem. 2000;275:28349–28352. doi: 10.1074/jbc.C000289200. [DOI] [PubMed] [Google Scholar]
- 34.Cooper MP, Machwe A, Orren DK, Brosh RM, Ramsden D, Bohr VA. Ku complex interacts with and stimulates the Werner protein. Genes Dev. 2000;14:907–912. [PMC free article] [PubMed] [Google Scholar]
- 35.Orren DK, Machwe A, Karmakar P, Piotrowski J, Cooper MP, Bohr VA. A functional interaction of Ku with Werner exonuclease facilitates digestion of damaged DNA. Nucleic Acids Res. 2001;29:1926–1934. doi: 10.1093/nar/29.9.1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hsu HL, Gilley D, Galande SA, Hande MP, Allen B, Kim SH, Li GC, Campisi J, Kohwi-Shigematsu T, et al. Ku acts in a unique way at the mammalian telomere to prevent end joining. Genes Dev. 2000;14:2807–2812. doi: 10.1101/gad.844000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Opresko PL, Otterlei M, Graakjaer J, Bruheim P, Dawut L, Kolvraa S, May A, Seidman MM, Bohr VA. The werner syndrome helicase and exonuclease cooperate to resolve telomeric D loops in a manner regulated by TRF1 and TRF2. Mol. Cell. 2004;14:763–774. doi: 10.1016/j.molcel.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 38.Oikawa S, Kawanishi S. Site-specific DNA damage at GGG sequence by oxidative stress may accelerate telomere shortening. FEBS Lett. 1999;453:365–368. doi: 10.1016/s0014-5793(99)00748-6. [DOI] [PubMed] [Google Scholar]
- 39.von Zglinicki T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002;27:339–344. doi: 10.1016/s0968-0004(02)02110-2. [DOI] [PubMed] [Google Scholar]
- 40.Machwe A, Ganunis R, Bohr VA, Orren DK. Selective blockage of the 3′→5′ exonuclease activity of WRN protein by certain oxidative modifications and bulky lesions in DNA. Nucleic Acids Res. 2000;28:2762–2770. doi: 10.1093/nar/28.14.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nick McElhinny SA, Snowden CM, McCarville J, Ramsden DA. Ku recruits the XRCC4-ligase IV complex to DNA ends. Mol. Cell. Biol. 2000;20:2996–3003. doi: 10.1128/mcb.20.9.2996-3003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Erzberger JP, Barsky D, Scharer OD, Colvin ME, Wilson DM., III Elements in abasic site recognition by the major human and Escherichia coli apurinic/apyrimidinic endonucleases. Nucleic Acids Res. 1998;26:2771–2778. doi: 10.1093/nar/26.11.2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nguyen LH, Barsky D, Erzberger JP, Wilson DM., III Mapping the protein-DNA interface and the metal-binding site of the major human apurinic/apyrimidinic endonuclease. J. Mol. Biol. 2000;298:447–459. doi: 10.1006/jmbi.2000.3653. [DOI] [PubMed] [Google Scholar]
- 44.Opresko PL, Laine JP, Brosh RM, Jr, Seidman MM, Bohr VA. Coordinate action of the helicase and 3′ to 5′ exonuclease of Werner syndrome protein. J. Biol. Chem. 2001;276:44677–44687. doi: 10.1074/jbc.M107548200. [DOI] [PubMed] [Google Scholar]
- 45.Karmakar P, Snowden CM, Ramsden DA, Bohr VA. Ku heterodimer binds to both ends of the Werner protein and functional interaction occurs at the Werner N-terminus. Nucleic Acids Res. 2002;30:3583–3591. doi: 10.1093/nar/gkf482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li B, Comai L. Requirements for the nucleolytic processing of DNA ends by the Werner syndrome protein-Ku70/80 complex. J. Biol. Chem. 2001;276:9896–9902. doi: 10.1074/jbc.M008575200. [DOI] [PubMed] [Google Scholar]
- 47.Wilson DM, III, Barsky D. The major human abasic endonuclease: formation, consequences and repair of abasic lesions in DNA. Mutat. Res. 2001;485:283–307. doi: 10.1016/s0921-8777(01)00063-5. [DOI] [PubMed] [Google Scholar]
- 48.Cheng X, Kelso C, Hornak V, de los Santos C, Grollman AP, Simmerling C. Dynamic behavior of DNA base pairs containing 8-oxoguanine. J. Am. Chem. Soc. 2005;127:13906–13918. doi: 10.1021/ja052542s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Robinson H, Gao YG, Bauer C, Roberts C, Switzer C, Wang AH. 2′-Deoxyisoguanosine adopts more than one tautomer to form base pairs with thymidine observed by high-resolution crystal structure analysis. Biochemistry. 1998;37:10897–10905. doi: 10.1021/bi980818l. [DOI] [PubMed] [Google Scholar]
- 50.Vu HM, Pepe A, Mayol L, Kearns DR. NMR-derived solution structure of a 17mer hydroxymethyluracil-containing DNA. Nucleic Acids Res. 1999;27:4143–4150. doi: 10.1093/nar/27.21.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cho BP, Kadlubar FF, Culp SJ, Evans FE. 15N nuclear magnetic resonance studies on the tautomerism of 8-hydroxy-2′-deoxyguanosine, 8-hydroxyguanosine, and other C8-substituted guanine nucleosides. Chem. Res. Toxicol. 1990;3:445–452. doi: 10.1021/tx00017a010. [DOI] [PubMed] [Google Scholar]
- 52.Cysewski P, Olinski R. Theoretical description of the coding potential of diamino-5-formamidopyrimidines. Z. Naturforschung. 1999;54:239–245. doi: 10.1515/znc-1999-3-414. [DOI] [PubMed] [Google Scholar]
- 53.La Francois CJ, Jang YH, Cagin T, Goddard WA, III, Sowers LC. Conformation and proton configuration of pyrimidine deoxynucleoside oxidation damage products in water. Chem. Res. Toxicol. 2000;13:462–470. doi: 10.1021/tx990209u. [DOI] [PubMed] [Google Scholar]
- 54.Rogstad KN, Jang YH, Sowers LC, Goddard WA., III First principles calculations of the pKa values and tautomers of isoguanine and xanthine. Chem. Res. Toxicol. 2003;16:1455–1462. doi: 10.1021/tx034068e. [DOI] [PubMed] [Google Scholar]
- 55.Venkateswarlu D, Leszczynski J. Tautomeric equilibria in 8-oxopurines: implications for mutagenicity. J. Comput. Aided Mol. Des. 1998;12:373–382. doi: 10.1023/a:1008067110965. [DOI] [PubMed] [Google Scholar]
- 56.Krishnamurthy N, Haraguchi K, Greenberg MM, David SS. Efficient removal of formamidopyrimidines by 8-oxoguanine glycosylases. Biochemistry. 2008;47:1043–1050. doi: 10.1021/bi701919u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.von Kobbe C, May A, Grandori C, Bohr VA. Werner syndrome cells escape hydrogen peroxide-induced cell proliferation arrest. FASEB J. 2004 doi: 10.1096/fj.04-1895fje. 04-1895fje. [DOI] [PubMed] [Google Scholar]
- 58.Kamath-Loeb AS, Shen JC, Loeb LA, Fry M. Werner syndrome protein. II. Characterization of the integral 3′→5′ DNA exonuclease. J. Biol. Chem. 1998;273:34145–34150. doi: 10.1074/jbc.273.51.34145. [DOI] [PubMed] [Google Scholar]
- 59.Machwe A, Xiao L, Orren DK. Length-dependent degradation of single-stranded 3′ ends by the Werner syndrome protein (WRN): implications for spatial orientation and coordinated 3′ to 5′ movement of its ATPase/helicase and exonuclease domains. BMC Mol. Biol. 2006;7:6. doi: 10.1186/1471-2199-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bjelland S, Seeberg E. Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat. Res. 2003;531:37–80. doi: 10.1016/j.mrfmmm.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 61.Mol CD, Hosfield DJ, Tainer JA. Abasic site recognition by two apurinic/apyrimidinic endonuclease families in DNA base excision repair: the 3′ ends justify the means. Mutat. Res. 2000;460:211–229. doi: 10.1016/s0921-8777(00)00028-8. [DOI] [PubMed] [Google Scholar]
- 62.Stevnsner T, Mortensen UH, Westergaard O, Bonven BJ. Interactions between eukaryotic DNA topoisomerase I and a specific binding sequence. J. Biol. Chem. 1989;264:10110–10113. [PubMed] [Google Scholar]
- 63.Zharkov DO, Ishchenko AA, Douglas KT, Nevinsky GA. Recognition of damaged DNA by Escherichia coli Fpg protein: insights from structural and kinetic data. Mutat. Res. 2003;531:141–156. doi: 10.1016/j.mrfmmm.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 64.Min J-H, Pavletich NP. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature. 2007;449:570–575. doi: 10.1038/nature06155. [DOI] [PubMed] [Google Scholar]
- 65.Choi JM, Kang SY, Bae WJ, Jin KS, Ree M, Cho Y. Probing the roles of active site residues in the 3′-5′ exonuclease of the Werner syndrome protein. J. Biol. Chem. 2007;282:9941–9951. doi: 10.1074/jbc.M609657200. [DOI] [PubMed] [Google Scholar]
- 66.Friedberg EC, Walker GC, Siede W. DNA Repair and Mutagenesis. Washington, D.C: ASM Press; 1995. [Google Scholar]
- 67.Oikawa S, Tada-Oikawa S, Kawanishi S. Site-specific DNA damage at the GGG sequence by UVA involves acceleration of telomere shortening. Biochemistry. 2001;40:4763–4768. doi: 10.1021/bi002721g. [DOI] [PubMed] [Google Scholar]