


Abstract
Transfected cell microarray is a promising method for accelerating the functional exploration of the genome, giving information about protein function in the living cell. The microarrays consist of clusters of cells (spots) overexpressing or silencing a particular gene product. The subsequent analysis of the phenotypic consequences of such perturbations can then be detected using cell-based assays. The focus in the present study was to establish an experimental design and a robust analysis approach for fluorescence intensity data, and to address the use of replicates for studying regulation of gene expression with varying complexity and effect size. Our analysis pipeline includes measurement of fluorescence intensities, normalization strategies using negative control spots and internal control plasmids, and linear regression (ANOVA) modelling for estimating biological effects and calculating P-values for comparisons of interests. Our results show the potential of transfected cell microarrays in studying complex regulation of gene expression by enabling measurement of biological responses in cells with overexpression and downregulation of specific gene products, combined with the possibility of assaying the effects of external stimuli. Simulation experiments show that transfected cell microarrays can be used to reliably detect even quantitatively minor biological effects by including several technical and experimental replicates.
INTRODUCTION
The application of microarray-based technology for molecule genetic analysis has revolutionized our ability to study some aspects of gene function. DNA microarrays have given us the possibility for high-throughput analysis of gene expression, and thus a technology for identifying genes that are potentially involved in particular cellular processes as well as in physiological and pathophysiological processes and conditions. However, DNA microarrays do not provide a direct analysis of the functions of the gene products within the living cell, and these functional analyses are often performed on a one by one gene basis. Ziauddin and Sabatini (1) established a method for miniaturization of cell-based functional studies called transfected cell microarrays. This technology allows spatially restricted transfection without the use of wells by immobilizing nucleic acids complexed with a transfection reagent in a gel, from which it is only accessible to nearby cells. Adherent cells growing on top of such printed spots will take up the nucleic acids deposited in the spot, while cells growing between the spots will not be transfected. Ziauddin and Sabatini (1) used this system for analysis of gene overexpression by printing cDNAs cloned in expression plasmids. Later this method has also been adopted for downregulation of gene expression using siRNAs or shRNAs (2–5). Thus, this experimental setup represents an array of collections of living cells, each overexpressing or silencing a specific gene product. By applying appropriate assays, the phenotypic consequences of hundreds or thousands of overexpressed or silenced genes can be detected simultaneously. Transfected cell microarrays have been used to identify genes involved in diverse cellular processes including apoptosis (6,7), chromosome segregation and nuclear structure (8), spindle formation (4), signal transduction and transcriptional regulation (9–12), secretory pathways (13,14), receptor binding (1,15) and binding to antibody fragments (16). Transfected cell microarrays have also been used to study subcellular localization of proteins (17–20), and can thus be applied in studies screening a large number of genes or compounds that may affect subcellular localization of given gene products.
The key advantages of all microarray technologies are the ability to conduct easy-to-handle, high or medium throughput studies with a small amount of reagents. Even though transfected cell microarrays are potentially a very powerful tool for screening and functional genomics, the widespread adaption of this technology has been relatively slow. One explanation might be that microarray procedures generally include many steps that have to be optimized in order to give reliable results. For transfected cell microarrays this includes e.g. array production, transfection efficiency, biological assays, data retrieval and statistical analysis.
A variety of algorithms for statistical analysis of DNA microarray high-throughput gene expression data have been reported over the last years [reviewed in (21)]. However, development of statistical methodology for high-throughput cell-based assays has lagged behind, and improvements in analysis methods for this type of data are needed. This includes the use of proper quality controls of the biological assay, normalization procedures of the data and statistical analysis protocols. Boutros et al. (22) and Hahne et al. (23) have contributed to this task by presenting statistical methods and software for the interpretation of data from high-throughput cell-based RNAi screens in 384-well microplates and cell-based assays with flow cytometric read-outs, respectively. In addition, statistical methods for analysis of high-throughput screening in drug discovery are under development [reviewed in (24)]. The use of transfected cell microarrays to analyse gene functions is still in its infancy, and most of the focus has been directed to optimization of the reverse transfection protocol and development of different output assays shown as proof-of-principles. More focus is needed on the quantitative analysis of the array-based data including e.g. the use of controls and replicates, normalization strategies and statistical analysis. Standardization of the analysis of transfected cell microarray data is much more complex than for traditional DNA microarrays. There is a huge variety in the assay outputs from transfected cell microarrays, with the possibility of measuring many parameters per spot or per cell, and the analysis is highly dependent on the biological assay applied. The analysis approaches of data from transfected cell microarrays can be divided into two groups depending on the level of resolution needed (5). In assays depending on single cell resolution, microscopes are used to acquire high-resolution readouts. Quantitative image analysis of fluorescence intensities down on a single-cell level, and approaches for classification of diverse cellular phenotypes using high-resolution images are under development (3,8,17,25–27). When single cell resolution is not needed, microarray scanners can be used to quickly provide a low-resolution image of the entire slide, and quantification of spot intensities can be obtained using software originally developed for traditional DNA microarray analysis (1,27).
Transfected cell microarray is a promising tool for high-throughput screening of gene functions. However, the possibility of printing many replicate spots on one array, as well as the possibility of multiplexing, makes it an attractive method also for more medium-scale studies addressing defined biological questions with fewer genes. We have focused on the use of transfected cell microarrays for medium-scale studies using fluorescent reporters and a laser scanner for obtaining fluorescence signal intensity values per spot. As has been acknowledged for DNA microarray analysis (21) and addressed for high-throughput screens in drug discovery (24), the inclusion of replicates is necessary to account for both technical and biological variation. The objective in the present study was to establish experimental and statistical approaches to enable a robust and reliable analysis of fluorescence intensity data, and to address the use of technical (replicate spots on one array) and experimental (the independent repetition of the reverse transfection experiment) replicates for studying regulation of gene expression with varying complexity and effect size. We performed three studies (summarized in Table 1) with increasing complexity, and with well-established biological effects, using fluorescent gene reporter plasmids, siRNAs and external stimulus treatment of cells. To deal with the replicate variation known to occur in all biological experiments (22,28), we used linear regression (ANOVA) modelling for estimating biological effects due to different conditions (the nucleic acids printed in the spots) and treatments (external stimuli added to the cells for the induction of gene expression), and for the calculation of P-values for comparisons of interests. For each study we performed a simulation experiment addressing the number of replicates necessary for detecting the biological effects with different effect sizes and variable degrees of complexity of the molecular mechanisms studied. The use of linear regression (ANOVA) modelling was then evaluated by comparing it with four other analysis approaches for transfection studies.
Table 1.
Studies performed on transfected cell microarrays
| Studies | Experimental replicates | Conditions | Treatment (stimulus) |
|---|---|---|---|
| 1. Dose siRNA Downregulation of EGFP using different concentrations of siRNA | 2 | pEGFP + pDsRed + siEGFP (0–30 ng/µl) pEGFP + pDsRed + siCAT (30 ng/µl) pCRE-Luc (negative control) | none |
| 2. NFκB Stimulation of reporter gene | 3 | pNFκB-EGFP pNFκB-EGFP + pDsRed pEGFP + pDsRed pCRE-Luc (negative control) | ±TNFαa |
| 3. ICER Transcriptional repressor-mediated inhibition of reporter gene | 4 | pCRE-EGFP + siCAT pCRE-EGFP + siICER pCRE-Luc (negative control) | ±tetracyclineb |
aActivation of promoter driving reporter gene.
bInduces gene expression of ICER transcriptional repressor.
ICER, inducible cAMP early repressor.
MATERIALS AND METHODS
Plasmids and siRNAs
The expression plasmids pEGFP-N1 and pDsRed-express-N1 were obtained from BD Bioscience Clontech. In these plasmids, the expression of the green (EGFP) or red (DsRed) fluorescent protein is driven by a cytomegalovirus (CMV) promoter. Reporter plasmids with promoters driven by CRE or NFκB elements were generated as follows: The gene encoding EGFP was prepared from pEGFP-N1 by digestion with NcoI and HpaI. The luciferase (luc) gene was removed from pCRE-Luc and pNFκB-Luc (Stratagene) by digestion with EcoNI and NcoI after creating a digestion site for NcoI around the start codon using the QuickChange Site-Directed Mutagenesis Kit from Stratagene. The fragment encoding EGFP was inserted into the plasmids to give pCRE-EGFP and pNFκB-EGFP. All the plasmids were transformed into E.coli and isolated with Plasmid Maxi Kit (Qiagen).
Synthetic siRNA specific to EGFP (siEGFP): sense, 5′-GCAAGCUGACCCUGAAGUUCAU-3′; antisense, 5′-GAACUUCAGGGUCAGCUUGCCG-3′ (3), specific to all ICER splice variants (siICER): sense, 5′-CAUUAUGGCUGUAACUGGATT-3′; antisense, 5′-UCCAGUUACAGCCAUAAUGGG-3′ (29). Control siRNA targeting CAT (chloramphenicol acetyl transferase) (siCAT): sense, 5′-GAGUGAAUACCACGACGAUUUC-3′; antisense, 5′-AAUCGUCGUGGUAUUCACUCCA-3′ (3). siEGFP and siCAT were obtained from The Biotechnology Centre, University of Oslo (30,31), and were annealed at 10 µM (∼0.14 µg/µl) in 10 mM Tris–HCl, pH 7.4. siICER were obtained from Qiagen and annealed at 20 µM (∼0.25 µg/µl) in siRNA suspension buffer (Qiagen).
Array printing and reverse transfection
In the present work, the protocol was based on the so-called lipid-DNA method reported by Ziauddin and Sabatini (1). The workflow includes making the printing solution, printing the arrays, incubate cells on top of the arrays and detect the resulting effects in the spots. (Illustrated in Supplementary Figure 1.) Below, the different steps in our version of the protocol with optimization efforts are described.
Printing solution
Several transfection reagents were tested (data not shown), and we found that the X-tremeGENE siRNA transfection reagent (Roche) gave good transfection efficiencies both for plasmids and siRNAs, and chose to use this reagent for all transfected cell microarray experiments in the present study. For printing the arrays, one major challenge is to find a good balance between high transfection efficiency and spatially confined spots to avoid cross-contamination between the spots. In order to optimize the reverse transfection protocol for HEK 293ind-ICER IIγ cells (see below) and X-tremeGENE transfection reagent, we investigated the effect of varying the concentrations of gelatine and sucrose in the printing solution. These reagents have been reported to influence both the transfection efficiency and spot integrity (6,27). Sucrose was observed to be specifically beneficial for obtaining high transfection efficiency when storing the arrays for several weeks before use (data not shown). Figure 1A and B show representative images of the observed effects of varying the concentrations of gelatine and sucrose. We observed that the transfection efficiency increased with increasing gelatine concentration (tested in the range 0.01–0.40%). However, an increased disturbance of the spatial definition of the spots was observed with increasing concentrations of gelatine or sucrose (tested in the range 0–100 mM). A combined effect of the concentrations of gelatine and sucrose was also observed, as low concentrations of gelatine allowed us to use higher concentrations of sucrose than with higher concentrations of gelatine before cells spread outside the spots. Based on several optimizing experiments, we found that 3 µl X-tremeGENE solution per microgram nucleic acid, 25 mM sucrose and 0.1% gelatine in the final printing solution reproducibly gave spatial restricted transfection with high transfection efficiency printing the arrays with both a pipette tip and a hand-held arrayer (see below).
Figure 1.

Effects of sucrose and gelatine concentrations on spot integrity and transfection efficiency. (A) Array printed with pDsRed (50 ng/µl) in a printing solution with different gelatine and sucrose concentrations. Scanning image of the whole array and magnifications of specific spots. (B) Array printed with pEGFP (50 ng/µl) in a printing solution with 25 mM sucrose and four different concentrations of gelatine. Top: Box plot of the fluorescence intensities in each spot (n = 32–34). Bottom: Scanning image showing squares of seven times five spots for the four gelatine concentrations. From left to right: 0.01, 0.05, 0.1 and 0.2% gelatine. The DNA-lipid-gelatine-sucrose solutions were printed manually with a 10 µl pipette tip (A) or by MicroCasterTM manual arrayer system (B).
In a 1.5 ml microcentrifuge tube, plasmid (1 µg/µl) and siRNA were mixed with growth medium without fetal calf serum (FCS), 0.5 µl 1.5 M sucrose and 3 µl X-tremeGENE per microgram nucleic acid to a final volume of 22.5 µl. After 15–20 min of incubation, 7.5 µl 0.4% gelatine (Type B, G9391, Sigma) was added to give 30 µl printing solution. The gelatine solution was prepared as described by Ziauddin and Sabatini (1). To achieve sufficient level of expression from the transfected plasmids, 25–50 ng/µl pEGFP-N1 or pDsRed-express-N1 and 50–75 ng/µl of CRE or NFκB reporter plasmids was used. For siRNA studies, 2–30 ng/µl siRNA in the final printing solution was used.
Array printing
The DNA-lipid-gelatine solution was arrayed onto UltraGAPSTM coated slides (Corning) at room temperature. The requirement of an expensive robotic arrayer for printing the microarrays can be an obstacle for many research groups to adopt this method in their lab. For small- and medium-scale studies it is possible to print the arrays using a small pipette tip (19), and we used a 10 µl pipette tip (Biosphere Filter Tips, type Gilson/Biohit, Sarstedt) giving spots of about 800 µm in diameter (about 15 nl sample per spot). We also used the hand-held microarrayer MicroCasterTM from Schleicher and Schuell, which consists of an arrayer tool containing eight pins and a slide holder with an indexing system to guide the spotting of up to 768 spots, each with a diameter of about 500 µm (about 6 nl sample per spot). For printing with the hand-held microarrayer, 15 µl of printing solution for each condition were added to wells in a 384-well plate. The eight pins were prepared and washed according to the manufacturer's instructions. After printing, the slides were dried for at least 1 h in room temperature before placed at 4°C together with a desiccant until use.
Cell line
HEK 293ind-ICER IIγ cells stably transfected with a gene encoding the ICER IIγ splice variant driven by a tetracycline-inducible promoter (29) were used in all transfected cell microarray experiments. The expression of ICER is at normal levels in the untreated condition, with an overexpression of ICER IIγ as a response to tetracycline. The cells were cultured at 37°C in a humidified 5% CO2 incubator in Dulbecco's modified Eagle's medium (DMEM) containing 4.5 g/l glucose (Gibco, Invitrogen) supplemented with 1% (v/v) penicillin-streptomycin (Gibco, Invitrogen), 0.1 mg/ml l-glutamine (Gibco, Invitrogen), 150 µg/ml Hygromycin B (Invitrogen), 15 µg/ml Blasticidin (Invitrogen) and 10% FCS (Euroclone).
Reverse transfection, treatment and fixation
Immediately before transfection, actively growing cells were trypsinized and resuspended in growth medium to desired density. The printed slides were placed in QuadriPERM plates (Vivascience) and overlaid with 3 × 106 cells in 8 ml medium. To be able to observe a treatment effect on the different conditions in the spots, it is necessary to use either two arrays or divide the array into separate wells. When using a cell culture accessory made of silicone to give two separate wells on the array (a modified version of FlexiPERM from Vivascience), each of the two wells was incubated with 1 × 106 cells in 2 ml medium. To stimulate expression from CRE and NFκB reporter plasmids, 10 µM forskolin (Sigma) and 20 ng/ml hrTNF-α (R&D Systems) was used, respectively. After 48 h of incubation, the slides were gently washed in PBS, and the cells were fixed (3.7% paraformaldehyde and 4% sucrose in PBS) for 20 min at room temperature. The slides were gently washed in PBS, and the nuclei were stained by incubating for 5 min with 500 µl 1 µl/ml DAPI (Invitrogen) in PBS per slide. The slides were gently washed three times with PBS before mounted with Mowiol 4-88 mounting medium pH 8.5 [6 g glycerol, 2.4 g Mowiol 4-–88 (Hoechst), 6 ml dH2O, 12 ml Tris–HCl buffer pH 8.5], and placed at 4°C overnight before image acquisition.
Image acquisition, data processing and statistical analysis
The data were analysed by the following steps:
Laser scanning to obtain a picture of the fluorescence intensities in the spots.
Quantification of spot intensities.
Log-transformation.
Normalization to negative control spots.
Linear regression models to explain the transformed and normalized fluorescence intensities in each spot based on data from more than one experimental replicate, resulting in estimated biological effects and calculated P-values for comparisons of interests.
Scanning
Transfected cell microarrays were scanned using Tecan's LS ReloadedTM scanner. The scanning images were obtained with a 6 µm resolution, and the EGFP and DsRed emission were visualized using lasers with wavelength 488 and 532 nm, and the filters 535/25 and 575/50 nm, respectively. For display, the scanning images were pseudocoloured and the levels were adjusted using Adobe Photoshop.
Quantification of spot intensities
Quantification of the level of fluorescence protein expression in each spot was performed using the GenePix software (Axon Instruments, Inc., Union City, CA, USA). Briefly, the mean greyscale values were measured within circles of diameter corresponding to 600 or 900 µm in the scanning image for arrays printed with MicroCasterTM or a 10 µl pipette tip, respectively. Features with visual defects were eliminated from the analysis (flagged spots). The percentage of flagged spots in the three studies presented in this work was <4%, and we did not observe any difference in the number of flagged spots printing with the 10 µl pipette tip or using MicroCasterTM.
A fluorescent microscope was used for confirming the results obtained from the laser scanner. Selected fluorescence microscope pictures were analysed in the open-source image analysis software CellProfiler (www.cellprofiler.org) (25), where the MeasureImageIntensity module was used to quantify the total image intensity in images of each spot. This quantification gave similar results as obtained using a laser scanner and GenePix software for quantification (an example is shown in Supplementary Figure 2).
Log-transformation and normalization to negative control spots
The green fluorescence protein (EGFP) was used to visualize the biological problem under study, while the red fluorescence protein (DsRed) was an internal control used for transfection efficiency normalization of the EGFP fluorescence intensity in each spot. The data were log-transformed (base 2) giving log-green or log-ratio (log-green minus log-red) intensity signals in each spot. The log-transformed data were normalized to the negative control spots (i.e. pCRE-Luc) by subtracting the median of the log-green or log-ratio intensities in the negative control spots from all the spots printed on the same array or in the same well on an array, i.e. as a means of background correction on the log scale. We used the median value as this is less influenced by outliers than the mean value. These log-transformed and normalized data were used further in the statistical analysis.
Plots of each experimental replicate on the original scale were constructed by first calculating the mean values and the upper and lower limits (±2 SD) for the log-transformed and normalized data and then transforming these values back to the original scale.
Linear regression method
We used linear regression models to explain the observed normalized and log-transformed spot intensities on the basis of the different conditions printed on the array (different printing solutions with plasmids and siRNAs), the different treatments (external stimulus added to the cells) and the different independent experiments (experimental replicates). Each condition was represented by several replicate spots per treatment (technical replicates) in each experimental replicate. Each study then consists of several experimental replicates, each with several replicate spots of each condition per treatment (technical replicates).
Let the number of experimental replicates be denoted ne, where we assume that ne ≥ 2. Further we have one or two treatments, nt. Denote by nc the number of conditions for every treatment and experimental replicate, where we assume that nc ≥ 2.
If there is only one treatment (i.e. no treatment effect to model), we fit the following linear regression model:
| 1 |
Here Yikl is either the normalized log-green or the normalized log-ratio signal in the lth spot in experimental replicate i for condition k. The overall level of all spots in all experimental replicates and for all conditions is called µ. The effect of the ith experimental replicate is denoted ei, i = 1, …, ne, and the effect of the kth condition is denoted ck, k = 1, …, nc. The term (ec)ik models the interaction between experimental replicate i and condition k. Finally, εikl denotes the remaining unexplained variation in the data, and we assume that εikl is normally distributed with mean 0 and variance σ2. We assume that this unexplained variation is independent between the spots. This linear regression model is the same as a two-way ANOVA model. For an introduction to the theory behind the linear regression models, we refer to (32,33).
When there are two treatments we add a treatment effect tj, j = 1, 2, to the previous model, and by including interaction terms between the treatment, experimental replicate and condition effects, the resulting model is given by
| 2 |
where Yijkl is the normalized log-green or normalized log-ratio signal in the lth spot in experimental replicate i for treatment j and condition k. Also here we assume that the unexplained variation, εijkl, is independent between the spots. This linear regression model is the same as a three-way ANOVA model.
From the linear regression models we estimate the treatment and condition effects and the difference between the different conditions within a treatment, or the difference between the two treatments for each condition.
In the estimation of the parameters, we use the following sum-to-zero constraints:
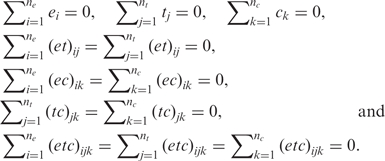 |
In a balanced design, the overall effect, µ, is estimated as the overall mean of all the observations, ei is the mean of the observations in experimental replicate i minus µ, tj is the mean of the observations in treatment j minus µ and ck is the mean of the observations in condition k minus µ. The estimated two and three-way interactions are estimated in a similar manner. In an unbalanced design, the estimates are slightly modified.
For each effect and comparison of effects of interest, we estimate parameters based on the log-transformed and normalized data. When results (plots, estimates) are presented on the original scale, all calculations are first performed on the log-scale and then transformed back to the original scale.
We compare conditions within or between treatments by testing the null hypothesis that their difference on log scale is 0. The test statistic used is the estimated difference between the conditions or treatments of interest divided by the estimated standard error of this difference. The standard error is found from the covariance matrix of the estimated effects, and the test statistic is t-distributed. The P-value is then calculated from the test statistic. An evaluation of our linear regression method is found in the Results and Discussion section.
All the analysis of the quantified spot intensities was done using the free software environment for statistical computing and graphics R. The R code is available upon request: <mettela@math.ntnu.no>.
Simulation experiments
We performed simulation experiments in order to evaluate the number of technical and experimental replicates necessary for detecting a known biological effect. Technical replicates were removed from the dataset and P-values for comparisons of interests were calculated based on the resulting dataset using the linear regression method presented above or four other analysis approaches (methods A–D) described in the Results and Discussion section and in Supplementary Data. The process of removing technical replicates from the dataset was repeated using all the experimental replicates present and, for some experiments, all combinations of at least two experimental replicates. The simulation experiments performed in this study are described in detail in the Results and Discussion section, and were performed using the R software.
RESULTS AND DISCUSSION
Measurement of feature intensity and choice of normalization strategies
Measurement of feature intensity
Biological effects are measured as exogenously expressed fluorescence protein in the cells localized in the spots of the array. It is therefore necessary to establish whether mean or median of the pixel intensities in each spot is the most suitable measure of the feature intensity representing the level of the fluorescent proteins. Evaluation of the distribution of pixel intensities in six spots printed with pEGFP showed that this distribution was skewed to the right (Figure 2A), in agreement with what others have reported for the fluorescence intensities in each cell within a spot (27). The fluorescence intensity varied substantially from pixel to pixel within one single spot (within-spot variability). As observed using a fluorescence microscope, this corresponded to a high variation in the amount of expressed fluorescence protein in each cell within one spot (data not shown). Most of the cells expressed the fluorescence protein with intensity in the lower range, while some cells expressed very high amounts of the fluorescence protein contributing to the uneven distribution of the pixel intensities in the spots. Since these high pixel intensities reflect true signals, we preferred to use the centre of gravity of the pixel intensity distribution. Therefore, we used the mean of the pixel intensities in each spot as a measure of the feature intensity for analysis of the effects of the various conditions and treatments.
Figure 2.

Evaluation of fluorescence intensities in each spot. (A) Density plots of pixel intensities from six spots printed with pEGFP (green lines). Mean (black lines) and median (red lines) of the pixel intensities in each spot. The scanning image shows the EGFP fluorescence intensity signal (green) in the six spots. The pixel intensities in each spot were acquired using the ‘Example save pixel values’ in the Report menu in GenePix software. This resulted in a text-file with 7884 pixel values for each spot. Plotting was done using the R software. (B) Spot-to-spot variability in 26 spots printed with pEGFP and pDsRed. EGFP, DsRed and ratio (EGFP normalized to DsRed) fluorescence intensity signal (mean of the pixel intensities) normalized to the mean of the signal from all the spots. The arrays were printed using MicroCasterTM.
Normalization using negative control spots and internal control plasmids
Evaluation of fluorescence intensity from spots printed with the non-fluorescent reporter plasmid pCRE-Luc revealed that these signals were higher than the signals from the cell layer between the spots. Using such spots as negative (or background) controls are valuable for the assessment of exogenous expression of fluorescence protein as spots expressing such proteins should yield higher intensity signals. We also observed that the intensity signal from spots printed with the non-fluorescent reporter plasmid pCRE-Luc varied between arrays. Thus, the use of these spots to normalize the data for differences in the global background signal on the arrays, allow comparisons of spot intensities between arrays. Based on these observations, spots printed with the non-fluorescent reporter plasmid pCRE-Luc were used as negative control spots throughout the present study. Similarly, Chang and colleagues (34) normalized the data to a negative control.
The transfection efficiency varies from spot to spot, and hence there is variability in the fluorescence intensity signal in spots printed with the same printing solution. This spot-to-spot variance creates high variability in the data obtained. Cotransfection with a plasmid, that gives constitutively active expression of a fluorescent protein, can be used to normalize for this variability (2,4,10,12). Cultivating cells on top of spots containing the two plasmids pEGFP and pDsRed yielded a good correlation between the two fluorescence intensities in each spot (correlation factor r = 0.97), and the variation in the data was reduced by normalizing the EGFP fluorescence intensity data to the DsRed fluorescence intensity data in each spot (Figure 2B). This illustrates that an internal control plasmid can be valuable in order to normalize for the spot-to-spot variability. Normalization to an internal control can also be seen as local background normalization in each spot.
Analysis of downregulation of reporter gene expression using siRNA (Dose siRNA study)
We first used the validated siRNA targeting EGFP (siEGFP) (3,27) in order to apply the linear regression model without a treatment effect [Equation (1)] for the analysis of data from two experimental replicates. pEGFP, pDsRed and different concentrations of siEGFP were cotransfected and the resulting EGFP fluorescence was analysed with and without normalization to DsRed in order to also evaluate the effects of using an internal control plasmid to correct for spot-to-spot variation. The setup and scanning image of the EGFP and the DsRed fluorescence intensity signal for one of the experimental replicates are shown in Figure 3A.
Figure 3.

Linear regression modelling without a treatment effect. Cells were cotransfected with pEGFP (30 ng/µl), pDsRed (30 ng/µl) and different concentrations of siEGFP (0–30 ng/µl). (A) Left diagram indicates the placement of the cell clusters for eight conditions. pEGFP, pDsRed and 0, 2.5, 5, 10, 15 or 30 ng/µl siEGFP. The non-fluorescent reporter pCRE-Luc (60 ng/µl) as a control of the background fluorescence in the spots (negctrl). pEGFP, pDsRed and 30 ng/µl siCAT as a siRNA control (ctrl). Middle diagram: Scanning image for EGFP-detection. Right diagram: Scanning image for DsRed-detection. (B) Illustration of the different terms in the linear regression model in Equation (1). The ratio fluorescence signal intensities (EGFP intensities normalized to DsRed intensities) from each spot (log-transformed and normalized to negative control spot intensities) in two experimental replicates (28–35 technical replicates after removing flagged spots) were used to fit the model. (a) The overall effect µ. (b) The overall effect µ and the experimental replicate effects e1 and e2. (c) The overall effect µ and the condition effects c1 through c8. (d) Combining µ, ei and ck. (e) Addition of the interaction effect term (ec)ik to the terms in (d). The observed data from each spot are shown together with the fitted model in (d) and (e) for experimental replicate 1 (black circles) and experimental replicate 2 (blue triangles). (C) Estimated effects of the different conditions are shown based on the EGFP fluorescence intensity data (green, left) or the EGFP fluorescence intensity data normalized to DsRed control plasmid intensities (ratio, right). The intensities are shown relative to 0 ng/µl siRNA, error bars are 95% CI. The arrays were printed using MicroCasterTM.
The linear regression modelling is explained in detail by looking specifically at the different terms in the model for the EGFP intensity data normalized to the DsRed intensities (ratio intensities) (Figure 3B). The first term in the model is the overall effect µ (Figure 3B-a), representing the level of all the spots in both experimental replicates. Adding an experimental replicate effect ei to the model, explains a factor difference in the fluorescence intensities between the two experimental replicates (Figure 3B-b). Even though each experimental replicate is performed under presumably identical circumstances, the absolute value of the detected fluorescence signal intensities will vary between experimental replicates, giving rise to the experimental replicate effect. The condition effect ck explains the differences between the eight conditions printed on the arrays (Figure 3B-c). The condition effect is added to the experimental effect to explain the observed condition effects in the two experimental replicates (Figure 3B-d). However, as shown below, the data is better described by including an interaction effect between condition and experimental replicate in the model (Figure 3B-e). The observed data from each spot illustrates the remaining unexplained variation in the data, εikl. The experimental replicate effect explains the same factor difference for all conditions between experimental replicates, which come from differences that affect all conditions in the same way. This can be a result of e.g. small differences in cell density and scanning settings. When the interaction effect between experimental replicate and condition is included, this opens the opportunity to manage distinct factor differences between experimental replicates for the various conditions. Such variations may arise from e.g. small differences in concentrations of the constituents of the printing solutions caused by the pipetting inaccuracy and from variations in the printing of the different conditions onto the array. The estimated effects of the k different conditions were then found as µ + ck (Figure 3B-c), while the estimated variance and P-values for comparisons of interests were calculated based on the fitted model in Figure 3B-e.
The estimated effect of siEGFP was a downregulation of the EGFP fluorescence of about 70–80% using 15–30 ng/µl siRNA (Figure 3C). The resulting 95% confidence intervals for the estimated effects were slightly smaller when normalizing to the DsRed fluorescence intensities (Figure 3C, right) than when only using the EGFP fluorescence intensities (Figure 3C, left). This shows that normalization to an internal control in each spot reduced the variation in the data obtained. It should be noted that when using EGFP fluorescence intensities alone, a significant difference (P < 0.01) was estimated between no siRNA (10) and control siRNA (ctrl) (Figure 3C, left). Using data normalized to the DsRed internal control plasmid, the linear regression method estimated no difference (P = 0.98) between the two conditions (Figure 3C, right). This further demonstrates the importance of using an internal control plasmid to correct for possible variations in the amount of transfected plasmid between different conditions printed on the array.
The inclusion of both technical and experimental replicates in transfected cell microarray experiments is important in order to achieve results with a high degree of confidence. However, the inclusion of more replicates than necessary for detecting biological effects under study is a waste of both reagents and labour time. It is therefore important to use an experimental design optimally suited to achieve significant analysis results with the smallest number of replicates possible. In order to evaluate how many technical replicates that was sufficient to detect the effect of different concentrations of siEGFP, we performed a simulation experiment by randomly removing technical replicates from each of the two experimental replicates. Without normalization to an internal control (Figure 4A), four technical replicates were sufficient to detect significant effects of 10–30 ng/µl siEGFP. Eight technical replicates resulted in a significant effect of the siRNA for all siRNA concentrations tested in this experiment (i.e. 2.5–30 ng/µl). When using data normalized to the internal control, two technical replicates were sufficient for detecting the downregulation of EGFP for all five siRNA concentrations tested (Figure 4B), showing again that normalization is valuable for reducing the variation in the data.
Figure 4.

Simulation experiment addressing the number of technical replicates necessary for detecting a significant effect (P < 0.01) of siEGFP. We randomly removed 4, 8, 12, 16, 20, 24 and 26 out of 28 technical replicates in each of two experimental replicates, used the linear regression model in Equation (1) to calculate P-values for certain comparisons based on the resulting dataset, repeated this 1000 times and recorded how many times the P-value was below 0.01. This was performed for the comparisons of either 2.5, 5, 10, 15 or 30 ng/µl siEGFP versus 0 ng/µl siRNA, and for 30 ng/µl siCAT (ctrl) versus 0 ng/µl siRNA. The simulation was done using the EGFP fluorescence intensity data (A), and the EGFP fluorescence intensity data normalized to DsRed control (ratio) (B).
These experiments showed that the results were highly reproducible using the optimized protocol for array production. The linear regression model presented fits the observed data and is a valuable tool for analysing data from more than one experimental replicate. Normalization of the data to an internal control plasmid in each spot resulted in reduced variation in data as observed by smaller confidence intervals in the estimated effects and fewer technical replicates necessary for obtaining significant effects of siEGFP.
Analysis of transcriptional induction using reporter gene and external stimulus (NFκB study)
Transfected cell microarrays can be used to study the expression of reporter genes under various conditions, thus enabling assaying large number of proteins of interest for their involvement in regulation of transcription via specific promoter elements (9–11). In such studies it is often of interest to add a stimulus to the cells. To develop a linear regression model suited for the analysis of effects from different cell treatments, we created a transfected cell microarray data set based on measurements of the stimulation of NFκB (Nuclear factor-kappa B) responsive promoter elements by tumour necrosis factor alpha (TNFα). NFκB is a well-known family of transcription factors involved in immune and inflammatory reactions (35), and is known to be activated by TNFα. To include the effects of an external stimulus in the linear regression (ANOVA) model, we added a treatment effect (t) to the model [Equation (2)].
By using this linear regression (ANOVA) model on data from three experimental replicates, the estimated effect of TNFα on NFκB-driven transcription from pNFκB-EGFP was a mean fold increase of 5.6 (Figure 5A). Cotransfection of pNFκB-EGFP with pDsRed yielded a mean TNFα stimulation effect of 4.7-fold increase based on EGFP (green) fluorescence intensity data (Figure 5B). This difference in the effect of TNFα on NFκB-driven transcription in spots with and without cotransfection with pDsRed was not significant (on 5% significance level calculated by a two-sample t-test using the Welch-approximation to the degree of freedom). Normalizing the EGFP fluorescence with the control plasmid DsRed fluorescence in each spot reduced the estimated mean TNFα stimulation effect to a 3.5-fold increase (Figure 5B), due to the fact that the CMV promoter in the DsRed control plasmid is also stimulated by TNFα. The CMV promoter driving expression from the plasmids pEGFP and pDsRed was stimulated by TNFα with a mean fold induction of about 1.5 (Figure 5C). The result is not surprising since the CMV promoter contains NFκB-binding sites (36). Thus, pDsRed is less suitable as an internal control plasmid in this system. However, since the promoter in pNFκB-EGFP was more strongly stimulated than the CMV promoter, the stimulation effect of TNFα was not abolished using the normalized data.
Figure 5.

Linear regression modelling with a treatment effect for analysis of transcriptional induction. Cells were transfected with pNFκB-EGFP (75 ng/µl), pNFκB-EGFP (75 ng/µl) and pDsRed (30 ng/µl), pEGFP (30 ng/µl) and pDsRed (30 ng/µl) or the negative control pCRE-Luc (60 ng/µl). The arrays were converted into two wells using a modified version of flexiPERM, and the cells in one of the wells were stimulated with TNFα (20 ng/ml) for 6 h to induce NFκB-driven transcription. The graphs show estimated effects based on data from three experimental replicates (4–6 technical replicates after removing flagged spots) using the linear regression model described in Equation (2). Fluorescence intensities from spots printed with (A) pNFκB-EGFP (green fluorescence intensity), (B) pNFκB-EGFP and pDsRed (green, red and ratio fluorescence intensity) or (C) pEGFP and pDsRed (green, red and ratio fluorescence intensity). Fluorescence intensities relative to the TNFα-untreated condition, error bars are 95% CI. *P < 0.01; significant difference from TNFα-untreated cells. The arrays were printed using a 10 µl pipette tip. The data from the three experimental replicates are shown in Supplementary Figure 3. (D) Simulation of the number of technical and experimental replicates necessary for obtaining a significant (P < 0.01) stimulation effect of TNFα on NFκB-driven transcription for the condition pNFκB-EGFP cotransfected with pDsRed. We removed 1, 2, 3 and 4 out of 6 technical replicates from each experimental replicate, repeated this 6, 15, 20 and 15 times, respectively (i.e. all possible permutations containing the desired number of spots), fitted the linear regression model in Equation (2) based on the resulting dataset, and recording the proportion of P-values below 0.01 for the effect of TNFα on NFκB-driven transcription. This was performed for all three experimental replicates and all combinations of two experimental replicates, and for both EGFP fluorescence intensity data (green, left) as well as for data after normalization with DsRed (ratio, right).
In order to evaluate the number of replicates necessary to detect the known stimulation effect on NFκB-driven transcription by TNFα, we performed a simulation experiment by removing replicates from the dataset (Figure 5D). Using only EGFP fluorescence intensities and all three experimental replicates, two technical replicates were sufficient to detect a significant effect of TNFα. The analysis further demonstrates that two experimental replicates were sufficient to identify a highly significant effect when three technical replicates were included. For the data normalized to DsRed fluorescence intensities, two technical replicates were sufficient both for three and two experimental replicates. Thus, even though normalization with DsRed weakens the estimated TNFα effect, due to TNFα–induced activation of the CMV promoter driving DsRed expression (Figure 5B), the ability to detect biological effects is enhanced due to reduced variation in the data.
These results showed that transfected cell microarray with fluorescent protein reporter genes can be used to measure the effect of an external stimulus treatment of the cells. An internal control plasmid may be valuable to reduce the variation in the data and thereby reduce the number of replicates necessary for detecting significant treatment effects. However, care has to be taken when normalizing the data to an internal control plasmid as the expression of the reporter gene from the plasmid can be affected by the treatment or the biological condition under study.
Analysis of function of transcriptional repressor using reporter gene, siRNAs and external stimulus (ICER study)
We further wanted to evaluate whether transfected cell microarrays and our analysis approach can be used to study more complex biological problems with smaller biological effects than in the two previous experiments described in this work. By use of luciferace reporter plasmids in a 96-well plate format, Misund et al. (29) have shown that overexpression of inducible cAMP early repressor splice variant IIγ (ICER IIγ) represses CRE-driven but not NFκB-driven transcription. ICER is transcribed from an intronic CRE-driven promoter (P2) in the cAMP responsive element modulator (CREM) gene, and functions as an effective repressor of CRE-mediated transcription, both its own and other cAMP responsive genes (37). (Illustrated in Supplementary Figure 4).
Since ICER may be involved in negative feed-back responses associated with growth regulation and tumorigenesis (38–41), it is of great interest to identify ICER target genes and molecular mechanisms involved in activating and modulating ICER repressing effects. Transfected cell microarrays may represent an attractive platform to test a high number of hypotheses related to such questions by using CRE-driven fluorescent protein reporter genes.
ICER-mediated repression of forskolin-induced CRE-driven transcription on transfected cell microarrays was assayed as reduced activation of the reporter plasmid pCRE-EGFP in cells overexpressing ICER IIγ. Overexpression of ICER IIγ was induced in HEK293ind-ICER IIγ cells by tetracycline (29). pCRE-EGFP was co-spotted with either a control siRNA targeting CAT (siCAT) or siRNA targeting ICER (siICER) in order to control that the transcriptional repression was indeed caused by ICER. The siICER used was shown in separate experiments to give a specific down-regulation of ICER IIγ expression both in a luciferase fusion reporter plasmid assay and in western blot (data shown in Supplementary Figure 4). An internal control plasmid was not used, as the CMV promoter in pDsRed contains CRE elements (36), and thus is expected to be regulated by the same activating and repressing molecular event as pCRE-EGFP. In preliminary experiments the effect we were looking for was observed to be smaller than the effects detected in the two previous studies. We, therefore, printed 36 technical replicates, and four experimental replicates were performed. In each of the experimental replicates, similar relative differences between the signals measured at the different conditions and treatments were observed, but the effect of ICER IIγ overexpression was small and the variation in the data was large (Figure 6A, left). When the linear regression (ANOVA) model in Equation (2) was used to estimate the effect of ICER IIγ overexpression based on all the data (Figure 6A, right), it could be shown that overexpression of ICER IIγ (+tet) resulted in a significant (P < 0.01) downregulation in CRE-driven EGFP expression of about 30%. In the presence of siICER, this repressing effect was reduced and was no longer significant (P = 0.11), illustrating that the observed effect was, indeed, caused by ICER IIγ. A control experiment was performed showing that overexpression of ICER IIγ (+tet) had no repressing effect on NFκB-driven transcription (Figure 6B).
Figure 6.

Linear regression modelling with a treatment effect for analysis of transcriptional repression. (A) Effect of ICER IIγ overexpression on forskolin-stimulated CRE-driven transcription. Duplicate arrays with spots printed with pCRE-EGFP (50 ng/µl) together with siCAT or siICER (25 ng/µl), and negative control spots (pCRE-Luc, 50 ng/µl) were overlaid with HEK293ind-ICER IIγ cells. The cells on one of the two arrays were treated with 1 µg/ml tetracycline (tet) for 20 h before fixation to induce overexpression of ICER IIγ, and both arrays were stimulated with 10 µM forskolin 6 h before fixation to induce CRE-driven expression. The EGFP fluorescence intensities are expressed relative to tetracycline untreated state for four experimental replicates (left). Mean ± 2SD (33–36 technical replicates after removing flagged spots). The resulting estimated effect of each condition and treatment based on all the data using the linear regression model in Equation (2) (right). Error bars are 95% CI. *P < 0.01; significant difference from tetracycline-untreated cells. (B) Effect of ICER IIγ overexpression on TNFα-stimulated NFκB-driven transcription. The experiment was performed as in A, with pCRE-EGFP substituted by pNFκB-EGFP, and stimulation with TNFα (20 ng/ml) instead of forskolin. EGFP fluorescence intensity relative to the tetracycline-untreated state estimated from three experimental replicates using the linear regression model in Equation (2). Error bars are 95% CI (24–36 technical replicates). The arrays were printed using a 10 µl pipette tip. (C) Simulation of the number of technical and experimental replicates necessary for obtaining a significant (P < 0.01) effect of ICER IIγ overexpression on CRE-driven transcription. We randomly removed 6, 12, 18, 24 and 30 out of 36 technical replicates in each experimental replicate, used the resulting datasets to fit the linear regression model in Equation (2), calculated the P-values for the effect of ICER IIγ overexpression for the condition pCRE-EGFP cotransfected with siCAT, repeated this 1000 times and recorded how many times the P-value was below 0.01. This was done for all four experimental replicates and all combinations of three and two experimental replicates.
A simulation experiment was then performed in order to evaluate the number of technical and experimental replicates necessary to detect the repressing effect of ICER IIγ on CRE-driven transcription. The effect of ICER IIγ overexpression on CRE-driven transcription was still significant (P < 0.01) for all combinations of three and two experimental replicates when all technical replicates were included in the analysis (data not shown). However, removing technical replicates from the dataset resulted in a reduced chance of obtaining a significant effect of ICER IIγ overexpression on CRE-driven transcription (Figure 6C). The simulation experiment indicated that we might have to include more than twelve technical replicates and three to four experimental replicates in order to detect the effect of ICER IIγ overexpression on CRE-driven transcription on transfected cell microarrays using the experimental setup presented here.
Evaluation of the linear regression method
In order to evaluate the use of our linear regression (ANOVA) models, we assessed the value of including the interaction effects in the models, and compared our analysis method with four other possible analysis approaches.
Interaction effects
The inclusion of interaction effects between experimental replicate and condition, between experimental replicate and treatment, and between experimental replicate, treatment and condition, will not influence the estimates of the effect sizes, but will provide improved estimates of the variability of the estimated effect sizes. For the three studies analysed we have compared the full model (all interaction effects included) with a reduced model (only marginal effects and interaction effect between condition and treatment included). We used a likelihood ratio test (42) to investigate the null hypothesis that the reduced model provides an equally good description of the data as the full model. The following results were found: in the analysis of the Dose siRNA study we fitted the model with and without the interaction effect of the condition and experimental replicate. The inclusion of this interaction effect gave a significant improvement to the model (P = 3E-53). For the NFκB study we compared the full model (all interaction effects included) with the model where only the marginal effects and the interaction effect of treatment and condition were present, and found the full model to be superior to the reduced model (P = 3E-8). The same was found for the ICER study (P = 1E-13). This means that by including the interaction effects in our models, we are able to provide a better description of the data than without the interaction effects.
Comparison with other analysis approaches
One common way to present data from biological assays is to show results from only one representative experimental replicate, even though the experiment has been repeated with similar results several times. Statistical analysis is thus performed using data from that experimental replicate alone. However, this results in loss of informative data and a potential selection bias. Using the data from all experimental replicates in the statistical analysis will give a more robust and objective analysis.
The linear regression method was compared with four other analysis approaches (denoted methods A–D), which may be used for analysing fluorescence intensities on transfected cell microarrays based on data from all experimental replicates. We used the data from the ICER study, and looked at the effect size of ICER IIγ overexpression (treatment with +tet) as compared to endogenous ICER IIγ expression (treatment with –tet) on CRE-driven EGFP expression, and calculated the P-values for this comparison using the different methods. The data used in the statistical analysis for methods A–D are:
Method A: Mean spot values for each condition and treatment from each experimental replicate.
Method B: Mean spot values for each condition and treatment normalized to the mean of the unstimulated state (endogenous ICER expression) for each condition in each experimental replicate.
Method C: Spot values from all experimental replicates.
Method D: Spot values from all experimental replicates normalized to the mean of the unstimulated state for each condition in each experimental replicate.
Methods A and B are two possible simplified strategies that only use mean values from each experimental replicate for estimating the effect size and calculating P-values. Method B differ from method A in that it includes a normalization step to achieve fold change values for each experimental replicate in order to correct for potential differences in absolute values that are often seen between experimental replicates (scale factor between the measured values in different experimental replicates on original scale). P-values for methods A and B are calculated using a two-sample two-sided t-test based on these means per experimental replicate and treatment for each condition separately. Methods C and D differ from methods A and B in that they include the variation in the technical replicates in the analysis by using all spot values, instead of only the mean values, from each experimental replicate as input to a two-sample, two-sided t-test to produce P-values. Method C simply uses all spot values from all experimental replicates, while in method D each spot value is first normalized to correct for differences in absolute values between the experimental replicates. For a mathematical description of the different methods we refer to Supplementary Data.
For all methods, the linear regression method and methods A–D, the estimated effect size (fold change) is approximately the same. The methods differ in the calculations of the estimated uncertainty of the estimated effect sizes, which is visible in the calculated P-values for the test that the log effect size equals zero, presented in Table 2. When we used method A or B we did not detect a significant effect of ICER IIγ overexpression on CRE-driven expression (P > 0.05) while, based on other studies (29), we expect ICER IIγ overexpression to result in repression of CRE-driven expression. Analyses using methods C, D and the linear regression method, revealed a significant treatment effect of ICER IIγ overexpression. We also compared the five methods by performing simulation experiments addressing the level of significance for the effect of ICER IIγ overexpression on CRE-driven transcription when removing technical replicates from the dataset (Figure 7, left). Using the mean-based methods (methods A and B), removing technical replicates from the dataset did still not give any significant effect of ICER IIγ overexpression. The linear regression method and methods C and D detected the repressing effect equally well even if 18 out of 36 technical replicates were removed from each experimental replicate. However, with fewer technical replicates, the linear regression method detected the effect more often than methods C and D. In the presence of siICER, as expected, none of the analysis methods gave a significant effect of ICER IIγ induction on CRE-driven transcription (Table 2 and Figure 7, right).
Table 2.
Estimated effect size and P-values for the effect of ICER IIγ overexpression compared to endogenous ICER IIγ overexpression on CRE-driven EGFP-expression calculated using five different methods (LM, A, B, C, D)
| Condition | Estimated effect sizea |
P-values |
||||
|---|---|---|---|---|---|---|
| LMb | Ac | Bc | Cc | Dc | ||
| pCRE-EGFP + siCAT | 0.71 | 5E-10 | 0.20 | 0.06 | 8.4E-7 | 4.4E-8 |
| pCRE-EGFP + siICER | 0.92 | 0.11 | 0.51 | 0.34 | 0.14 | 0.14 |
aOn original scale.
bLM, Linear regression method described in Materials and Methods section (Equation 2).
cMethods A–D are described mathematically in Supplementary Data.
Figure 7.

Evaluation of five different analysis methods by simulation experiments. From the ICER study data, we randomly removed 6, 12, 18, 24 and 30 out of 36 technical replicates in each of four experimental replicates, and used the resulting datasets to calculate P-values for the effect of ICER IIγ overexpression using different analysis methods. For each analysis method, this was repeated 1000 times, and the number of P-values below 0.01 was recorded. The graphs show the effect on CRE-driven transcription for pCRE-EGFP cotransfected with siCAT (left), and pCRE-EGFP cotransfected with siICER (right).
These results show the importance of including all available data in the analysis when aiming at detecting small biological effects. The linear regression method and the methods using all spot values in the analysis detected the small biological effect of ICER IIγ overexpression on CRE-driven EGFP expression, where the mean-based methods could not. In addition, when reducing the number of technical replicates in the dataset, the linear regression method was shown to detect the small biological effect more often than methods C and D. This is due to a reduced variation in the estimated effects using the linear regression method. While methods C and D only use the information in one condition at a time for estimating the variation in the data, the linear regression method uses the whole dataset.
CONCLUDING REMARKS
Transfected cell microarray is a versatile, efficient and cost-reducing technology (43) that can contribute to further understanding of gene functions and regulatory mechanisms governing gene expression by enabling measurements of biological responses in cells with overexpression or downregulation of specific gene products combined with the possibility of assaying effects of external stimuli. We showed the importance of including a sufficient number of replicates and of using all available data in the statistical analysis in order to reliably detect biological effects. Simulation experiments may be valuable for estimating the number of replicates necessary for detecting biological effects of interest, based on data from positive and negative controls for the assay being performed. As the analysis of transfected cell microarrays is in its infancy, no state-of-the-art coherent statistical methodology is currently established. Our linear regression (ANOVA) models have the advantage of providing the research community with a general framework that enables simultaneous use of measurements from all spots and all experimental replicates in a study. Our method takes into account both marginal and interaction effects of treatment, condition and experimental replicate, and reports valid estimates of effect sizes, i.e. fold changes, and significance thereof.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank Hallgeir Bergum and Atle Van Beelen Granlund at the Norwegian Microarray Consortium (NMC) for valuable input and help in the work of establishing transfected cell microarrays in our lab. We also want to thank Marit Otterlei at the Department of Cancer Research and Molecular Medicine, Norwegian University of Science and Technology (NTNU), for valuable discussion, and Anne Kristensen and Kari Slørdahl at the Department of Cancer Research and Molecular Medicine, NTNU, for help with cell cultivation. Funding was provided by The Research Council of Norway; The Norwegian Cancer Society; The Cancer Fund at St Olavs Hospital, Trondheim, Norway; Liaison Committee between the Central Norway Regional Health Authority (RHA) and NTNU. Funding to pay the Open Access publication charges for this article was provided by Liaison Committee between RHA and NTNU.
Conflict of interest statement. None declared.
REFERENCES
- 1.Ziauddin J, Sabatini DM. Microarrays of cells expressing defined cDNAs. Nature. 2001;411:107–110. doi: 10.1038/35075114. [DOI] [PubMed] [Google Scholar]
- 2.Kumar R, Conklin DS, Mittal V. High-throughput selection of effective RNAi probes for gene silencing. Genome Res. 2003;13:2333–2340. doi: 10.1101/gr.1575003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mousses S, Caplen NJ, Cornelison R, Weaver D, Basik M, Hautaniemi S, Elkahloun AG, Lotufo RA, Choudary A, Dougherty ER, et al. RNAi microarray analysis in cultured mammalian cells. Genome Res. 2003;13:2341–2347. doi: 10.1101/gr.1478703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silva JM, Mizuno H, Brady A, Lucito R, Hannon GJ. RNA interference microarrays: high-throughput loss-of-function genetics in mammalian cells. Proc. Natl Acad. Sci. USA. 2004;101:6548–6552. doi: 10.1073/pnas.0400165101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wheeler DB, Carpenter AE, Sabatini DM. Cell microarrays and RNA interference chip away at gene function. Nat. Genet. 2005;37(Suppl):S25–S30. doi: 10.1038/ng1560. [DOI] [PubMed] [Google Scholar]
- 6.Mannherz O, Mertens D, Hahn M, Lichter P. Functional screening for proapoptotic genes by reverse transfection cell array technology. Genomics. 2006;87:665–672. doi: 10.1016/j.ygeno.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 7.Palmer E, Miller A, Freeman T. Identification and characterisation of human apoptosis inducing proteins using cell-based transfection microarrays and expression analysis. BMC Genomics. 2006;7:145. doi: 10.1186/1471-2164-7-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neumann B, Held M, Liebel U, Erfle H, Rogers P, Pepperkok R, Ellenberg J. High-throughput RNAi screening by time-lapse imaging of live human cells. Nat. Methods. 2006;3:385–390. doi: 10.1038/nmeth876. [DOI] [PubMed] [Google Scholar]
- 9.Webb BL, Diaz B, Martin GS, Lai F. A reporter system for reverse transfection cell arrays. J. Biomol. Screen. 2003;8:620–623. doi: 10.1177/1087057103259324. [DOI] [PubMed] [Google Scholar]
- 10.Redmond TM, Ren X, Kubish G, Atkins S, Low S, Uhler MD. Microarray transfection analysis of transcriptional regulation by cAMP-dependent protein kinase. Mol. Cell. Proteomics. 2004;3:770–779. doi: 10.1074/mcp.M400018-MCP200. [DOI] [PubMed] [Google Scholar]
- 11.Tian L, Wang P, Guo J, Wang X, Deng W, Zhang C, Fu D, Gao X, Shi T, Ma D. Screening for novel human genes associated with CRE pathway activation with cell microarray. Genomics. 2007;90:28–34. doi: 10.1016/j.ygeno.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 12.Pannier AK, Ariazi EA, Bellis AD, Bengali Z, Jordan VC, Shea LD. Bioluminescence imaging for assessment and normalization in transfected cell arrays. Biotechnol. Bioeng. 2007;98:486–497. doi: 10.1002/bit.21477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erfle H, Simpson JC, Bastiaens PI, Pepperkok R. siRNA cell arrays for high-content screening microscopy. Biotechniques. 2004;37:454–458. doi: 10.2144/04373RT01. 460, 462. [DOI] [PubMed] [Google Scholar]
- 14.Simpson JC, Cetin C, Erfle H, Joggerst B, Liebel U, Ellenberg J, Pepperkok R. An RNAi screening platform to identify secretion machinery in mammalian cells. J. Biotechnol. 2007;129:352–365. doi: 10.1016/j.jbiotec.2006.12.027. [DOI] [PubMed] [Google Scholar]
- 15.Mishina YM, Wilson CJ, Bruett L, Smith JJ, Stoop-Myer C, Jong S, Amaral LP, Pedersen R, Lyman SK, Myer VE, et al. Multiplex GPCR assay in reverse transfection cell microarrays. J. Biomol. Screen. 2004;9:196–207. doi: 10.1177/1087057103261880. [DOI] [PubMed] [Google Scholar]
- 16.Delehanty JB, Shaffer KM, Lin B. Transfected cell microarrays for the expression of membrane-displayed single-chain antibodies. Anal. Chem. 2004;76:7323–7328. doi: 10.1021/ac049259g. [DOI] [PubMed] [Google Scholar]
- 17.Conrad C, Erfle H, Warnat P, Daigle N, Lorch T, Ellenberg J, Pepperkok R, Eils R. Automatic identification of subcellular phenotypes on human cell arrays. Genome Res. 2004;14:1130–1136. doi: 10.1101/gr.2383804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu YH, Vanhecke D, Lehrach H, Janitz M. High-throughput subcellular protein localization using cell arrays. Biochem. Soc. Trans. 2005;33:1407–1408. doi: 10.1042/BST0331407. [DOI] [PubMed] [Google Scholar]
- 19.Hu YH, Warnatz HJ, Vanhecke D, Wagner F, Fiebitz A, Thamm S, Kahlem P, Lehrach H, Yaspo ML, Janitz M, et al. Cell array-based intracellular localization screening reveals novel functional features of human chromosome 21 proteins. BMC Genomics. 2006;7:155. doi: 10.1186/1471-2164-7-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palmer E, Freeman T. Investigation into the use of C- and N-terminal GFP fusion proteins for sub-cellular localisation studies using reverse transfection microarrays. Comp. Funct. Genomics. 2004;5:342–353. doi: 10.1002/cfg.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allison DB, Cui X, Page GP, Sabripour M. Microarray data analysis: from disarray to consolidation and consensus. Nat. Rev. Genet. 2006;7:55–65. doi: 10.1038/nrg1749. [DOI] [PubMed] [Google Scholar]
- 22.Boutros M, Bras LP, Huber W. Analysis of cell-based RNAi screens. Genome Biol. 2006;7:R66. doi: 10.1186/gb-2006-7-7-r66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hahne F, Arlt D, Sauermann M, Majety M, Poustka A, Wiemann S, Huber W. Statistical methods and software for the analysis of high throughput reverse genetic assays using flow cytometry readouts. Genome Biol. 2006;7:R77. doi: 10.1186/gb-2006-7-8-r77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malo N, Hanley JA, Cerquozzi S, Pelletier J, Nadon R. Statistical practice in high-throughput screening data analysis. Nat. Biotechnol. 2006;24:167–175. doi: 10.1038/nbt1186. [DOI] [PubMed] [Google Scholar]
- 25.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pepperkok R, Ellenberg J. High-throughput fluorescence microscopy for systems biology. Nat. Rev. Mol. Cell. Biol. 2006;7:690–696. doi: 10.1038/nrm1979. [DOI] [PubMed] [Google Scholar]
- 27.Baghdoyan S, Roupioz Y, Pitaval A, Castel D, Khomyakova E, Papine A, Soussaline F, Gidrol X. Quantitative analysis of highly parallel transfection in cell microarrays. Nucleic Acids Res. 2004;32:e77. doi: 10.1093/nar/gnh074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruijter JM, Thygesen HH, Schoneveld OJ, Das AT, Berkhout B, Lamers WH. Factor correction as a tool to eliminate between-session variation in replicate experiments: application to molecular biology and retrovirology. Retrovirology. 2006;3:2. doi: 10.1186/1742-4690-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Misund K, Steigedal TS, Laegreid A, Thommesen L. Inducible cAMP early repressor splice variants ICER I and IIgamma both repress transcription of c-fos and chromogranin A. J. Cell. Biochem. 2007;101:1532–1544. doi: 10.1002/jcb.21267. [DOI] [PubMed] [Google Scholar]
- 30.Amarzguioui M, Holen T, Babaie E, Prydz H. Tolerance for mutations and chemical modifications in a siRNA. Nucleic Acids Res. 2003;31:589–595. doi: 10.1093/nar/gkg147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amarzguioui M, Prydz H. An algorithm for selection of functional siRNA sequences. Biochem. Biophys. Res. Commun. 2004;316:1050–1058. doi: 10.1016/j.bbrc.2004.02.157. [DOI] [PubMed] [Google Scholar]
- 32.Rosner B. Fundamentals of Biostatistics, chapter 12.6. 6th edn. CA: Thomson-Brooks/Cole Belmont; 2006. [Google Scholar]
- 33.Walpole RE, Myers RH, Myers SL, Ye K. Probability and Statistics for Engineers and Scientists, chapter 14.4. 8th edn. 2007. Pearson Prentice Hall, Upper Saddle River. [Google Scholar]
- 34.Chang FH, Lee CH, Chen MT, Kuo CC, Chiang YL, Hang CY, Roffler S. Surfection: a new platform for transfected cell arrays. Nucleic Acids Res. 2004;32:e33. doi: 10.1093/nar/gnh029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baeuerle PA, Baltimore D. NF-kappa B: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 36.He B, Weber GF. Synergistic activation of the CMV promoter by NF-kappaB P50 and PKG. Biochem. Biophys. Res. Commun. 2004;321:13–20. doi: 10.1016/j.bbrc.2004.06.101. [DOI] [PubMed] [Google Scholar]
- 37.Molina CA, Foulkes NS, Lalli E, Sassone-Corsi P. Inducibility and negative autoregulation of CREM: an alternative promoter directs the expression of ICER, an early response repressor. Cell. 1993;75:875–886. doi: 10.1016/0092-8674(93)90532-u. [DOI] [PubMed] [Google Scholar]
- 38.Rosenberg D, Groussin L, Jullian E, Perlemoine K, Bertagna X, Bertherat J. Role of the PKA-regulated transcription factor CREB in development and tumorigenesis of endocrine tissues. Ann. N Y Acad. Sci. 2002;968:65–74. doi: 10.1111/j.1749-6632.2002.tb04327.x. [DOI] [PubMed] [Google Scholar]
- 39.Ohtsubo H, Ichiki T, Miyazaki R, Inanaga K, Imayama I, Hashiguchi Y, Sadoshima J, Sunagawa K. Inducible cAMP early repressor inhibits growth of vascular smooth muscle cell. Arterioscler. Thromb. Vasc. Biol. 2007;27:1549–1555. doi: 10.1161/ATVBAHA.107.145011. [DOI] [PubMed] [Google Scholar]
- 40.Steigedal TS, Bruland T, Misund K, Thommesen L, Laegreid A. Inducible cAMP early repressor suppresses gastrin-mediated activation of cyclin D1 and c-fos gene expression. Am. J. Physiol. Gastrointest. Liver Physiol. 2007;292:G1062–G1069. doi: 10.1152/ajpgi.00287.2006. [DOI] [PubMed] [Google Scholar]
- 41.Ruchaud S, Seite P, Foulkes NS, Sassone-Corsi P, Lanotte M. The transcriptional repressor ICER and cAMP-induced programmed cell death. Oncogene. 1997;15:827–836. doi: 10.1038/sj.onc.1201248. [DOI] [PubMed] [Google Scholar]
- 42.Johnson RA, Wichern DW. Applied Multivariate Statistical Analysis, Section 7.4. 6th edn. 2007. Pearson Prentice Hall, Upper Saddle River. [Google Scholar]
- 43.Starkuviene V, Pepperkok R, Erfle H. Transfected cell microarrays: an efficient tool for high-throughput functional analysis. Expert Rev. Proteomics. 2007;4:479–489. doi: 10.1586/14789450.4.4.479. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.