

Abstract
A novel mode of regiochemical control over the allylic [1,3]-transposition of silyloxy groups catalyzed by Re2O7 has been developed. This strategy relies on a cis-oriented vinyl boronate, generated from the Alder ene reaction of homoallylic silyl ethers and alkynyl boronates, to trap out the allylic hydroxyl group. The resulting cyclic boronic acids are excellent partners for cross coupling reactions. High chirality transfer is observed for the rearrangement of enantioenriched allylic silyl ethers.
In the context of manipulating synthetic intermediates, the efficient [1,3]-transposition of allylic heteroatom functionalities is an important goal in synthetic chemistry.1 One of the most direct and atom-economical methods for this transformation is a transition metal-catalyzed rearrangement of an allylic hydroxyl group.2 Although the rhenium catalyst-based rearrangement developed by Osborn and coworkers3 is the most efficient method, it generally suffers from low regioselectivity, giving equilibrium mixtures of two isomers (eq 1). Efforts to control the regioselectivity have focused on introducing stereoelectronic biasing elements around the allylic double bond2c such as
(1).

(2).
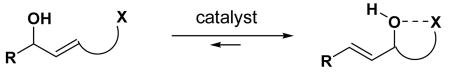
conjugation or by taking advantage of the steric environment of the allylic hydroxyl group.2a,c,4 To further improve the scope of this rearrangement, we envisioned a new approach where the equilibrium can be shifted toward the desired direction by introducing certain functionality within the molecule that can interact with the [1,3]-transposed hydroxyl group (eq 2).
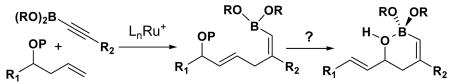
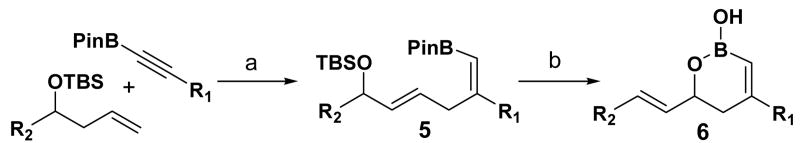
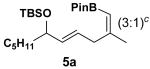
In search of a suitable platform that allows us to test this concept, we became interested in boron's unique affinity for hydroxyl group. The cis-vinyl boronate generated from the ruthenium-catalyzed Alder ene reaction5 between homoallylic alcohol derivatives and alkynyl boronates nicely sets the stage for testing the subsequent allylic [1,3]-transposition, after removal of the hydroxyl protecting group (eq 3). Herein we report a successful implementation of a two-step protocol for an efficient synthesis of cyclic vinyl boronic acids via a novel mode of regiochemical control in the allylic [1,3]-transposition induced by boronate trapping of hydroxyl and silyloxy groups.
(3).
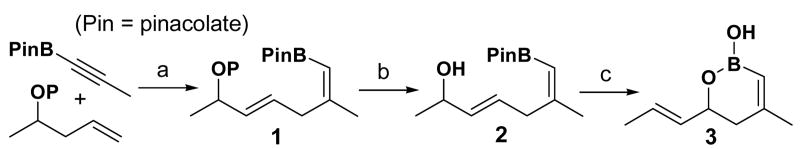
Having recognized the critical role of the protecting group in the Alder ene reaction as well as in the deprotection step, particularly due to the unstable nature of the boronate both under basic and acidic conditions, we tested several common protecting groups on the substrate homoallylic alcohol (Table 1). First, an ethyl carbonate and an acetate group were used (entries 1 and 2), which gave a good yield of the Alder ene reaction product 1a and 1b, respectively. However, deprotection of these protecting groups under typical mildly basic conditions gave only low yield of free alcohol 2. Again, ethereal protecting group MOM is suitable for the first step but its removal under acidic conditions led to rapid protodeboronation (entry 3). To avoid both basic and acidic aqueous conditions for the deprotection, the Troc (trichloroethyloxycarbonyl) protecting group was used. Although the deprotection was nicely accomplished in Zn/AcOH as expected, the first coupling step gave low yield of 1d probably due to the ionization of the allylic C–O bond with the highly activated Troc group.6 A silyl protecting group such as TBS (1e) was found to be problematic for its removal in the presence of the boronate functionality (entry 5).
Table 1.
Protecting Group Effect for Alder Ene Reaction of Borylated Alkynes–Deprotection–Allylic Transposition
 | |||||
|---|---|---|---|---|---|
| entry | P = | yield 1 (%) | b: deprotection | yield 2 (%) | yield 3 (%)d |
| 1 | CO2Et (a) | 68 | K2CO3/MeOH | 25 | 17 |
| 2 | Ac (b) | 70 | LiOH/MeOH | 30 | 20 |
| 3 | CH2OMe (c) | 67 | HF/pyr | – e | – |
| 4 | CO2CH2CCl3 (d) | 33 | Zn/AcOH | 95 | 31 |
| 5 | TBS (e) | 77 | TBAF | – f | – |
With 10 mol % of RuCp(CH3CN)3PF6 at 25 °C.
Deprotection conditions.
Ph3SiOReO3 or Re2O7 in CH2Cl2, 25 °C.
Three-step yield.
Protodeboronation was observed.
Decomposition was observed.
Having allylic alcohol 2 in hand, we next tried the allylic [1,3]-transposition. The rearrangement was found to be very efficient to give an excellent yield of the singly allylic [1,3]-transposed product in <5 min using either Ph3SiOReO37 or Re2O7 at room temperature. Surprisingly, the identity of the rearranged product is not the expected vinyl boronate with the transposed hydroxyl group but instead the cyclic vinyl boronic acid 3.8 Despite the efficient allylic transposition, the significance of this overall transformation was compromised by unacceptably low yields for the three-step sequence caused by the protecting group incompatibility in the first two steps.
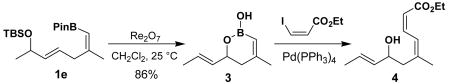
At this juncture, we turned our attention to the possibility of direct rearrangement of the protected form of hydroxyl functionality. Based on examples of the allylic [1,3]-transposition of silyl ethers3b,9 in combination with their efficient preparation, we chose 1e as the model substrate for the rearrangement. Gratifyingly, treatment of 1e with Re2O7 (CH2Cl2, 25 °C, 2 h) provided silyl group-free product, which was identical to vinyl boronic acid 3, the structure of which was confirmed after conversion to 4 via Suzuki coupling (eq 4).10
(4).
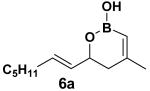
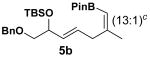
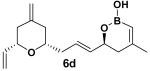
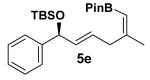
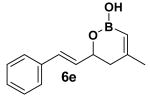
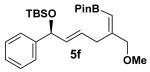
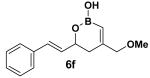
Encouraged by this observation, a variety of cis-vinyl boronates were synthesized via the Alder-ene reaction in order to test the generality of the rearrangement (Table 2). Substrates 5a–d were obtained in good yield and moderate selectivity for the Z-isomer.11 Under typical conditions (Re2O7; CH2Cl2 or Et2O, 25°C), these substrates provided the rearrangement products 6a–d in excellent yields.12 Unexpectedly, products 5e and 5f were not obtained from the Alder-ene reaction. Instead, the reaction directly lead to the [1,3]-transposed products 6e and 6f in 43% and 54% yields respectively (entries 5 and 6).10,13 This tandem Alder ene reaction followed by allylic transposition is probably due to the activated nature of the benzylic silyl ether toward the cationic ruthenium catalyst employed.6
Table 2.
Allylic [1,3]-Transposition of Silyl Ethers
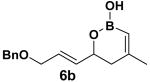
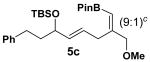
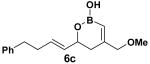
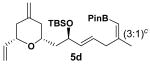
With 5 mol % of RuCp(CH3CN)3PF6 in acetone at 25 °C.
With 2.5 mol% Re2O7 in CH2Cl2 or ether at 25 °C.
Mixtures of boronate stereoisomers.
Based on recovered starting material.
Racemic mixture.
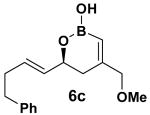
The rearrangement of 5d renders the important issue of chirality transfer during the reaction (entry 4). Because the assessment of the diastereomeric ratio of 6d14 was not trivial, enantioenriched substrates 5b and 5c were tested. Interestingly, when the rearrangement was performed in CH2Cl2, significant loss of stereochemical information was observed for 6b and 6c (Table 3). However, when the reaction was run in ether, the stereochemical integrity was preserved.15 The efficiency of chirality transfer from 5b to 6b and 5c to 6c was determined by analysis of the Mosher ester derivates after conversion to 7b and 7c.
Table 3.
Chirality Transfer for Allylic [1,3]-Transposition
| substrate | e.r. | product | solvent | yield (%) | e.r. |
|---|---|---|---|---|---|
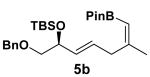
|
99:1 |
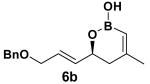
|
CH2Cl2 | 68 | 84:16 |
| Et2O | 72 | 99:1 | |||
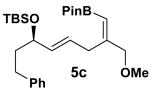
|
80:20 |
|
CH2Cl2 | 64 | 50:50 |
| Et2O | 70 | 80:20 |
The cyclic vinyl boronic acids 6a–f were found to be excellent coupling partners in Suzuki couplings.14,16 Compounds 6a–c and 6e–f underwent coupling with cis-ethyl-β-iodoacrylate to give the corresponding coupled products 7a–c and 7e–f, representative examples of which are illustrated in Scheme 1.
Scheme 1.

Suzuki Coupling of Cyclic Vinyl Boronic Acids
In conclusion, we have developed a highly efficient protocol for the synthesis of cyclic vinyl boronic acid. This novel mode of regiochemical control in the allylic [1,3]-transposition was achieved by using the oxygen affinity of a cis-oriented vinyl boronate to trap out the hydroxyl and silyloxy group. Application of this consecutive Alder ene reaction followed by allylic transposition to natural products synthesis is underway.
Supplementary Material
General procedures and characterization of represented compounds. This material is available free of charge via the internet at http://pubs.acs.org
Acknowledgments
We thank NIH (CA106673) for financial support of this work as well as the NSF and NIH for NMR and Mass Spectrometry instrumentation. E. C. H. thanks Abbott Laboratories for an Abbott fellowship. D. L. is a fellow of the Alfred P. Sloan foundation.
References
- 1.For epoxidation/reduction, see: Wharton PS, Bohlen DH. J Org Chem. 1961;26:3615–3616.Nicolaou KC, Duggan ME, Ladduwahetty T. Tetrahedron Lett. 1984;25:2069–2072.For [3,3]-sigmatropic rearrangements, see: Overman LE. Angew Chem Int Ed. 1984;23:579–586.Lutz RP. Chem Rev. 1984;84:205–247.For [2,3]-sigmatropic rearrangements, see: Evans DA, Andrews GC. Acc Chem Res. 1974;7:147–155.Reich HJ. J Org Chem. 1975;40:2570–2572.
- 2.Takai K, Nozaki H, Oshima K, Okazoe T, Matsubara S. Bull Chem Soc Jpn. 1985;58:844–849.Narasaka K, Kusama H, Hayashi Y. Tetrahedron. 1992;48:2059–2068.Gordon MS, Jensen JH, Espenson JH, Jacob J. Organometallics. 1998;17:1835–1840.Bellemin-Laponnaz S, Le Ny JP, Dedieu A. Chem Eur J. 1999;5:57–64.For a review, see: Le Ny JP, Bellemin-Laponnaz S. C R Chimie. 2002:217–224.
- 3.(a) Bellemin-Laponnaz S, Gisie H, Le Ny JP, Osborn JA. Angew Chem Int Ed. 1997;36:976–978. [Google Scholar]; (b) Bellemin-Laponnaz S, Gisie H, Le Ny JP, Osborn JA. Tetrahedron Lett. 2000;41:1549–1552. [Google Scholar]
- 4.Morrill C, Grubbs RH. J Am Chem Soc. 2005;127:2842–2843. doi: 10.1021/ja044054a. [DOI] [PubMed] [Google Scholar]
- 5.Hansen EC, Lee D. J Am Chem Soc. 2005;127:3252–3253. doi: 10.1021/ja0424629.For a review of ruthenium-catalyzed reactions, see: Trost BM, Federiksen MU, Rudd MT. Angew Chem Int Ed. 2005;44:6630–6666. doi: 10.1002/anie.200500136.
- 6.For π-allylruthenium complex formation from allylic carbonates or chlorides, see: Pregosin PS, Fernandez I, Breher F, Hermatschweiler R. Organometallics. 2006;25:1440–1447.
- 7.The more soluble catalyst Ph3SiOReO3 gave lower conversion in the current reaction. For synthesis and structure of this catalyst, see: Schoop T, Roesky HW, Noltemeyer M, Schmidt HG. Organometallics. 1993;12:571–574.
- 8.For related compounds, see: Falck JR, Bondlela M, Venkataraman SK, Srinivas D. J Org Chem. 2001;66:7148–7150. doi: 10.1021/jo015838z.Micalizio GC, Schreiber SL. Angew Chem Int Ed. 2002;41:3272–3276. doi: 10.1002/1521-3773(20020902)41:17<3272::AID-ANIE3272>3.0.CO;2-C.Jean Zhou Q, Worm K, Dolle RE. J Org Chem. 2004;69:5147–5149. doi: 10.1021/jo049343w.
- 9.Herrmann WA, Wojtczak WA, Artus GRJ, Kuhn FE, Mattner MR. Inorg Chem. 1997;36:465–471. [Google Scholar]
- 10.The cyclic boronic acids could be identified by crude 1H NMR but complete characterization was only achieved after derivitization via Suzuki coupling.
- 11.Higher Z/E ratios had been observed previously for alkenes with no branching substituents in the homoallylic position (ref 5).
- 12.The E-isomer could be recovered as a mixture of allylic silyl ethers regioisomers.
- 13.Complete racemization of 6e was observed.
- 14.6d represents the C4–C16 subunit of (-)-zampanolide and (-)-dactylolide. Details of its use in total synthesis will be reported in a future article.
- 15.Chirality transfer has been observed for cyclic allylic alcohols, see: Troste BM, Toste FD. J Am Chem Soc. 2000;122:11262–11263.Acyclic allylic alcohols require low temperatures (-78 °C) for chirality transfer (ref 4).
- 16.Frank SA, Chen H, Kunz RK, Schnaderbeck MJ, Roush WR. Org Lett. 2000;2:2691–2694. doi: 10.1021/ol0062446.For a review, see: Miyaura N, Suzuki A. Chem Rev. 1995;95:2457–2483.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
General procedures and characterization of represented compounds. This material is available free of charge via the internet at http://pubs.acs.org