

Abstract
The importance of hormone therapy in affording protection against the sequelae of global ischemia in postmenopausal women remains controversial. Global ischemia arising during cardiac arrest or cardiac surgery causes highly selective, delayed death of hippocampal CA1 neurons. Exogenous estradiol ameliorates global ischemia-induced neuronal death and cognitive impairment in male and female rodents. However, the molecular mechanisms by which estrogens intervene in global ischemia-induced apoptotic cell death are unclear. Here we show that estradiol acts via the classical estrogen receptors, the IGF-I receptor and the ERK/MAPK signaling cascade to protect CA1 neurons in ovariectomized female rats and gerbils. We demonstrate that global ischemia promotes early dephosphorylation and inactivation of ERK1 and the transcription factor cAMP-response element binding protein (CREB), subsequent downregulation of the anti-apoptotic protein Bcl-2, a known gene target of estradiol and CREB, and activation of caspase-3. Estradiol treatment increases basal phosphorylation of both ERK1 and ERK2 in hippocampal CA1 and prevents ischemia-induced dephosphorylation and inactivation of ERK1 and CREB, downregulation of Bcl-2 and activation of the caspase death cascade. Whereas ERK/MAPK signaling is critical to CREB activation and neuronal survival, the impact of estradiol on Bcl-2 levels is ERK-independent. These findings support a model whereby estradiol acts via the classical estrogen receptors and IGF-I receptors, which converge on activation of ERK/MAPK signaling and CREB to promote neuronal survival in the face of global ischemia.
Keywords: estrogen; IGF-I; MAP, kinase signaling; neuronal, death; apoptosis; global, ischemia
Introduction
Global brain ischemia, arising during cardiac arrest, open heart surgery, profuse bleeding or induced experimentally in animals via bilateral carotid artery occlusion, causes selective, delayed neuronal death and delayed neurological deficits (reviewed in (1). Pyramidal neurons in the hippocampal CA1 are particularly vulnerable, whereas interneurons in this cell layer and pyramidal neurons in other hippocampal subfields survive. Histological evidence of CA1 pyramidal neuron degeneration is not observed until 2–3 days after global ischemia in rats or 3–4 days in gerbils (2–5). Although the mechanisms underlying ischemia-induced death are as yet unclear, the substantial delay between insult and onset of death provides the opportunity to examine molecular events that destine these neurons to die.
Estradiol-17β, the primary estrogen secreted by the ovaries, acts on neurons to increase spine density and synapse number (6–8) and NMDA receptor NR1 subunit expression (9) and potentiates kainate-elicited currents in CA1 pyramidal neurons (10, 11). Moreover, estrogens afford neuroprotection in experimental models of global and focal ischemia (12–15) and ameliorate the cognitive deficits associated with ischemic cell death (16, 17). Although the cellular sites mediating these actions are unclear, estrogen receptors-α and -β are expressed in the hippocampus where they could subserve the neuroprotective actions of estradiol (18).
Crosstalk between estradiol and trophic factors such as IGF-I is implicated in the cellular actions of estradiol. Estradiol and IGF-I act synergistically in neurons to regulate synaptic remodeling, neuronal differentiation and neuronal survival (19). IGF-I receptors are critical to estradiol protection of hilar neurons from seizure-induced injury (20). In the brain, estradiol and IGF-I activate ERK/MAPK (19), a well-characterized intracellular signaling cascade implicated in neuronal plasticity and survival (13, 21, 22). Upon stimulation with estradiol, estradiol receptor-α and IGF-I receptor form a macromolecular signaling complex, which recruits and activates downstream kinases including MAPK (23–25). ERK/MAPK signaling culminates in phosphorylation and activation of nuclear transcription factors such as cAMP-response element binding protein (CREB), which regulate target genes important to neuronal survival and protection. CREB targets implicated in neuronal survival include the anti-apoptotic protein Bcl-2 and brain-derived neurotrophic factor (BDNF) (21).
The present study sought to identify molecular targets, especially intracellular signaling cascades, which mediate estradiol neuroprotection in global ischemia. We show that estradiol acts via classical estrogen receptors, IGF-I receptors and ERK/MAPK signaling to promote neuronal survival after transient global ischemia. Global ischemia promotes dephosphorylation and inactivation of ERK1 and its target, the transcription factor CREB, followed by downregulation of Bcl-2 and activation of the caspase death cascade. Estradiol markedly attenuates ischemia-induced dephosphorylation and inactivation of ERK1 and CREB, downregulation of Bcl-2 and activation of caspase-3. Pharmacological blockade of ERK reverses the effects of estradiol on both ERK and CREB phosphorylation, but not on Bcl-2. Thus, estradiol and IGF-I coordinately activate ERK/MAPK signaling and its target CREB, thereby promoting neuronal survival in the face of global ischemia.
Materials and Methods
Animals
Age-matched adult female Mongolian gerbils weighing 60–80 g (Tumblebrook Farms, Wilmington, MA) and female Sprague-Dawley rats weighing 100–150 g (Charles River, Wilmington, DE) at the time of ischemic insult were maintained in a temperature and light-controlled environment with a 14 hr light/10 hr dark cycle and were treated in accordance with the principles and procedures of the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Protocols were approved by the Institutional Animal Care and Use Committee of the Albert Einstein College of Medicine.
Ovariectomy and estradiol pellet implantation
Female Mongolian gerbils were ovariectomized and female Sprague-Dawley rats were ovariohysterectomized under halothane anesthesia (4% for induction, followed by 1% for maintenance). Seven days later, pellets containing estradiol-17β (gerbils: 0.36 mg/pellet, 60 days sustained release; rats: 0.05 mg/pellet, 21-day sustained release; Innovative Research of America, Inc.; Sarasota, FL) or placebo were inserted subcutaneously beneath the dorsal surface of the neck. Estradiol pellets were present for 14 days before ischemia and remained in place until sacrifice at 1 hr to 2 days after ischemia for molecular studies or 7 days after ischemia for histological analysis.
Global ischemia
Experiments were performed in rats, with the exception of the estrogen receptor and IGF-I receptor antagonist treatments, which were performed in gerbils. Gerbils offer an advantage compared with rats in that they lack posterior communicating arteries, structures that in humans and rats complete the circle of Willis and permit collateral blood flow via the vertebral arteries. Thus, global ischemia can be produced in gerbils by the relatively simple procedure, bilateral occlusion of the carotid arteries. However, many of the available antibodies used in the present study recognize rat (but not gerbil) proteins with high affinity and good signal-to-noise. Therefore, we carried out most experiments in rats using 4-vessel occlusion (see below).
Fourteen days after pellet implantation, gerbils were fasted overnight and anesthetized with halothane (4% for induction, followed by 1% for maintenance) delivered by mask in a mixture of N2/02 (70:30) by means of a Vapomatic anesthetic vaporizer (CWE Inc., Ardmore, PA). Gerbils were subjected to global ischemia by temporary occlusion of both carotid arteries for 5 min with nontraumatic aneurism clips followed by reperfusion as described (26) or to sham operation. Sham-operated animals were subjected to the same anesthesia and surgical procedures as animals subjected to global ischemia, except that the carotid arteries were not occluded. Rats were subjected to global ischemia by four-vessel occlusion as described (27). In brief, 13 days after pellet implantation, rats were fasted overnight and anesthetized with halothane as described above for gerbils. The vertebral arteries were subjected to electrocauterization, the common carotid arteries were exposed and isolated with a 3-0 silk thread, and the wound was sutured. Twenty-four hours later, the animals were anesthetized again, the wound was reopened and both carotid arteries were occluded for 10 min with nontraumatic aneurism clips, followed by reperfusion. Arteries were visually inspected to ensure adequate reflow. Sham-operated rats were subjected to the same anesthesia and surgical procedures as animals subjected to global ischemia (vertebral artery coagulation and carotid artery exposure), except that the carotid arteries were not occluded.
In all cases, anesthesia was discontinued immediately after initiation of carotid artery occlusion. Body temperature was monitored and maintained at 37.5 ± 0.5 °C with a rectal thermistor and heat lamp until recovery from anesthesia. Animals that failed to show complete loss of the righting reflex and dilation of the pupils from 2 min after occlusion was initiated until the end of occlusion and the rare animals that exhibited obvious behavioral manifestations (abnormal vocalization when handled, generalized convulsions, hypoactivity) or loss of > 20% body weight by 3–7 days were excluded from the study. After reperfusion, arteries were visually inspected to ensure adequate flow. Sixty gerbils and 175 rats were subjected to global ischemia. There were 7 deaths in gerbils and 3 deaths in rats due to respiratory arrest. Another 2 gerbils and 18 rats were excluded from the study because they failed to show neurological signs of ischemia (no loss of consciousness or incomplete dilation of the pupils during occlusion). Rats and gerbils that died from respiratory arrest or failed to show neurological signs of ischemia were not concentrated in either the placebo or estradiol-treated groups.
ICI 182,780, JB-1 and PD98059 intracerebroventricular administration
Halothane-anesthetized animals were injected with the broad-spectrum estrogen receptor antagonist ICI 182,780 (100 μg, Tocris; Ellisville, MO), the competitive IGF-I receptor antagonist JB-1 (10 μg, Bachem Inc.; Budendorf, Switzerland) or the ERK/MAPK inhibitor PD98059 (3 μg, Calbiochem, La Jolla, CA) or vehicle (10 μl of 50% DMSO, saline or 10% DMSO, respectively) by unilateral injection into the right lateral ventricle at a flow rate of 5 μl/min immediately after global ischemia (14 days after pellet implantation) or sham surgery and again 12 hr later. Intracerebroventricular (icv) injections of 75% DMSO have no obvious harmful effects (28). Animals were positioned in a Kopf small animal stereotaxic frame with the incisor bar lowered 3.3 ± 0.4 mm below horizontal zero. A stainless steel cannula (28 gauge) was lowered stereotaxically into the right lateral ventricle to a position defined by the following coordinates: 0.4 mm posterior to bregma, 1.2 mm lateral to bregma, 2.6 mm below the skull surface (gerbils) or 0.92 mm posterior to bregma, 1.2 mm lateral to bregma, 3.6 mm below the skull surface (rats) according to the atlas of Paxinos and Watson (29).
Histological analysis
Neuronal cell loss was assessed by histological examination of toluidine blue-stained brain sections at the level of the dorsal hippocampus from animals sacrificed at 7 days after ischemia (21 days after pellet implantation) or sham operation as described (27). Animals were deeply anesthetized with pentobarbital (50 mg/kg, ip), and blood was collected by cardiac puncture for assay of plasma estradiol levels (see below); this was followed by transcardiac perfusion with ice cold 4% paraformaldehyde in PBS (0.1 M, pH 7.4). Brains were removed and immersed in fixative (4°C overnight). Coronal sections (15 μm) were cut at the level of the dorsal hippocampus (3.3 to 4.0 mm posterior from bregma) with an electronic cryotostat (Thermo Electron Corporation, Pittsburgh, PA), and every fourth section was collected and stained with toluidine blue. The number of surviving pyramidal neurons per 250-μm length of the medial CA1 pyramidal cell layer was counted bilaterally in 4 sections per animal as described (30) under a light microscope at 40X magnification. Cell counts from the right and left hippocampus on each of the four sections were averaged to provide a single value (number of neurons/250 μm length) for each animal.
Serum estradiol assay
Tubes containing whole blood were placed on ice (10 min) and centrifuged at 300 X g for 5 min. Serum was collected and stored (−20°C) until analyzed. Serum hormone levels were measured by fluoroimmunoassay using the DELPHIA estradiol assay (Perkin Elmer Life Sciences; Turku, Finland). All assays were performed in duplicate, and the mean value reported. The sensitivity of detection is 13 pg/ml. The inter- and intra-assay coefficients of variance are 10.1% and 4.1%, respectively.
Antibodies
The following antibodies were used in this study: 1) Bcl-2, an affinity purified polyclonal antibody raised against a peptide mapping within the amino terminus of Bcl-2 of human origin (1:100 for immunolabeling, 1:1000 for Western, Santa Cruz Biotechnology, Inc., Santa Cruz, CA); 2) anti-phospho MAPK (p-ERK1/2) mouse monoclonal antibody, which recognizes ERK1 and ERK2 that are phosphorylated on both a threonine and a tyrosine residue (clone 12D4, 1:5000, Upstate Biotechnology, Inc., Lake Placid, NY); 3) anti-MAPK1/2 (ERK1/2) rabbit polyclonal antibody, which recognizes total ERK1/ERK2 (1:5000, Upstate Biotechnology, Inc.); 4) anti-ERK5 and phospho-ERK5 (p-Thr218/p-Tyr220) rabbit polyclonal antibodies (ERK5, 1:500; p-ERK5, 1:200; Cell Signaling Technology); 5) anti-phospho-CREB (p-CREB) rabbit polyclonal antibody, which recognizes CREB phosphorylated at serine 133 (1:3000; Upstate Biotechnology, Inc.); 6) anti-CREB rabbit polyclonal antibody, which recognizes total CREB (1:3000; Upstate Biotechnology, Inc.); 7) anti-β-actin mouse monoclonal antibody, which recognizes an epitope located within the N-terminal domain of the β-isoform of actin (1:20000; Sigma, Saint Louis, MI); 8) anti-histone H3 rabbit polyclonal antibody, which recognizes endogenous histone H3 protein only (1:1000; Cell Signaling Technology, Inc., Beverly, MA). Secondary antibodies for Westerns were horseradish peroxidase (HRP)-conjugated donkey anti-rabbit IgG (1:5000, Amersham, Buckinghamshire, England, UK) for polyclonal antibodies, or sheep anti-mouse IgG (1:2500, Amersham) for monoclonal antibodies.
Western blot analysis
For quantification of protein abundance in the hippocampal CA1, whole-cell lysates (ERK1/2, pERK1/2), cytosolic (Bcl-2) and nuclear (CREB, p-CREB) fractions isolated from the micro-dissected hippocampal CA1 subfield of experimental and sham animals were prepared at 1, 3, 12, 24 and 48 hr after reperfusion. Proteins were separated by SDS-PAGE and subjected to Western blot analysis as described (27). Protein concentration was determined by BCA protein assay kit (Pierce, Rockford, IL). Aliquots of protein (40–70 μg) were dissolved in Laemmli sample buffer (0.025 M Tris-HCI, 5% glycerol, 1% SDS, 0.5% PBS, 0.1 M dithiothreitol, 2.5 mM β-mercaptoethanol, 1 mM PMSF, 0.5 mM NaHNO3 buffer, pH 6.8), loaded on 10% polyacrylamide gels, subjected to electrophoresis and transferred to nitrocellulose membranes for immunolabeling with antibodies to p-ERK1/2, ERK1/2, p-ERK5, ERK5, p-CREB, CREB or Bcl-2. After reaction, membranes were treated with enhanced chemiluminescence reagents (ECL, Amersham Life Science) and apposed to XAR-5 X-ray film (Eastman Kodak Co., Rochester, NY). Membranes were re-probed with anti-β-actin antibody as a loading control. In Western experiments to monitor p-CREB, the purity of the nuclear and cytosolic fractions was routinely monitored by re-probing membranes with anti-histone H3 antibody (nuclear marker; Fig. 4c). Whereas histone H3 labeling was strong in the nuclear fractions, it was absent in all cytosolic fractions examined.
Fig. 4. Estradiol acts via MAPK signaling to prevent ischemia-induced CREB dephosphorylation.
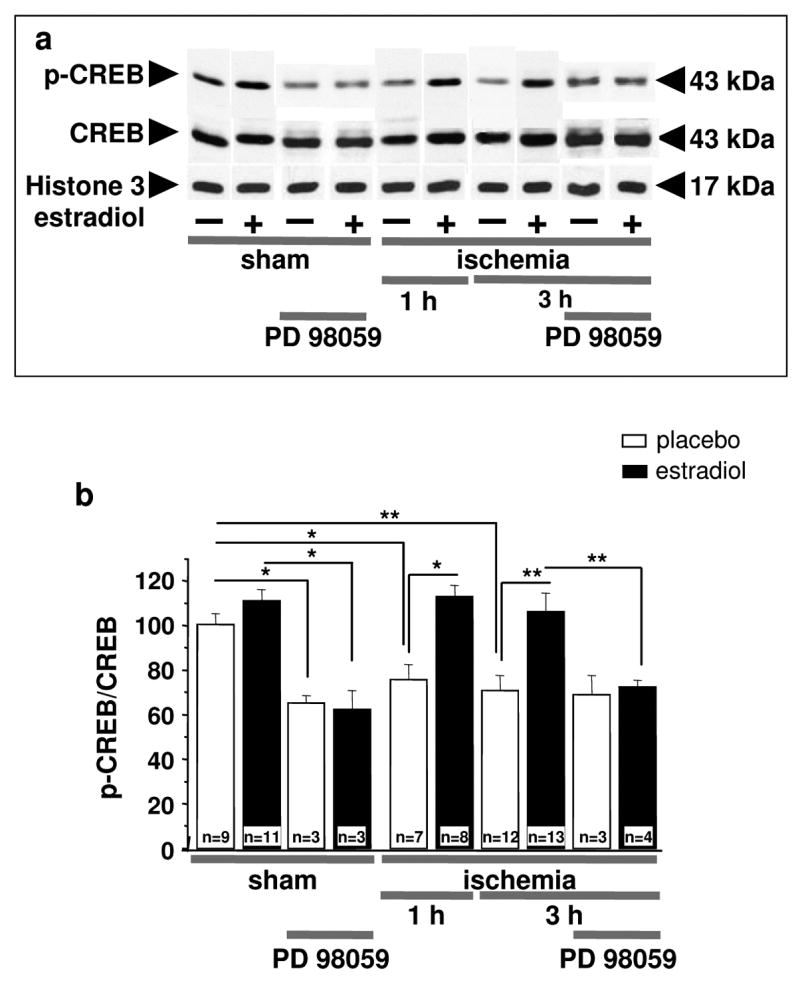
Representative Western blots (a) and relative p-CREB abundance (b) in the nuclear fraction of CA1 of placebo- and estradiol-treated rats subjected to sham operation or ischemia at 1 and 3 hr after surgery. Subsets of animals were injected with PD98059 at 0 h and sacrificed at 3 h after surgery. Westerns were probed with antibodies to p-CREB, CREB and histone 3 (nuclear marker). Ischemia promoted dephosphorylation of CREB in the nuclei of CA1 neurons. Estradiol maintained the phosphorylated, activated state of CREB in the CA1 of ischemic rats. PD98059 blocked the ability of estradiol to maintain p-CREB levels in post-ischemic CA1. Values for experimental animals were normalized to the corresponding sham value. **, P < 0.01.*, P < 0.05.
To quantitate protein abundance, bands on Western blots were analyzed with a Scan Jet 4-C computing densitometer using NIH IMAGE 1.61 software. Band densities for p-CREB or p-ERK1 and p-ERK2 were corrected for variations in loading and normalized to the corresponding band densities for total CREB or total ERK1 and ERK2, respectively; normalized means were expressed as a percentage of the corresponding value for control (sham-operated) animals. Band densities for Bcl-2 were expressed as a percentage of the corresponding value for control animals. Statistical comparisons were assessed by analysis of variance (ANOVA), followed by the Newman-Keuls test. Because of the large number of treatment groups, which included two surgical conditions (sham vs. ischemia), two hormone treatments (estradiol vs. placebo), multiple time points after surgery, and two drugs (PD98059 vs. vehicle), it was not always possible to run samples for all conditions on a single gel. Therefore, to enable comparisons from experiment to experiment, band densities for all samples on a given gel were normalized to the band density for a sample from an animal treated with placebo, subjected to sham operation and killed 1 hr after surgery (“control”). Each gel included at least one sample from such a control animal to enable comparisons of data across different experiments, and a different control animal was prepared for each experiment.
Immunolabeling
Animals were deeply anesthetized with pentobarbital (50 mg/kg, i.p.) and perfused transcardially with 4% paraformaldehyde in phosphate buffer (0.1 M, pH 7.4) at 24 hr and 48 hr after ischemia or sham operation (n = 3 independent experiments for each time point and treatment group). Brains were removed, postfixed (2 hr at 4°C), frozen and cut into sections (40 μm) in the coronal plane of the dorsal hippocampus (3.3 to 4.0 mm posterior from bregma. Free-floating sections were blocked in 10% normal serum, 5% bovine serum albumin and 0.01% saponin in PBS (2 hr at room T) and processed for immunolabeling with anti-Bcl-2 polyclonal antibody, overnight at 4°C, followed by biotinylated goat anti-rabbit IgG (1:200; Vector Laboratories, Burlingame, CA). Sections were then incubated with avidin peroxidase complex (ABC kit, Vector Laboratories, 1 hr at room T), followed by 3–3′-diaminobenzidine (DAB, Vector Labs). Images were viewed through a Nikon inverted microscope ECLIPSE TE300 and images acquired with a SPOT RT CCD-cooled camera with Diagnostic Software version 3.0 (Diagnostic Instruments, Inc. Sterling Heights, MI).
Caspase activity assay
Caspase activity assays were performed on fresh frozen brain sections using an APO LOGIXTM carboxyfluorescein (FAM) caspase detection kit (Cell Technology, Minneapolis, MN) according to manufacturer’s instructions. FAM-DEVD-FMK is a carboxy-fluorescein analog of zDEVD-fluoromethyl ketone (FMK), a broad-spectrum cysteine protease inhibitor that enters cells and irreversibly binds activated caspases (31–33). FAM-DEVD-FMK exhibits higher affinity for caspase-3 than for caspase-8, caspase-7, caspase-10 or caspase-6 (34) and exhibits much lower affinity for the calpains than for caspases; thus, at 5 μM FAM-DEVD is a relatively selective inhibitor of caspase-3. Moreover, FAM-DEVD-FMK labeling of CA1 neurons correlates well with caspase-3 activation, as assessed by Western blot analysis. In this study we therefore refer to FAM-DEVD-FMK labeling as indicative of caspase-3 activity. In brief, animals were deeply anesthetized with pentobarbital (50 mg/kg, i.p.) and killed by decapitation at 24 hr after ischemia or sham operation (control). Brains were removed, frozen and cut into sections (18 μm) in the coronal plane of the dorsal hippocampus. Brain sections (3 per animal) were labeled with 5 μM FAM-DEVD-FMK (1 hr, 37°C), washed three times with 1X Working Dilution Wash Buffer and viewed under a Nikon ECLIPSE TE-300 fluorescent microscope equipped with an image analysis system at an excitation wavelength of 488 nm and emission wavelength of 565 nm. Images were acquired with a SPOT RT CCD-cooled camera with Diagnostic Software version 3.0. For quantitation of caspase-3 activity, the fluorescence intensity within the entire hippocampal CA1 cell layer of the images was analyzed using NIH Image 1.61. The mean fluorescence intensity of CA1 in the right and left hemisphere from each of the three sections was averaged to provide a single value for each animal.
Statistical analysis
The results were expressed as mean ± SEM. Data analysis was performed using PHAR/PCSv 4.2. Statistical comparisons were made between groups using a one-way ANOVA followed by Newman-Keuls posthoc analysis (neuron counts, immunoblots and caspase-3 activity). T-test was used for the serum estradiol data. Differences were considered significant at P < 0.05.
Results
Estradiol acts via the classic estrogen receptors and IGF-I receptor to protect CA1 neurons
The intracellular signaling pathways that mediate the neuroprotective actions of estradiol in hippocampal neurons subjected to global ischemia are not well delineated. To address this issue, we treated ovariectomized gerbils with estradiol or placebo for 14 days, subjected animals to sham operation or global ischemia, and administered the broad-spectrum estrogen receptor antagonist ICI 182,780 (ICI, 100 μg in 50% DMSO, icv) or vehicle (50% DMSO) into the lateral ventricles at 0 and 12 hr after ischemia or sham surgery. Global ischemia induced extensive death of pyramidal cells in the hippocampal CA1 evident at 7 days (P < 0.01 vs. sham animals); the few remaining pyramidal neurons were severely damaged and appeared pyknotic (compare Fig. 1b,g with Fig. 1a,f, Fig. 1k). Qualitative analysis revealed that neuronal death was specific in that little or no cell loss occurred in the nearby CA2 or transition zone, CA3 or dentate gyrus (Fig. 1b). Estradiol reduced the loss of CA1 neurons by approximately 60% (P < 0.01 vs. ischemia, Fig. 1c,h,k). Plasma estradiol levels at the time of death were 15.4 ± 1.4 pg/ml in the placebo group and 136.35 ± 12.9 pg/ml in the estradiol group. The estrogen receptor antagonist ICI 182,780 did not detectably alter the appearance or number of surviving neurons in placebo or estradiol-treated animals subjected to sham surgery (see Supplementary Fig. 1), but ICI 182,780 abrogated the neuroprotective action of estradiol in CA1, assessed at 7 days after ischemia (P < 0.01 vs. estradiol, Fig. 1d,i,k). Moreover, the vehicle for ICI 182,780 had no effect on surviving neurons in either sham-operated or ischemic animals (Fig. 1k and Supplementary Fig. 1). These findings indicate that estradiol protection of hippocampal neurons requires activation of the classical estrogen receptors-α and/or -β, and is similar to our findings with female rats (30).
Fig. 1. The estrogen receptor antagonist ICI 182,780 and IGF-I receptor antagonist JB-1 block estradiol protection.
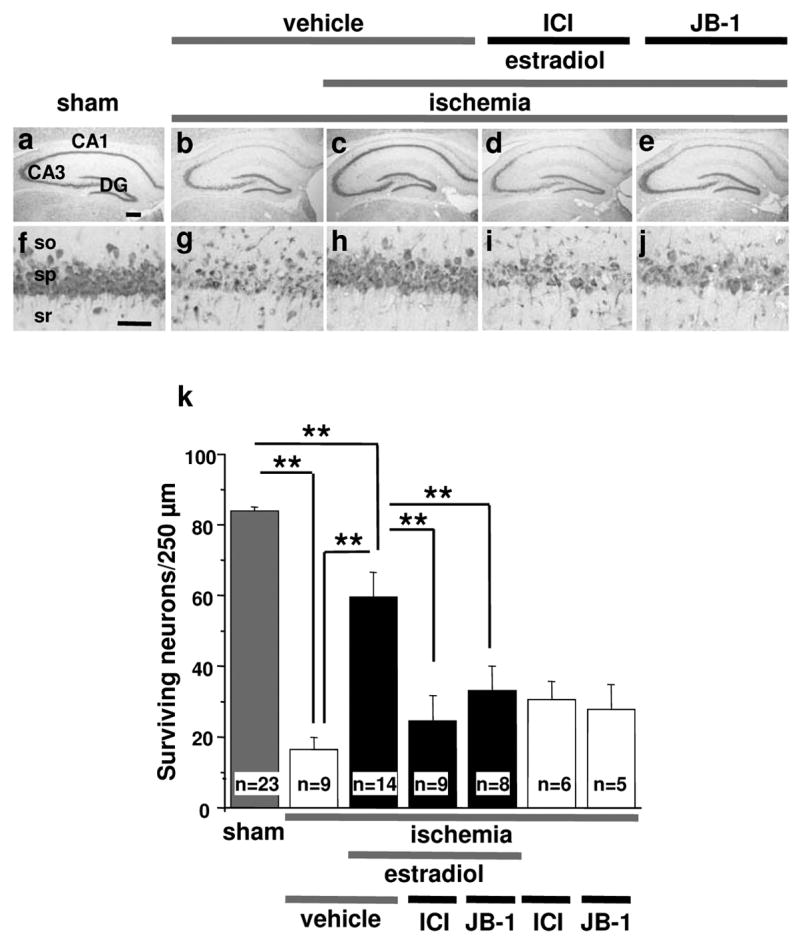
Ovariectomized female gerbils were treated with estradiol or placebo for 14 days and subjected to global ischemia or sham operation. Animals received the estrogen receptor antagonist ICI 182,780 (ICI, 100 μg in 50% DMSO), the IGF-I receptor antagonist JB-1 (10 μg in saline) or vehicle (50% DMSO or saline) icv at 0 and 12 hr after ischemia. ICI 182,780 and JB-1 did not affect neuronal survival in ischemic (two rightmost bars in k) or sham animals (Supplementary Fig. 1), but greatly reduced estradiol protection, assessed at 7 days after surgery (d,i,e,j,k). Neither vehicle significantly altered neuronal survival vs. no treatment in placebo- or estradiol-treated animals subjected to sham operation. Therefore, vehicle and control data were pooled for purposes of illustration (grey bar labeled “sham”; see grey bars in Supplementary Fig. 1 for different groups). Similarly, neither vehicle significantly altered neuronal survival vs. no treatment in placebo-treated animals subjected to global ischemia. Thus, these data were also pooled (white bar labeled “ischemia+vehicle”; see white bars in Supplementary Fig. 1 for different groups). so, stratum oriens; sp, stratum pyramidale; sr, stratum radiatum. Scale bars: lower magnification, 400 μm; higher magnification, 60 μm. (**, P < 0.01).
To examine a possible role for brain IGF-I receptor activation in estradiol protection of CA1 neurons, we administered the highly selective and potent IGF-I receptor antagonist JB-1 (10 μg in saline) or vehicle (saline) icv at 0 and 12 h after reperfusion. JB-1 is a peptide analogue of IGF-I corresponding to the recognition motif or “D domain” within the carboxy-terminus of IGF-I that binds the IGF-I receptor. JB-1 effectively blocks IGF-I receptor-mediated autophosphorylation and cellular proliferation (35). JB-1 did not detectably alter the number or appearance of neurons in placebo-treated animals subjected to global ischemia (Supplementary Fig. 1), but abolished estradiol protection of CA1 neurons, assessed at 7 days after ischemia (P < 0.01 vs. estradiol, Fig. 1e,j,k). Saline and JB-1 alone had no effect on surviving neurons in either sham or ischemic animals (Fig. 1k, see also Fig. 1 legend and Supplementary Fig. 1). These findings indicate that brain IGF-I receptors are critical to estradiol protection of gerbil hippocampal neurons in global ischemia.
MAPK signaling is critical to estradiol protection of CA1 neurons
Because estradiol and IGF-I are known upstream regulators of ERK/MAPK signaling in hippocampal neurons (19, 36), we next examined a possible role for MAPK signaling in estradiol protection. Many of the available antibodies used in the present study recognize rat (but not gerbil) antigens with high affinity and good signal-to-noise. Thus, these experiments were conducted in rats. Ovariectomized female rats pretreated with estradiol or placebo for 14 days were subjected to global ischemia or sham operation, and the MAPK kinase (MEK) inhibitor PD98059 or vehicle was administered into the lateral ventricle at 0 and 12 hr after surgery. Global ischemia induced extensive death of pyramidal cells in the hippocampal CA1 at 7 days post-ischemia (P < 0.01 vs. sham; compare Fig. 2d,k with Fig. 2a,h; Fig. 2o). As observed for gerbils, estradiol did not detectably alter the appearance or number of CA1 neurons in sham-operated rats (Fig. 2b,i,o), but greatly reduced the ischemia-induced neuronal loss (P < 0.01 vs. ischemia, Fig. 2f,m,o). Plasma estradiol levels at the time of death were 20.6 ± 1.3 pg/ml in the placebo group and 58.1 ± 3.8 pg/ml in the estradiol group. The MEK inhibitor PD98059 did not detectably alter the number or appearance of surviving neurons in sham-operated rats (Fig. 2c,j,o), but abrogated the neuroprotective action of estradiol in the hippocampal CA1 (P < 0.01 vs. estradiol alone, Fig. 2g,n,o). In contrast, PD98059 did not affect neuronal survival in placebo-treated animals subjected to sham surgery (Fig. 2c,j,o) or ischemia (Fig. 2e,I,o). These findings indicate that PD98059 administered icv is neither toxic nor protective in the global ischemia model. Consistent with this, PD98059 does not impair locomotion, and its inhibitory actions on hormone-dependent behaviors are readily reversible (37, 38). Together, these findings indicate that ERK/MAPK signaling is critical to estradiol protection of hippocampal neurons in a rat model of global ischemia.
Fig. 2. The ERK/MAP kinase inhibitor PD98059 attenuates estradiol protection.
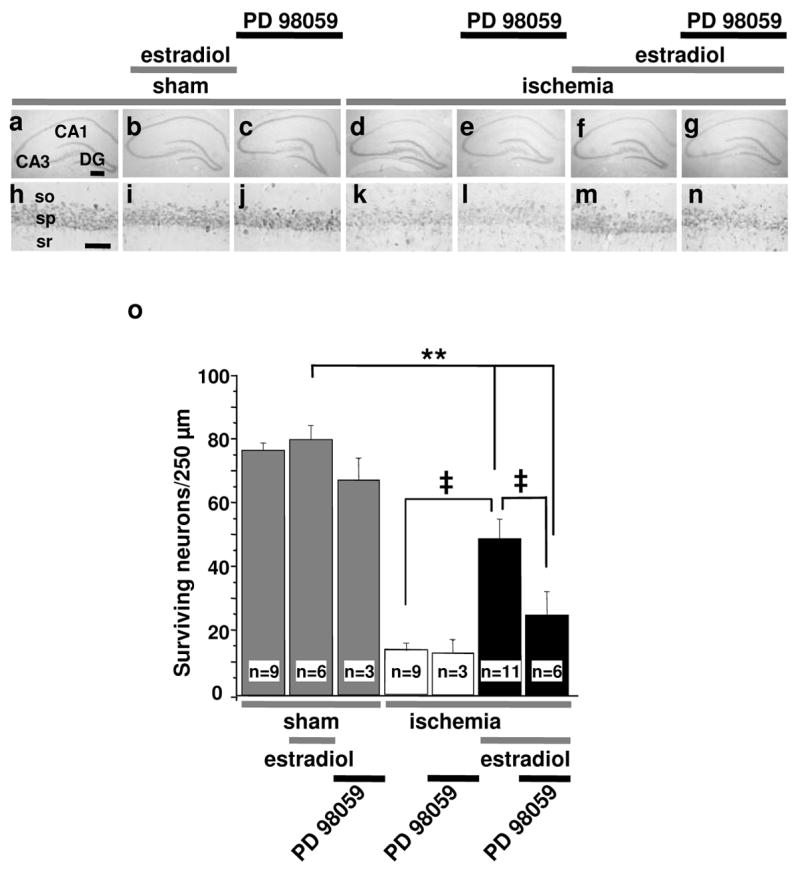
Ovariectomized female rats were treated with estradiol or placebo for 14 days and subjected to global ischemia (white and black bars) or sham operation (grey bars). Animals received PD98059 (3 μg) or vehicle (10% DMSO) icv at 0 and 12 hr after ischemia. Global ischemia induced extensive death of pyramidal cells in the hippocampal CA1 at 7 days post-ischemia (P < 0.01 vs. sham). PD98059 greatly reduced estradiol protection, assessed at 7 days after surgery. Abbreviations as in Fig. 1. Scale bars: lower magnification, 400 μm; higher magnification, 60 μm. **, P < 0.01 vs. all sham groups; ‡, P < 0.01 ischemia+estradiol vs. ischemia or ischemia+estradiol+PD98059.
Estradiol prevents dephosphorylation and inactivation of ERK1 in post-ischemic CA1 neurons
Because PD98059 administered at 0 and 12 hr after ischemia blocked the protective actions of estradiol, we reasoned that ischemia might inactivate ERK/MAPK in the first few hours after insult. To examine the effects of ischemia and estradiol on MAPK activity, we subjected rats that had been treated with estradiol or placebo for 14 days to global ischemia or sham operation and examined ERK1/2 and p-ERK1/2 abundance in CA1 at 1, 3 and 24 hr after reperfusion. Global ischemia significantly reduced phosphorylation of ERK1 in CA1, evident at 1 hr after ischemia (reduction to 64% of control; P < 0.05; Fig. 3a,b); at 3 hr, p-ERK1 levels were not significantly different from controls (Fig. 3a,b). Estradiol significantly increased ERK1 and ERK2 phosphorylation in sham-operated animals (ERK1, increase to ~200% of control, P < 0.01, Fig. 3a,b; ERK2, increase to ~142% of control, P < 0.05, Fig. 3a,c) and prevented the early ischemia-induced dephosphorylation of ERK1 (Fig. 3a,b). Whereas p-ERK1 was reduced at 1 hr after ischemia, it was increased significantly in the CA1 at 24 hr after ischemia vs. sham-operated animals (Fig. 3d). In contrast, global ischemia did not significantly alter ERK2 phosphorylation at any times examined (Fig. 3a,c,e).
Fig. 3. Estradiol prevents ischemia-induced dephosphorylation of ERK1 in CA1.
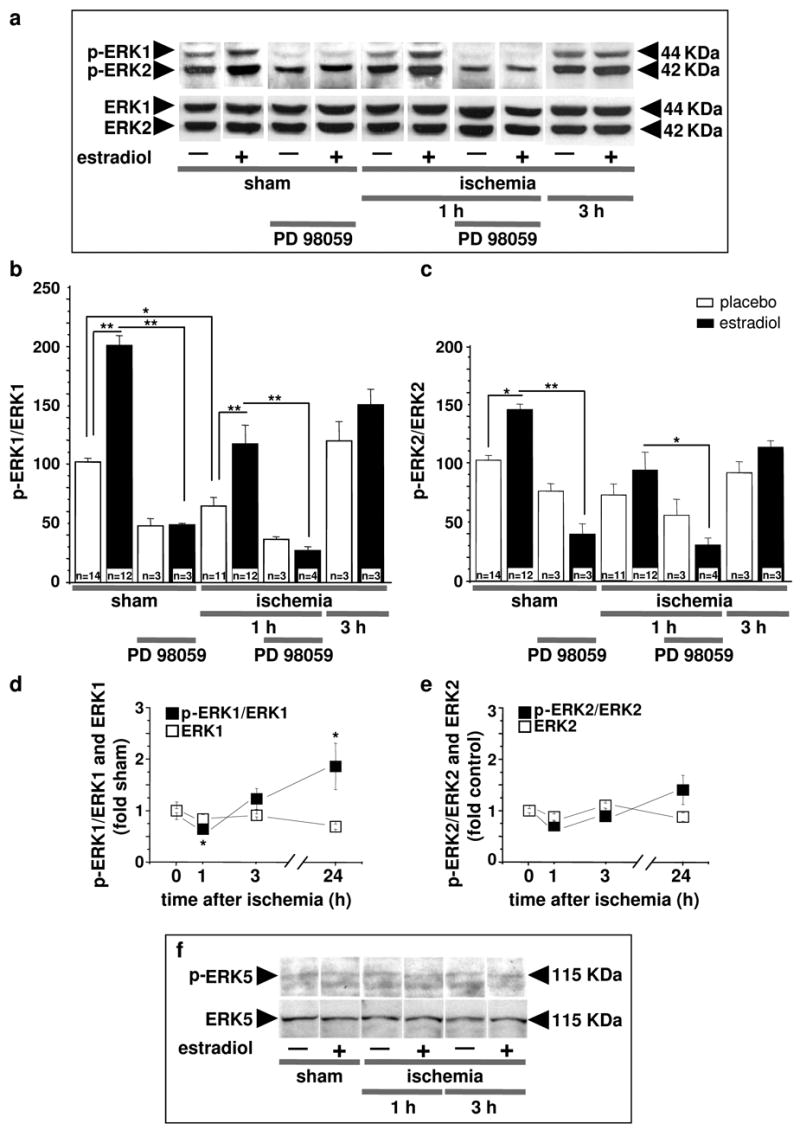
Representative Western blots (a) and relative abundance of p-ERK1 (b) and p-ERK2 (c) in CA1 whole-cell lysates from placebo- and estradiol-treated rats subjected to sham surgery or ischemia at 1, 3, or 24 hr after surgery. Subsets of animals were injected with PD98059 at 0 h and sacrificed at 1 hr after surgery. Westerns were probed with antibodies to p-ERK1/2 and ERK1/2. Ischemia induced dephosphorylation of ERK1 (a,b), but not ERK2 (a,c). Estradiol significantly enhanced ERK1 (b) and ERK2 (c) phosphorylation in shams and maintained levels of p-ERK1 (b) in ischemic rats. PD98059 blocked the estradiol-induced increase in phosphorylation of ERK1 (a,b) in control (sham) CA1. PD98059 also reversed the ability of estradiol to maintain ERK1 phosphorylation in post-ischemic CA1 (a,b). ERK1 dephosphorylation was maximal at 1 hr after ischemia; by 24 hr, ERK1 phosphorylation was increased relative to that of sham animals (d). In the absence of estradiol treatment, ERK2 and pERK2 levels were unchanged at all times examined after ischemia (e). Neither ischemia nor estradiol treatment altered levels of ERK5 or pERK5 in CA1 at 1 or 3 hr after ischemia (f); ERK5 data are representative of 4–5 animals per group. For panels b–e, values for ischemic rats were normalized to the corresponding sham value. *, P < 0.05; **, P < 0.01.
Because PD98059 at high concentrations can also inhibit MEK5 and activation of ERK5 (39), we assessed whether ERK5 phosphorylation was modified by ischemia or estradiol. Neither global ischemia nor estradiol significantly altered abundance or phosphorylation of ERK5 at 1 or 3 hr after insult (Fig. 3f). These findings indicate that global ischemia induces dephosphorylation of ERK1 (but not ERK2 or 5) in the early postischemic period and that estradiol maintains p-ERK1 at higher levels than would otherwise be observed in the post-ischemic period. PD98059 administered immediately after reperfusion blocked the estradiol-induced increase in phosphorylation of ERK1 (Fig. 3a,b) and ERK2 (Fig. 3a,c) in sham-operated animals and reversed the ability of estradiol to maintain ERK1 phosphorylation in post-ischemic CA1, assessed at 1 hr after surgery (Fig. 3a,b). These findings demonstrate that PD98059 infused into the ventricles reaches the hippocampal CA1 and effectively inhibits its target MEK1/2.
Estradiol prevents ischemia-induced dephosphorylation and inactivation of CREB in CA1
A well-characterized downstream target of ERK/MAPK signaling is the transcription factor CREB, which promotes transcription of a number of pro-survival genes. ERK/MAPK rapidly induces sustained phosphorylation of CREB at serine 133 (40). To examine whether estradiol regulates phosphorylation and activation of CREB, we subjected estradiol- and placebo-treated rats to global ischemia or sham operation and examined CREB and p-CREB abundance in CA1 at 1 and 3 hr after reperfusion. Global ischemia induced a significant decrease in p-CREB, with no significant change in total CREB abundance in the nuclear fraction of CA1 (decrease by ~25% at 1 hr, P < 0.05 vs. sham-operated animals; decrease by ~40% at 3 hr; P < 0.01 vs. sham-operated animals, Fig. 4a,b). Estradiol prevented the ischemia-induced dephosphorylation and inactivation of CREB, evident at both 1 and 3 hr after ischemia (P < 0.05 vs. placebo-treated, ischemic animals at 1 hr, P < 0.01 vs. placebo-treated ischemic animals at 3 hr; Fig. 4a,b). Thus, estradiol blocks insult-induced dephosphorylation and inactivation of CREB in the vulnerable CA1. PD98059 administered immediately after ischemia blocked the ability of estradiol to maintain p-CREB levels in post-ischemic CA1, assessed at 3 hr after surgery. These findings suggest a causal role for ERK/MAPK signaling in the ability of estradiol to maintain CREB phosphorylation and activation in post-ischemic CA1.
Estradiol attenuates ischemia-induced decrease of Bcl-2 protein expression in CA1 neurons
Bcl-2 is an anti-apoptotic factor implicated in preservation of the integrity of the mitochondrial outer membrane, and the bcl-2 gene is a downstream target of estradiol (41, 42) and CREB (for review, see (43, 44). Estradiol can also act indirectly to regulate Bcl-2 gene expression by promoting CREB phosphorylation (42, 45). To examine the effects of estradiol pretreatment and ischemia on Bcl-2 protein expression, we subjected estradiol- and placebo-treated animals to global ischemia or sham operation and examined Bcl-2 abundance in CA1 at later times (12, 24 or 48 hr) after reperfusion. Global ischemia induced a marked decrease in Bcl-2 expression in pyramidal neurons of the hippocampal CA1, evident at 24 and 48 hr, times prior to onset of neuronal death, as revealed by immunolabeling (Fig. 5e–h). Estradiol enhanced Bcl-2 immunolabeling in CA1 neurons of sham-operated animals (Fig. 5a–d) and blocked the ischemia-induced downregulation of Bcl-2 in CA1 neurons of ischemic animals (Fig. 5 i–l). Because the loss of CA1 pyramidal neurons in rats subjected to 10 min of global ischemia is not detectable until 2–3 days after ischemia (26), it is unlikely that the reduced Bcl-2 immunolabeling at 12 and 24 hr simply reflects pyramidal cell death. To assess the effects of ischemia and estradiol on Bcl-2 protein abundance, protein samples from the CA1 were subjected to Western blot analysis (Fig. 5m). Global ischemia induced a marked (~50%) decrease in Bcl-2 protein abundance in CA1, evident at 12 hr (P < 0.05 vs. placebo-treated, sham animals). Bcl-2 was modestly, but not significantly, decreased as late as 24 and 48 hr after ischemia (Fig. 5m,n). Estradiol blocked the ischemia-induced downregulation of Bcl-2 in CA1 at 12 hr (P < 0.05 vs. ischemia at 12 hr), with no detectable decline in Bcl-2 as late as 24 or 48 hr after reperfusion (Fig. 5m,n).
Fig. 5. Estradiol prevents ischemia-induced downregulation of Bcl-2 in CA1.
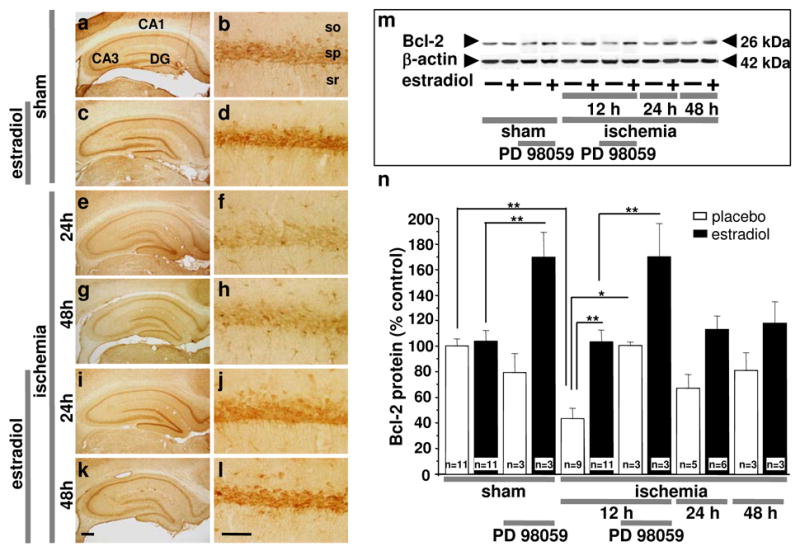
Bcl-2 immunolabeling in the CA1 pyramidal cell layer in brain sections at the level of the dorsal hippocampus from ovariectomized rats subjected to sham operation (a–d) or global ischemia at 24 hr (e, f and i, j) and 48 hr (g, h and k, l) after reperfusion. Data are typical of a minimum of three animals per time point and treatment group. Estradiol attenuated the ischemia-induced decrease in Bcl-2 in CA1 neurons. Abbreviations as in Fig. 1. Scale bars: lower magnification, 400 μm; higher magnification, 60 μm. Representative Western blots (m) and relative abundance of Bcl-2 (n) in the cytosolic fraction of CA1 from placebo- and estradiol-treated rats subjected to sham operation or global ischemia at 12, 24 and 48 hr after surgery. Subsets of animals were injected with PD98059 at 0 h and sacrificed at 12 hr after surgery. Westerns were probed with antibodies to Bcl-2 and β-actin (loading control). Global ischemia caused downregulation of Bcl-2 at 12 hr. Estradiol prevented the ischemia-induced downregulation of Bcl-2. PD98059 did not block the ability of estradiol to maintain Bcl-2 levels, and increased Bcl-2 abundance in the CA1 of estradiol-treated sham and ischemic animals (m,n). Band densities for experimental animals were normalized to the corresponding values for control animals. *, P < 0.05.
Because the MAPK inhibitor PD98059 blocks the neuroprotective actions of estradiol, we next determined whether ERK/MAPK signaling is causally related to the effects of estradiol on Bcl-2 protein abundance. PD98059 administered immediately after ischemia did not prevent estradiol maintenance of Bcl-2 abundance in CA1 (Fig. 5m,n), even though this drug blocked the ability of estradiol to promote CA1 pyramidal cell survival (Fig. 2). Interestingly, PD98059 alone significantly increased Bcl-2 in the CA1 of estradiol- (but not placebo) treated sham and ischemic animals (Fig. 5m,n). Therefore, while estradiol appears to maintain CREB signaling in the post-ischemic CA1 via the ERK/MAPK pathway (Fig. 4), the positive effects of estradiol on Bcl-2 expression are evident even when this pathway is blocked, and the effects of estradiol and PD908059 on Bcl-2 may be additive. Hence our findings are consistent with a model whereby estradiol acts on the Bcl-2 gene in a MAPK-independent manner to sustain Bcl-2 gene expression at normal levels in post-ischemic CA1 neurons.
Estradiol blocks ischemia-induced activation of caspase-3 activity in CA1 neurons
Injurious stimuli such as global ischemia disrupt the integrity of the mitochondrial membrane, leading to the release of cytochrome c and activation of caspase-3, a “terminator” caspase implicated in the execution step of apoptosis (for review, see (1)). Global ischemia promotes cleavage of the biologically inactive precursor procaspase-3 to generate activated caspase-3 (46); ischemia-induced caspase-3 activity is maximal at 24 hr after insult (47). To directly measure caspase-3 functional activity after ischemia, we labeled brain sections with FAM-DEVD-FMK, a fluorescein-tagged analogue of the caspase inhibitor zDEVD-FMK, at 24 hr. FAM-DEVD-FMK enters cells and binds irreversibly to catalytically active caspase-3, and thus provides a fluorescent indicator of the abundance of active caspase-3. In brain sections from control animals, caspase activity was low (Fig. 6a,b). Global ischemia induced a dramatic, six-fold increase in caspase activity in the hippocampal CA1, evident at 24 hr (P < 0.01 vs. placebo-treated, sham animals; Fig. 6c,d,g). The increase in caspase activity was specific in that it was not observed in the resistant CA3 or dentate gyrus. Estradiol pretreatment (14 days) completely blocked the ischemia-induced elevation of caspase-3 activity in CA1 (Fig. 6e,f,g).
Fig. 6. Estradiol blocks ischemia-induced caspase-3 activity in CA1.
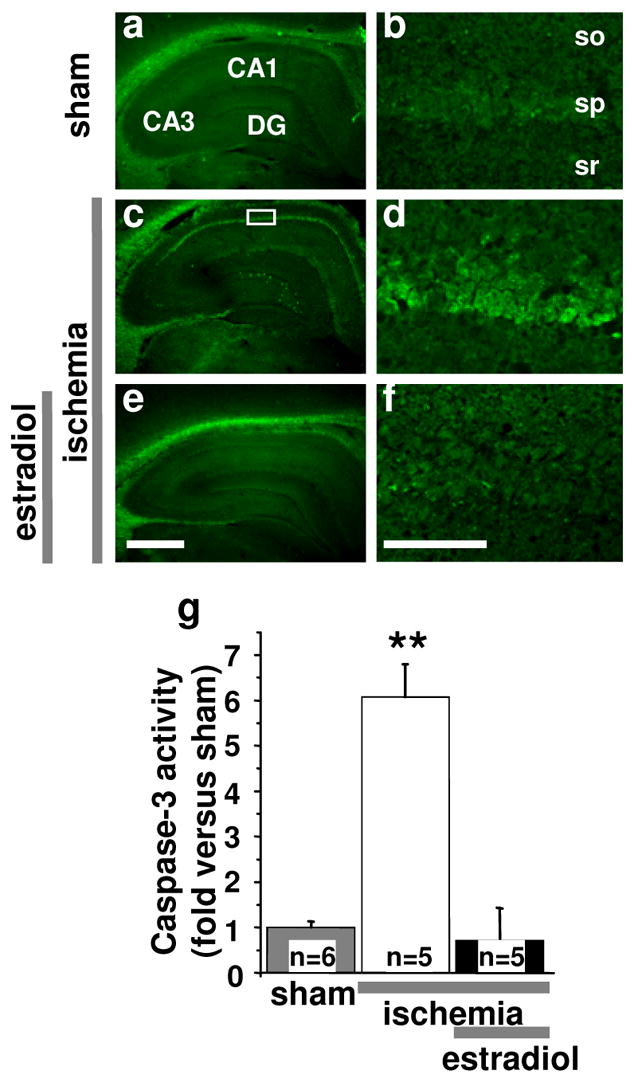
Representative brain sections at the level of the dorsal hippocampus from control (a,b) and experimental animals subjected to global ischemia (c–f) labeled with FAM-DEVD-FMK, a cell-permeant, irreversible inhibitor of caspase-3. In control brain caspase-3 activity was undetectable (a,b,g). Global ischemia activated caspase-3 in CA1 neurons, evident at 24 hr (c,d,g). Estradiol prevented ischemia-induced caspase-3 activity (e,f,g). Abbreviations as in Fig. 1. Scale bars: lower magnification, 400 μm; higher magnification, 50 μm. **, P < 0.01 vs. sham and ischemia+estradiol.
Discussion
Estradiol at levels used for hormone therapy in women ameliorates hippocampal injury in animals subjected to global ischemia (46, 48, 49). Here we report the novel observations that estradiol acts in vivo via classical estrogen receptors, IGF-I receptors and ERK/MAPK signaling to promote survival of post-ischemic hippocampal CA1 neurons in ovariectomized rodents. Ischemia promotes dephosphorylation and inactivation of ERK1 and the transcription factor CREB (a known ERK target) in the first few hours after ischemia, subsequent downregulation of the anti-apoptotic protein Bcl-2 and activation of caspase-3. Estradiol prevents ischemia-induced dephosphorylation and inactivation of ERK1 and CREB, downregulation of Bcl-2 and caspase activation. ERK/MAPK signaling is critical to CREB activation and neuronal survival, in that administration of the MEK inhibitor PD98059 blocks the ability of estradiol to maintain ERK1 and CREB phosphorylation and to promote survival of CA1 pyramidal neurons in the post-ischemic hippocampus. In contrast, the impact of estradiol on Bcl-2 abundance is ERK-independent, consistent with the concept that the activated estrogen receptor interacts with enhancers such as cyclic AMP response elements or Sp1 sites on the bcl-2 gene to regulate bcl-2 gene expression (see (42)). These findings support a model whereby estradiol acts via classical estrogen receptors and IGF-I receptors (Fig. 7), which converge on ERK/MAPK signaling and CREB to promote neuronal survival in the face of ischemic insults. Although estradiol neuroprotection has previously been shown to require concurrent IGF-I signaling in other animal models (e.g., see 19,20), a recent clinical study demonstrating a positive correlation between the level of circulating IGF-I and better functional outcomes in ischemic stroke suggests that adequate levels of IGF-I receptor activity may be relevant to the human situation (see (50)).
Fig. 7. Hypothesized scheme of estradiol protection against global ischemia-induced cell death.
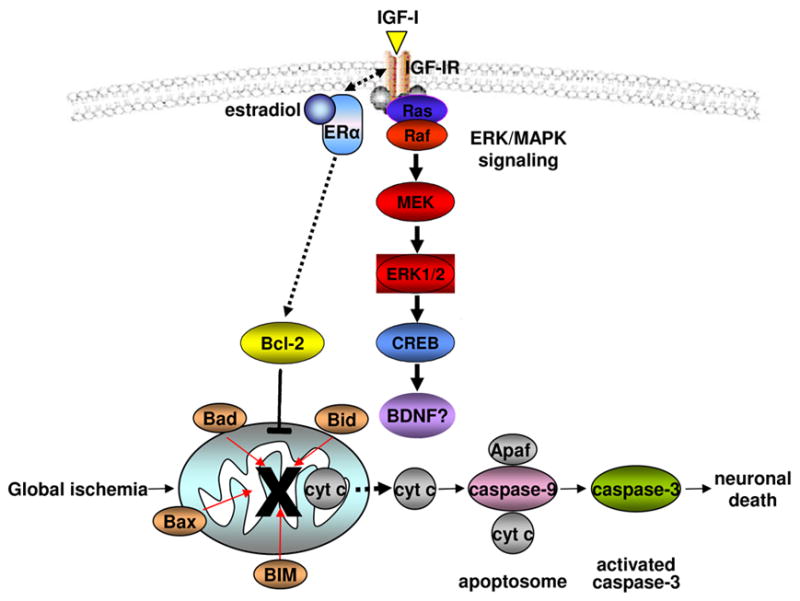
Upon stimulation with estradiol, the estrogen receptor (ER)α (not ERβ (23)) forms a macromolecular signaling complex with the IGF-I receptor in the plasma membrane; the complex recruits and activates downstream kinases including ERK/MAPK (24, 25). ERK/MAPK signaling culminates in phosphorylation and activation of nuclear transcription factors such as CREB, which regulate target genes important to neuronal survival. CREB targets implicated in neuronal survival include BDNF and the anti-apoptotic protein Bcl-2 (43). Bcl-2 promotes the integrity of the mitochondrial outer membrane and halts the caspase death cascade, enabling neurons to survive. Estradiol maintains ERK and CREB activation and opposes ischemia-induced downregulation of Bcl-2. Data in the present study are consistent with a direct action of estradiol on Bcl-2 gene expression rather than maintenance of Bcl-2 levels by ERK/MAPK and CREB signaling.
Whereas estradiol neuroprotection in animal models of focal and global ischemia is well established (12–15), but see (51), the therapeutic potential of estrogens in ameliorating the sequelae of cardiac arrest (global ischemia) or stroke (focal ischemia) in humans remains controversial. The Women’s Health Initiative Memory Study indicates that hormone treatment initiated many years after menopause does not protect against dementia or cognitive decline in elderly women (52, 53). Moreover, postmenopausal treatment with estrogen plus progestin impairs global cognitive performance in some subjects (53). One explanation for these unanticipated outcomes might be the long (years) duration of the interval between cessation of ovarian function and initiation of hormone therapy (see (54)); therefore, the significance of carefully controlled estradiol studies in animals has increased (55).
Estrogen receptor/IGF-I receptor interactions in estradiol neuroprotection
Our study provides new insight into intracellular signaling pathways that mediate estradiol neuroprotection in an in vivo model of global ischemia. The estrogen receptor antagonist ICI 182,780 abrogated estradiol protection in gerbils (present study) and rats (30), documenting a critical role for classical estrogen receptors in protection of hippocampal neurons from global ischemia (6, 8). The fact that both estrogen receptor-α and β-selective agonists produce neuroprotection (30, 49) suggests that either receptor can mediate estradiol protection in global ischemia. In contrast, studies involving mice with targeted gene deletions indicate that estrogen receptor-α, but not -β, mediates estradiol protection in focal ischemia (56). Moreover, we cannot rule out a role for GPR30, a G protein-coupled receptor implicated in estradiol regulation of epidermal growth factor receptor signaling. GPR30 binds estradiol with nanomolar affinity, and ICI 182,780 inhibits estradiol signaling mediated by GRP30 (57, 58). Thus, estradiol might promote neuronal survival by different mechanisms in the global and focal ischemia paradigms.
The finding that the competitive IGF-I receptor antagonist JB-1 also blocks estradiol protection documents a role for IGF-I receptor signaling in preservation of hippocampal neurons after global ischemia. This finding is consistent with evidence that estrogen receptor-α and IGF-I receptors are functionally linked (24, 25, 59). Activation of estrogen/IGF-I signaling is implicated in protection of hilar neurons from seizure-induced injury (60), protection of retinal neurons against light-induced degeneration (61), synaptic remodeling of hypothalamic neurons in vivo (62) and estradiol-dependent reproductive behaviors and neuroendocrine responses (63). Recent studies have begun to reveal the molecular underpinnings of this interdependence. Estrogen receptor-α and IGF-I receptors form a macromolecular signaling complex that recruits downstream signaling molecules such as Shc, phosphatidylinositol 3-kinase (PI3K) and insulin receptor substrate-1 (23). Although estrogen receptors also interact with epidermal growth factor receptors and HER-2/neu in peripheral reproductive tissues and tumor cells (57, 64), the relevance to neurons is unclear.
The present experiments were performed in rats, with the exception of ICI and JB-1 experiments, which were in gerbils. Although not confirmed in rats, a role for IGF-I receptors in estradiol protection against ischemic damage is likely to apply across species. First, experiments employing JB-1 show that IGF-I receptors are critical to estradiol protection against seizure-induced damage of hippocampal neurons (20) and light-induced damage of retinal neurons in rats (61). Second, there are strong commonalities between the gerbil and rat models of global ischemia (1). In both species, cell death occurs primarily in the hippocampal CA1, where pyramidal neurons (but not interneurons or astrocytes) die. In both species, histological evidence of cell death is not detectable until at least 48 hr. Moreover, ischemia promotes activation of the same death cascades, translocation of the same proteins and alterations in expression of the same genes, albeit with a somewhat slower time course in gerbils (26, 27, 47). Finally, it is unlikely that there are species differences in the actions of the IGF-I antagonist JB-1, which was developed to target human IGF-I receptors (35).
ERK/MAPK signaling is critical to neuronal survival
ERK/MAPK signaling is a well known downstream target of estradiol and IGF-I in neurons (65). We show that estradiol pretreatment promotes phosphorylation and activation of ERK1/2 in CA1 of sham-operated animals and blocks ischemia-induced dephosphorylation of ERK1. Furthermore, administration of the ERK/MAPK antagonist PD98059 after ischemia reverses estradiol-dependent ERK1/2 phosphorylation and neuroprotection. These findings implicate ERK/MAPK signaling in protection of hippocampal neurons and are consistent with observations that ERK/MAPK mediates estradiol neuroprotection in models of quinolinic acid (66) and glutamate excitotoxicity (67), β-amyloid toxicity (68) and oxidative stress (69). In contrast, PD98059 protects neurons in focal ischemia (70–72). Thus, ERK/MAPK signaling can promote neuronal apoptosis in some cases (73).
Although the PD98059 experiments suggest a role for ERK1 signaling in estradiol protection, we cannot rule out a role for other targets of PD98059 such as MEK5 and/or cyclooxygenase (39). However, we believe that these molecules are unlikely to mediate estradiol neuroprotection. First, estradiol clearly promotes phosphorylation and activation of ERK1/2, but not ERK5. Second, PD98059 reverses the effects of estradiol on ERK1/2 phosphorylation in both sham-operated and ischemic females. Third, PD98059 exhibits a higher affinity for MEK1/2 than for MEK5; thus, at the concentration used in the present study PD98059 may not inhibit ERK5. Moreover, ischemia does not alter ERK5 abundance or activity in CA1 ((74), present study), providing strong evidence for target specificity. Fourth, although estradiol regulates cyclooxygenase-2 expression, it does not regulate enzyme activity (75). Finally, PD98059 is a standard pharmacological agent for inhibiting ERK1/2 signaling in hippocampal neurons in vivo and in vitro (76–80). Behavioral studies involving PD98059 and another ERK inhibitor (U0126) report essentially identical results (37, 76). While our findings implicate ERK/MAPK signaling as a necessary cellular mediator of estradiol protection, this pathway is not necessarily sufficient for neuronal survival. In particular, our results do not rule out an important role for PI3K, another downstream mediator of estradiol/IGF-I protection (59, 65, 81).
ERK/MAPK signaling is critical to estradiol-dependent CREB activation
The ERK/MAPK cascade can promote neuronal survival by a number of mechanisms including phosphorylation and activation of CREB, which drives transcription of prosurvival genes such as BDNF; phosphorylation and inactivation of the proapoptotic protein BAD (40); and activation of glycogen synthase kinase-3β, which inhibits Wnt signaling (82). Present findings that estradiol maintains CREB phosphorylation at serine 133 in the face of ischemia and that PD98059 reverses estradiol-dependent CREB phosphorylation indicate a causal relation between ERK/MAPK signaling and CREB activation. Estradiol binding to estrogen receptor-α can also activate CREB by promoting metabotropic glutamate receptor-mediated stimulation of MAPK (83) or by inducing cyclic AMP synthesis (42, 45).
ERK/MAPK signaling is not critical to estradiol-dependent regulation of Bcl-2 expression
Our finding that estradiol maintains levels of the anti-apoptotic protein Bcl-2 after global ischemia is consistent with studies implicating Bcl-2 in estradiol protection in focal ischemia (84, 85) and in vitro models of cell death (67, 69). Although these paradigms target different neurons and induce death via different mechanisms, regulation of Bcl-2 may be a common mechanism of estradiol neuroprotection. However, it is clear that maintaining Bcl-2 is not sufficient to rescue CA1 neurons from ischemia-induced cell death. We found that the MEK inhibitor PD98059 potentiated the effects of estradiol on Bcl-2 expression, but completely blocked the effects of estradiol on cell survival and pERK1. Estradiol can increase bcl-2 gene expression by CREB activation in other systems (42)(45), but our finding that PD98059 does not reverse (and may even potentiate) regulation of Bcl-2 levels by estradiol is consistent with a MAPK-independent action of estradiol on bcl-2 gene expression. The loss of CA1 neurons in rats subjected to 10 min of global ischemia is not detectable until 2–3 days after ischemia (26); therefore, it is unlikely that the reduced Bcl-2 immunoreactivity detected at 12 and 24 hr after ischemia simply reflects neuronal cell death. However, it is possible that cell death could contribute to the reduction of Bcl-2 observed at 48 hr.
Injurious stimuli such as global ischemia disrupt the functional integrity of the outer mitochondrial membrane, releasing cytochrome c and Smac/DIABLO, which activate the caspase death cascade (47, 86–88). Pro-apoptotic Bcl-2 family members are thought to participate in formation and activation of large, multiconductance channels that mediate cytochrome c release (1). Anti-apoptotic family members such as Bcl-2 preserve the mitochondrial integrity and block caspase activation in the face of injury. Present findings suggest that estradiol promotes neuronal survival, at least in part, by sustaining levels of Bcl-2. Accordingly, acute estradiol prevents ischemia-induced translocation of cytochrome c from mitochondria to the cytosol (89). However, the current findings also suggest that maintenance of Bcl-2 expression is not sufficient to mediate estradiol protection of CA1 neurons, as Bcl-2 protein abundance was sustained in estradiol-treated, ischemic females injected with the MEK inhibitor PD98059. Therefore, it is likely that estradiol protects neurons from apoptotic death by regulating the activity of multiple cell survival pathways, and that interference with any one of these signaling pathways may lead to delayed cell death.
Summary
The present study shows that estradiol acts via estrogen receptors, IGF-I receptors and ERK/MAPK signaling to promote CA1 neuronal survival after global ischemia. Ischemia promotes early dephosphorylation and inactivation of ERK1 and its target CREB, followed by downregulation of Bcl-2 and caspase activation. Estradiol attenuates dephosphorylation of ERK1 and CREB, downregulation of Bcl-2 and activation of caspase-3. These findings identify intracellular signaling pathways that mediate estradiol protection and reveal mechanisms by which estradiol protects hippocampal neurons from global ischemia-induced degeneration. These findings suggest that estradiol and IGF-I act in a coordinate manner to promote activation of ERK/MAPK signaling and its target CREB and thereby promote neuronal survival in the face of ischemic insults.
Supplementary Material
Acknowledgments
The authors thank Ms. Roodland Regis, Monique Bryan, Adrianna Latuszek-Barrantes, Tovaghgol Adel and Mr. Nicolas Bamat for excellent technical assistance.
Supported by NIH grant NS045693 (to R.S.Z and A.M.E.), American Heart Association Development Award 0335285N (to T.J.M.) and the F.M. Kirby Program in Neuroprotection and Repair.
Footnotes
Disclosure statement : T.J.-M. has nothing to declare. R.S.Z. received lecture fees from Novo Nordisk. A.M.E. consulted for Pfizer, Inc.
Publisher's Disclaimer: “This is an un-copyedited author manuscript copyrighted by The Endocrine Society. This may not be duplicated or reproduced, other than for personal use or within the rule of “Fair Use of Copyrighted Materials” (section 107, Title 17, U.S. Code) without permission of the copyright owner, The Endocrine Society. From the time of acceptance following peer review, the full text of this manuscript is made freely available by The Endocrine Society at http://www.endojournals.org/. The final copy edited article can be found at http://www.endojournals.org/. The Endocrine Society disclaims any responsibility or liability for errors or omissions in this version of the manuscript or in any version derived from it by the National Institutes of Health or other parties. The citation of this article must include the following information: author(s), article title, journal title, year of publication and DOI.”
Reference List
- 1.Zukin RS, Jover T, Yokota H, Calderone A, Simionescu M, Lau CG. Molecular and Cellular Mechanisms of Ischemia-induced Neuronal Death. In: Mohr JP, Choi D, Grotta JC, Weir B, Wolf PA, editors. Stroke: Pathophysiology, Diagnosis, and Management. Harcourt; Orlando: 2004. pp. 829–854. [Google Scholar]
- 2.Chen J, Zhu RL, Nakayama M, Kawaguchi K, Jin K, Stetler RA, Simon RP, Graham SH. Expression of the apoptosis-effector gene, Bax, is up-regulated in vulnerable hippocampal CA1 neurons following global ischemia. J Neurochem. 1996;67:64–71. doi: 10.1046/j.1471-4159.1996.67010064.x. [DOI] [PubMed] [Google Scholar]
- 3.Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- 4.Rosenbaum DM, D'Amore J, Llena J, Rybak S, Balkany A, Kessler JA. Pretreatment with intraventricular aurintricarboxylic acid decreases infarct size by inhibiting apoptosis following transient global ischemia in gerbils. Ann Neurol. 1998;43:654–660. doi: 10.1002/ana.410430515. [DOI] [PubMed] [Google Scholar]
- 5.Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- 6.Murphy DD, Segal M. Regulation of dendritic spine density in cultured rat hippocampal neurons by steroid hormones. J Neurosci. 1996;16:4059–4068. doi: 10.1523/JNEUROSCI.16-13-04059.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pozzo-Miller LD, Inoue T, Murphy DD. Estradiol increases spine density and NMDA-dependent Ca2+ transients in spines of CA1 pyramidal neurons from hippocampal slices. J Neurophysiol. 1999;81:1404–1411. doi: 10.1152/jn.1999.81.3.1404. [DOI] [PubMed] [Google Scholar]
- 8.Woolley CS, McEwen BS. Estradiol regulates hippocampal dendritic spine density via an N-methyl-D-aspartate receptor-dependent mechanism. J Neurosci. 1994;14:7680–7687. doi: 10.1523/JNEUROSCI.14-12-07680.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gazzaley AH, Weiland NG, McEwen BS, Morrison JH. Differential regulation of NMDAR1 mRNA and protein by estradiol in the rat hippocampus. J Neurosci. 1996;16:6830–6838. doi: 10.1523/JNEUROSCI.16-21-06830.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moss RL, Gu Q. Estrogen: mechanisms for a rapid action in CA1 hippocampal neurons. Steroids. 1999;64:14–21. doi: 10.1016/s0039-128x(98)00092-0. [DOI] [PubMed] [Google Scholar]
- 11.Gu Q, Moss RL. Novel mechanism for non-genomic action of 17 beta-oestradiol on kainate-induced currents in isolated rat CA1 hippocampal neurones. J Physiol. 1998;506(Pt 3):745–754. doi: 10.1111/j.1469-7793.1998.745bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amantea D, Russo R, Bagetta G, Corasaniti MT. From clinical evidence to molecular mechanisms underlying neuroprotection afforded by estrogens. Pharmacol Res. 2005;52:119–132. doi: 10.1016/j.phrs.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 13.Behl C. Oestrogen as a neuroprotective hormone. Nat Rev Neurosci. 2002;3:433–442. doi: 10.1038/nrn846. [DOI] [PubMed] [Google Scholar]
- 14.Garcia-Segura LM, Azcoitia I, DonCarlos LL. Neuroprotection by estradiol. Prog Neurobiol. 2001;63:29–60. doi: 10.1016/s0301-0082(00)00025-3. [DOI] [PubMed] [Google Scholar]
- 15.Wise P. Estradiol exerts neuroprotective actions against ischemic brain injury: insights derived from animal models. Endocrine. 2003;21:11–15. doi: 10.1385/endo:21:1:11. [DOI] [PubMed] [Google Scholar]
- 16.Gulinello M, Lebesgue D, Jover-Mengual T, Zukin RS, Etgen AM. Acute and chronic estradiol treatments reduce memory deficits induced by transient global ischemia in female rats. Horm Behav. 2006;49:246–260. doi: 10.1016/j.yhbeh.2005.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Plamondon H, Morin A, Charron C. Chronic 17beta-estradiol pretreatment and ischemia-induced hippocampal degeneration and memory impairments: A 6-month survival study. Horm Behav. 2006 doi: 10.1016/j.yhbeh.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 18.McEwen B. Estrogen actions throughout the brain. Recent Prog Horm Res. 2002;57:357–384. doi: 10.1210/rp.57.1.357. [DOI] [PubMed] [Google Scholar]
- 19.Cardona-Gomez GP, Mendez P, DonCarlos LL, Azcoitia I, Garcia-Segura LM. Interactions of estrogen and insulin-like growth factor-I in the brain: molecular mechanisms and functional implications. J Steroid Biochem Mol Biol. 2002;83:211–217. doi: 10.1016/s0960-0760(02)00261-3. [DOI] [PubMed] [Google Scholar]
- 20.Azcoitia I, Sierra A, Garcia-Segura LM. Neuroprotective effects of estradiol in the adult rat hippocampus: interaction with insulin-like growth factor-I signalling. J Neurosci Res. 1999;58:815–822. doi: 10.1002/(sici)1097-4547(19991215)58:6<815::aid-jnr8>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 21.Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol. 2004;14:311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 22.Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- 23.Kahlert S, Nuedling S, van Eickels M, Vetter H, Meyer R, Grohe C. Estrogen receptor alpha rapidly activates the IGF-1 receptor pathway. J Biol Chem. 2000;275:18447–18453. doi: 10.1074/jbc.M910345199. [DOI] [PubMed] [Google Scholar]
- 24.Mendez P, Azcoitia I, Garcia-Segura LM. Estrogen receptor alpha forms estrogen-dependent multimolecular complexes with insulin-like growth factor receptor and phosphatidylinositol 3-kinase in the adult rat brain. Brain Res Mol Brain Res. 2003;112:170–176. doi: 10.1016/s0169-328x(03)00088-3. [DOI] [PubMed] [Google Scholar]
- 25.Song RX, Barnes CJ, Zhang Z, Bao Y, Kumar R, Santen RJ. The role of Shc and insulin-like growth factor 1 receptor in mediating the translocation of estrogen receptor alpha to the plasma membrane. Proc Natl Acad Sci USA. 2004;101:2076–2081. doi: 10.1073/pnas.0308334100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Calderone A, Jover T, Mashiko T, Noh KM, Tanaka H, Bennett MV, Zukin RS. Late calcium EDTA rescues hippocampal CA1 neurons from global ischemia-induced death. J Neurosci. 2004;24:9903–9913. doi: 10.1523/JNEUROSCI.1713-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calderone A, Jover T, Noh K-M, Tanaka H, Yokota H, Lin Y, Grooms S, Regis R, Bennett MV, Zukin RS. Ischemic insults de-repress the gene silencer rest in neurons destined to die. J Neurosci. 2003;23:2112–2121. doi: 10.1523/JNEUROSCI.23-06-02112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blevins JE, Stanley BG, Reidelberger RD. DMSO as a vehicle for central injections: tests with feeding elicited by norepinephrine injected into the paraventricular nucleus. Pharmacol Biochem Behav. 2002;71:277–282. doi: 10.1016/s0091-3057(01)00659-1. [DOI] [PubMed] [Google Scholar]
- 29.Paxinos GaWC. The rat brain in stereotaxic coordinates. Academic Press; San Diego: 1998. [Google Scholar]
- 30.Miller NR, Jover T, Cohen HW, Zukin RS, Etgen AM. Estrogen can act via estrogen receptor alpha and beta to protect hippocampal neurons against global ischemia-induced cell death. Endocrinology. 2005;146:3070–3079. doi: 10.1210/en.2004-1515. [DOI] [PubMed] [Google Scholar]
- 31.Amstad PA, Yu G, Johnson GL, Lee BW, Dhawan S, Phelps DJ. Detection of caspase activation in situ by fluorochrome-labeled caspase inhibitors. Biotechniques. 2001;31:608–10. 612, 614. doi: 10.2144/01313pf01. passim. [DOI] [PubMed] [Google Scholar]
- 32.Bedner E, Smolewski P, Amstad P, Darzynkiewicz Z. Activation of caspases measured in situ by binding of fluorochrome-labeled inhibitors of caspases (FLICA): correlation with DNA fragmentation. Exp Cell Res. 2000;259:308–313. doi: 10.1006/excr.2000.4955. [DOI] [PubMed] [Google Scholar]
- 33.Smolewski P, Bedner E, Du L, Hsieh TC, Wu JM, Phelps DJ, Darzynkiewicz Z. Detection of caspases activation by fluorochrome-labeled inhibitors: Multiparameter analysis by laser scanning cytometry. Cytometry. 2001;44:73–82. doi: 10.1002/1097-0320(20010501)44:1<73::aid-cyto1084>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 34.Garcia-Calvo M, Peterson EP, Leiting B, Ruel R, Nicholson DW, Thornberry NA. Inhibition of human caspases by peptide-based and macromolecular inhibitors. J Biol Chem. 1998;273:32608–32613. doi: 10.1074/jbc.273.49.32608. [DOI] [PubMed] [Google Scholar]
- 35.Pietrzkowski Z, Wernicke D, Porcu P, Jameson BA, Baserga R. Inhibition of cellular proliferation by peptide analogues of insulin-like growth factor 1. Cancer Res. 1992;52:6447–6451. [PubMed] [Google Scholar]
- 36.Bi R, Broutman G, Foy MR, Thompson RF, Baudry M. The tyrosine kinase and mitogen-activated protein kinase pathways mediate multiple effects of estrogen in hippocampus. Proc Natl Acad Sci USA. 2000;97:3602–3607. doi: 10.1073/pnas.060034497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Etgen AM, Acosta-Martinez M. Participation of growth factor signal transduction pathways in estradiol facilitation of female reproductive behavior. Endocrinology. 2003;144:3828–3835. doi: 10.1210/en.2003-0157. [DOI] [PubMed] [Google Scholar]
- 38.Gonzalez-Flores O, Shu J, Camacho-Arroyo I, Etgen AM. Regulation of lordosis by cyclic 3′,5′-guanosine monophosphate, progesterone, and its 5alpha-reduced metabolites involves mitogen-activated protein kinase. Endocrinology. 2004;145:5560–5567. doi: 10.1210/en.2004-0823. [DOI] [PubMed] [Google Scholar]
- 39.English JM, Cobb MH. Pharmacological inhibitors of MAPK pathways. Trends Pharmacol Sci. 2002;23:40–45. doi: 10.1016/s0165-6147(00)01865-4. [DOI] [PubMed] [Google Scholar]
- 40.Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- 41.Garcia-Segura LM, Cardona-Gomez P, Naftolin F, Chowen JA. Estradiol upregulates Bcl-2 expression in adult brain neurons. Neuroreport. 1998;9:593–597. doi: 10.1097/00001756-199803090-00006. [DOI] [PubMed] [Google Scholar]
- 42.Dong L, Wang W, Wang F, Stoner M, Reed JC, Harigai M, Samudio I, Kladde MP, Vyhlidal C, Safe S. Mechanisms of transcriptional activation of bcl-2 gene expression by 17beta-estradiol in breast cancer cells. J Biol Chem. 1999;274:32099–32107. doi: 10.1074/jbc.274.45.32099. [DOI] [PubMed] [Google Scholar]
- 43.Polster BM, Fiskum G. Mitochondrial mechanisms of neural cell apoptosis. J Neurochem. 2004;90:1281–1289. doi: 10.1111/j.1471-4159.2004.02572.x. [DOI] [PubMed] [Google Scholar]
- 44.Yuan J, Lipinski M, Degterev A. Diversity in the mechanisms of neuronal cell death. Neuron. 2003;40:401–413. doi: 10.1016/s0896-6273(03)00601-9. [DOI] [PubMed] [Google Scholar]
- 45.Kanda N, Watanabe S. 17beta-estradiol inhibits oxidative stress-induced apoptosis in keratinocytes by promoting Bcl-2 expression. J Invest Dermatol. 2003;121:1500–1509. doi: 10.1111/j.1523-1747.2003.12617.x. [DOI] [PubMed] [Google Scholar]
- 46.Jover T, Tanaka H, Calderone A, Oguro K, Bennett MV, Etgen AM, Zukin RS. Estrogen protects against global ischemia-induced neuronal death and prevents activation of apoptotic signaling cascades in the hippocampal CA1. J Neurosci. 2002;22:2115–2124. doi: 10.1523/JNEUROSCI.22-06-02115.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tanaka H, Yokota H, Jover T, Cappuccio I, Calderone A, Simionescu M, Bennett MV, Zukin RS. Ischemic preconditioning: neuronal survival in the face of caspase-3 activation. J Neurosci. 2004;24:2750–2759. doi: 10.1523/JNEUROSCI.5475-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Plahta WC, Clark DL, Colbourne F. 17beta-estradiol pretreatment reduces CA1 sector cell death and the spontaneous hyperthermia that follows forebrain ischemia in the gerbil. Neuroscience. 2004;129:187–193. doi: 10.1016/j.neuroscience.2004.07.037. [DOI] [PubMed] [Google Scholar]
- 49.Shughrue PJ, Merchenthaler I. Estrogen prevents the loss of CA1 hippocampal neurons in gerbils after ischemic injury. Neuroscience. 2003;116:851–861. doi: 10.1016/s0306-4522(02)00790-x. [DOI] [PubMed] [Google Scholar]
- 50.Bondanelli M, Ambrosio MR, Onofri A, Bergonzoni A, Lavezzi S, Zatelli MC, Valle D, Basaglia N, Degli Uberti EC. Predictive value of circulating insulin-like growth factor I levels in ischemic stroke outcome. J Clin Endocrinol Metab. 2006;91:3928–3934. doi: 10.1210/jc.2006-1040. [DOI] [PubMed] [Google Scholar]
- 51.Harukuni I, Hurn PD, Crain BJ. Deleterious effect of beta-estradiol in a rat model of transient forebrain ischemia. Brain Res. 2001;900:137–142. doi: 10.1016/s0006-8993(01)02278-8. [DOI] [PubMed] [Google Scholar]
- 52.Espeland MA, Rapp SR, Shumaker SA, Brunner R, Manson JE, Sherwin BB, Hsia J, Margolis KL, Hogan PE, Wallace R, Dailey M, Freeman R, Hays J. Conjugated equine estrogens and global cognitive function in postmenopausal women: Women's Health Initiative Memory Study. JAMA. 2004;291:2959–2968. doi: 10.1001/jama.291.24.2959. [DOI] [PubMed] [Google Scholar]
- 53.Rapp SR, Espeland MA, Shumaker SA, Henderson VW, Brunner RL, Manson JE, Gass ML, Stefanick ML, Lane DS, Hays J, Johnson KC, Coker LH, Dailey M, Bowen D. Effect of estrogen plus progestin on global cognitive function in postmenopausal women: the Women's Health Initiative Memory Study: a randomized controlled trial. JAMA. 2003;289:2663–2672. doi: 10.1001/jama.289.20.2663. [DOI] [PubMed] [Google Scholar]
- 54.Sherwin BB. Estrogen and memory in women: how can we reconcile the findings? Horm Behav. 2005;47:371–375. doi: 10.1016/j.yhbeh.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 55.Morrison JH, Brinton RD, Schmidt PJ, Gore AC. Estrogen, menopause, and the aging brain: how basic neuroscience can inform hormone therapy in women. J Neurosci. 2006;26:10332–10348. doi: 10.1523/JNEUROSCI.3369-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dubal DB, Zhu H, Yu J, Rau SW, Shughrue PJ, Merchenthaler I, Kindy MS, Wise PM. Estrogen receptor alpha, not beta, is a critical link in estradiol-mediated protection against brain injury. Proc Natl Acad Sci USA. 2001;98:1952–1957. doi: 10.1073/pnas.041483198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Filardo EJ. Epidermal growth factor receptor (EGFR) transactivation by estrogen via the G-protein-coupled receptor, GPR30: a novel signaling pathway with potential significance for breast cancer. J Steroid Biochem Mol Biol. 2002;80:231–238. doi: 10.1016/s0960-0760(01)00190-x. [DOI] [PubMed] [Google Scholar]
- 58.Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. 2005;146:624–632. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- 59.Cardona-Gomez GP, Mendez P, Garcia-Segura LM. Synergistic interaction of estradiol and insulin-like growth factor-I in the activation of PI3K/Akt signaling in the adult rat hypothalamus. Brain Res Mol Brain Res. 2002;107:80–88. doi: 10.1016/s0169-328x(02)00449-7. [DOI] [PubMed] [Google Scholar]
- 60.Azcoitia I, Fernandez-Galaz C, Sierra A, Garcia-Segura LM. Gonadal hormones affect neuronal vulnerability to excitotoxin-induced degeneration. J Neurocytol. 1999;28:699–710. doi: 10.1023/a:1007025219044. [DOI] [PubMed] [Google Scholar]
- 61.Yu X, Rajala RV, McGinnis JF, Li F, Anderson RE, Yan X, Li S, Elias RV, Knapp RR, Zhou X, Cao W. Involvement of insulin/phosphoinositide 3-kinase/Akt signal pathway in 17 beta-estradiol-mediated neuroprotection. J Biol Chem. 2004;279:13086–13094. doi: 10.1074/jbc.M313283200. [DOI] [PubMed] [Google Scholar]
- 62.Cardona-Gomez GP, Trejo JL, Fernandez AM, Garcia-Segura LM. Estrogen receptors and insulin-like growth factor-I receptors mediate estrogen-dependent synaptic plasticity. Neuroreport. 2000;11:1735–1738. doi: 10.1097/00001756-200006050-00027. [DOI] [PubMed] [Google Scholar]
- 63.Quesada A, Etgen AM. Functional interactions between estrogen and insulin-like growth factor-I in the regulation of alpha 1B-adrenoceptors and female reproductive function. J Neurosci. 2002;22:2401–2408. doi: 10.1523/JNEUROSCI.22-06-02401.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pietras RJ. Interactions between estrogen and growth factor receptors in human breast cancers and the tumor-associated vasculature. Breast J. 2003;9:361–373. doi: 10.1046/j.1524-4741.2003.09510.x. [DOI] [PubMed] [Google Scholar]
- 65.Ronnekleiv OK, Kelly MJ. Diversity of ovarian steroid signaling in the hypothalamus. Front Neuroendocrinol. 2005;26:65–84. doi: 10.1016/j.yfrne.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 66.Kuroki Y, Fukushima K, Kanda Y, Mizuno K, Watanabe Y. Neuroprotection by estrogen via extracellular signal-regulated kinase against quinolinic acid-induced cell death in the rat hippocampus. Eur J Neurosci. 2001;13:472–476. doi: 10.1046/j.0953-816x.2000.01409.x. [DOI] [PubMed] [Google Scholar]
- 67.Singer CA, Figueroa-Masot XA, Batchelor RH, Dorsa DM. The mitogen-activated protein kinase pathway mediates estrogen neuroprotection after glutamate toxicity in primary cortical neurons. J Neurosci. 1999;19:2455–2463. doi: 10.1523/JNEUROSCI.19-07-02455.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fitzpatrick JL, Mize AL, Wade CB, Harris JA, Shapiro RA, Dorsa DM. Estrogen-mediated neuroprotection against beta-amyloid toxicity requires expression of estrogen receptor alpha or beta and activation of the MAPK pathway. J Neurochem. 2002;82:674–682. doi: 10.1046/j.1471-4159.2002.01000.x. [DOI] [PubMed] [Google Scholar]
- 69.Mize AL, Shapiro RA, Dorsa DM. Estrogen receptor-mediated neuroprotection from oxidative stress requires activation of the mitogen-activated protein kinase pathway. Endocrinology. 2003;144:306–312. doi: 10.1210/en.2002-220698. [DOI] [PubMed] [Google Scholar]
- 70.Alessandrini A, Namura S, Moskowitz MA, Bonventre JV. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc Natl Acad Sci USA. 1999;96:12866–12869. doi: 10.1073/pnas.96.22.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Namura S, Iihara K, Takami S, Nagata I, Kikuchi H, Matsushita K, Moskowitz MA, Bonventre JV, Alessandrini A. Intravenous administration of MEK inhibitor U0126 affords brain protection against forebrain ischemia and focal cerebral ischemia. Proc Natl Acad Sci USA. 2001;98:11569–11574. doi: 10.1073/pnas.181213498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vartiainen N, Goldsteins G, Keksa-Goldsteine V, Chan PH, Koistinaho J. Aspirin inhibits p44/42 mitogen-activated protein kinase and is protective against hypoxia/reoxygenation neuronal damage. Stroke. 2003;34:752–757. doi: 10.1161/01.STR.0000057813.31798.1F. [DOI] [PubMed] [Google Scholar]
- 73.Cheung EC, Slack RS. Emerging role for ERK as a key regulator of neuronal apoptosis. Sci STKE. 2004;2004:E45. doi: 10.1126/stke.2512004pe45. [DOI] [PubMed] [Google Scholar]
- 74.Wang RM, Zhang QG, Li CH, Zhang GY. Activation of extracellular signal-regulated kinase 5 may play a neuroprotective role in hippocampal CA3/DG region after cerebral ischemia. J Neurosci Res. 2005;80:391–399. doi: 10.1002/jnr.20433. [DOI] [PubMed] [Google Scholar]
- 75.Ospina JA, Brevig HN, Krause DN, Duckles SP. Estrogen suppresses IL-1beta-mediated induction of COX-2 pathway in rat cerebral blood vessels. Am J Physiol Heart Circ Physiol. 2004;286:H2010–H2019. doi: 10.1152/ajpheart.00481.2003. [DOI] [PubMed] [Google Scholar]
- 76.Miller CA, Marshall JF. Molecular substrates for retrieval and reconsolidation of cocaine-associated contextual memory. Neuron. 2005;47:873–884. doi: 10.1016/j.neuron.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 77.Guirland C, Suzuki S, Kojima M, Lu B, Zheng JQ. Lipid rafts mediate chemotropic guidance of nerve growth cones. Neuron. 2004;42:51–62. doi: 10.1016/s0896-6273(04)00157-6. [DOI] [PubMed] [Google Scholar]
- 78.Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 79.Bailey CH, Kaang BK, Chen M, Martin KC, Lim CS, Casadio A, Kandel ER. Mutation in the phosphorylation sites of MAP kinase blocks learning-related internalization of apCAM in Aplysia sensory neurons. Neuron. 1997;18:913–924. doi: 10.1016/s0896-6273(00)80331-1. [DOI] [PubMed] [Google Scholar]
- 80.Martin KC, Michael D, Rose JC, Barad M, Casadio A, Zhu H, Kandel ER. MAP kinase translocates into the nucleus of the presynaptic cell and is required for long-term facilitation in Aplysia. Neuron. 1997;18:899–912. doi: 10.1016/s0896-6273(00)80330-x. [DOI] [PubMed] [Google Scholar]
- 81.Zheng WH, Quirion R. Insulin-like growth factor-1 (IGF-1) induces the activation/phosphorylation of Akt kinase and cAMP response element-binding protein (CREB) by activating different signaling pathways in PC12 cells. BMC Neurosci. 2006;7:51. doi: 10.1186/1471-2202-7-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Goodenough S, Schleusner D, Pietrzik C, Skutella T, Behl C. Glycogen synthase kinase 3beta links neuroprotection by 17beta-estradiol to key Alzheimer processes. Neuroscience. 2005;132:581–589. doi: 10.1016/j.neuroscience.2004.12.029. [DOI] [PubMed] [Google Scholar]
- 83.Boulware MI, Weick JP, Becklund BR, Kuo SP, Groth RD, Mermelstein PG. Estradiol activates group I and II metabotropic glutamate receptor signaling, leading to opposing influences on cAMP response element-binding protein. J Neurosci. 2005;25:5066–5078. doi: 10.1523/JNEUROSCI.1427-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dubal DB, Shughrue PJ, Wilson ME, Merchenthaler I, Wise PM. Estradiol modulates bcl-2 in cerebral ischemia: a potential role for estrogen receptors. J Neurosci. 1999;19:6385–6393. doi: 10.1523/JNEUROSCI.19-15-06385.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Alkayed NJ, Goto S, Sugo N, Joh HD, Klaus J, Crain BJ, Bernard O, Traystman RJ, Hurn PD. Estrogen and Bcl-2: gene induction and effect of transgene in experimental stroke. J Neurosci. 2001;21:7543–7550. doi: 10.1523/JNEUROSCI.21-19-07543.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ouyang YB, Tan Y, Comb M, Liu CL, Martone ME, Siesjo BK, Hu BR. Survival- and death-promoting events after transient cerebral ischemia: phosphorylation of Akt, release of cytochrome C and Activation of caspase-like proteases. J Cereb Blood Flow Metab. 1999;19:1126–1135. doi: 10.1097/00004647-199910000-00009. [DOI] [PubMed] [Google Scholar]
- 87.Sugawara T, Fujimura M, Morita-Fujimura Y, Kawase M, Chan PH. Mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient global cerebral ischemia. J Neurosci. 1999;19:RC39. doi: 10.1523/JNEUROSCI.19-22-j0002.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sugawara T, Noshita N, Lewen A, Gasche Y, Ferrand-Drake M, Fujimura M, Morita-Fujimura Y, Chan PH. Overexpression of copper/zinc superoxide dismutase in transgenic rats protects vulnerable neurons against ischemic damage by blocking the mitochondrial pathway of caspase activation. J Neurosci. 2002;22:209–217. doi: 10.1523/JNEUROSCI.22-01-00209.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bagetta G, Chiappetta O, Amantea D, Iannone M, Rotiroti D, Costa A, Nappi G, Corasaniti MT. Estradiol reduces cytochrome c translocation and minimizes hippocampal damage caused by transient global ischemia in rat. Neurosci Lett. 2004;368:87–91. doi: 10.1016/j.neulet.2004.06.062. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.