

Abstract
Isopentenyl diphosphate isomerase (IDI) catalyzes the interconversion of isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP). This is an essential step in the mevalonate entry into the isoprenoid biosynthetic pathway. The isomerization catalyzed by type I IDI involves protonation of the carbon-carbon double bond in IPP or DMAPP to form a tertiary carbocation, followed by deprotonation. Diene analogs for DMAPP (E-2-OPP and Z-2-OPP) and IPP (4-OPP) were synthesized and found to be potent active-site directed irreversible inhibitors of the enzyme. X-ray analysis of the E·I complex between E. coli IDI and 4-OPP reveals the presence of two isomers that differ in the stereochemistry of the newly formed C3-C4 double bond in the hydrocarbon chain of the inhibitor. In both adducts C5 of the inhibitor is joined to the sulfur of C67. In these structures the methyl group formed upon protonation of the diene moiety in 4-OPP is located near E116, implicating that residue in the protonation step.
The isomerization of isopentenyl diphosphate (IPP) to dimethylallyl diphosphate (DMAPP) is a crucial activation step in isoprenoid biosynthesis, during which IPP is converted to its highly electrophilic isomer. These two molecules are the building blocks used to construct over 35,000 isoprenoid compounds found in nature. In Archaea, Eukaryota, and some Bacteria, IPP is synthesized from acetyl-CoA by the mevalonate (MVA) pathway and is the exclusive product from the phosphorylation and decarboxylation of the key intermediate mevalonic acid.1,2 IPP is required for biosynthesis of DMAPP in the MVA pathway, and IPP isomerase is essential.3 In most bacteria and plant chloroplasts, IPP and DMAPP are synthesized from pyruvate and D-glyceraldehyde phosphate by the methylerythritol phosphate (MEP) pathway.4 The final step produces both compounds during the reduction of hydroxydimethylallyl diphosphate. IPP isomerase activity is often found in organisms that utilize the MEP pathway, but the enzyme is not essential.
Two convergently evolved IPP isomerases are known.5,6 The type I IPP isomerase was discovered in the late 1950’s7 and is found in Eukaryota and Bacteria. The enzyme requires a divalent metal and catalyzes the antarafacial isomerization of IPP and DMAPP8 by a protonation-deprotonation mechanism9 (see Scheme 1). During the reaction, the pro-R hydrogen (blue) is removed from C(2) of IPP, and a proton from solvent (red) is added to the re-face at C4 of the double bond to generate the E-methyl group at C(3).
Scheme 1.
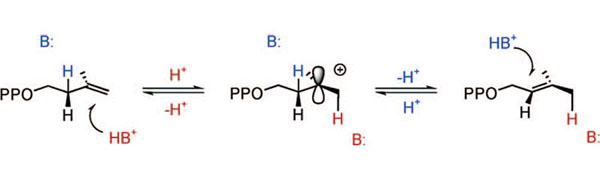
Protonation-Deprotonation Mechanism for Isomerization of IPP and DMAPP
Type II IPP isomerase is found in Archaea and in Bacteria.10 Typically, a given bacterium has only one form of IPP isomerase, but, in at lease one instance, the type I and type II enzymes co-exist.11,12 Type II IPP isomerase requires reduced flavin and divalent metal cofactors for activity. The stereochemistry of the reaction is similar to that catalyzed by the type I enzyme,13.14 although the facial selectivity for proton addition at C(4) has not yet been determined.
Several lines of evidence support the protonation-deprotonation mechanism shown in Scheme 1 for type I IPP isomerase. These include proton exchange experiments,15 substituent effects on the rate of the reaction,9,16 tight binding of transition-state analogues,9 and irreversible inhibition by mechanism-based inhibitors.9 Covalent modification of active site residues by the irreversible inhibitors and site-directed mutagenesis of the modified amino acids implicate two amino acids, C67 and E116 (Escherichia coli numbering) in the proton transfer steps.17 X-ray analysis of E. coli IPP isomerase shows that these amino acids are located on opposite sides of the active site.18 E116 is also part of a hexacoordinate binding site for a divalent metal. In the structure of the enzyme containing N,N-2-(dimethylamino)ethyl diphosphate, a transition-state analogue for the putative tertiary cationic intermediate where C(3) is replaced by a positively charged N-H ammonium moiety, one of the carboxylate oxygens in the E116 side chain is coordinated to the H-N+ unit and the other to the divalent metal. In another structure, where the enzyme was inactivated with the epoxide derivative of IPP, the oxirane ring had been opened to give a primary alcohol at C(4) with concomitant formation of a thioether bond between C(3) of the inhibitor and the sulfhydryl moiety of C67.17 The X-ray structures of the enzyme·inhibitor complexes also contained a second divalent metal, which was coordinated to non-bridging oxygens at P(1) and P(2) of the diphosphate moiety, the side chain carboxylate of E87, and the amide carbonyl oxygen in C67.
Replacement of the active-site cysteine in yeast IPP isomerase by serine produced a modest two-fold increase in KM but reduced kcat by ~104.17 The related alanine mutant was inactive. Likewise, substitution of the active site glutamate with glutamine or valine gave inactive proteins. Interestingly, an X-ray structure of the C67A mutant of E. coli IPP isomerase, which had been treated with an epoxy analogue of IPP, showed that the protein was covalently modified.19 In this case the oxirane ring had opened to form an ester linkage between C(3) of the inhibitor and the side chain carboxylate moiety of E116. Thus, the “inactive” protein retained the ability to activate and open the oxirane ring, suggesting a role for E116 in the protonation step of the isomerization reaction.
Diene analogues of oxidosqualene have been used to intercept carbocationic intermediates as allylic cations, which react with active site nucleophiles and abort the cascade of cyclization reactions leading to the tetracyclic skeleton polycyclic triterpenes.20, 21 We thought that similar analogues of IPP and DMAPP might be potent mechanism-based inhibitors of IPP isomerase by creating an electrophilic center in the substrate analogues that is susceptible to alkylation. We now report the synthesis of two diene analogues of DMAPP and a diene analogue of IPP, their irreversible inactivation of type I IPP isomerase from Schizosaccharomyces pombe, and an X-ray structure of E. coli IPP isomerase covalently modified by the IPP analogue that provides insights about the protonation step.
EXPERIMENTAL SECTION
(E)-3-Methyl-2,4-pentadien-1-ol (E-2-OH)
A solution of 1.0 g (10.4 mmol) of (E)-3-methyl-2-penten-4-yn-1-ol (E-1-OH) in 10.0 mL of anhydrous ethyl alcohol was reduced using 0.148 g of Lindlar catalyst. The catalyst was removed by filtration, solvent was removed by rotary evaporation, and the resulting yellow oil was purified by flash chromatography on silica gel (3/2 = hexane/diethyl ether) followed by distillation at 50–55°C (Kugelrohr, 3 mmHg) (lit.22 bp 74 °C at 14 mmHg) to give 0.801 g (77%) of a colorless oil;22, 23 1H NMR (C6D6) _ 0.37 (1H, s (broad)), 1.53 (3H, s), 4.40 (2H, dd, JH,H = 6.6 Hz), 4.96 (1H, d, JH,H = 10.5 Hz), 5.09 (1H, dd, JH,H = 17.4 Hz), 5.52 (1H, d, JH,H = 6.6 Hz), 6.33 40 (1H, dd, JH,H = 17.4, 10.8 Hz); 13C NMR (CDCl3) _ 12.2, 59.4, 112.6, 132.4, 135.8, 141.7.
(E)-5-Bromo-3-methylpenta-1,3-diene (E-2-Br)
To a solution of 45 mg (0.42 mmol) of E-2-OH in CH2Cl2 were added 182 mg (0.55 mmol) of CBr4 and 144 mg (0.55 mmol) of triphenylphosphine. After 3 h at room temperature, pentane was added, the resulting suspension was filtered, and solvent was removed at reduced pressure. The residue was purified by distillation at 40 °C (Kugelrohr, 3 mmHg) to give 49 mg (62%) of a colorless oil;241H NMR (CDCl3) _ 1.85 (3H, d, JH,H = 1.5 Hz), 4.13 (2H, d, JH,H = 8.4 Hz), 5.12 (1H, d, JH,H = 10.8 Hz), 5.30 (1H, d, JH,H = 17.1 Hz), 5.38 (1H, d, JH,H = 10.8 Hz), 5.78 (1H, dd, JH,H = 17.4, 10.8 Hz).
(Z)-3-Methyl-2,4-pentadien-1-ol (Z-2-OH)
Following the procedure described for E-2-OH, 2.36 g (24.5 mmol) (Z)-3-methyl-2-pentene-4-yn-1-ol (Z-1-OH) was reduced using 0.40 g of Lindlar catalyst to give 1.67 g (75 %) of a colorless oil;22, 23 1H NMR (C6D6) _ 0.76 (1H, s (broad), 1.72 (3H, s), 4.21 (2H, t, JH,H = 6.6 Hz), 5.01 (1H, d, JH,H = 11.1 Hz), 5.13 (1H, dd, JH,H = 17.3, 1.0 Hz), 5.57 (1H, d, JH,H = 6.6 Hz), 6.68 (1H, dd, JH,H = 17.4, 10.8 Hz); 13C NMR (CDCl3) _ 20.1, 58.5, 115.2, 130.6, 134.0, 134.6.
(Z)-5-Chloro-3-methylpenta-1,3-diene (Z-2-Cl)
To a solution of 0.367 g (2.75 mmol) of NCS in 10 mL of CH2Cl2 at −30 °C was added 1.86.4 mg (3.0 mmol) of dimethyl sulfide. The mixture was allowed to warm to 0 °C for 5 min and then cooled to −40 °C before 246 mg (2.51 mmol) of Z-2-OH was added. The suspension was allowed to warm to 0 °C and stirred for 2 h. The resulting solution was washed with saturated NaCl, solvent was removed at reduced pressure, and the resulting yellow oil was distilled at 35 °C (Kugelrohr, 4 mmHg) to give 0.175 g (60%) of a colorless oil;25 1H NMR (NMR (CDCl3) _ 1.72 (3H, s) 4.21 (2H, d, JH,H = 6.6 Hz), 5.01 (1H, d, JH,H = 11.1 Hz), 5.31 (1H, dd, JH,H = 17.3, 1.0 Hz), 5.57 (1H, d, JH,H = 6.6 Hz), 6.68 (1H, dd, JH,H = 17.4, 10.8 Hz); 13C NMR (CDCl3) _ 20.1, 58.5, 115.2, 130.6, 134.0, 134.6.
3-Methylene-4-penten-1-ol (4-OH)
Hexane was removed from 8.4 mL (17 mmol) of a solution of butyl lithium (2.0 M in hexane) before a solution containing 170 mg (16.8 mmol) of diisopropylamine, 1.68 mL of (1.68 mmol) of potassium tert-butoxide (1.0 M in THF), and 1.50 g (16.8 mmol) of 4-methylenetetrahydropyran (3) in 20 mL of THF (precooled at −75 °C) was added. After 2 h at −50 °C, the reaction was quenched with saturated potassium dihydrogen phosphate, and extracted with 30/70 diethyl ether/hexane. Solvent was removed at reduced pressure (bp 68–71 °C at 22 mmHg) to give 0.75 g (50%) of a colorless oil;23 1H NMR (CDCl3) _ 0.80 (1H, broad s, 2.52 (2H, t, JH,H = 6.6 Hz), 3.77 (2H, dt, JH,H = 6.3 Hz), 509–5.30 (4H, m), 6.40 (1H, ddd, JH,H = 18.0, 10.5, 0.6 Hz); 13C NMR (CDCl3) _ 34.8, 61.1, 114.0, 117.8, 138.5, 142.8.
3-Methylene-4-pentenyl tosylate (4-OTs)
To a solution of 93 mg (0.50 mmol) of p-toluenesulfonyl chloride and 73 mg (0.60 mmol) of 4-(N,N-dimethylamino)pyridine (DMAP) in 4 mL of CH2Cl2 was added 47 mg (0.48 mmol) of 4-OH. After 2 h, the mixture was poured into hexane, and the resulting precipitates were removed by filtration. Solvent was removed at reduced pressure, and the residue was chromatographed on silica gel (40/60 = diethyl ether/hexane) to give 113 mg (92%) of a clear colorless oil; IR (neat) 3100, 3043, 3015, 2959, 2917, 1650, 1603, 1358, 1309, 1293, 1230, 1188, 1176, 1096, 970 cm−1; UV (CHCl3) _max 222 (_ 18,600), 197 (_13,800) nm; 1H NMR (CDCl3) _ 2.45 (3H, s), 2.59 (2H, td, JH,H = 7.2, 0.9 Hz), 4.14 (2H, t, JH,H = 7.2 Hz), 5.0–5.15 (4H, m), 6.29 (1H, dd, JH,H = 17.6, 10.5 Hz), 7.35 (1H, d, JH,H = 8.4 Hz), 7.79 (1H, d, JH,H = 8.4 Hz); 13C NMR (CDCl3) _ 21.9, 31.1, 68.9, 114.0, 118.6, 128.0, 129.9, 130.0, 133.2, 138.0, 140.7, 144.9; HRMS (EI at 70 eV) calcd for C13H16O3S 252.0816, found 252.0797.
General procedure for synthesis of diphosphates
The procedure of Davission and coworkers26 was followed with the exception that HPLC was used in the final purification step instead of flash chromatography on cellulose. To a solution of 1.5–3.0 equiv of tris(tetra-n-butylammonium) hydrogen pyrophosphate in CH3CN was added a solution of tosylate, chloride, or bromide (0.25–0.50 mM) in CH3CN. The resulting mixture was allowed to stir for 2–3 h at room temperature. Solvent was removed at reduced pressure, and the resulting residue was dissolved in a minimal volume of 25 mM NH4HCO3 (ion-exchange buffer). The clear solution was passed through DOWEX AG 50W-X8 (100–200 mesh, ammonium form) that had been equilibrated with two column volumes of the ion exchange buffer at a flow rate of one column volume/15 min. The resulting clear solution was concentrated by rotary evaporation and lyophilized. The residue was then transferred to a centrifuge tube and dissolved in 100 mM NH4HCO3. The resulting solution was extracted two or three times with a 1:1 mixture of CH3CN and isopropanol. CH3CN and isopropanol were removed at reduced pressure and the aqueous layer was lyophilized. The residue was by HPLC, and the purified diphosphate was stored at −78 °C.
(E)-3-Methyl-2,4-pentadienyl diphosphate (E-2-OPP)
A solution of 90 mg (0.92 mmol) of E-2-Br in CH3CN was treated with 2.74g (3.00 mmol) of tris(tetra-n-butyl-ammonium) hydrogren pyrophosphate. The ammonium form of the diphosphate was purified by HPCL on an Asahipak ODP-90 HPLC column with gradient elution (A = 100 mM NH4HCO3, solvent B = CH3CN; 0–20 min (A) 20–35 min 1:1 (A/B), 35–45 min, 1:1 (A/B)); to give 120 mg (42%) of a white solid; UV (25 mM NH4HCO3) _max 228 (_ 14,800), 197 (_ 10,600) nm; 1H NMR (D2O/ND4OD, pH = 8.0) _ 1.60 (3H, s), 4.40 (2H, dd, JH,H = JH,P = 6.6 Hz), 4.94 (1H, d, JH,H = 11.1 Hz), 5.26 (1H, d, JH,H = 17.7 Hz), 5.53 (1H, t, JH,H = 6.6 Hz), 6.29 (1H, dd, JH,H = 17.7, 10.8 Hz); 13C NMR (D2O/ND4OD, pH = 8.0) _ 14.1, 65.3 (d, JC,P = 5.2 Hz), 112.7, 129.9 (d, JC,P = 7.9 Hz), 140.9, 143.2; 31P NMR (D2O/ND4OD, pH = 8.0) _ −1.90 (1P, d, JP,P = 22 Hz), −4.76 (1P, d, JP,P = 22 Hz); HRMS ((−)-FAB) calcd for C6H11P2O7 256.9980, found 256.9966.
(Z)-3-Methyl-2,4-pentadienyl diphosphate (Z-2-OPP)
Following procedure described for E-2-OPP, 63 mg (0.54 mmol) of Z-2-Cl was treated with 0.901 g (1.0 mmol) of tris(tetra-n-butylammonium) hydrogen pyrophosphate to give 87 mg (52 %) of a white solid; UV (25 mM NH4HCO3) _max 229 (_ 8,9000), 196 (_ 6,800), nm; 1H NMR (D2O/ND4OD, pH = 8.0) _ 1.72 (3H, s), 4.44 (2H, dd, JH,H = JH,P = 7.0 Hz), 5.13 (1H, d, JH,H = 11.1 Hz), 5.25 (1H, d, JH,H = 17.4 Hz), 5.50 (1H, t, JH,H = 7.3 Hz), 6.71 (1H, dd, JH,H = 17.2, 10.9 Hz); 13C NMR (D2O/ND4OD, pH = 8.0) _ 21.6, 64.2 (d, JC,P = 5.2 Hz), 119.3, 127.7 2 (d, JC,P = 7.8 Hz), 135.6, 140.1; 31P NMR (D2O/ND4OD, pH = 8.0) _ −1.83 (1P, d, JP,P = 21 Hz, P1), −4.83 (1P, d, JP,P = 21 Hz, P2), HRMS ((−)-FAB) calcd for C6H11P2O7 256.9980, found 256.9959.
3-Methylene-4-pentenyl diphosphate (4-OPP)
Following the procedure described for E-2-OPP, 230 mg (1.10 mmol) of 4-OTs was treated with 2.98 g (3.30 mmol) of tris(tetra-n-butyl) ammonium pyrophosphate to give 147 mg (43 %) of a white solid; UV (25 mM NH4HCO3) _max 224 (_ 16,600) nm; 1H NMR (D2O/ND4OD, pH = 8.0) _ 2.51 (1H, dd, JH,H = 18.0, 10.5 Hz), 3.98 (2H, dd, JH,H = 7.2 Hz), 5.03–5.31 (4H, m), 6.37 (1H, dd, JH,H = 18.0, 10.5 Hz); 13C NMR (D2O/ND4OD, pH = 8.0) _ 34.5 (d, JC,P = 5.6 Hz), 67.7 (d, JC,P = 4.8 Hz), 116.9, 120.5, 141.1, 145.3; 31P NMR (D2O/ND4OD, pH = 8.0) _ −0.46 (1P, d, JP,P = 22 Hz), −4.63 (1P, d, JP,P = 22 Hz); HRMS ((−)-FAB) calcd for C6H11P2O7 256.9980, found 256.9972.
Assay for IPP isomerase
Schizosaccharomyces pombe27 and Escherichia coli28 IPP isomerase for were obtained as described previously. The activity of the enzymes was measured by the acidliability protocol9 in a total volume 200 _l of 50 mM HEPES buffer, pH 7.0, containing10 mM MgCl2, 200 mM KCl, 0.5 mM DTT, 1 mg/ml BSA, 400 _M [1-14C] IPP (10 _Ci/_mol). The mixture equilibrated at 37 °C and initiated by addition of 10 _l of a solution of enzyme in assay buffer. Addition of a His6 tag to E. coli IPP isomerase did not significantly alter its kinetic properties.
Time dependent inactivation of the enzyme was measured as follows. Assay buffer containing varying concentrations of inhibitor was equilibrated at 37 °C before a 20-fold higher concentration of enzyme than required for the standard assay was added to a final volume of 200 _l. Samples (10 _l) were removed at different times and added to assay buffer containing 400 _M [14C]IPP (10 _Ci/_mol) to a final volume of 200 _l.
Crystallization of E. coli IPP isomerase
Crystals of recombinant (C-terminal His-tagged) E. coli IPP isomerase were grown at room temperature by the hanging drop method as described by Oudjama et al.29. Briefly, protein (10 mg/ml) was equilibrated against a reservoir containing PEG2000 (16%), ammonium sulfate (100 mM) and MnCl2 (10 mM) buffered with Tris/maleate at pH 5.5. Complexes with 4-OPP were obtained by soaking crystals of the enzyme in a (25 mM) solution of the inhibitor in Tris/maleate (100 mM, pH 5.5), PEG2000 (16%), ammonium sulfate (100 mM), MnCl2 (10 mM), and glycerol (25 %). These conditions were identical to those used to crystallize the wild type enzyme. After soaking, a crystal was flash frozen and diffraction data were collected with a Mar345 imaging plate system from Marresearch equipped with Osmic optics and running on an FR591 rotating anode generator. Diffraction data were processed with the MarFLM suite. In the presence of metal, the protein crystallizes, with two molecules in the asymmetric unit, in orthorhombic space group P212121 with cell parameters a = 69.3, b = 72.6 and c = 92.5 Å. A data set was recorded at 2.05 Å resolution. Refinement was performed with the program Shelxl97 using the structure of unliganded IPP isomerase (PDB reference code : 1hxz) as starting model.
RESULTS
Synthesis of Diene Analogues of IPP and DMAPP
The syntheses of DMAPP analogues E-2-OPP and Z-2-OPP and IPP analogue 4-OPP are shown in Scheme 2.
Scheme 2.

Synthesis of Diphosphates E-2-OPP, Z-2-OPP, and 4-OPP.
The E- and Z-isomers of diene analogues for DMAPP (E-2-OH and Z-2-OH) were prepared from commercially available isomeric acetylenic alcohols E-1-OH and Z-1-OH by selective hydrogenation of the triple bond using Lindlar catalyst. Initially both alcohols were converted to the corresponding bromides. However, the yield for the Z-isomer was substantially lower, and the less reactive chloride was prepared instead. The halides were phosphorylated by treatment with tris-(tetra-n-butylammonium)hydrogen pyrophosphate to give E- and Z-2-OPP. The diene analogue for IPP was synthesized from 4-methylene tetrahydropyran. The pyran ring was opened upon treatment with lithium diisopropyl amide (LDA)/potassium t-butoxide. Alcohol 4-OH was converted to the corresponding tosylate, followed by treatment with inorganic pyrophosphate to give 4-OPP.
Kinetic Studies
Preliminary experiments indicated that S. pombe and E. coli IPP isomerase was irreversibly inactivated upon incubation with the diene analogues of IPP and DMAPP. The kinetic constants for inactivation of the S. pombe enzyme were measured by incubation of the protein with varying concentrations of the inhibitors. Samples were removed over a period of 30 minutes, diluted 20-fold into buffer containing [14C]IPP, and the activity of the enzyme was measured. In a control experiment, the enzyme retained >97% of its activity over a period of 1 hour when incubated under similar conditions without addition of an inhibitor. In each case concentration-dependent first order inactivation was observed (Figure 1). The rate constants for inactivation (kinact) were determined from a semilogarithmic plot of residual enzyme activity versus time. The rate constants for inhibition at saturating levels of the inhibitors (kI) and the inhibitor constants (KI) were determined from replots of kinact versus the reciprocal of the inhibitor concentrations. The values for kI and KI are given in Table 1. All of the diene analogues were potent inhibitors of the enzyme. E-2-OPP was the most efficient inhibitor as measured by kI/KI. Although Z-2-OPP was ~10 times more reactive than E-2-OPP, its KI was substantially higher. 4-OPP was only slightly less efficient than E-2-OPP. Both of the DMAPP analogues were alternate substrates for chain elongation by avian FPP synthase, with steady state kinetic constants similar to those for DMAPP (data not shown); however, neither compound inactivated the enzyme.
Figure 1.
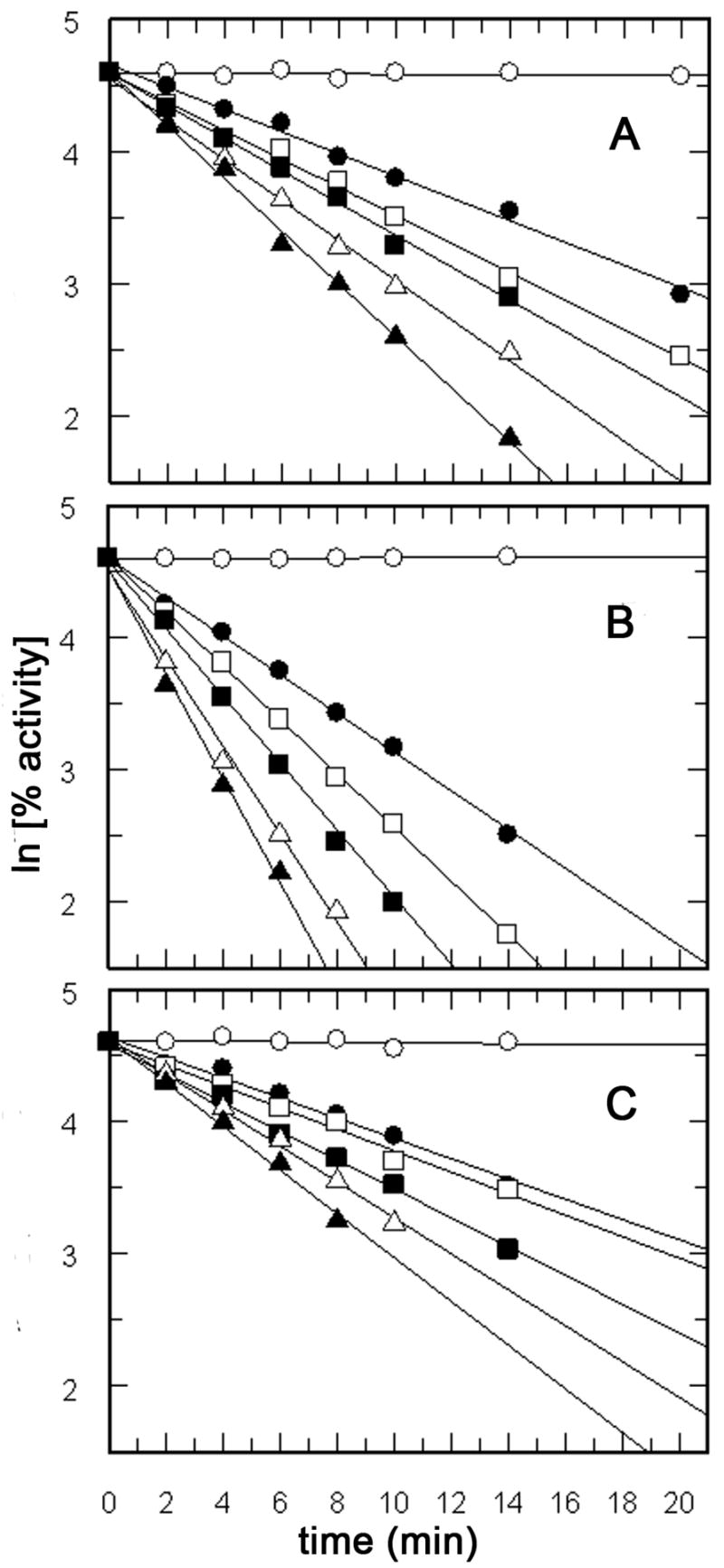
Plots of ln [% inactivation] versus time for incubation of IPP isomerase with E-2-OPP, Z-2-OPP, and 4-OPP. Panel A, [E-2-OPP] = 0 (_), 0.4 (_), 0.5 (_), 0.7 (_), 1.0 (Δ), and 2.5 (_) μM. Panel B, [Z-2-OPP] = 0 (_), 15 (_), 20 (_), 30 (_), 40 (Δ), and 50 (_) μM. Panel C, [4-OPP] = 0 (_), 10 (_), 12 (_), 15 (_), 20 (Δ), and 25 (_) μM.
Table 1.
Kinetic Constants for Irreversible Inhibition of S. pombe IPP Isomerase by E-2-OPP, Z-2-OPP, and 4-OPP.
| Inhibitor | kI (s−1) |
KI (μM) |
kI/KI (s−1 μM−1) |
|---|---|---|---|
| E-2-OPP | 17 ± 1.2 | 0.91 ± 0.08 | 19 |
| Z-2-OPP | 186 ± 11 | 84 ± 7 | 2.2 |
| 4-OPP | 43 ± 5.1 | 3.2 ± 0.2 | 13 |
Crystallographic structure of the E. coli 4-OPP·IPP isomerase complex
The E. coli The quality of the crystallographic data and the refinement are summarized in Table 2. Unambiguous electron density in the initial Fourier difference (Fo-Fc) maps (contoured at 3σ) confirmed the presence of the inhibitor within the active site of the protein. Simulated-annealing omit maps of the refined models confirmed the placement on the inhibitor with a distinct geometry for both molecules in the asymmetric unit. Electron density is observed consistent with formation of a covalent bond between the sulfur of Cys67 in the active site and C5 in 4-OPP and a new carbon-carbon double bond between C3 and C4. Interestingly, in one protein molecule, the stereochemistry of the double bond in the covalently attached diene inhibitor is Z while in the other it is E. The position of the diphosphate group in 4-OPP is similar in both geometries. A second metal binds to non-bridging oxygens in the diphosphate moiety and the stabilizing roles of Lys21, Arg51, Lys55, Glu83 and Glu87 are confirmed.
Table 2.
Statistics of data collection and structure refinement.
| IPP isomerase · 4-OPP | |
|---|---|
| Crystal data | |
| Space group | P212121 |
| Cell dimensions (Å) | 68.633 |
| 70.681 | |
| 90.773 | |
| Subunits per asymmetric unit | 2 |
| Data set | |
| Wavelength (Å) | 1.54179 |
| Highest resolution (Å) | 2.03 |
| Total reflections | 55557 |
| Unique reflections | 26732 |
| Observed reflections (I > 2σ(I)) | 23693 |
| Completeness (%) a | 93.3 (93.2) |
| Rmerge (%) a | 8.0 (27.5) |
| I/σ(I) | 6.2 |
| Refinement | |
| Resolution range (Å) | 10.0-2.0 |
| Number of protein atoms | 2831 |
| Number of ligand atoms | 52 |
| Number of water molecules | 85 |
| Rcryst (observed/all data) (%) | 18.8/22.1 |
| Rfree b (%) | 23.9 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.007 |
| Bond angles (Å) | 0.022 |
| Ramachadran plot c | |
| Core Region (%) | 95.8 |
| Inner core region (%) | 72.8 |
Statistics for the highest resolution shell (2.08-1.97 Å) are given in parenthesis
Free test subset represents 10% of total unique reflections
As defined by SHELXPRO29, Gly and Pro residues excluded.
DISCUSSION
The substrates for chain elongation,30,31 isomerization,9 and cyclization reactions31 in isoprenoid metabolism are processed by reactions that proceed through discrete carbocationic intermediates. A common feature of these reactions is the addition of an electrophile (R3+) to a carbon-carbon double bond to generate a tertiary cation as illustrated in Scheme 3. While the carbocationic intermediates are sufficiently electrophilic to alkylate any of the nucleophiles typically found in enzyme active sites, enzymes that process carbocations typically do not suffer inactivation during catalysis. However, if the structure of the intermediate is altered in a manner that changes the location of the positive charge in the active site, alkylation is commonly seen. For example, diene analogues of oxidosqualene (R2 = −CH=CH2), which generate allylic cations during cyclization, irreversible inactivate oxidosqualene cyclase20 and squalene-hopene cyclase21 during turnover. We reasoned that the IPP and DMAPP diene analogues 2-E-OPP, 2-Z-OPP, and 4-OPP were likely candidates for irreversible inhibition of IPP isomerase by a related mechanism. In this case, an allylic cation is generated by protonation of the diene in an active site known to contain carboxylate and thiol nucleophiles. All three dienes were potent time-dependent irreversible inhibitors of S. pombe and E.coli IPP isomerase.
Scheme 3.
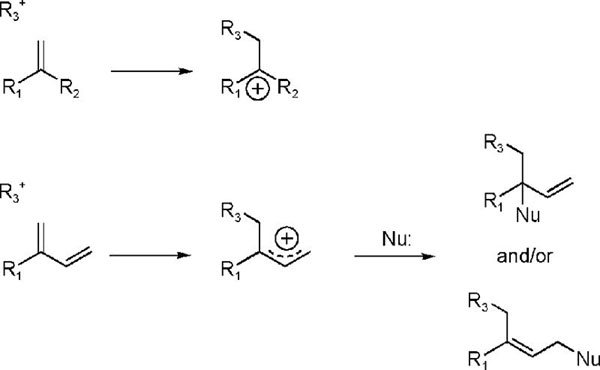
Electrophilic Additions to Carbon-Carbon Double Bonds in Substrates and Diene Mechanism Based Inhibitors.
The isomeric forms of covalently bound 4-OPP are shown in Figure 2. The structures mostly differ in the stereochemistry of the C3-C4 double bond and the conformation of the hydrocarbon chain of the bound analogue. The most likely mechanism for inactivation involves protonation of the methylene group in 4-OPP to give isomeric allylic cations, which span C3, C4, and C5, followed by alkylation of the thiol group in C67 (Scheme 4). Since the barrier to rotation about the C3-C4 partial double bond in the allylic cation should exceed 10 kcal/mol,32 we conclude that 4-OPP is bound in s-cis and s-trans conformations in the E·I complex prior to protonation.
Figure 2.
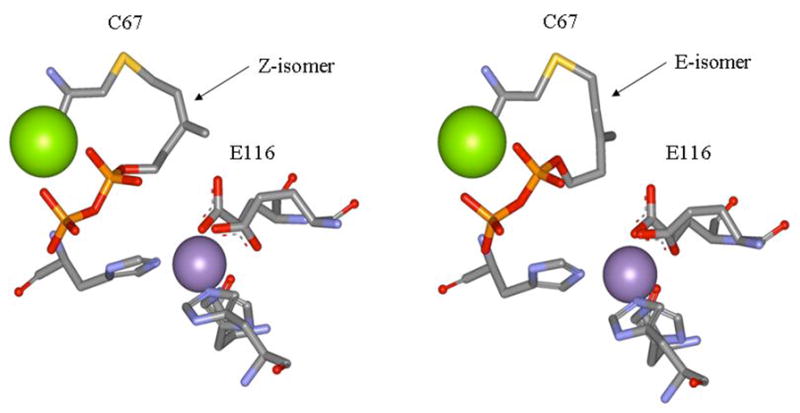
Structures of the isomeric IPP isomerase ·4-OPP complexes
Scheme 4.
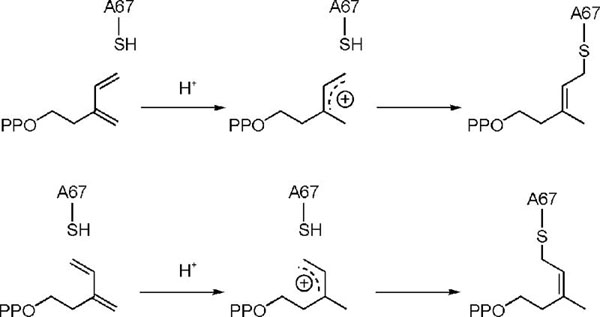
Mechanism for Inhibition of Type I IPP Isomerase by 4-OPP.
One of the carboxyl oxygens in the side chain of E116 is coordinated with a tightly bound divalent metal ion in type I IPP isomerase. The other carboxyl oxygen points toward the substrate in the binding pocket. The distance between the non-coordinated oxygen and the newly formed methyl group in the Z-adduct is 2.80 Å and is the only amino that could protonate the diene to give the structure that was seen. In the E-adduct, the methyl group is rotated to increase the distance to 3.98 Å, but again the non-coordinated oxygen of E116 is the only obvious source for the proton. This oxygen is also strongly hydrogen-bonded to the positively charged nitrogen in the tightly bound transition state analogue N,N-dimethyl-2-aminoethyl diphosphate.18 Thus, E116 is a likely active site acid in the protonation-deprotonation mechanism for type I IPP isomerase. Coordination with the divalent metal would enhance the acidity of the carboxyl group, although the state of protonation of that residue in the resting enzyme has not been established. In addition, the tertiary carbocationic intermediate would be stabilized by the carboxylate moiety following the protonation step.
In summary, diene analogues of IPP and DMAPP are potent active-site directed irreversible inhibitors of type I IPP isomerase. During the inhibition reaction, the IPP analog 4-OPP is protonated at its C3 methylene to generate an allylic cation that alkylates the thiol group in C67 to form a primary thioether. An X-ray structure of inactivated E. coli IPP isomerase shows that the methyl group generated by protonation is located near E116, thereby implicating that residue in the protonation step of the isomerization reaction.
Acknowledgments
This project was supported by NIH Grant GM 25521 (CDP) and an IISN Grant from the FNRS (Belgium). The authors thank Y. Oudjama and V. Durisotti for purification of the E.coli enzyme and crystallization assays.
Footnotes
Publisher's Disclaimer: This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2-3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of the PI. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
References
- 1.Poulter CD, Rilling H. Biosynthesis of Isoprenoid Compounds. Wiley; New York: 1981. [Google Scholar]
- 2.Koyama T, Ogura K. Comprehensive Natural Product Chemistry. Pergmon; Oxford: 1999. [Google Scholar]
- 3.Mayer MP, Hahn FM, Stillman DJ, Poulter CD. Yeast. 1992;8:743–748. doi: 10.1002/yea.320080907. [DOI] [PubMed] [Google Scholar]
- 4.Rodríguez-Concepción M, Boronat A. Plant Phys. 2002;130:1079–1089. doi: 10.1104/pp.007138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson MS, Muehlbacher M, Street IP, Proffitt J, Poulter CD. J Biol Chem. 1989;264:19169–19175. [PubMed] [Google Scholar]
- 6.Kandea K, Kuzuyama T, Takagi M, Hayakawa Y, Seto H. Proc Natl Acad Sci USA. 2001;98:932–937. doi: 10.1073/pnas.020472198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agranoff BW, Eggerer H, Henning U, Lynen F. J Am Chem Soc. 1959;81:1254–55. [Google Scholar]
- 8.Clifford K, Cornforth JW, Mallaby R, Philips RT. Chem Commun. 1971:1599–1600. [Google Scholar]
- 9.Muehlbacher M, Poulter CD. Biochemistry. 1988;27:7315–7328. doi: 10.1021/bi00419a021. [DOI] [PubMed] [Google Scholar]
- 10.Rohdich F, Bacher A, Eisenreich W. Bioorg Chem. 2004;32:292–308. doi: 10.1016/j.bioorg.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 11.Kuzuyama T, Seto H. Nat Prod Rept. 2003;20:171–183. doi: 10.1039/b109860h. [DOI] [PubMed] [Google Scholar]
- 12.Laupitz R, Hecht S, Amslinger S, Zepeck F, Kaiser J, Richter G, Schramek N, Steinbacher S, Huber R, Arigoni D, Bacher A, Eisenreich W, Rohdich R. Eur J Biochem. 2004;271:2658–2669. doi: 10.1111/j.1432-1033.2004.04194.x. [DOI] [PubMed] [Google Scholar]
- 13.Laupitz R, Grawert T, Rieder C, Zepeck F, Bacher A, Arigoni D, Rohdich F, Eisenreich W. Chem Biodiversity. 2004;1:1367–1376. doi: 10.1002/cbdv.200490099. [DOI] [PubMed] [Google Scholar]
- 14.Barkley SJ, Desai SV, Poulter CD. Org Lett. 2004;6:5019–5021. doi: 10.1021/ol0477273. [DOI] [PubMed] [Google Scholar]
- 15.Street IP, Christensen DJ, Poulter CD. J Am Chem Soc. 1990;112:8577–8578. [Google Scholar]
- 16.Reardon JE, Abeles RH. Biochemistry. 1986;25:5609–5616. doi: 10.1021/bi00367a040. [DOI] [PubMed] [Google Scholar]
- 17.Street IP, Coffman HR, Baker JA, Poulter CD. Biochemistry. 1994;33:4212–4217. doi: 10.1021/bi00180a014. [DOI] [PubMed] [Google Scholar]
- 18.Wouters J, Oudjama Y, Barkley SJ, Tricot C, Stalon V, Droogmans L, Poulter CD. J Biol Chem. 2003;278:11903–11908. doi: 10.1074/jbc.M212823200. [DOI] [PubMed] [Google Scholar]
- 19.Wouters J, Oudjama Y, Stalon V, Droogmans L, Poulter CD. Proteins. 2004;54:216–221. doi: 10.1002/prot.10573. [DOI] [PubMed] [Google Scholar]
- 20.Xiao XY, Prestwich GD. J Am Chem Soc. 1991;113:9673–9674. [Google Scholar]
- 21.Ceruti M, Rocco F, Viola F, Balliano G, Milla P, Arpicco S, Cattel L. J Med Chem. 1998;41:540–554. doi: 10.1021/jm970534j. [DOI] [PubMed] [Google Scholar]
- 22.Burger BV, Plessis du E, Garbers CF, Pachler KGR. J Chem Soc Perkin Trans I. 1973:584–587. [Google Scholar]
- 23.Margot C, Rizzolio M, Scholosser M. Tetrahedron. 1990;46:2411–2424. [Google Scholar]
- 24.Clark KB, Culshaw PN, Griller D, Lossing FP, Martinho Simoes JA, Walter JC. J Org Chem. 1991;56:5535–5539. [Google Scholar]
- 25.Mayr H, Heilman W. Tetrahedron. 1986;42:6657–6662. [Google Scholar]
- 26.Davisson VJ, Woodside AB, Neal TR, Stremler KE, Muehlbacher M, Poulter CD. J Org Chem. 1989;51:4768–4779. [Google Scholar]
- 27.Hahn FM, Poulter CD. J Biol Chem. 1995;270:11298–11303. doi: 10.1074/jbc.270.19.11298. [DOI] [PubMed] [Google Scholar]
- 28.Oudjama Y, Derbecq V, Sainz G, Clantin B, Tricot C, Stalon V, Villeret V, Droogmans L. Acta Crystallogr Sect D Biol Crystallogr. 2001;57:287–288. doi: 10.1107/s0907444900017571. [DOI] [PubMed] [Google Scholar]
- 29.Sheldrick GM, Schneider TR. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 30.Erickson H, Poulter CD. J Am Chem Soc. 2003;23:6885–6888. doi: 10.1021/ja034520g. [DOI] [PubMed] [Google Scholar]
- 31.Liang PH, Ko TP, Wang A. Eur J Biochem. 2002;269:3339–3354. doi: 10.1046/j.1432-1033.2002.03014.x. [DOI] [PubMed] [Google Scholar]
- 32.Allinger NL, Siefert JH. J Am Chem Soc. 1975;97:752–760. [Google Scholar]