

Abstract
The proposed structures of jenamidines A, B, and C (1−3) were revised to jenamidines A1/A2, B1/B2, and C (8-10). Jenamidines A1/A2 (8) were synthesized from activated proline derivative 43 by conversion to 26 in two steps and 50% overall yield. Acylation of 26 with acid chloride 38d gave 39d, which was deprotected with TFA and then mild base to give 8 in 45% yield from 26. (−)-trans-2,5-Dimethylproline ethyl ester (49) was prepared by the enantioselective Michael reaction of ethyl 2-nitropropionate (51) and methyl vinyl ketone (50) using modified dihydroquinine 60 as the catalyst. Further elaboration converted 49 to natural (+)-NP25302 (12). A Wittig reaction of proline NCA (76) with ylide 79 gave 72 as a 9/1 E/Z mixture in 27% yield completing a one step formal synthesis of SB-311009 analogues.
Introduction
Structure Revision
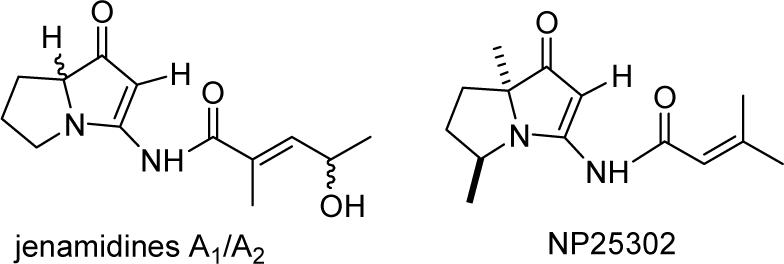
Sattler and co-workers reported the isolation of jenamidines A, B, and C (1−3) from the culture broth of Streptomyces sp. (strain HKI0297) in 2003 (see Figure 1).1 Jenamidine A inhibits proliferation of the chronic myeloid leukemic cell line K-562 with a GI50 of 1.9 μg/mL. The structures were determined by HRMS, IR, UV and a series of NMR spectroscopic experiments. One of the most striking features of these structures is that the ketone in jenamidine A is in a different position than in jenamidines B and C.
FIGURE 1.
Proposed structures of jenamidines A, B, and C.
Our examination suggested that the aminal hydrogen H9a in jenamidine A should absorb further downfield than the observed value of δ 3.94 and that carbons C7 and C9 of 1, which are adjacent to a ketone, should absorb further downfield than the observed values of δ 27.5 and 28.8, respectively.2 In addition, one of the H9's absorbs at δ 1.53, which is further upfield than expected for a proton next to a ketone. The methylene carbons of the analogous compound N-acetyl-4-piperidinone absorb between δ 40.6−44.9 and the methylene protons absorb between δ 2.4−2.6 and 3.7−4.0.3
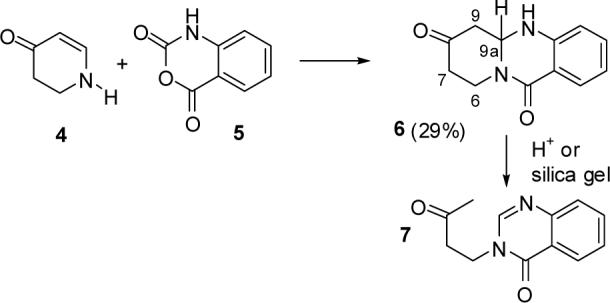
These expectations were confirmed by the synthesis of tricycle 6, which is a good model for the piperidinone moiety of the proposed structure of jenamidine A (see Scheme 1). Reaction of 2,3-dihydro-4-pyridinone (4)4 with isatoic anhydride (5) and Et3N in THF for 8 h in a sealed tube at 80 °C provided 29% (65% based on recovered 4) of the surprisingly unstable tricyclic piperidinone 6. Analysis of the NMR spectral data of 6 confirmed our doubts regarding the structure proposed for jenamidine A. H9a of 6, which is adjacent to two nitrogens, absorbs at δ 5.12 (dd, J = 3.7, 9.2 Hz). The methylene groups adjacent to the ketone absorb between δ 2.4 and 2.6. The three CH2 carbons absorb at δ 47.8, 40.9 and 39.7. These data are consistent with those expected for this structure.5 Treatment of 6 with dilute acid resulted in a facile retro-Mannich reaction to give 76 quantitatively. Partial conversion of 6 to 7 occurred during flash chromatography on silica gel suggesting that structure 1 would not survive the isolation protocol for jenamidine A.
SCHEME 1.
Synthesis of Model 6
These observations suggested that the three methylene carbons of jenamidine A might be part of a pyrrolidine ring with the ketone elsewhere in the molecule. Eventually, we considered the unusual ketene aminals 8, 9, and 10 as possible structures for jenamidines A, B, and C (see Figure 2). A literature search established that two compounds containing the identical ring system have been isolated. The structure of bohemamine (11) was determined in 1980 by X-ray crystallography.7 NP25302 (12), the deoxy analogue of bohemamine was recently reported8 and shown to inhibit the adhesion of HL-60 cells to CHO-ICAM-1 cells with an IC50 of 24 μM. The 1H and 13C NMR spectral data for 12 correspond well with those reported for jenamidine A, aside from the expected differences resulting from the methyl groups and differing side chains.2a The NMR spectral data for the jenamidine A side chain correspond well with those reported for ethyl 4-hydroxy-2E-methylpenten-2-oate ester.9 The unusual UV absorption at 326 nm (log ε 3.32) in the jenamidines is probably due to the N-acyl vinylogous urea10 and is also present in bohemamine (335 nm)7 and NP25302 (334 nm).8
FIGURE 2.
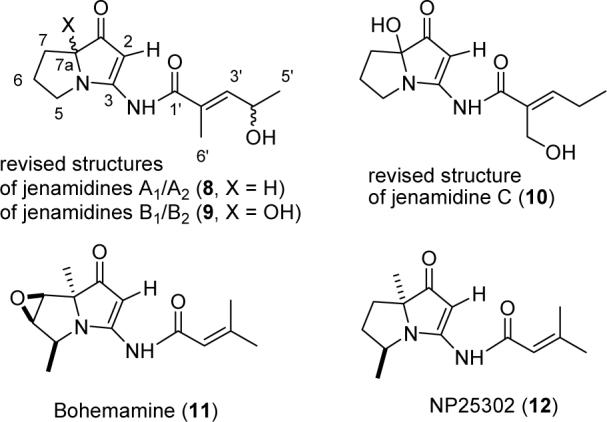
Bohemamine, NP25302, and revised structures of jenamidines A1/A2, B1/B2, and C.
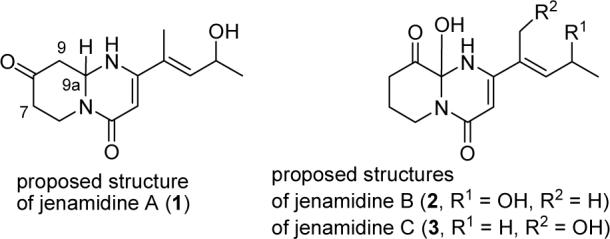
Careful analysis of the 1H and 13C NMR spectra indicated that jenamidine A is a mixture of two compounds.2a The 13C NMR spectrum contains doubled peaks between 0.02 and 0.1 ppm apart for all carbons except C5, C6, and C6′. Similarly, the 1H NMR spectrum showed two peaks separated by 0.04 ppm around δ 5.65 for H2. Jenamidine A was therefore renamed jenamidines A1/A2 because the natural product is a pair of diastereomers. Presumably, epimerization of the ring fusion hydrogen α to the ketone occurs readily. Jenamidine B is renamed jenamidines B1/B2 because the NMR spectra indicate that this compound is also a mixture of diastereomers.2a The third member of the family, which has only one stereocenter, remains jenamidine C, most likely as a mixture of enantiomers because the optical rotation of jenamidine C (10) ([α]22D +1.8) is lower than that of jenamidines A1/A2 (8) and jenamidines B1/B2 (9) ([α]22D +6.8 and +8.4, respectively).1
These structures are also biosynthetically reasonable. Jenamidines B1/B2 (9) can be formed by hydroxylation of jenamidines A1/A2 (8), while jenamidine C (10) can be formed by bis hydroxylation of jenamidines A1/A2 precursor 13, which lacks the side chain hydroxy group (see Scheme 2).
SCHEME 2.
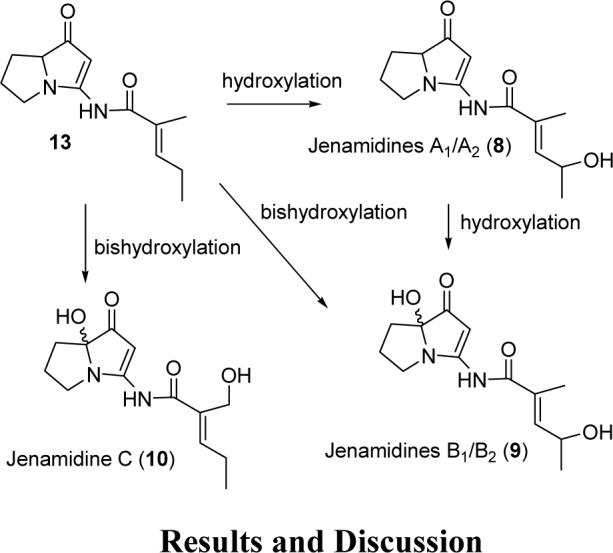
Possible Late Steps in the Biosynthesis of Jenamidines A1/A2, B1/B2, and C
Results and Discussion
Synthesis of Jenamidines A1/A2 (8)
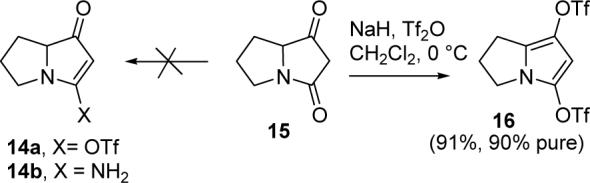
We now turned to the synthesis of jenamidines A1/A2 (8), which required the development of new methods for the preparation of the novel N-acyl vinylogous urea in the right hand ring. A wide variety of approaches proved to be unsuccessful. For instance, we attempted to convert the known keto lactam 1511 to enol triflate 14a, which should undergo Pdcatalyzed amidation as recently reported for related systems (see Scheme 3).12 Unfortunately, reaction of 15 with NaH and Tf2O gave only the unstable pyrrole bis triflate 16.13 Use of excess NaH and Tf2O gave crude (90% pure) 16 in 91% yield, which was isolated in only 17% yield. Although we could cleanly couple 2-methyl-2-butenamide14 with the enol triflate prepared from dimedone, initial attempts at Pd-catalyzed couplings of amides with 16 were unsuccessful. Attempted preparation of vinylogous urea 14b by reaction of keto lactam 15 with NH3 gave complex mixtures that did not contain 14b.15
SCHEME 3.
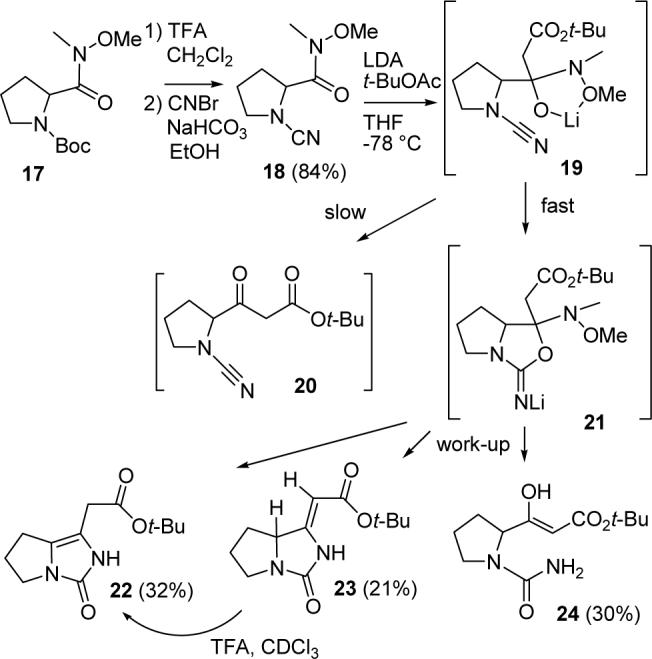
We then turned to the preparation of a vinylogous urea by the addition of an enolate to a cyanamide. Deprotection of the Boc group of Weinreb amide 1711a with TFA in CH2Cl2 and reaction of the liberated amine with CNBr16 and NaHCO3 in EtOH afforded cyanamide 18 in 84% yield (see Scheme 4). Addition of methylmagnesium bromide gave the methyl ketone, which could not be cyclized to give 14b. Cyanamide 18 was then treated with the lithium enolate of tert-butyl acetate in an attempt to form keto ester 20, which we hoped would cyclize to form the desired vinylogous urea. However, we obtained a mixture of 22 (32%), 23 (21%), and 24 (30%). Presumably, the stabilized tetrahedral intermediate 19 cyclized to the cyanamide to form bicyclic intermediate 21 more rapidly than it lost N-methoxymethylamine to give keto ester 20. Work up provided urea keto ester 24, which underwent cyclodehydration to give 22 and 23. The double bond stereochemistry of 23 was established by an NOE between the alkene and ring fusion hydrogens; the chemical shift of the alkene hydrogen, δ 5.19 is consistent with that expected for the (Z)-isomer.17 Imidazolone 22 is the thermodynamic product because treating a solution of 23 in CDCl3 with one drop of TFA cleanly isomerized 23 to 22.
SCHEME 4.
The Weinreb amide appeared to be a poor choice of electrophile because the tetrahedral intermediate 19 was too stable and cyclized to give 21 more rapidly than it decomposed to give keto ester 20. A simple ester should be better because the tetrahedral intermediate will decompose rapidly to give 20, which could then cyclize to give the desired product 26. Fortunately, this proved to be the case. Acid catalyzed esterification of proline18 and cyanation16 with CNBr and NaHCO3 in EtOH gave the known cyanamide 25 (see Scheme 5).19 Cyanamide methyl ester 25 was added to a solution of the lithium enolate of tert-butyl acetate (2.3 equiv) in THF at −45 °C.20 The solution was stirred for 1 h at −45 °C to give keto ester 20, treated with 1.2 equiv of LHMDS in THF, and stirred at 25 °C for 2 h to give the desired product 26 in 27% yield. Byproduct 28 was formed in 24% yield by the addition of the enolate to the cyanamide to give 27, which then cyclized to the methyl ester to form the alkylidene imidazolidinedione 28.21 The stereochemistry of the double bond was established by an NOE between the alkene proton and the methylene group. The methyl ester of 25 is less electrophilic than the Weinreb amide of 17 so that the enolate added to both the methyl ester and the cyanamide.
SCHEME 5.
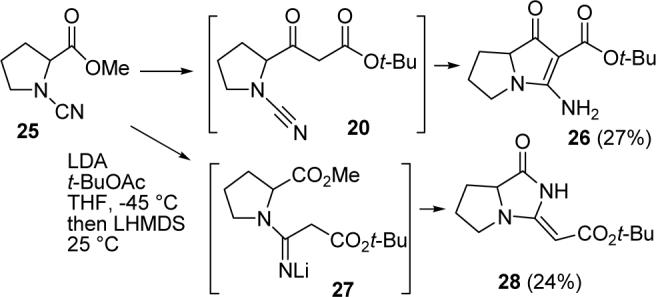
Synthesis of Intermediate 26
Vinylogous urea 26 has the ring system of jenamidines A1/A2 with an additional tert-butyl carboxylate, which we hoped we could remove by hydrolysis and decarboxylation either before or after the introduction of the side chain amide. Reaction of 26 with 9:1 CH2Cl2/TFA effected hydrolysis, but did not provide the desired vinylogous urea 14b.
Acylation of 26 with 2.5 equiv of NaH and 2.2 equiv of tigloyl chloride for 2 h afforded a mixture of the desired product amide 29 and the bis-acylated product pyrrole 30 (see Scheme 6). Stirring the crude mixture in 9:1 CH2Cl2/TFA for 15 h effected hydrolysis of the tert-butyl esters of 29 and 30 and the enol ester of 30 and decarboxylation to give jenamidines A1/A2 model 31 in 69% overall yield from 26. The spectral data of the ring portion of 31 correspond very closely to those of the natural product, supporting the assignment of 8 as the structure of jenamidines A1/A2.
SCHEME 6.
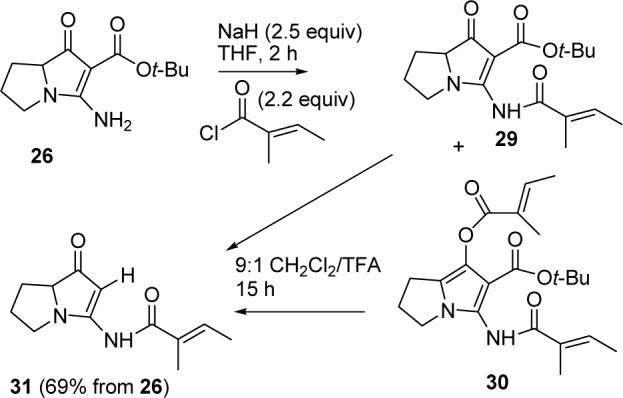
Synthesis of Jenamidine A1/A2 Model 31
The side chain was then prepared by a modification of Adam's procedure for the ethyl ester.9 Ylide 3222 was prepared from tert-butyl 2-bromopropionate by conversion of the bromide to an iodide, reaction of the iodide with triphenylphosphine, and treatment of the phosphonium salt with aqueous NaOH (see Scheme 7). Aldehyde 3323 was prepared from S-methyl lactate by first protecting the alcohol as the TBDMS ether and then reducing the ester with DIBALH. Reaction of aldehyde 33 with ylide 32 in CH2Cl2 for 2 h provided α,β unsaturated ester 349 in 67% yield. Due to the incompatibility of the TBDMS group with the formation of the acid chloride, the alcohol was deprotected with pyr•HF in THF to give alcohol 35 in 99% yield. Initially we chose to protect the alcohol as an acetate ester. Reaction of 35 with AcCl, DMAP, and pyridine in THF gave 36a in 99% yield, which was deprotected in 9:1 CH2Cl2/TFA to give acetoxy acid 37a in 99% yield. Stirring 37a in oxalyl chloride gave crude acid chloride 38a, which was used without purification.
SCHEME 7.
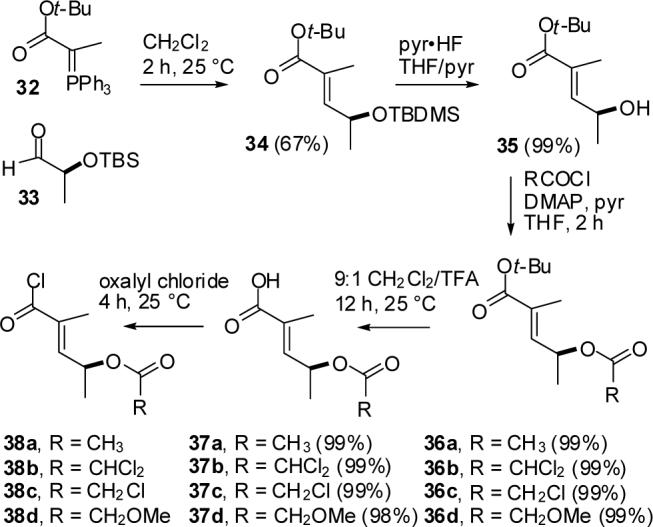
Synthesis of Side-chain Acid Chlorides
Reaction of vinylogous urea 26 with NaH and acid chloride 38a followed by hydrolysis and decarboxylation with 9:1 CH2Cl2/TFA as described above for the preparation of 31 gave jenamidines A1/A2 acetate (39a) in 84% yield (see Scheme 8). To our surprise, hydrolysis using KOH in methanol/water or K2CO3 in MeOH for 12 h at 25 °C afforded primarily 14b. The “amide” and acetate were cleaved at similar rates. Since the nitrogen of the “amide” of 39a is part of a vinylogous urea, the “amide” is actually a vinylogous acyl urea. Acyl ureas are rapidly hydrolyzed in basic methanol.24 Vinylogous urea 14b is polar and hard to work with; initial attempts to reacylate it were unsuccessful. This is ironic since 14b was our initial target. The tert-butyl ester of 26, which we thought was undesirable, turns out to facilitate handling and acylation, and can then be easily removed with TFA in CH2Cl2.
SCHEME 8.
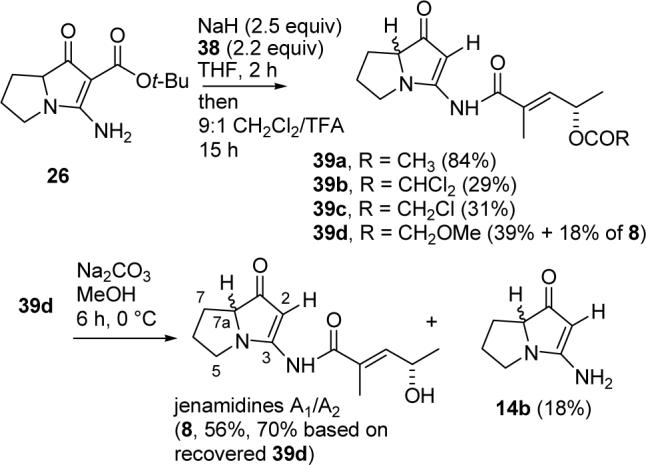
Synthesis of Jenamidines A1/A2
Initial attempts at milder or enzymatic selective cleavage of the acetate of 39a were unpromising. Acid-labile protecting groups were appealing since they would be cleaved by the 9:1 CH2Cl2/TFA used for the hydrolysis of the tert-butyl esters. Unfortunately, most acid-labile protecting groups are not compatible with the formation of acid chloride 38. The TBDMS group has been used with mixed anhydrides,25 but acylation of 26 with mixed anhydrides formed from tiglic acid proceeded in significantly lower yield than with the acid chloride. Unfortunately, application of Wissner's procedure26a for making TBDMS ether-containing acid chlorides to the acid prepared by hydrolysis of 34 converted the allylic OTBDMS group to an allylic chloride.
We then examined more base labile ester protecting groups. Dichloroacetate, chloroacetate, and methoxyacetate esters are hydrolyzed 10,000, 700 and 20 times faster then acetate esters, respectively.27 Alcohol 35 was protected with dichloroacetyl chloride, chloroacetyl chloride, or methoxyacetyl chloride to give 36b-d, respectively, in good yield. Acidic hydrolysis proceeded smoothly to give 37b-d, which were transformed into the corresponding acid chlorides 38b-d.
Reaction of 38b with vinylogous urea 26 gave only 29% of jenamidine A1/A2 dichloroacetate (39b), but the deprotection proceeded smoothly with NaHCO3 in MeOH at 0 °C for 30 min to give jenamidines A1/A2 (8) in quantitative yield. The dichloroacetate could be selectively cleaved, but was too unstable for the coupling and decarboxylation reactions. Reaction of chloroacetate 38c with 26 gave 39c in a still unacceptable 31% yield, which could also be cleaved by NaHCO3 in MeOH for 1 h at 25 °C to give 8 cleanly.
The best compromise was the methoxyacetate protecting group. Acylation of 26 with acid chloride 38d, hydrolysis of the tert-butyl ester and decarboxylation with 9:1 CH2Cl2/TFA gave crude jenamidines A1/A2 methoxy acetate (39d) in ∼70% yield based on analysis of the 1H NMR spectrum. Flash chromatography provided pure 39d in 39% yield and jenamidines A1/A2 (8) in 18% yield. Since 8 was not present in the crude product, hydrolysis occurred during chromatography. Pure 39d was treated with K2CO3 in methanol for 6 h at 0 °C to give 8 in 56% yield (70% based on recovered 39d), recovered 39d in 20% yield, and 14b resulting from cleavage of the amide in 18% yield. A more efficient procedure involved hydrolysis of crude 39d with K2CO3 in MeOH at 0 °C for 24 h to give jenamidines A1/A2 in 45% overall yield from 26, 39d in 11% overall yield from 26, and traces (<5%) of 14b.
The spectral data of synthetic jenamidines A1/A2 (8) are identical to those of the natural product thereby confirming the revised structure we proposed. Both synthetic and natural jenamidines A1/A2 are a 1:1 mixture of diastereomers. Even though 26 was prepared from (S)-proline and 33 was prepared from (S)-methyl lactate, we obtained 8 as a mixture of diastereomers. The ring fusion hydrogen is readily epimerized and this stereocenter is lost in the formation of the bis acylated intermediate analogous to 30, which will give a mixture of diastereomers on hydrolysis. In the 1H NMR spectrum of 8 in CD3OD, the ring fusion hydrogen, H7a, integrates for only ∼0.5, suggesting that partial deuterium exchange has occurred. In the 13C NMR spectrum, C2 and C7 absorb as four peaks since a separate peak is observed for the H7a and D7a isomer of each diastereomer.28
The optical rotation of synthetic 8, [α]D +4.2, is very similar to that of the natural product, [α]D +6.8.1 Therefore natural jenamidines A1/A2 (8) could also be a mixture of isomers at the ring fusion and the (S) isomer on the side chain. However, since both rotations are for mixtures of diastereomers, it is also possible that the natural product is a mixture of isomers on the side chain.
The three-step sequence from vinylogous urea 26 and acid chloride 38d to jenamidines A1/A2 (8) proceeded in 45% yield, which was acceptable given the instability of the amide linkage in base. The one-pot preparation of 26 from cyanamide 25 provided adequate quantities of material, but the 27% yield left room for improvement. Coupling of various N-acetyl amino acid derivatives 40 with ethyl cyanoacetate had been reported to give 41, which cyclized on treatment with 8% HCl in EtOH at reflux to provide 42 in 18−51% overall yield (see Scheme 9).29 We examined variants of this procedure because the acid-catalyzed cyclization used to convert 41 to 42 is not compatible with the tert-butyl ester of 26.
SCHEME 9.
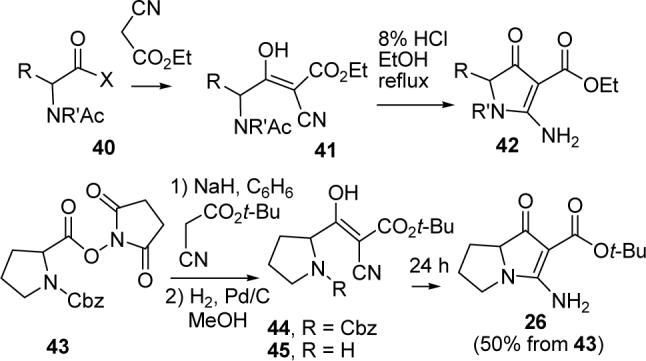
Improved Synthesis of 26
Reaction of Cbz-proline N-hydroxysuccinimide ester (43) with the enolate of tert-butyl cyanoacetate and NaH in benzene for 3 h gave crude 44, which was hydrogenated (1 atm) over 10% Pd/C in MeOH for 2 h to provide 45 as complex mixture of keto/enol tautomers. Fortunately, crude 45 cyclized on standing for 1 d to give 26 in 50% overall yield from 43. Using this sequence, which has not been fully optimized, jenamidines A1/A2 (8) are now available from commercially available 43 in 5 steps and 23% overall yield.
Synthesis of NP25302 (12)
We now turned to applying what we had learned in the synthesis of jenamidines A1/A2 (8) to the synthesis of NP25302 (12). Acylation of 46 followed by acid-catalyzed hydrolysis and decarboxylation should be straightforward because the problematic side chain alcohol of jenamidines A1/A2 is not present in NP25302 (see Scheme 10). Vinylogous urea 46 can be prepared by addition of the enolate of tert-butyl acetate to cyanamide 47 or by the addition of tert-butyl cyanoacetate to 48. Both 47 and 48 should be readily accessible from trans-2,5-dimethylproline ethyl ester (49).
SCHEME 10.
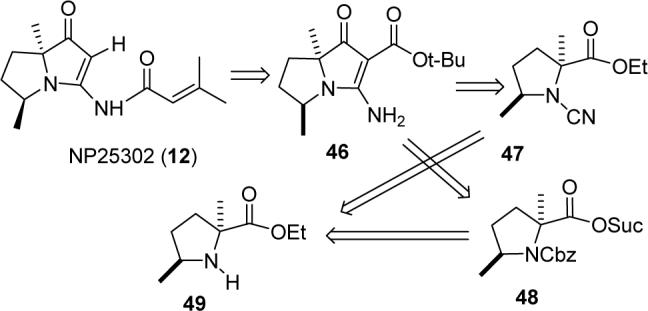
Retrosynthesis of NP25302
The challenging portion of this synthesis is the efficient and stereospecific preparation of 49. 2,5-Dimethylproline has been prepared, but the stereochemistry was not addressed.30 Feringa reported the Yb(OTf)3-catalyzed Michael addition of ethyl 2-nitropropionate (51) to methyl vinyl ketone (50) to give 52 in 99% yield (see Scheme 11).31 Hydrogenation (35 psi) of 52 over Pd/C for 3 d gave a 2:1 mixture of hydroxylamines 53 and 54 in 69% yield.31 We decided to reexamine this hydrogenation in an attempt to improve the stereoselectivity and complete the reduction to give 49.
SCHEME 11.
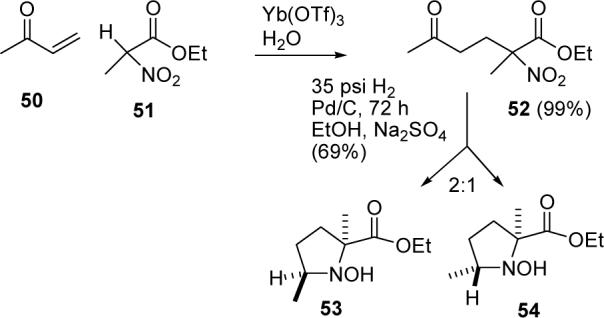
Feringa's Synthesis of 53 and 54
Reaction of 50 and nitro ester 51 with 0.1 equiv of DABCO in CH2Cl2 gave 52 in 98% yield (see Scheme 12). Reduction under Feringa's conditions31 gave a 2:1 mixture of 53 and 54 as reported. The stereoselectivity problem was eventually overcome by developing a two-step route that gave 53 selectively. Reductive cyclization of 52 over Pd/C for 15 h at 1 atm of H2 provided nitrone 55. Further reduction over PtO2 for 8 h at 1 atm of H2 gave a >20:1 mixture of hydroxylamines 53 and 54 in quantitative yield. The stereochemistry of 53 was confirmed by an NOE observed between the proton α to the nitrogen and both methyl groups, showing that H-5 and Me-2 are on the same face of the proline ring. Presumably, hydrogen approaches from the less sterically hindered side of the proline ring with the quaternary methyl rather than the carboethoxy group. It is not clear why the selectivity achieved with Pt is so much better than that obtained with Pd.
SCHEME 12.
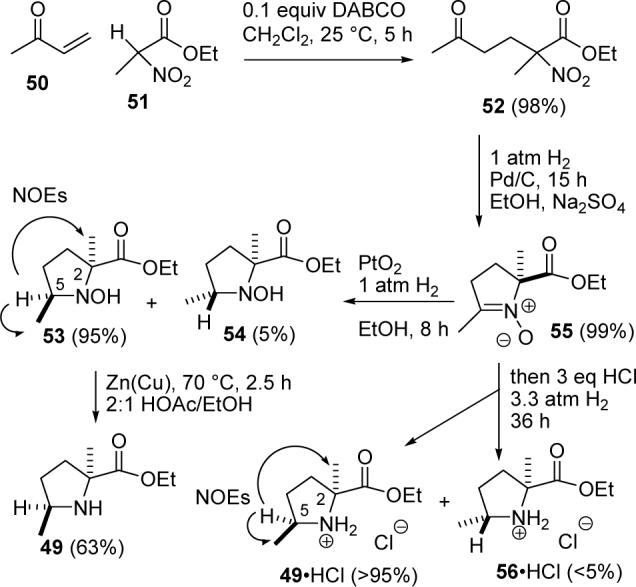
Synthesis of trans-2,5-Dimethylproline Ethyl Ester (49)
There are numerous reports of the reduction of nitro ketones to pyrrolidines rather than N-hydroxy pyrrolidines.32 We suspect that steric hindrance retards hydrogenolysis of 53 and 54. The N-OH bond of 53 can be reduced by stirring with Zn(Cu) in refluxing acetic acid to give the required trans-2,5-dimethylproline ethyl ester (49). The NMR spectra of 49 and 53 are very similar, but the IR spectra show the expected peaks for an OH in 53 at 3446 cm−1 (neat) and for an NH in 49 at 3345 cm−1 (neat).33
The need for three separate steps to reduce 52 to 49 was unappealing. We thought that addition of acid might accelerate the hydrogenation of 55 over Pd. This led to the development of a one-pot reductive cyclization of 52 that gave 49 stereospecifically. Hydrogenation (1 atm) of 52 as before over 10% Pd/C with Na2SO4 in EtOH gave nitrone 55. Concentrated hydrochloric acid (3 equiv) was added and the hydrogenation was continued at 3.3 atm for 36 h to provide amine 49•HCl with >20:1 stereoselectivity. An NOE observed between the proton α to the nitrogen and both methyl groups in 49•HCl confirmed the trans relationship of the methyl groups. Hydrogenation of 52 in the presence of HCl, without first reducing to the nitrone at neutral pH, was not as clean.
The conversion of 49 to 46 could be carried out via N-hydroxysuccinimide ester 48 (analogously to the conversion of 43 to 26) or cyanamide 47 (analogously to the conversion of 25 to 26). The first route was much more efficient for the preparation of jenamidines A1/A2 intermediate 26. However, we chose to use cyanamide 47 for two reasons. First, it takes only one step to make 47 from 49; at least three steps will be needed to prepare 48. Second, the two methyl groups in cyanamide 47 should decrease the reactivity of the cyanamide more than that of the ester so that enolate addition should occur selectively to the ester. The low yield of 26 from cyanamide 25 resulted from addition of the enolate to the ester and cyanamide at similar rates.
Reaction of 49•HCl with excess NaHCO3 and CNBr in EtOH for 2 h at 25 °C gave cyanamide 47 in 75% overall yield from nitro keto ester 52 (see Scheme 13). Addition of 47 to the lithium enolate of tert-butyl acetate at −45 to 25 °C in THF gave β-keto ester 57 as a mixture of keto/enol tautomers as the major product instead of the expected bicyclic product 46. The two methyl groups prevent the addition of the enolate to the cyanamide, but they also retard the cyclization of 57 to give the desired and expected product 46. This cyclization failed under a variety of conditions, but eventually was efficiently accomplished by treatment with t-BuOK in t-BuOH in a sealed tube at 135 °C for 15 h toafford bicyclic vinylogous urea 46 in 49% yield from cyanamide 47.
SCHEME 13.
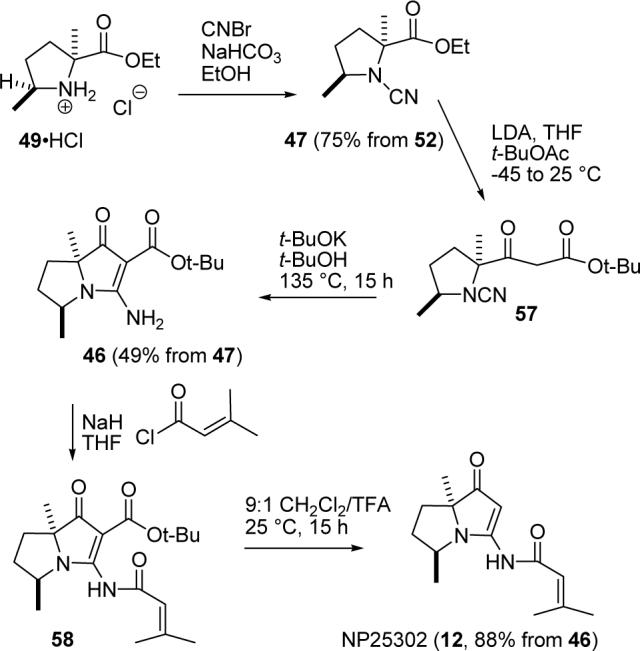
Completion of the Synthesis of (±)-NP25302 (12)
The completion of the synthesis was now trivial. Acylation of 46 with 3-methyl-2-butenoyl chlorideand NaH in THF at 25 °C for 2 h gave 58, which was stirred in 9:1 CH2Cl2/TFA for 15 h to give (±)-NP25302 (12) in 88% yield from 46.
Enantioselective Synthesis of Natural (+)-NP25302 (12)
Enantioselective synthesis of (+)-NP25302 will require carrying out an enantioselective Michael reaction of ethyl 2-nitropropionate (51) and methyl vinyl ketone (50) to give optically pure 52. Feringa prepared 52 in 72% ee using Al-Li-2,2′-dihydroxybinaphthyl, prepared in situ from LAH and 2.45 equiv of (R)-BINOL, as the asymmetric catalyst.34 Deng and coworkers have developed bifunctional cinchona alkaloids such as 59 that efficiently catalyze enantioselective Michael additions to α,β-unsaturated carbonyl compounds.35 The modified dihydroquinine 60 was synthesized analogously to 5935b from hydrocupreine36 by protection of the phenol with TIPSCl and imidazole in DMF in 95% yield, arylation of the alcohol with 4,6-dichloro-2,5-diphenylpyrimidine and KOH in toluene at 115 °C for 1 h, and deprotection of the TIPS group with aqueous HF in CH3CN to give 60 in 50% overall yield (see Figure 3).
FIGURE 3.
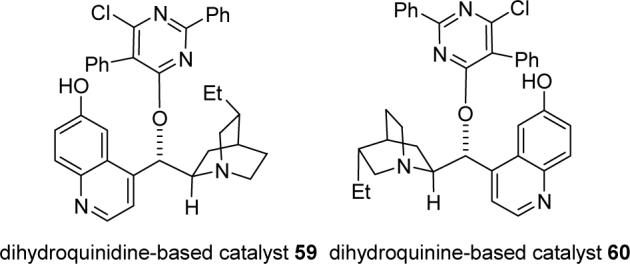
Dihydroquinidine-based catalysts 59 and 60.
Michael addition of 51 to 50 with 10 mol% of catalysts 59 and 60 at −20 °C for 3 days gave quantitative yields of (−)-52 (80% ee) and (+)-52 (90% ee), respectively (see Scheme 14). Reaction with 59 at 23 °C was complete in 5 h, but the enantioselectivity dropped to 66%. At −50 °C, the reaction with 59 proceeded too slowly and did not go to completion. The enantioselectivity was determined by chiral HPLC analysis of the ethylene glycol ketal derived from 52 as described by Feringa.34
SCHEME 14.
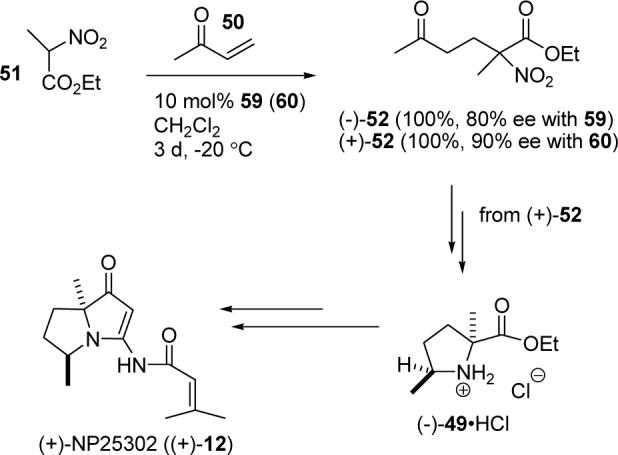
Synthesis of (+)-NP25302
Carrying out the sequence developed in the racemic series starting from (−)-52 (80% ee) gave (+)-49 and (−)-NP25302 (80% ee) with [α]22D −60 (c 1.0, MeOH) while the natural product has [α]22D +115 (c 1.1, MeOH). The synthesis of natural (+)-NP25302 (90% ee) with [α]22D +70 (c 1.0, MeOH) was completed from (+)-52 (90% ee) via (−)-49.
Although chiral HPLC established the enantiomeric excess of 52, we still needed to assign the absolute configuration. We thought that this could be best carried out at the stage of the dimethylproline derivative 49. Initial attempts to form a crystalline salt from (−)-49 with (+)-camphor-10-sulfonic acid for x-ray crystallography failed, although this approach has been used to determine the absolute configuration of a 2-methylproline ester.37 Hoye has prepared the diastereomeric Mosher amides 61 and 62 from (2R,5R)-2,5-dimethylpyrrolidine and showed that the NMR spectra are remarkably different as indicated on the structures (see Figure 4).38 Conformational studies indicated that the dominant conformation has the trifluoromethyl group syn to the carbonyl as shown. One methyl group, presumably that adjacent to the carbonyl, absorbed at δ 1.30 and 1.32 in the two diastereomers. As expected, the methyl group adjacent to the phenyl group in 62 (δ 0.10) is shifted upfield 0.99 ppm from the methyl group in 61 (δ 1.09). Similarly, the methine hydrogen in 61 adjacent to the phenyl group (δ 3.19) is shifted upfield 1.24 ppm from the methine hydrogen in 62 (δ 4.43).
FIGURE 4.
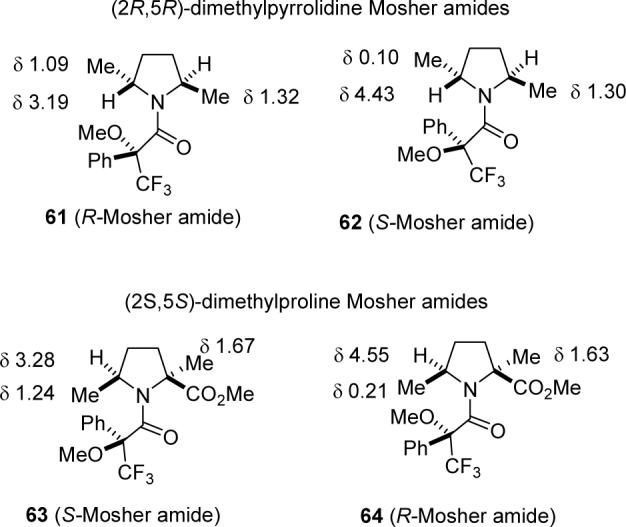
1H NMR spectral data of Mosher amides 61-64.
There were some concerns regarding the application of this procedure to determining the absolute stereochemistry of (−)-49. The amine component of Hoye's Mosher amides 61 and 62 is C2-symmetric so that there is only one conformer. Although Mosher amides 63 and 64 can exist as two conformers, we expected that the conformer with the smaller carbonyl group adjacent to the more hindered side bearing the ester group would be much more stable so that the shielding effects on the secondary methyl group and methine hydrogen should be similar to those observed in 61 and 62. Acylation of (−)-49 with both R- and S-Mosher acid chlorides39 gave the corresponding S- and R-Mosher amides, 63 and 64, respectively. The shielding effects are almost identical to those observed in the pseudo-enantiomeric amides 61 and 62, establishing that the (−)-49 obtained from catalyst 60 is the (2S,5S) isomer and that natural (+)-NP25302 (12) is the (4S,7S) isomer. Synthetic 12 with 90% ee has [α]D +70 indicating that optically pure material should have [α]D +78, rather than +115 as reported. This is not due to degradation of the 90% ee of 52 during the reductive cyclization to give 49 because the NMR spectrum of crude Mosher amides 63 and 64 prepared from (−)-49 indicated the presence of a 20:1 mixture of isomers (90% ee).
Approaches toward the synthesis of jenamidines B1/B2 (9)
Jenamidines B1/B2 (9) differ from jenamidines A1/A2 (8) by the presence of an additional hydroxy group on the ring fusion. Attempted oxidation of jenamidines A1/A2 acetate (39a) under a variety of conditions gave a complex mixture of products as expected for a functionally dense, base-labile compound. Attempted hydroxylation of 26 was equally unsuccessful as was epoxidation of the N-acetyl enol acetate of 26 prepared with 2 equiv of AcCl and 2 equiv of THF at 25 °C.
α-Acetoxylation of ketones can be achieved with Mn(OAc)3 and Pb(OAc)4.40,41 Reaction of 26 with Mn(OAc)3 was not promising, but reaction with 2 equiv of Pb(OAc)4 in refluxing benzene for 9 h proceeded cleanly, but with modest material balance (45%) to give a compound in 39% yield whose structure was eventually established to be 65 based on 1H and 13C NMR, IR and mass spectral data (see Scheme 15). The 1H NMR spectrum showed NH2 protons at δ 8.28 and 7.95. The absorption for H4 at δ 6.04 (dd, 1, J = 6.1, 6.7) is consistent with a proton adjacent to a nitrogen and an acetate.42,43 We expected that acetoxylation at C4 should occur from the convex face to give 65 with the acetates cis. This was supported by molecular mechanics calculations. MMX calculations with conformational searching using PCMODEL predicted vicinal coupling constants for H4 (dd, J = 8.0, 7.1) for 65, which correspond well with the observed values, and (dd, J = 6.7, 2.8) for the other diastereomer with the acetates trans, which do not correspond well with the observed values. An NOE was observed between H4 and H6α as expected for a calculated distance of 3 Å in 65. The calculated distance between H4 and the closest H6 in the other isomer is 4 Å, which is too far for an NOE to be observed.
SCHEME 15.
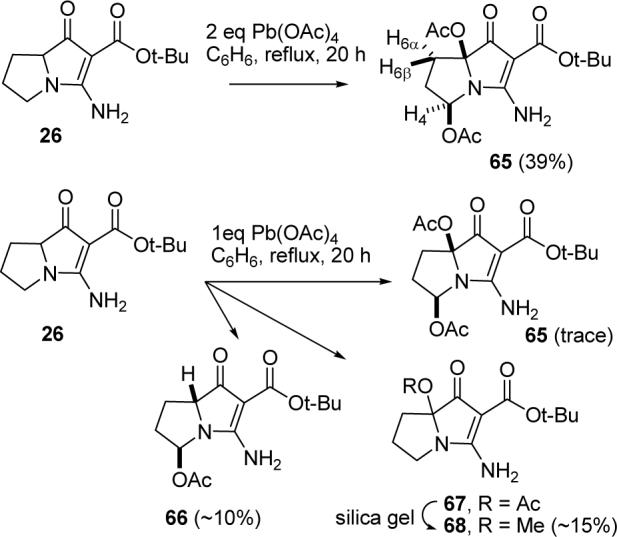
Oxidative Acetoxylation of 26
We hoped that oxidation of 26 with only 1 equiv of Pb(OAc)4 would occur selectively at the ring fusion to give 67. Unfortunately, oxidation of 26 with 1 equiv of Pb(OAc)4 in refluxing benzene gave a 3:2 mixture of the desired monoacetate 67, and the undesired regioisomer 66 and traces of bis acetate 65. A similar reaction at room temperature was selective for the undesired regioisomer giving a 1:5 mixture of 67 and 66. Slow flash chromatography using 39:1 CH2Cl2/MeOH as eluent afforded pure 66, but the acetate of the desired regioisomer 67 exchanged with methanol to give 68. The relative stereochemistry of 66 was not established, but it is tentatively assigned assuming that oxidation occurs on the less hindered convex face. The facile oxidation adjacent to the amide nitrogen was unexpected because we thought that the position adjacent to the ketone should be more easily oxidized. However, there is limited precedent for oxidation adjacent to an amide nitrogen with Pb(OAc)443 and electrochemical oxidation adjacent to amide nitrogens occurs readily.44 Complex mixtures were obtained from Pb(OAc)4 oxidation of the 29/30 mixture or 31 and attempts to prepare jenamidines B1/B2 analogues from 68 were not promising. In conclusion, acetoxylation with Pb(OAc)4 at the ring fusion could be accomplished to give 67, but acetoxylation adjacent to the nitrogen occurred at a competitive rate to give 66 and bis acetate 65 indicating that application of this method to the synthesis of jenamidines B1/B2 (9) will not be straightforward.
Formal synthesis of SB-311009 analogues. We isolated 23 from an unsuccessful approach to jenamidines A1/A2. While looking for analogous compounds to help us confirm the structure of 23, we came across SB-253514 (70), which was isolated by Readshaw and coworkers from the culture broths of Pseudomonas fluorescens strain DSM11579 in 2000.45 The glycosidic linkage was hydrolyzed enzymatically to give the alcohol SB-311009 (69) (see Scheme 16).45c SB-253514 is a potent and selective inhibitor (IC50 = 51 nM) of lipoprotein-associated phospholipase A2 (LpPLA2), the enzyme responsible for the conversion of phosphatidylcholine to lysophosphatidylcholine and oxidized fatty acids during the conversion of low density lipoprotein to its oxidized form.45a Pinto prepared 74, in which R is a variety of long alkyl chains, and found that these analogues were 2−4 times more biologically active than 69 or 70.46 A five-step sequence converted N-Boc-proline (71) to the unsaturated 2-trimethylsilylethyl ester 72 in 7% overall yield. Deprotection of the 2-trimethylsilylethyl ester with TBAF gave acid 73 in 100% yield, which was coupled with an amine using EDCI to give 74.
SCHEME 16.
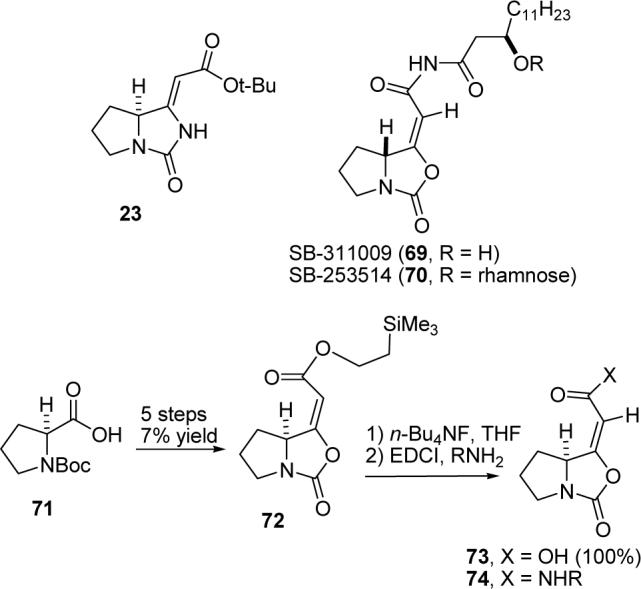
Pinto's Synthesis of SB-311009 Analogue 74
We thought that 72 and related esters could be prepared in a single step by a Wittig reaction between the N-carboxyanhydride (NCA) of proline (76) and a stabilized ylide such as 77 or 79 (see Scheme 17). Bateson reported that the Wittig reaction of the NCA of the cyclohexylidene aminal of homoserine with ylide 77 in EtOAc gave the enoate in 81% yield.47 To the best of our knowledge, this is the only report of a Wittig reaction on an NCA although Wittig reactions on succinic and phthalic anhydrides have been extensively studied.48
SCHEME 17.
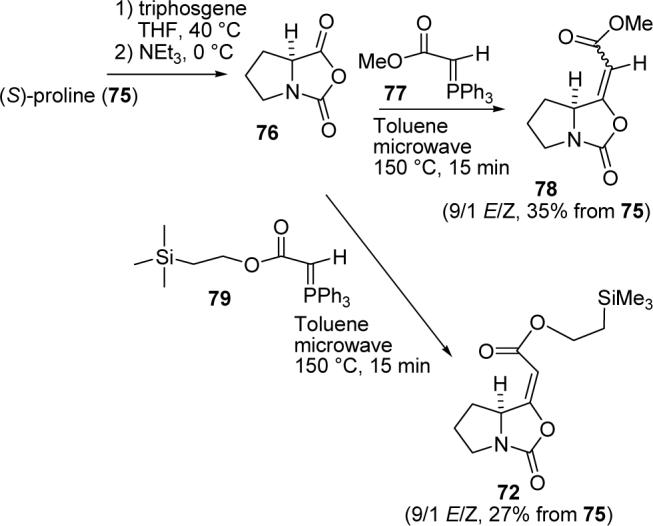
One Step Synthesis of Intermediate 72
Proline-NCA (76) was prepared by reaction of proline (75) with triphosgene at 40 °C in THF followed by the addition of triethylamine at 0 °C.49 Proline-NCA is very unstable because nucleophiles initiate polymerization,50 so freshly prepared 76 in THF was used for all reactions. Reaction of 76 with ylide 77 in EtOAc following Bateson's procedure gave no 78. Eventually we found that heating 76 and 77 in toluene at reflux provided 78 as a >9:1 E/Z mixture in a highly variable 10−20% yield from proline. Fortunately, reaction of proline NCA (76) with 77 in toluene at 150 °C in a microwave oven for 15 min (75 psi) reproducibly afforded enoate 78 as a >9:1 E/Z mixture of isomers in 35% yield. The alkene hydrogen absorbs at δ 5.66 in the E-isomer and δ 5.05 in the Z-isomer as observed by Pinto for the isomers of 72.
Hydrolysis of the methyl ester of 78 to give acid 73 could not be accomplished under a variety of typical hydrolysis conditions. This is consistent with the observation that cleavage of the glycosidic linkage of SB-253514 (70) to give SB-311009 (69) could not be achieved with either acid or base, possibly due to the instability of the cyclic carbamate.45c The cleavage could only be achieved enzymatically.45c
We therefore decided to prepare 2-trimethylsilylethyl ester 72 since Pinto had hydrolyzed this to give acid 73 with TBAF.46 The Wittig reaction of 76 with ylide 7951 in toluene at 150 °C for 15 min in a microwave oven gave Pinto's intermediate 72 in 27% overall yield from proline as a >9:1 E/Z mixture of stereoisomers with spectra identical to those provided by Dr. Pinto. This completes a formal synthesis of SB-311009 analogues, which is a significant improvement over the earlier procedure which required 5 steps and proceeded in 7% yield.
In conclusion, the proposed structures of jenamidines (1−3) have been revised to 8-10. The revised structural assignment has been confirmed by the synthesis of jenamidines A1/A2 (8). The first total synthesis of natural (+)-NP25302 (12) was completed both stereospecifically and enantioselectively. Pb(OAc)4 oxidation of jenamidines A1/A2 intermediate 26 gave access to jenamidines B1/B2 precursors, but indicated that oxidation α to the amide occurred at a rate comparable to oxidation α to the ketone. A one-step formal synthesis of SB311009 analogues was completed via a Wittig reaction with proline-NCA to give 72.
Experimental Section
Preparation of 26 from Cbz-Proline N-Hydroxysuccinimide Ester (43)
tert-Butyl cyanoacetate (0.68 mL, 4.8 mmol) was added dropwise to a solution of NaH (160 mg, 4.0 mmol, 60% dispersion in mineral oil) in 8 mL of dry benzene at rt. The resulting slurry was stirred for 1 h and treated with 43 (554 mg, 1.6 mmol) in 2 mL of dry benzene. The mixture was stirred for an additional 3 h at 25 °C, quenched with H2O, and extracted with Et2O. The aqueous layer was acidified with 2 M HCl resulting in the formation of a white precipitate, which dissolved upon addition of CH2Cl2. The layers were separated and the aqueous layer was extracted with CH2Cl2. The combined CH2Cl2 extracts were dried over MgSO4 and concentrated to give 44.
A solution of crude 44 in 25 mL of MeOH was stirred with 10% Pd/C (170 mg, 0.16 mmol) under 1 atm of H2 at 25 °C for 2 h. The mixture was filtered through Celite and concentrated to give crude 45, which cyclized on standing for 24 h at 25 °C to give crude 26. Flash chromatography on silica gel (95:5 EtOAc/MeOH) gave 190 mg (50%) of 26: mp 200−203 °C; [α]22D −62.2 (c 1.1, MeOH); UV (MeOH) λmax nm (log ε) 211(4.17), 241 (4.21), 264 (4.11); 1H NMR (CDCl3) 8.00 (br, 1, NH), 5.42 (br, 1, NH), 3.87 (dd, 1, J = 6.7, 9.8), 3.34−3.42 (m, 1), 3.20−3.28 (m, 1), 2.06−2.26 (m, 3), 1.54−1.60 (m, 1), 1.55 (s, 9); 13C NMR (CDCl3) 193.1, 174.0, 164.9, 90.5, 79.9, 69.7, 46.6, 28.5 (3 C), 27.6, 26.7; IR (KBr) 3475, 3077, 1714, 1638; HRMS (DEI) calcd for C12H18N2O3 (M+) 238.1317, found 238.1315.
tert-Butyl (4S)-4-[[(tert-Butyl)dimethylsilyl]oxy]-2-methylpent-2E-enoate (34)
A solution of (1-tert-butoxycarbonylethylidene)triphenylphosphorane (32)22 (1.9 g, 4.7 mmol) in 18 mL of dry CH2Cl2 was added to 2-[[(tert-butyl)dimethylsilyl]oxy]-propanal (33)23 (730 mg, 3.8 mmol) in 8 mL of dry CH2Cl2. The mixture was stirred at 25 °C for 2 h and quenched with 25 mL of H2O. The layers were separated and the organic layer was dried over MgSO4 and concentrated. Flash chromatography on silica gel (98:2 hexanes/EtOAc) yielded 770 mg (67%) of 34: 1H NMR (CDCl3) 6.57 (br d, 1, J = 8.5), 4.59 (dq, 1, J = 8.5, 6.7), 1.78 (d, 3, J = 1.2), 1.49 (s, 9), 1.22 (d, 3, J = 6.7), 0.88 (s, 9), 0.05 (s, 3), 0.04 (s, 3); 13C NMR (CDCl3) 167.3, 144.7, 126.8, 80.1, 66.0, 28.0 (3 C), 25.8 (3C), 23.4, 18.1, 12.5, −4.7, −4.8; IR (neat) 1710; HRMS (DCI/NH3) calcd for C16H36NO3Si (MNH4+) 318.2464, found 318.2455.
tert-Butyl (4S-4-Hydroxy-2-methylpent-2E-enoate (35)
Silyl ether 34 (300 mg, 1.0 mmol) in 2 mL of THF was added to 18 mL of a 1.4 M solution of pyridine•HF (5 mL pyridine•(HF)x, 20 mL THF, 20 mL pyridine). The mixture was stirred for 4 h in an Eppendorf tube, slowly quenched with saturated sodium bicarbonate solution, and extracted with EtOAc. The combined organic extracts were dried over MgSO4 and concentrated to yield 185 mg (99%) of pure 35: 1H NMR (CDCl3) 6.58 (br d, 1, J = 8.3), 4.67 (dq, 1, J = 8.3, 6.1), 1.83 (d, 3, J = 1.5), 1.49 (s, 9), 1.31 (d, 3, J = 6.1); 13C NMR (CDCl3) 167.2, 143.0, 129.0, 80.5, 64.9, 28.0 (3 C), 22.6, 12.6; IR (neat) 3421, 1707; HRMS (DCI/NH3) calcd for C10H22NO3 (MNH4+) 204.1600, found 204.1606.
tert-Butyl (S)-4-Methoxyacetoxy-2-methylpent-2E-enoate (36d)
Methoxyacetyl chloride (0.15 mL, 1.6 mmol) was added slowly to a solution of alcohol 35 (185 mg, 0.99 mmol), pyridine (0.17 mL, 2.12 mmol), and DMAP (12 mg, 0.1 mmol) in 15 mL of dry THF under N2 resulting in a white precipitate. The mixture was stirred for 2 h at 25 °C, filtered, diluted with EtOAc, washed successively with 2 M HCl, water, saturated sodium bicarbonate solution, and water, dried over MgSO4, and concentrated to give 255 mg (99%) of pure 36d: 1H NMR (CDCl3) 6.51 (br d, 1, J = 8.9), 5.73 (dq, 1, J = 8.9, 6.1), 4.03 (s, 2), 3.45 (s, 3), 1.88 (d, 3, J = 1.5), 1.49 (s, 9), 1.36 (d, 3, J = 6.1); 13C (CDCl3) 169.4, 166.6, 137.6, 131.3, 80.7, 69.8, 68.2, 59.3, 27.9 (3 C), 19.7, 12.8; IR (neat) 1756, 1709; HRMS (DCI/NH3) calcd for C13H26NO5 (MNH4+) 276.1811, found 276.1809.
(S)-4-Methoxyacetoxy-2-methylpent-2E-enoic acid (37d)
A solution of ester 36d (255 mg, 0.98 mmol) in 5 mL of 9:1 CH2Cl2:TFA was stirred for 12 h at 25 °C. The mixture was diluted with H2O and extracted with CH2Cl2. The combined organic extracts were dried over MgSO4 and concentrated to give 194 mg (98%) of pure 37d: 1H NMR (CDCl3) 11.0 (br s, 1, OH), 6.74 (d, 1, J = 8.8), 5.74 (dq, 1, J = 8.8, 6.4), 4.05 (s, 2), 3.45 (s, 3), 1.93 (s, 3), 1.38 (d, 3, J = 6.4); 13C NMR (CDCl3) 172.8, 169.5, 141.3, 129.0, 69.7, 68.1, 59.3, 19.4, 12.4; IR (neat) 3187, 1753, 1696; HRMS (DCI/NH3) calcd for C9H18NO5 (MNH4+) 220.1185, found 220.1189.
Conversion of 37d to the Acid Chloride
A solution of acid 37d (194 mg, 0.96 mmol) in 8 mL of oxalyl chloride was stirred for 4 h at 25 °C under N2. Excess oxalyl chloride was removed under reduced pressure to give (S)-4-methoxyacetoxy-2-methylpent-2E-enoyl chloride (38d), which was used immediately: 1H NMR (CDCl3) 6.98 (d, 1, J = 8.6), 5.73 (dq, 1, J = 8.6, 6.7), 4.06 (s, 2), 3.46 (s, 3), 2.00 (s, 3), 1.42 (d, 3, J = 6.7).
Jenamidines A1/A2 (8), Jenamidines A1/A2 4'-Methoxyacetate (39d), and 3-Amino-5,6,7,7a-tetrahydro-1H-pyrrolizin-1-one (14b)
Vinylogous urea 26 (106 mg, 0.44 mmol) and NaH (60% dispersion in mineral oil, 45 mg, 1.1 mmol) were stirred in 10 mL of dry THF under N2 for 10 min. Acid chloride 38d (0.96 mmol) in 3 mL of dry THF was added and the resulting solution was stirred for 2 h, quenched with brine, and extracted with CH2Cl2. The combined organic extracts were dried over MgSO4 and concentrated to give 196 mg of a mixture of mono and bis acylated products. A solution of the mixture in 10 mL of 9:1 CH2Cl2:TFA was stirred for 15 h, neutralized with saturated sodium bicarbonate solution, saturated with NaCl and extracted with EtOAc. The combined organic extracts were dried over MgSO4 and concentrated to afford jenamidines A1/A2 4'-methoxyacetate (39d) as the major product and no jenamidines A1/A2 (8), as expected. Flash chromatography on silica gel (97:3 CH2Cl2/MeOH to 90:10 CH2Cl2/MeOH) gave 55 mg (39%) of 39d, followed by 20 mg (18%) of a mixture of 8 and other decomposition products.
A solution of a second batch of crude 39d (prepared as before from 96 mg (0.40 mmol) of 26, 42 mg (1.0 mmol) of NaH, and 0.96 mmol of 38d followed by TFA hydrolysis) was stirred in 10 mL of MeOH containing 10 drops of H2O and 200 mg of Na2CO3 at 0 °C for 24 h. The mixture was filtered through silica gel and concentrated to give a 4:1:1 mixture of 8, 39d, and 14d. Flash chromatography on silica gel (95:5 CH2Cl2/MeOH) gave 14 mg (11% from 26) of 39d and 45 mg (45% from 26) of 8, while 14b was recovered along with a complex mixture of products upon flushing the silica gel with MeOH.
Data for 39d as a 1:1 mixture of diastereomers: 1H NMR (CD3OD) 6.345 (br d, 1 × 0.5, J = 8.0), 6.340 (br d, 1 × 0.5, J = 8.0), 5.72−5.80 (m, 1), 5.64 (s, 1), 4.07 (s, 2 × 0.5), 4.06 (s, 2 × 0.5), 3.95 (dd, 1, J = 8.8, 8.8), 3.40−3.47 (m, 1), 3.42 (s, 3 × 0.5), 3.41 (s, 3 × 0.5), 3.20−3.26 (m, 1), 2.10−2.25 (m, 3), 1.994 (s, 3 × 0.5), 1.991 (s, 3 × 0.5), 1.50−1.60 (m, 1), 1.41 (d, 3 × 0.5, J = 6.1), 1.40 (d, 3 × 0.5, J = 6.1); 13C NMR (CD3OD) 204.7, 173.4, (171.39, 171.37), (169.4, 169.3), (138.04, 138.00), (134.20, 134.18), 93.9, 70.7, 70.5, (69.4, 69.3), (59.54, 59.52), 49.3, 28.8, 27.5, 19.8, (13.29, 13.26); IR (neat) 1744, 1702, 1636, 1563, 1506; HRMS (DCI/NH3) calcd for C16H23N2O5 (MH+) 323.1607, found 323.1609.
Data for jenamidines A1/A2 (8) as a 1:1 mixture of diastereomers: [α]22D 4.2 (c 0.6, MeOH), lit.1 [α]22D 6.8 (c 0.7, MeOH); UV λmaxMeOH nm (log ε) 240 (4.29), 282 (3.84), 3.26 (4.02); 1H NMR (CD3OD) 6.37 (d, 1, J = 7.9), 5.67 (s, 1 × 0.5), 5.63 (s, 1 × 0.5), 4.67 (dq, 1, J = 6.5, 7.9), 3.92−3.98 (m, 1), 3.42−3.49 (m, 1), 3.18−3.26 (m, 1), 2.08−2.24 (m, 3), 1.92 (s, 3), 1.50−1.60 (m, 1), 1.31 (d, 3 × 0.5, J = 6.5), 1.29 (d, 3 × 0.5, J = 6.5); 13C NMR (CD3OD) (204.69, 204.57), (173.61, 173.48), (169.84, 169.82), (143.67, 143.65), (131.62, 131.60), (93.82, 93.80, 93.76, 93.75), (70.77, 70.67), (65.31, 65.25), 49.29, 28.80, (27.51, 27.47, 27.40, 27.37), (22.68, 22.64), 12.92; IR (neat) 2973, 1699, 1634, 1557, 1504, 1404, 1244, 1139, 1059; HRMS (DCI/NH3) calcd for C13H19N2O3 (MH+) 251.1396, found 251.1394. The 1H and 13C NMR spectrum are identical to those of the natural product.1,2a H7a is approximately 50% exchanged with deuterium. C2 (93.7−93.8) and C7 (27.3−27.5) absorb as four peaks since the carbon for each diastereomer absorbs separately for the proton and deuterium isomers due to the isotope shift.28
Pure 39d (30 mg, 0.093 mmol) was stirred in 2 mL of MeOH containing 5 drops of H2O and 50 mg of Na2CO3 at 0 °C for 6 h. The mixture was filtered through silica gel and concentrated. Flash chromatography on silica gel (90:10 CH2Cl2/MeOH) gave 6 mg (20%) of recovered 39d, followed by 13 mg (56%, 70% based on recovered 39d) of 8 and 2.5 mg (18%) of 14b.
Data for 14b: 1H NMR (CD3OD) 4.50 (s, 1), 3.90 (dd, 1, J = 6.7, 9.1), 3.28−3.35 (m, 1), 3.12−3.20 (m, 1), 2.05−2.25 (m, 3), 1.40−1.48 (m, 1); 13C NMR (CD3OD) 197.5, 178.5, 72.1, 47.7, 29.3, 28.2 (C-2 was not observed because H-2 exchanged with D); HRMS (DEI) calcd for C7H10N2O (M+) 138.0793, found 138.0789.
Ethyl trans-2,5-Dimethyl-2-pyrrolidinecarboxylate (49)
A solution of 52 (886 mg, 4.08 mmol) in 16 mL of EtOH containing 10% Pd on activated carbon (430 mg, 0.41 mmol) and Na2SO4 (580 mg, 4.08 mmol) was stirred under 1 atm of H2 at 25 °C for 20 h to give nitrone 55. Concd HCl (1 mL) was added and the mixture was stirred in a Parr Shaker under 50 psi of H2 at 25 °C for 36 h. The mixture was filtered through Celite and concentrated to give 49•HCl containing <5% of the diastereomer 56 •HCl, which was used without purification in the next step: 1H NMR (CDCl3) 11.0 (br s, 1, NH), 8.7 (br s, 1, NH), 4.32 (q, 2, J = 7.0), 4.10−4.20 (m, 1), 2.45−2.51 (m, 1), 2.22−2.30 (m, 1), 2.05−2.15 (m, 1), 1.88 (s, 3), 1.60−1.72 (m, 1), 1.59 (d, 3, J = 6.7), 1.36 (t, 3, J = 7.0). The 1H NMR spectrum of 49•HCl in CDCl3 containing excess NaHCO3 to convert it back to the free amine is identical to that of 49 prepared by Zn(Cu) reduction of hydroxylamine 53.
(−)-49·HCl was prepared analogously from (+)-52: [α]22D −44.5 (c 0.6, MeOH).
Ethyl 1-Cyano-2,5-dimethyl-2-pyrrolidinecarboxylate (47)
NaHCO3 (1.7 g, 20.4 mmol) and CNBr (520 mg, 4.9 mmol) were added to a solution of 49•HCl (prepared from 4.08 mmol of 52) in 20 mL of EtOH and the resulting mixture was stirred for 2 h at 25 °C. H2O was added and the solution was stirred for 15 min and extracted with CH2Cl2. The combined organic extracts were dried over MgSO4 and concentrated to give 599 mg (75% from 52) of cyanamide 47: 1H NMR (CDCl3) 4.18−4.27 (m, 2), 3.82 (ddd, 1, J = 6.1, 6.1, 9.1), 2.40−2.46 (m, 1) 2.02−2.10 (m, 1), 1.75−1.84 (m, 1), 1.61 (s, 3), 1.50−1.60 (m, 1), 1.38 (d, 3, J = 6.1), 1.31 (t, 3, J = 7.0); 13C NMR (CDCl3) 172.9, 114.5, 68.0, 61.8, 58.3, 37.1, 32.0, 23.0, 19.5, 14.0; IR (neat) 2208, 1737; HRMS (EI) Calcd for C10H16N2O2 (M+) 196.1212, found 196.1206.
(−)-47 was prepared analogously from (−)-49•HCl: [α]22D −34.3 (c 0.6, MeOH).
tert-Butyl 1-Cyano-2,5-dimethyl-β-oxo-2-pyrrolidinepropanoate (57)
tert-Butyl acetate (0.84 mL, 6.2 mmol) was added dropwise over a period of 10 min to a freshly prepared solution of LDA (5.4 mmol in 14 mL of THF) at −45 °C under N2. The solution was stirred for 15 min and treated with cyanamide 47 (467 mg, 2.4 mmol) in 5 mL of dry THF. The mixture was stirred for 1 h at −45 °C and 2 h at 25 °C, quenched with H2O, and extracted with CH2Cl2. The combined organic extracts were dried over MgSO4 and concentrated to give crude 57, which was used without purification. An analytical sample was prepared by flash chromatography on silica gel (95:5 CH2Cl2/MeOH) to give pure 57 as a 2:1 keto/enol mixture: 1H NMR (CDCl3) 5.25 (s, 1 × 0.33), 3.75−3.86 (m, 1), 3.62 (d, 1 × 0.66, J = 15.9), 3.55 (d, 1 × 0.66, J = 15.9), 2.54 (ddd, 1 × 0.66, J = 2.4, 6.7, 13.4), 2.38 (ddd, 1 × 0.33, J = 1.8, 6.7, 12.8), 1.95−2.05 (m, 1), 1.65−1.80 (m, 1), 1.60 (s, 3 × 0.33), 1.54 (s, 3 × 0.66), 1.40−1.55 (m, 1), 1.51 (s, 9 × 0.33), 1.48 (s, 9 × 0.66), 1.40 (d, 3 × 0.33, J = 6.1), 1.38 (d, 3 × 0.66, J = 6.1); 13C NMR (CDCl3) 203.1, 177.1, 172.6, 166.0, 114.7, 114.6, 89.7, 82.2, 81.5, 73.4, 67.4, 59.8, 58.9, 45.3, 37.2, 35.8, 31.8, 31.6, 28.1 (3 C × 0.33), 27.9 (3 C × 0.66), 23.7, 21.9, 19.5, 19.4; IR (neat) 2207, 1742, 1716; HRMS (ES) Calcd for C14H23N2O3 (MH+) 267.1709, found 267.1704.
tert-Butyl 3-Amino-5,7a-dimethyl-5,6,7-trihydro-1-oxo-1H-pyrrolizine-2-carboxylate (46)
Crude 57 (prepared from 2.4 mmol of 47) was stirred in 10 mL of t-BuOH with 3.5 mL of a 1.0 M solution of t-BuOK in t-BuOH under N2 in a sealed tube submerged in a 135 °C oil bath. After 15 h, the mixture was cooled, diluted with H2O and extracted with CH2Cl2. The combined organic extracts were dried over MgSO4 and concentrated. Flash chromatography on silica gel (95:5 CH2Cl2/MeOH) gave 310 mg (49% over 2 steps) of 46: mp 247−249 °C; 1H NMR (CDCl3) 8.20 (br s, 1, NH), 5.98 (br s, 1, NH), 3.94−4.04 (m, 1), 2.36−2.47 (m, 1), 1.74−1.92 (m, 2), 1.66−1.73 (m, 1), 1.55 (s, 9), 1.33 (d, 3, J = 6.7), 1.31 (s, 3); 13C NMR (CDCl3) 195.8, 170.6, 166.2, 90.7, 79.9, 75.3, 53.9, 35.3, 29.2, 28.5 (3 C), 24.4, 17.3; IR (neat) 3454, 1705, 1635; HRMS (ES) Calcd for C14H23N2O3 (MH+) 267.1709, found 267.1696.
(−)-46 was prepared analogously from (−)-47: [α]22D −21.3 (c 0.3, MeOH).
(±)-NP25302 (12)
Vinylogous urea 46 (150 mg, 0.56 mmol) and NaH (60% dispersion in mineral oil, 40 mg, 1.0 mmol) were stirred in 10 mL of dry THF under N2 at 25 °C for 10 min. A solution of 3,3-dimethylacryloyl chloride (95 mg, 0.80 mmol) in 2 mL of dry THF was added and the resulting mixture was stirred for 2 h, quenched with H2O, and extracted with CH2Cl2. The combined organic extracts were dried over MgSO4 and concentrated to give 233 mg of crude 58, which was stirred in 15 mL of a 9:1 CH2Cl2/TFA solution for 15 h at 25 °C. The mixture was neutralized with saturated sodium bicarbonate solution, saturated with NaCl, and extracted with EtOAc. The combined organic extracts were dried over MgSO4 and concentrated to give crude NP25302. Flash chromatography on silica gel (95:5 CH2Cl2/MeOH) gave 122 mg (88%) of pure; (±)-NP25302 (12): mp 193−195 °C; UV (MeOH) λmax nm (log ε) 252 (4.09), 282 (3.82), 334 (3.73); 1H NMR( CDCl3) 10.38 (br s, 1, NH), 6.01 (s, 1), 5.80 (s, 1), 4.05−4.13 (m, 1), 2.41−2.52 (m, 1), 2.21 (s, 3), 1.90 (s, 3), 1.77−1.88 (m, 2), 1.64−1.71 (m, 1), 1.34 (s, 3), 1.17 (d, 3, J = 6.5); 13C NMR (CDCl3) 205.5, 167.3, 164.2, 158.2, 117.6, 93.5, 74.8, 54.8, 35.4, 28.2, 27.8, 24.6, 20.4, 17.4; IR (KBr) 3296, 3206, 2974, 1710, 1641, 1572, 1507, 1140; HRMS (ES) Calcd for C14H21N2O2 (MH+) 249.1603, found 249.1593. The 1H and 13C NMR spectra are identical to those of an authentic sample.8
(+)-NP25302 (12) (90 mg) was prepared analogously from (−)-46: mp 227−230 °C, lit.8 mp 229−230 °C; [α]22D +70 (c 1.0, MeOH), lit.8 [α]22D +115.5 (c 1.1, MeOH).
(−)-NP25302 (12) (25 mg) was prepared analogously from (−)-52 via (+)-49, (+)-47, and (+)-46: mp 224−227 °C; [α]22D −60 (c 1.0, MeOH).
[2-Oxo-2-[2-(Trimethylsilyl)ethoxy]ethyl]triphenylphosphonium bromide was prepared as previously described.51 A solution of 2-(trimethylsilyl)ethanol (143 μL, 1.0 mmol) and triethylamine (140 μL, 1 mmol) in 1 mL of dry CH2Cl2 was added to a solution of bromoacetyl bromide (87 μL, 1 mmol) in 2 mL of dry CH2Cl2. The mixture was stirred overnight, diluted with water and extracted with CH2Cl2. The combined organic extracts were washed successively with 2 M HCl and brine, dried over MgSO4, and concentrated. The resulting oil was stirred with triphenylphosphine (525 mg, 2 mmol) in EtOAc for 24 h at rt. The precipitated salt was collected and used in the next step.
2-(Trimethylsilyl)ethyl (2-E)-[7aS)-Tetrahydro-3-oxo-1H,3H-pyrrolo[1,2-c]oxazol-1-ylidene]-acetate (72)
Triethylamine (0.11 mL, 0.80 mmol) was added to the above phosphonium salt (400 mg, 0.80 mmol) in 2 mL of toluene in a microwave tube. The mixture was stirred for 30 min under N2 to generate ylide 78. A solution of 76 in 2 mL of dry THF (prepared from 0.52 mmol of proline) was then added. The mixture was heated at 150 °C at 100 psi for 15 min in a microwave. After cooling, the mixture was concentrated to give crude 72 as a 9:1 mixture of the E and Z products. Flash chromatography on silica gel (CH2Cl2) gave 3 mg of a 6:1 mixture of (E)-72 and (Z)-72, followed by 39 mg (27%) of pure (E)-72: [α]22D -141 (c 0.9, MeOH); UV (MeOH) λmax nm (log ε) 212 (4.00), 238 (4.12); 1H NMR (CDCl3) 5.64 (d, 1, J = 1.8), 4.92 (ddd, 1, J = 7.3, 7.3, 1.8), 4.15−4.28 (m, 2), 3.69 (ddd, 1, J = 8.0, 8.0, 11.3), 3.26−3.35 (m, 1), 2.59−2.67 (m, 1), 2.05−2.19 (m, 2), 1.55−1.66 (m, 1), 0.99−1.04 (m, 2), 0.05 (s, 9); 13C NMR 166.4, 165.3, 156.8, 95.9, 64.2, 62.6, 45.9, 30.3, 26.3, 17.3, −1.5 (3 C); IR (neat) 1805, 1713, 1667; HRMS (CI) Calcd for C13H22NO4Si (MH+) 284.1318, found 284.1313. The 1H NMR spectral data for (E)-72 are identical to those provided by Dr. Ivan Pinto.46
The 1H NMR peaks for (Z)-72 in the mixture are identical to those in an NMR spectrum of (Z)-72 provided by Dr. Pinto:46 1H NMR (CDCl3) 5.05 (d, 1, J = 1.5), 4.35−4.45 (m, 1), 4.15−4.25 (m, 2), 3.60−3.75 (m, 1), 3.20−3.30 (m, 1), 2.00−2.30 (m, 3), 1.60−1.70 (m, 1), 0.99−1.05 (m, 2), 0.05 (s, 9).
Supplementary Material
Acknowledgment
We thank the NIH (GM50151) for generous financial support. We thank Dr. Isabel Sattler for copies of the 1H, 13C, and 2D NMR spectra of jenamidines A1/A2 (8), Dr. Satoshi Takamatsu for copies of the 1H and 13C NMR spectra of NP25302 (12), and Dr. Ivan Pinto for copies of the 1H NMR spectra of the (E)- and (Z)-isomers of 72. We thank Prof. Li Deng for communicating unpublished results on the conjugate addition of 51 to 50 with cinchona alkaloid 59.
Footnotes
Supporting Information Available. Details of experimental procedures and characterization for all of compounds not reported in the experimental section; copies of 1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org
References and Notes
- 1.Hu J-F, Wunderlich D, Thiericke R, Dahse H-M, Grabley S, Feng X-Z, Sattler I. J. Antibiot. 2003;56:747–754. doi: 10.7164/antibiotics.56.747. [DOI] [PubMed] [Google Scholar]
- 2.For preliminary communication of portions of this work see: Snider BB, Duvall JR, Sattler I, Huang X. Tetrahedron Lett. 2004;45:6725–6727. Snider BB, Duvall JR. Org. Lett. 2005;7:4519–4522. doi: 10.1021/ol0518784.
- 3.Pouchert CJ, Behnke J. The Aldrich Library of 13C and 1H FT NMR Spectra. Edition 1 Vol. 1. Aldrich; Milwaukee, WI: 1993. p. 1251c. [Google Scholar]
- 4.a Raucher S, Macdonald JE. Synth. Commun. 1980;10:325–331. [Google Scholar]; b Haider A, Cornuz G, Wyler H. Helv. Chim. Acta. 1975;58:1287–1292. [Google Scholar]
- 5.For the preparation of analogous compounds with the NH replaced by O or S see: Hamley P, Tinker A. Chem. Abstr. 2000;132:137406a. PCT Int. Appl. WO 00 06,576, 2000.
- 6.Singh H, Deep K. Tetrahedron. 1984;40:4937–4939. [Google Scholar]
- 7.Doyle TW, Nettleton DE, Balitz DM, Moseley JE, Grulich RE, McCabe T, Clardy J. J. Org. Chem. 1980;45:1324–1326. [Google Scholar]
- 8.Zhang Q, Schrader KK, ElSohly HN, Takamatsu S. J. Antibiot. 2003;56:673–681. doi: 10.7164/antibiotics.56.673. [DOI] [PubMed] [Google Scholar]
- 9.Adam W, Renze J, Wirth T. J. Org. Chem. 1998;63:226–227. [Google Scholar]
- 10.Ostercamp DL. J. Org. Chem. 1970;35:1632–1641. [Google Scholar]
- 11.a Murray A, Proctor GR, Murray PJ. Tetrahedron. 1996;52:3757–3766. [Google Scholar]; b Galeotti N, Poncet J, Chiche L, Jouin P. J. Org. Chem. 1993;58:5370–5376. [Google Scholar]
- 12.Wallace DJ, Klauber DJ, Chen C.-y., Volante RP. Org. Lett. 2003;5:4749–4752. doi: 10.1021/ol035959g. [DOI] [PubMed] [Google Scholar]
- 13.For the 1H NMR data of several similar pyrroles see: Barluenga J, Tomás M, Kouznetsov V, Suárez-Sobrino A, Rubio E. J. Org. Chem. 1996;61:2185–2190. Padwa A, Dean DC, Zhi L. J. Am. Chem. Soc. 1992;114:593–601.
- 14.Miyata O, Shinada T, Ninomiya I, Naito T, Date T, Okamura K, Inagaki S. J. Org. Chem. 1991;56:6556–6564. [Google Scholar]
- 15.Vinylogous urea 14b was later obtained as a byproduct during the hydrolysis of jenamidine A1/A2 esters 39. Spectral analysis indicated that it was not present in the mixture of products obtained from 15 and NH3.
- 16.a Snider BB, O'Hare SM. Tetrahedron Lett. 2001;42:2455–2458. [Google Scholar]; b Rydzewski RM, Bryant C, Oballa R, Wesolowski G, Rodan SB, Bass KE, Wong DH. Bioorg. Med. Chem. 2002;10:3277–3284. doi: 10.1016/s0968-0896(02)00173-6. [DOI] [PubMed] [Google Scholar]
- 17.In a related system, the alkene hydrogen of the (Z)-isomer absorbs at δ 5.07, whereas the alkene hydrogen of the (E)-isomer absorbs at δ 5.61: Bacchi A, Chiusoli GP, Costa M, Gabriele B, Righi C, Salerno G. Chem. Commun. 1997:1209–1210. Chiusoli GP, Costa M, Gabriele B, Salerno G. J. Mol. Catal. 1999;143:297–310.
- 18.a Montiel-Smith S, Cervantes-Mejía V, Dubois J, Guénard D, Guéritte F, Sandoval-Ramírez J. Eur. J. Org. Chem. 2002:2260–2264. [Google Scholar]; b Dietrich E, Lubell WD. J. Org. Chem. 2003;68:6988–6996. doi: 10.1021/jo034739d. [DOI] [PubMed] [Google Scholar]
- 19.Prasit P, Falgueyret J-P, Oballa R, Rydzewski R, Okamoto O. Chem. Abstr. 2001;135:318414j. doi: 10.1021/jm0003440. PCT Int. Appl. WO 01 07,7073, 2001. [DOI] [PubMed] [Google Scholar]
- 20.For similar syntheses of keto esters from esters see: Honda Y, Katayama S, Kojima M, Suzuki T, Izawa K. Org. Lett. 2002;4:447–449. doi: 10.1021/ol017163s. Honda Y, Katayama S, Kojima M, Suzuki T, Izawa K. Tetrahedron Lett. 2003;44:3163–3166.
- 21.For similar compounds see: Zhao M-X, Wang MX, Huang Z-T. Tetrahedron. 2002;58:1309–1316. Ceder O, Stenhede U. Acta Chem. Scand. 1973;27:2221–2223.
- 22.Giner J-L. Tetrahedron Lett. 2002;43:5457–5459. [Google Scholar]
- 23.a Hirama M, Shigemoto T, Itô S. J. Org. Chem. 1987;52:3342–3346. [Google Scholar]; b Massad SK, Hawkins LD, Baker DC. J. Org. Chem. 1983;48:5180–5182. [Google Scholar]
- 24.Rachina V, Blagoeva I. Synthesis. 1982:967–968. [Google Scholar]
- 25.a Hartmann B, Kanazawa AM, Deprés J-P, Greene AE. Tetrahedron Lett. 1991;32:5077–5080. [Google Scholar]; b Mori K, Matsushima Y. Synthesis. 1995:845–850. [Google Scholar]; c Saito N, Yamauchi R, Kubo A. Heterocycles. 1991;32:1203–1214. [Google Scholar]
- 26.a Wissner A, Grudzinskas CV. J. Org. Chem. 1978;43:3972–3974. [Google Scholar]; b Vidya R, Eggen M, Nair SK, Georg GI, Himes RH. J. Org. Chem. 2003;68:9687–9693. doi: 10.1021/jo0302197. [DOI] [PubMed] [Google Scholar]
- 27.Kocieński PJ. Protecting Groups. 3rd ed. Georg Thieme Verlag; Stuttgart: 2005. pp. 333–337. [Google Scholar]
- 28.For discussion of deuterium-induced 13C NMR shifts see: Morales-Rios MS, Cervantes-Cuevas H, Salgado-Escobar I, Joseph-Nathan P. Magn. Reson. Chem. 1999;37:243–245. Dziembowska T, Hansen PE, Rozwadowski Z. Prog. Nucl. Magn. Reson. Spectrosc. 2004;45:1–29.
- 29.a Igglessi-Markopoulou O, Sandris C. J. Heterocycl. Chem. 1982;19:883–890. [Google Scholar]; b Sauvé G, Le Berre N, Zacharie B. J. Org. Chem. 1990;55:3002–3004. [Google Scholar]; c Detsi A, Micha-Screttas M, Igglessi-Markopoulou O. J. Chem. Soc., Perkin Trans. 1. 1998:2443–2449. [Google Scholar]; d Gola A, Samartzi E, Bardakos V, Petroliagi M, Igglessi-Markopoulou O, Markopoulos J, Barkley JV. J. Heterocycl. Chem. 2000;37:681–686. [Google Scholar]; e Petroliagi M, Igglessi-Markopoulou O. J. Heterocycl. Chem. 2001;38:917–922. [Google Scholar]; f Detsi A, Gavrielatos E, Adam M-A, Igglessi-Markopoulou O, Markopoulos J, Theologitis M, Reis H, Papadopoulos M. Eur. J. Org. Chem. 2001:4337–4342. [Google Scholar]; g Hamilakis S, Tsolomitis A. Synth. Commun. 2003;33:4313–4319. [Google Scholar]; h Detsi A, Emirtzoglou P, Prousis K, Nikolopoulos AN, Skouridou V, Igglessi-Markopoulou O. Synlett. 2004:353–355. [Google Scholar]
- 30.Wright WB, Jr., Brabander HJ, Greenblatt EN, Day IP, Hardy RA., Jr. J. Med. Chem. 1978;21:1087–1089. doi: 10.1021/jm00208a017. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 31.Keller E, Feringa BL. Synlett. 1997:842–844. [Google Scholar]
- 32.For examples of pyrrolidine formation by reduction of nitro ketones see: Stevens RV, Lee AWM. J. Chem. Soc., Chem. Commun. 1982:102–103. Coda AC, Desimoni G, Invernizzi AG, Righetti PP, Seneci PF, Tacconi G. Gazz. Chim. Ital. 1985;115:111–117. Turner MJ, Luckenbach LA, Turner EL. Synthetic Commun. 1986;16:1377–1385. Ali Sk. A., Wazeer MIM. Tetrahedron. 1993;49:4339–4354. Halland N, Hazell RG, Jørgensen KA. J. Org. Chem. 2002;67:8331–8338. doi: 10.1021/jo0261449. Stylianakis I, Kolocouris A, Kolocouris N, Fytas G, Foscolos GB, Padalko E, Neyts J, De Clercq E. Bioorg. Med. Chem. Lett. 2003;13:1699–1703. doi: 10.1016/s0960-894x(03)00231-2.
- 33.The OH of ethyl N-hydroxy-N-benzyl-2-aminobutanoate absorbs at 3572 cm-1 (CHCl3) whereas the NH of ethyl N-benzyl-2-aminobutanoate absorbs at 3337 cm-1: Caddick S, Afonso CAM, Candeias SX, Hitchcock PB, Jenkins K, Murtagh L, Pardoe D, Santos AG, Treweeke NR, Weaving R. Tetrahedron. 2001;57:6589–6605. Miyabe H, Ueda M, Naito T. J. Org. Chem. 2000;65:5043–5047. doi: 10.1021/jo000318+.
- 34.Keller E, Veldman N, Spek AL, Feringa BL. Tetrahedron: Asymmetry. 1997;8:3403–3413. [Google Scholar]
- 35.a Wu F, Li H, Hong R, Deng L. Angew. Chem., Int. Ed. 2006;45:947–950. doi: 10.1002/anie.200502658. [DOI] [PubMed] [Google Scholar]; b Wu F, Hong R, Khan J, Liu X, Deng L. Angew. Chem., Int. Ed. 2006;45:4301–4305. doi: 10.1002/anie.200600867. [DOI] [PubMed] [Google Scholar]
- 36.Heidelberger M, Jacobs WA. J. Am. Chem. Soc. 1919;41:817–833. [Google Scholar]
- 37.Stadler P, Troxler F, Hofmann A. Chem. Abstr. 1967;67:82297x. Neth. Appl. 6609202. [Google Scholar]
- 38.Hoye TR, Renner MK. J. Org. Chem. 1996;61:2056–2064. and 2006, 71, 1754. Hoye TR, Renner MK. J. Org. Chem. 1996;61:8489–8495. For an application see: Pettersen D, Ahlberg P. Tetrahedron: Asymmetry. 2005;16:2075–2080.
- 39.Ward DE, Rhee CK. Tetrahedron Lett. 1991;32:7165–7166. [Google Scholar]
- 40.a Dunlap NK, Sabol MR, Watt DS. Tetrahedron Lett. 1984;25:5839–5842. [Google Scholar]; b Demir AS, Reis Ö, Igdir AC. Tetrahedron. 2004;60:3427–3432. [Google Scholar]
- 41.For a review see: Demir AS, Jeganathan A. Synthesis. 1992:235–247.
- 42.a Bohlmann F, Zdero C, Berger D, Suwita A, Mahanta P, Jeffrey C. Phytochemistry. 1979;18:79–93. [Google Scholar]; b Bascop S-I, Laronze J-Y, Sapi J. Synthesis. 2002:1689–1694. [Google Scholar]
- 43.Sammes PG, Weedon AC. J. Chem. Soc., Perkin Trans. 1. 1979:3053–3060. [Google Scholar]
- 44.a Shono T, Matsumura Y, Tsubata K. J. Am. Chem. Soc. 1981;103:1172–1176. [Google Scholar]; b Barrett AGM, Pilipauskas D. J. Org. Chem. 1991;56:2787–2800. [Google Scholar]
- 45.a Thirkettle J, Alvarez E, Boyd H, Brown M, Diez E, Hueso J, Elson S, Fulston M, Gershater C, Morata ML, Perez P, Ready S, Sanchez-Puelles JM, Sheridan R, Stefanska A, Warr S. J. Antibiot. 2000;53:664–669. doi: 10.7164/antibiotics.53.664. [DOI] [PubMed] [Google Scholar]; b Busby DJ, Copley RCB, Hueso JA, Readshaw SA, Rivera A. J. Antibiot. 2000;53:670–676. [PubMed] [Google Scholar]; c Thirkettle J. J. Antibiot. 2000;53:733–735. doi: 10.7164/antibiotics.53.733. [DOI] [PubMed] [Google Scholar]
- 46.Pinto IL, Boyd HF, Hickey DMB. Bioorg. Med. Chem. Lett. 2000;10:2015–2017. doi: 10.1016/s0960-894x(00)00395-4. [DOI] [PubMed] [Google Scholar]
- 47.a Bateson JH, Fell SCM, Kaura AC, Southgate R. J. Chem. Soc., Perkin Trans. 1. 1992:1577–1581. [Google Scholar]; b Bateson JH, Kaura AC, Southgate R. Tetrahedron Lett. 1991;32:2065–2068. [Google Scholar]
- 48.Murphy PJ, Lee SE. J. Chem. Soc., Perkin Trans. 1. 1999:3049–3066. [Google Scholar]
- 49.a Xavier LC, Mohan JJ, Mathre DJ, Thompson AS, Carroll JD, Corley EG, Desmond R. Org. Synth. 1997;74:50–71. [Google Scholar]; b Mathre DJ, Jones TK, Xavier LC, Blacklock TJ, Reamer RA, Mohan JJ, Turner Jones ET, Hoogsteen K, Baum MW, Grabowski EJJ. J. Org. Chem. 1991;56:751–762. [Google Scholar]
- 50.Kricheldorf HR. α-Aminoacid-N-Carboxy-Anhydrides and Related Heterocycles. Springer-Verlag; Berlin: 1987. [Google Scholar]
- 51.a Zhu X-F, Henry CE, Wang J, Dudding T, Kwon O. Org. Lett. 2005;7:1387–1390. doi: 10.1021/ol050203y. [DOI] [PubMed] [Google Scholar]; b Norris JL, Porter NA, Caprioli RM. Anal. Chem. 2005;77:5036–5040. doi: 10.1021/ac050460g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.