

Summary
The flagellar proteins FlgN and FliT have been proposed to act as substrate-specific export chaperones, facilitating incorporation of the enterobacterial hook-associated axial proteins (HAPs) FlgK/FlgL and FliD into the growing flagellum. In Salmonella typhimurium flgN and fliT mutants, the export of target HAPs was reduced, concomitant with loss of unincorporated flagellin into the surrounding medium. Gel filtration chromatography of wild-type S. typhimurium cell extracts identified stable pools of FlgN and FliT homodimers in the cytosol, but no chaperone–substrate complexes were evident. Nevertheless, stable unique complexes were assembled efficiently in vitro by co-incubation of FlgN and FliT with target HAPs purified from recombinant Escherichia coli. The sizes of the chaperone–substrate complexes indicated that, in each case, a chaperone homodimer binds to a substrate monomer. FlgN prevented in vitro aggregation of FlgK monomers, generating a soluble form of the HAP. Recombinant polypeptides spanning the potentially amphipathic C-terminal regions of FlgN or FliT could not complement in trans the chaperone deficiency of the respective flgN and fliT mutants, but efficient flagellar assembly was restored by homodimeric translational fusions of these domains to glutathione S-transferase, which bound FlgK and FlgL like the wild-type FlgN. These data provide further evidence for the substrate-specific chaperone function of FlgN and FliT and indicate that these chaperones comprise common N- and C-terminal domains mediating homodimerization and HAP substrate binding respectively. In support of this view, the flgN mutation was specifically complemented by a hybrid chaperone comprising the N-terminal half of FliT and the C-terminal half of FlgN.
Introduction
During assembly of flagella on the bacterial cell surface, structural proteins are exported via a flagellum-specific export pathway and polymerize to form contiguous substructures. The long helical filament comprises ≈20 000 monomers of flagellin (FliC). This is attached to a flexible hook (FlgE) extending from the cell surface (Aizawa, 1996) by two hook-associated proteins (HAPs) FlgK and FlgL (Jones et al., 1990; Fahrner et al., 1994). The filament is capped at its extremity by another HAP, FliD, which acts as a nucleation point for FliC monomers to polymerize into the growing filament (Vonderviszt et al., 1998). Assembly is sequential; FlgK and FlgL rings must be present before FliD can polymerise, and the FliD cap must be in place before FliC can be incorporated (Kubori et al., 1992).
Comparatively little is known about the export of structural proteins through the hollow core of the flagellum to their sites of oligomerization (Homma et al., 1990; Ruiz et al., 1993; Minamino and Macnab, 1999). Owing to the small diameter of the export channel, proteins destined for incorporation into the growing flagellum are thought to be exported in a partially unfolded state, implying that premature oligomerization in the cytosol must be prevented. We have shown previously that the small flagellar proteins FlgN and FliT bind in vitro to immobilized HAPs FlgK/FlgL and FliD, respectively (Fraser et al., 1999), specifically to the HAP C-terminal amphipathic domains believed to interact with and provide stable quaternary interactions between flagellum substructures. We suggested that FlgN and FliT might function as chaperones facilitating the export of specific flagellar structural components, most probably acting as ‘bodyguards’ to prevent premature interaction of HAPs (Fraser et al., 1999; Bennett and Hughes, 2000). This would be analogous to one of the mechanisms proposed for the function of chaperones aiding the related type III export of virulence effector proteins (Ménard et al., 1994; Woestyn et al., 1996). Although primary sequence identity is low or not evident among the substrate-specific flagellar and virulence type III export chaperones, they are all small and have similar characteristics (Wattiau et al., 1996; Gygi et al., 1997; Fraser et al., 1999; Bennett and Hughes, 2000). Indeed, the putative flagellar chaperones are arguably progenitors of this family. To date, studies have focused primarily on the chaperone substrates (Elliot et al., 1999; Fraser et al., 1999), and less is known about the chaperones themselves. In this study, we investigate further the nature and action of Salmonella typhimurium FlgN and FliT and provide further evidence of their function as HAP chaperones.
Results
The phenotype of S. typhimurium flgN and fliT mutants: inability to export specific flagellar hook-associated axial proteins
S. typhimurium flgN and fliT chromosomal null mutants were constructed by insertional inactivation (see Experimental procedures). Disruption of the appropriate loci was confirmed by Southern blotting of putative mutant DNA with the wild-type (WT) flgN and fliT genes and by immunoblotting mutant cell lysates with anti-FlgN or anti-FliT antisera (not shown). The mutant phenotypes were assessed (Fig. 1A). The flgN mutant was unable to swarm over the surface of 0.6% agar or to swim through semi-solid (0.3%) LB agar during overnight colony growth. Equivalent overnight colonies of the fliT mutant indicated that, in contrast, this mutation had not affected swimming motility and only marginally decreased the capacity to swarm (Fig. 1A). However, monitoring colony expansion at 60 min intervals over 16 h revealed that, although the growth rate of the swimming colony was the same as that of the WT (3.4 mm h−1), swarming was reduced from 3.9 mm h−1 to 2.1 mm h−1 (not shown). That the disability was seen only in swarming is consistent with Salmonella swarm cells needing an ≈ fivefold higher concentration of surface flagella than during swimming (Harshey and Matsuyama, 1994).
Fig. 1.
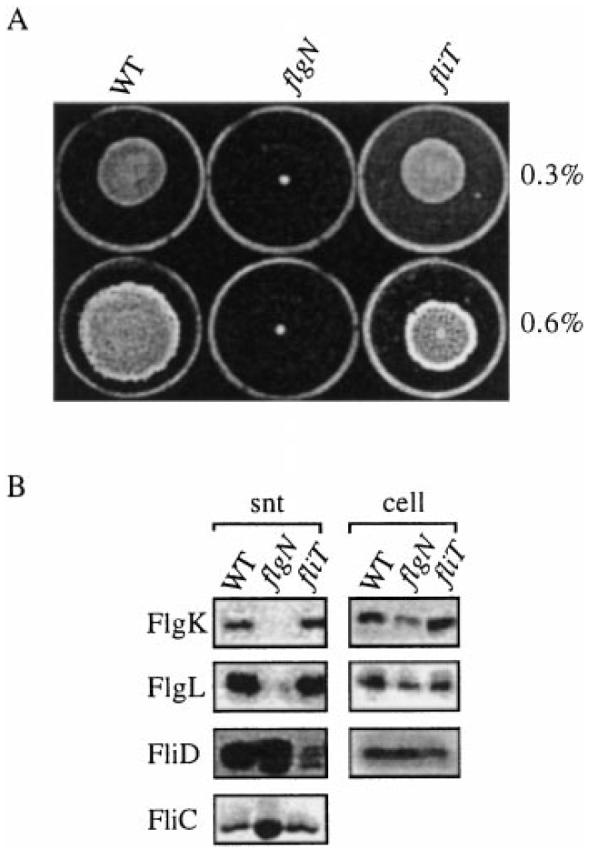
Phenotypes of the S. typhimurium flgN and fliT null mutants.
A. Swimming and swarming motility (assessed on 0.3% and 0.6% LB agar respectively) of WT S. typhimurium SJW1103 and the derived flgN and fliT mutants. Stationary phase broth cultures were centrally inoculated onto the agar plates and incubated at 37°C for 16 h.
B. Flagellar axial proteins in the supernatant (snt) and total cell lysate (cell) from late exponential phase cultures of WT and the flgN and fliT mutants were harvested and separated by SDS–(12%)PAGE. They were detected by immunoblotting with anti-FlgK, anti-FlgL and anti-FliD antisera or by Coomassie blue staining (FliC).
Immunoblotting of flagellar axial proteins in the late exponential culture supernatants of the mutants (Fig. 1B, snt) indicated that the flgN mutation had all but abolished export of FlgK and FlgL. Similarly, in the fliT mutant, export of FliD was reduced ≈20-fold. Parallel blots of the total cell fraction (i.e. without supernatant; Fig. 1B, cell) showed that substrate did not accumulate intracellularly in the absence of chaperone; indeed, concentrations of the FlgK, FlgL and FliD substrates were ≈20–50% that of wild type. In both mutants, but most obviously in the flgN mutant, the HAP export defect impaired efficient incorporation of other axial components of the flagellum, causing leaching into the medium of unpolymerized low-molecular-weight filament FliC (and also FliD in the flgN mutant), as reported in an flgN mutant of Proteus mirabilis (Gygi et al., 1997). Efficient Salmonella HAP export is therefore specifically dependent upon the two putative chaperones.
FlgN and FliT accumulate in the cytoplasm as stable homodimers
The cellular location of FlgN and FliT was determined by fractionating wild-type S. typhimurium cells. Both proteins localized, apparently exclusively, to the cytosol (Fig. 2A). Gel filtration was used to ascertain the sizes of FlgN and FliT and also to identify chaperone–substrate complexes (Fig. 2B). Soluble proteins from late exponential phase cultures (A600 1.2), corresponding to peak expression of FlgN and FliT (not shown), were isolated and loaded onto a pre-equilibrated Superose 12 10/30 HR gel filtration column. Eluted fractions were immunoblotted with anti-FlgN or anti-FliT antisera. Neither FlgN nor FliT was detected in fractions corresponding to their predicted monomeric molecular weights (≈15 kDa and ≈14 kDa respectively). Each eluted as a single peak at ≈30 kDa, too small in each case to be a chaperone–substrate complex (the substrates being ≈32–58 kDa), but corresponding to twice the monomeric weights. This suggests that stable pools of chaperone homodimers accumulate in the cytosol. This view was substantiated by measurement of chaperone half-lives after spectinomycin arrest of translation during late exponential growth (not shown). These were 20 min (± 2 min) for both FlgN and FliT.
Fig. 2.
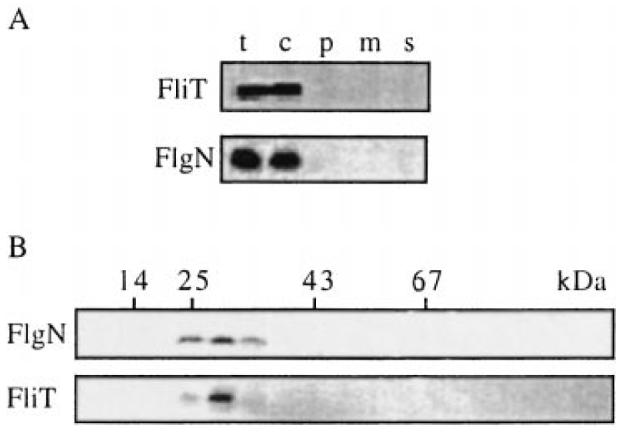
Characterization of the cellular pools of FlgN and FliT.
A. Location. Late exponential phase cultures of WT S. typhimurium were harvested, and cells were fractionated. Proteins from total cells (t), culture supernatant (s) and the cytosolic (c), periplasmic (p) and membrane (m) fractions were separated by SDS–(15%)PAGE and immunoblotted with anti-FlgN or anti-FliT antisera.
B. Size. Cytoplasmic cell extracts were loaded onto a calibrated Superose 12 10/30 HR column and developed by FPLC. Eluted fractions were immunoblotted with anti-FlgN or anti-FliT antisera. Peak fractions of eluting size markers are indicated (BSA, 67 kDa; ovalbumin, 43 kDa; chymotrypsinogen, 25 kDa; ribonuclease A, 14 kDa).
In vitro chaperone-mediated inhibition of HAP aggregation
As no complex assembly was detected in the cell lysates, an in vitro assay was developed to demonstrate the postulated chaperone–HAP interactions. To this end, both chaperones and HAPs were expressed with in frame N-terminal hexahistidine affinity tags. IPTG-induced Escherichia coli BL21(DE3) carrying plasmids pETN and pETT generated soluble His(6)FlgN and His(6)FliT chaperone proteins, which were purified to homogeneity using nickel nitrilotriacetic acid (NTA) affinity chromatography under native conditions according to the manufacturer's instructions (not shown). In parallel, IPTG induction of expression from the recombinant pET15b (Fraser et al., 1999) constructs pETK and pETD generated His(6)FlgK and His(6)FliD HAPs, which were purified from inclusion bodies using nickel NTA chromatography under denaturing conditions (not shown).
Although His(6)FliD refolded to a soluble form upon 1:100 dilution with 20 mM Tris-HCl, pH 7.4, the His(6)FlgK protein did not. This was exploited to assess whether the HAP aggregation could be prevented by FlgN. The insoluble His(6)FlgK was refolded by 1:100 dilution into a solution of His(6)FlgN and incubated for 20 min at 20°C (room temperature). Partitioning of FlgN and FlgK to the soluble and insoluble fractions by ultracentrifugation was assayed by SDS–PAGE and immunoblotting of the pellet and supernatant. The results (Fig. 3) show that His(6)FlgK alone remained approximately 100% insoluble in the refolding buffer, whereas His(6)FlgN was completely soluble. Incubation of His(6)FlgK with a twofold molar excess of His(6)FlgN resulted in a marked reduction in the proportion of the HAP in the pellet, with densitometric analysis of scanned immunoblots indicating that ≈90% of His(6)FlgK was soluble. The effect was less pronounced when the molar ratio of chaperone to substrate was reversed to give an excess of substrate (Fig. 3, right), with only ≈20% of His(6)FlgK soluble.
Fig. 3.
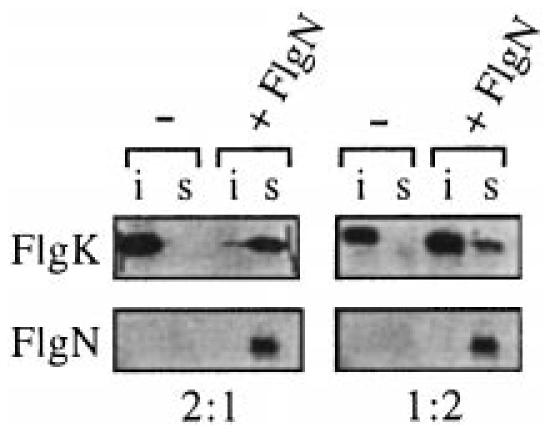
In vitro complex formation of FlgN and FliT with substrate HAPs. The solubility of purified His(6)FlgK (top) either alone (−) or with His(6)FlgN at a 2:1 or 1:2 molar ratio (+FlgN). After centrifugation at 607 000 g for 30min, proteins in the supernatant (s) or insoluble pellet (i) were immunoblotted with anti-FlgN or anti-FlgK antiserum. The corresponding location of His(6)FlgN is shown at the bottom.
In vitro assembly of chaperone–HAP complexes
Gel filtration chromatography through a pre-equilibrated Sephacryl S200 HR column was carried out to estimate the size of the chaperone–substrate complexes that were implied by the substrate solubility assay. Immunoblotting of the eluted fractions with anti-FlgN antisera showed that purified His(6)FlgN alone (−K) eluted as a single peak of ≈34 kDa (Fig. 4A(i), top). This is consistent with the assembly of stable homodimers, as indicated earlier by the gel filtration of WT S. typhimurium cell extracts. The equivalent elution fractions of soluble protein from the 2:1 His(6)FlgN–His(6)FlgK mixture (+K) were identically immunoblotted (Fig. 4A(i), bottom) and indicate that all the chaperone was located in a single complex of ≈90 kDa. This would be compatible with a dimer of His(6)FlgN (≈34 kDa) binding to a monomer of His(6)FlgK (≈60 kDa). This possibility was supported by anti-FlgK immunoblots of the same fractions. After incubation with the chaperone (+N), the HAP was identified at ≈90 kDa with the His(6)FlgN (Fig. 4A(ii), bottom), whereas without the chaperone (−N), His(6)FlgK formed a high-molecular-weight aggregate that eluted in the void volume (Fig. 4-A(ii), top).
Fig. 4.

In vitro complex formation of FlgN and FliT with substrate HAPs. Gel filtration chromatography of the following purified chaperone and HAP substrate proteins, either alone or after coincubation at a 1:2 molar ratio (substrate–chaperone). Elution fractions from a calibrated Sephacryl S200-HR column were separated by SDS–(15%)PAGE and immunoblotted as indicated. Molecular weight markers are in kDa (see Fig. 2); V0, column void volume.
A. (i) His(6)FlgN alone (−K) and after incubation with His(6)FlgK (+K).Immunoblotted with anti-FlgN antiserum.
(ii) His(6)FlgK alone (−N) and after incubation with His(6)FlgN (+N). [A(ii) bottom shows the same fractions as A(i) bottom]. Immunoblotted with anti-FlgK antiserum.
B. (i) His(6)FliT alone (−D) and after incubation with His(6)FliD (+D). Immunoblotted with anti-FliT antiserum.
(ii) His(6)FliD alone (−T) and after incubation with His(6)FliD (+T). [B(ii) bottom shows the same fractions as B(i) bottom]. Immunoblotted with anti-FliD antiserum.
To assess whether FliD is similarly able to form a stable complex with its chaperone in the absence of other flagellar components, the purified soluble ≈52 kDa His(6)FliD was diluted into a solution containing His(6)FliT, and the proteins were analysed as above. The purified His(6)FliT alone (–D) eluted as a single peak of ≈30 kDa (Fig. 4B(i), top), again indicating a soluble chaperone homodimer, in agreement with the earlier gel filtration of Salmonella cell lysates. After incubation with His(6)FliD, the chaperone was detected as a single ≈90 kDa species (Fig. 4B(i), bottom). Immunoblotting of the same fractions with anti-FliD antisera (+T; Fig. 4B(ii), bottom) confirmed that His(6)FliD co-eluted with His(6)FliT, suggesting that, as with FlgN, a FliT homodimer binds to a HAP substrate monomer. Again, a single complex was evident with, in this case, a trace of uncomplexed chaperone at ≈30 kDa (this trace was not always seen in repeat experiments, and its presence may not necessarily indicate that FliT has a lower substrate binding affinity than that of FlgN). In the absence of the chaperone (–T), the His(6)FliD eluted as a single peak of ≈50 kDa (Fig. 4B(ii), top), corresponding to a monomer (i.e. it did not aggregate or self-assemble into its pentameric form). No complexes were formed when the chaperones were incubated with the ‘wrong’ HAP(s) (not shown).
Chaperone domains mediating dimerization and substrate interaction
The C-terminal regions of FlgN and FliT contain heptad repeats of hydrophobic residues that are predicted to form amphipathic helices spanning residues 74–114 (FlgN) and 60–98 (FliT). These structures are predicted in many of the virulence chaperones (Wattiau et al., 1996). FlgN is also predicted to contain an N-terminal leucine zipper-like motif (between residues 1–30) with a high probability (> 0.99) of coiled-coil formation (Lupas et al., 1991), a feature commonly involved in protein dimerization (Vinson et al., 1989). To ascertain the contribution of each predicted domain to chaperone function, recombinant derivatives were constructed (Fig. 5A), so that the C-terminal putative substrate-binding domain of each of the two chaperones was generated either alone (in the pBAD plasmid vector) or as a translational fusion (in the pGEX vector) to glutathione-S-transferase (GST), a protein that forms homodimers (Yan et al., 1995).
Fig. 5.
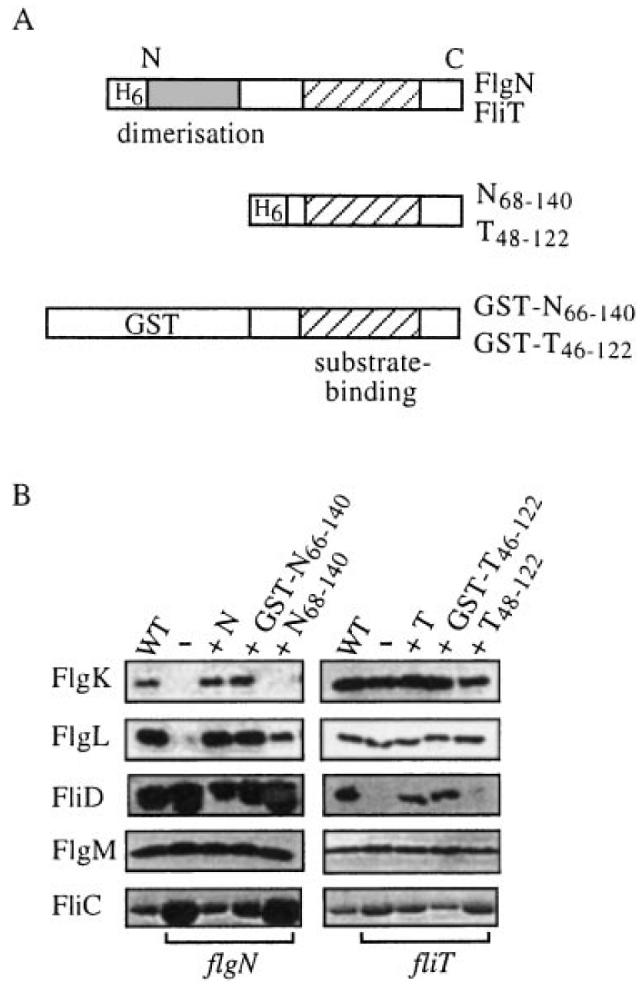
Complementation of flgN and fliT mutants by chaperone C-terminal regions.
A. Representations of (top) FliT and FlgN indicating putative N-terminal dimerization (shaded) and C-terminal substrate-binding (hatched) domains; (middle) the FlgN and FliT N-terminally histidine-tagged (H6) polypeptides; and (bottom) GST translational fusions. The C-terminal amphipathic helix is predicted to span amino acids 74–114 in FlgN and residues 60–98 in FliT. The predicted FlgN leucine zipper is located between residues 1 and 30.
B. Flagellar proteins collected from the supernatants of late exponential phase flgN (left) or fliT (right) mutant cultures alone (–) or expressing either His-tagged FlgN or FliT (labelled N and T) or the respective GST fusion or the C-terminal polypeptide. The same proteins exported by WT S. typhimurium are shown in each case in the leftmost columns. Proteins separated by SDS–(12%)PAGE were immunoblotted with anti-FlgK, anti-FlgL, anti-FliD or anti-FlgM antisera or by Coomassie blue staining (FliC).
After induction of protein synthesis, gel filtration of cell lysates confirmed that the GST fusions were dimeric and that the C-terminal polypeptides were monomeric (not shown). The C-terminal polypeptides His(6)FlgN68–140 and His(6)FliT48–122 were unable to recover swimming or swarming motility in the respective Salmonella flgN or fliT mutants, but flgN cells expressing GST-N66–140 and fliT cells expressing GST-T46–122 exhibited wild-type swarming and motility (not shown; colonies were indistinguishable from those shown in Fig. 1). Supernatant proteins from late exponential cultures of the flgN and fliT mutants, their transformants and the wild type were immunoblotted with antisera against the three HAPs and, as a control, the regulatory anti-sigma factor FlgM exported by the flagella pathway (Fig. 5B). The filament protein FliC was assayed by Coomassie blue staining.
All the strains secreted the same level of FlgM. In the flgN mutant (Fig. 5B, left), the dimeric fusion GST-N66–140, like the FlgN chaperone (+N), increased the export of FlgK and FlgL to WT levels. Both reduced the loss of unincorporated axial proteins FliC and FliD. Similarly (Fig. 5B, right), like FliT, the dimeric GST-T46–122 recovered FliD export and reduced FliC leaching by the fliT mutant. FliC and FliD released by the fliT mutant expressing His(6)FliT48–122 remained ≈ threefold and ≈20-fold higher than wild type, showing that the distal incorporation of these proteins into the growing flagellum was still disabled, and His(6)FlgN68–140 was unable to restore the export of FlgK. Surprisingly, it did partially restore export of FlgL (≈20% of WT). These data strongly indicate that the C-terminal domain binds the substrate, and this was confirmed by incubating soluble GST-N66–140 from S. typhimurium with nitrocellulose-immobilized cell lysates of E. coli overexpressing one of FlgK, FlgL or FliD. These affinity blots (Fig. 6) showed that the fusion specifically binds to FlgK and FlgL to a degree comparable with that of the wild-type FlgN.
Fig. 6.
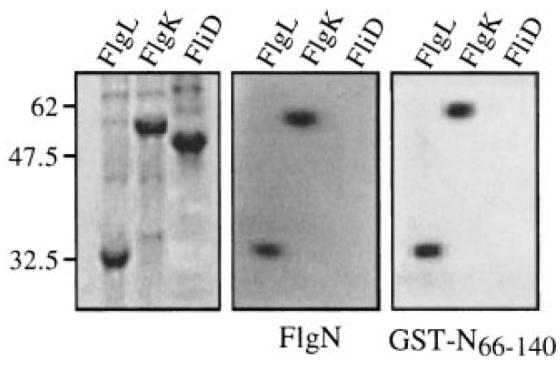
Binding of GST-N66–140 to FlgL and FlgK. Late exponential phase cultures of IPTG-induced E. coli BL21 (DE3) carrying the T7 recombinant derivatives pETL (FlgL), pETK (FlgK) or pETD (FliD) were lysed in urea–SDS loading buffer. Proteins were separated by SDS–PAGE (15%) and either stained with Coomassie brilliant blue (left) or transferred onto nitrocellulose membrane and incubated overnight with purified FlgN (middle) or GST-N66–140 (right). Bound proteins were detected by immunoblotting with anti-FlgN polyclonal antisera. Molecular weight markers are in kDa.
To consolidate these results, a hybrid chaperone, comprising the substrate-binding domain of FlgN and the dimerization domain of FliT, was assayed for its ability to complement the flgN mutation. The hybrid contained the first 50 N-terminal amino acids of fliT fused to residues 66–140 of flgN (Fig. 7A, see Experimental procedures) and was generated from plasmid pBAD18. Immunoblotting of cell lysates from the transformed S. typhimurium flgN mutant with anti-FlgN antisera confirmed arabinose-induced production of the fusion polypeptide, T-N, slightly larger (≈16 kDa) than the WT FlgN (Fig. 7B). Immunoblotting of the culture supernatant from late exponential phase cultures revealed (Fig. 7C) that this restored export of its HAP substrate FlgK to the WT level. Export of the other substrate FlgL was restored, but only to ≈30% of WT, correlating with a reduced loss of unincorporated FliC and FliD. The T-N hybrid chaperone had no complementing effect on the fliT mutant (not shown).
Fig. 7.
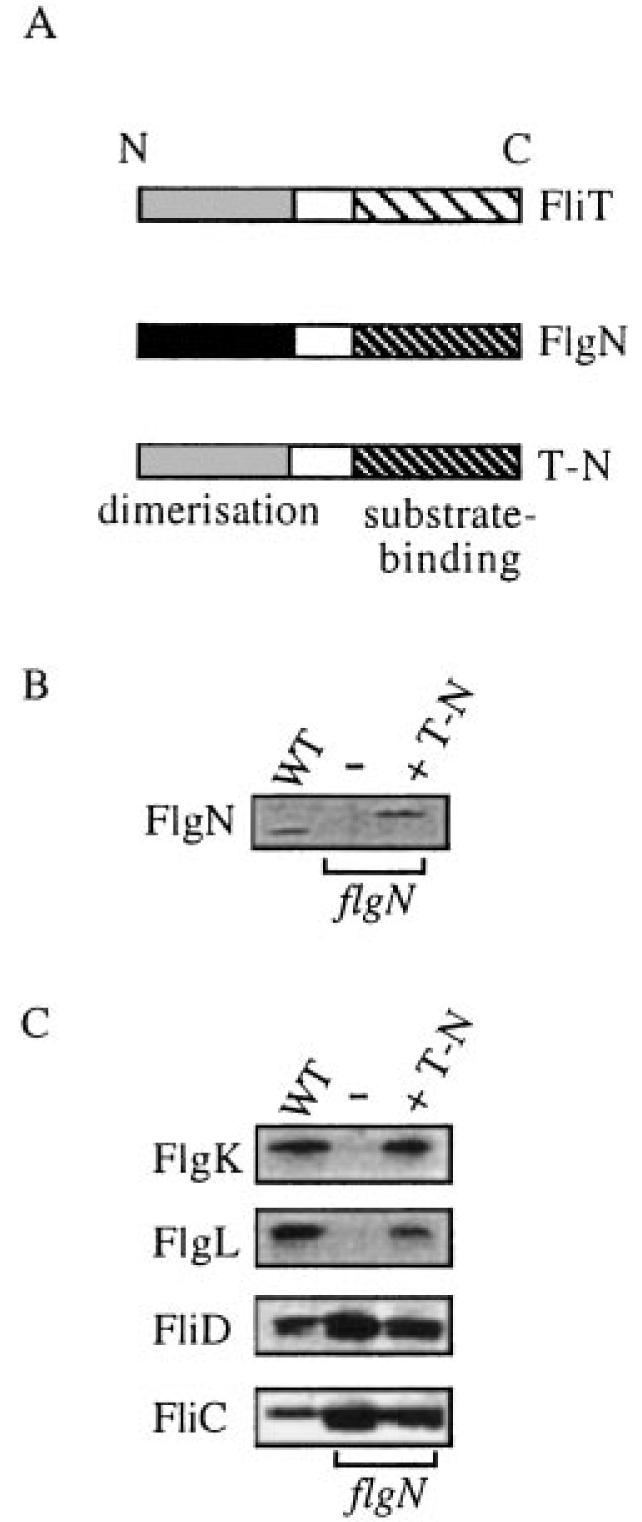
Complementation of the flgN mutant by a hybrid chaperone.
A. Representation of the hybrid chaperone (T-N) comprising the N-terminal domain of FliT fused to the C-terminal domain of FlgN.
B. Expression of the hybrid T-N chaperone. Late exponential phase wild-type S. typhimurium and flgN mutant cells alone (–) or expressing T-N (+T-N) were harvested, and cellular proteins were separated by SDS–(15%)PAGE and immunoblotted with anti-FlgN antisera.
C. Supernatant proteins from late exponential phase cultures of S. typhimurium WT and flgN mutant cells alone (–) or expressing the T-N hybrid chaperone (+T-N), separated by SDS–(12%)PAGE and immunoblotted with anti-FlgK, anti-FlgL or anti-FliD antisera or by Coomassie blue staining (FliC).
Discussion
We provide in vivo and in vitro evidence for the action of FlgN and FliT as substrate-specific flagellar chaperones in HAP export. S. typhimurium flgN and fliT mutants were specifically attenuated in the export of the HAPs FlgK/FlgL and FliD, respectively, and showed reduced incorporation of other flagellar axial components as a result, most obviously the filament subunit FliC. The flgN mutant could neither swim nor swarm, whereas the fliT defect reduced swarming, which requires more flagella than swimming (Harshey and Matsuyama, 1994). That the fliD mutation completely prevents filament assembly and swimming (Yokoseki et al., 1995) serves to emphasize the facilitating role of the chaperones.
FlgN and FliT located, seemingly exclusively, to the cytosol, where they formed stable pools. Virulence-related type III export chaperones have been similarly localized (Ménard et al., 1994), although CesD, a chaperone facilitating secretion of the E. coli EspB and EspD virulence effectors, is reported to associate with the inner membrane (Wainwright and Kaper, 1998). Our data do not preclude contact of chaperones or chaperone–substrate complexes with the membrane export apparatus; indeed, such contact may be essential, but they may indicate that such interactions are unstable. S. typhimurium cytosolic FlgN and FliT (≈15 kDa and ≈14 kDa respectively) both eluted as a single peak of ≈30 kDa, indicating that they are homodimers in vivo. No other elution peaks were detected, indicating that chaperone–HAP complexes are short-lived and that the HAPs are fed rapidly into the export machinery. It would seem advantageous for the cell to maintain a high ratio of chaperone to HAP substrate, so that the substrate would be bound immediately after its synthesis and the equilibrium would be pushed towards substrate export. Indeed, immunoblots of wild-type cell extracts (in parallel with purified proteins) indicate a large excess of FlgN, ≈200-fold, over FlgK (not shown). Immunoblots of flgN and fliT mutant cell lysates revealed that there was no marked accumulation of unchaperoned substrate; indeed, levels were reduced ≈ two- to fourfold. This may reflect the degradation of aggregated substrate.
As no chaperone–substrate complexes were detected in vivo, we established an in vitro assay using recombinant proteins affinity purified from E. coli to demonstrate interaction and possibly a concomitant effect on HAP behaviour. Purified His(6)FlgN and His(6)FliT formed homodimers in solution, in agreement with the behaviour of native cytosolic FlgN and FliT and of purified type III export chaperone SycE (Wattiau and Cornelis, 1993). The recombinant flagellar chaperones formed unique complexes with purified cognate HAPs in vitro, in each case of ≈90 kDa. This demonstrated stable and specific interaction and indicated a 2:1 stoichiometry of chaperone to HAP substrate in the complexes. This agrees with emerging evidence from the related virulence systems, as SycE homodimers bind YopE monomers in vitro (Cheng and Schneewind, 1999), and the EPEC intimin receptor Tir is reported to form a large multimeric complex with its chaperone CesT, possibly comprising a chaperone homotetramer and a substrate dimer (Abe et al., 1999). This in vitro approach also showed that purified FlgN was able to prevent aggregation of FlgK as, when refolded in the presence of its chaperone (at a 2:1 molar ratio of chaperone–substrate), ≈90% of the otherwise insoluble HAP was soluble (the effect was less powerful at a 1:2 ratio, compatible with the postulated stoichiometry of the complex). Under the in vitro conditions used in this study, FliD did not aggregate or self-assemble into its final pentameric form (Ikeda et al., 1996). It was therefore not possible to test whether FliT prevented the oligomerization of FliD. Nevertheless, it is reasonable to suppose that FliT performs an analogous function to FlgN. The combined in vitro data provide strong evidence of chaperone activity and are compatible with both FlgN and FliT acting as bodyguards, inhibiting premature self- interaction of their HAP substrates before export, as has been proposed for the Shigella IpgC chaperone, which prevents IpaB and IpaC virulence protein oligomerization (Ménard et al., 1994). Recently Karlinsey et al. (2000) have provided genetic evidence of an additional role for FlgN in flagellar protein export, proposing that FlgN acts to couple the translation of the anti-σ28 FlgM to type III secretion. This interpretation would indicate a multifactor- ial role for these small proteins in type III export, especially as the authors speculate that FlgN may do the same for FlgK and FlgL.
Most type III export chaperones, whether needed for virulence or motility, are predicted to contain a C-terminal amphipathic helix (Bennett and Hughes, 2000), and it has been speculated that this predicted structure could mediate interaction with cognate substrate proteins. Neither of the monomeric FlgN and FliT C-terminal polypeptides spanning the predicted helix were able to restore HAP export by the flgN and fliT mutants, but export was restored by dimeric translational fusions of GST to the same C-terminal regions. These data indicated that, in each chaperone, a C-terminal domain does indeed determine substrate binding, and this was confirmed by affinity blotting of FlgK and FlgL to GST-N66–140. The data suggest that full activity requires an N-terminal dimerization domain that does not form part of the substrate binding interface. FlgN has an N-terminal leucine zipper-like motif that might mediate dimerization (Vinson et al., 1989), but FliT does not. Nevertheless, the ability of a FliT–FlgN hybrid chaperone to restore HAP export to the flgN mutant suggests that FlgN and FliT have a common and closely related organization. As flagellar chaperones are possibly the progenitors of virulence-related type III chaperones, this could be a general feature of the family, compatible with the observation that a fusion containing a C-terminally truncated SycE is unable to bind its substrate YopE (Frithz-Linsten et al., 1995). Although N- and C-terminal GST fusions of full-length SycE are able to bind YopE, preventing intracellular aggregation and degradation of the substrate, they are unable to restore secretion to a sycE mutant (Cheng and Schneewind, 1999). This apparent discrepancy can be readily explained as, in contrast to flagellar chaperone binding to a substrate C-terminal domain (Fraser et al., 1999; Bennett and Hughes, 2000), binding of GST–SycE to the YopE N-terminus could interfere with substrate recognition by the type III export apparatus.
The mechanistic detail of flagellum assembly is poorly understood. Chaperone–substrate complexes might enter the pathway via events analogous to those proposed for the involvement of the homotetrameric chaperone SecB in the general secretory pathway (GSP). SecB binds cytosolic polypeptides and interacts with an inner membrane translocase via binding to the membrane-associated SecA ATPase (Economou, 2000). This interaction allows release of the export substrate from the chaperone, and ATP binding by SecA promotes both translocation of the substrate and displacement of SecB (Economou, 2000). Without SecB, the GSP is less efficient, and precursor protein folds into an export-incompetent conformation (Kumamoto and Gannon, 1988). Flagellar chaperone–substrate complexes could interact similarly with cytosolic and membrane-associated export components, including FliI, a homologue of the β catalytic subunit of the F0F1 ATPase (Fan and Macnab, 1996). Like SecB, released flagellar chaperone might then undergo several cycles of substrate binding and release. As the proteins involved in the early stages of flagellum assembly are known, the postulated interactions can now be examined.
Experimental procedures
Bacteria and recombinant DNA manipulation
Bacteria were grown at 37°C in Luria–Bertani (LB), 2× TY broth or on LB agar plates, supplemented with ampicillin (100 μg ml−1), chloramphenicol (25 μg ml−1) or spectinomycin (50 μg ml−1) as necessary. DNA manipulation and electroporation were carried out as described previously (Sambrook et al., 1989) using recA1 E. coli XL1 Blue (Stratagene). Plasmids and oligonucleotides are listed in Tables 1 and 2. Wild-type flgN and fliT genes were polymerase chain reaction (PCR) amplified (Perkin-Elmer thermal cycler) from S. typhimurium SJW1103 chromosomal DNA (1–10 ng) using 2.5 U of native Pfu DNA polymerase (Stratagene) and the primers N1/N2 and T1/T2 (50 pmol each) in native Pfu buffer (Stratagene) containing 0.25 mM each dNTP. Amplified DNA was purified using Wizard PCR Preps (Promega), digested with NdeI and BamHI and ligated to NdeI–BamHI-digested T7 expression vector pET15b (Studier et al., 1990; Novagen), creating pETN and pETT (Table 1). DNA sequences encoding FlgN and FliT C-terminal domains were amplified using primer pairs N3/N2 or T3/T2 and ligated into NdeI–BamHI-digested pET15b to create pETNΔ1–67 and pETTΔ1–47. Inserted fragments were excised from pET15b by digestion with XbaI–HindIII and ligated into the arabinose-inducible expression vector pBAD18 (Guzman et al., 1995), generating pBADflgN, pBADfliT, pBADNΔ1–67 and pBADTΔ1–47. GST fusions were created by PCR amplification of DNA encoding the C-terminal domains of FlgN or FliT using appropriate primer pairs and insertion into the BamHI site of pGEX-1 (Smith and Johnson, 1988). The IPTG-inducible Ptac promoter controlled the expression of recombinant proteins from pGEX-1 derivatives. To create the hybrid chaperone, codons 1–50 of fliT were amplified by PCR using primers T1 and T5 and inserted into pET15b, generating pETTΔ51–122. DNA encoding the FlgN C-terminal domain (amino acids 68–140) was excised from pGSTΔ1–67 and inserted into the BamHI site of pETTΔ51–122, generating pETT-N (Fig. 5A). The resulting translational fusion was excised from pET15b and ligated into pBAD18, generating pBADT-N.
Table 1.
Plasmids.
| Plasmid | Description | Reference |
|---|---|---|
| pET11c | Ampr T7 expression vector | Studier et al. (1990) |
| pET15b | Ampr T7 expression vector, 6×His translational fusion | Studier et al. (1990) |
| pBAD18 | Ampr PBAD expression vector | Guzman et al. (1995) |
| pCVD442 | Ampr, suicide vector, R6K ori | Donnenberg and Kaper (1991) |
| pGEX-1 | Ampr Ptac expression vector | Smith and Johnson (1988) |
| pBluescript-II SK | Ampr | Stratagene |
| pETflgN | pET11c PlacT7flgN (flgN 1–420) | Fraser et al. (1999) |
| pETfliT | pET11c PlacT7fliT (fliT 1–366) | Fraser et al. (1999) |
| pETN | pET15b PlacT7flgN (flgN 1–420, amplified using N1 and N2) | This study |
| pETT | pET15b PlacT7fliT (fliT 1–366, amplified using T1 and T2) | This study |
| pETK | pET15b PlacT7flgK (flgK 1–1656) | Fraser et al. (1999) |
| pETL | pET15b PlacT7flgL (flgL 1–951) | Fraser et al. (1999) |
| pETD | pET15b PlacT7fliD (fliD 1–1398) | Fraser et al. (1999) |
| pBADflgN | PBADflgN (flgN 1–420) | This study |
| pBADNΔ1–67 | PBADflgNΔ1–67 (flgN 204–420, amplified using N3 and N2) | This study |
| pGSTNΔ1–65 | PtacflgNΔ1–65 (flgN 198–420, amplified using N3 and N2) | This study |
| pBADfliT | PBAD fliT (fliT 1–366) | This study |
| pBADTΔ1–47 | PBAD fliTΔ1–47 (fliT 144–366, amplified using T3 and T2) | This study |
| pGSTTΔ1–45 | PtacfliTΔ1–45 (fliT 144–366, amplified using T4 and T2) | This study |
| pETTΔ51–122 | PtacfliTΔ51–122 (fliT 1–150, amplified using T1 and T5) | This study |
| pETT-N | PtacfliTΔ51–122flgNΔ1–65 (fliT 1–150::flgN 198–420) | This study |
| pBADT-N | PBADfliTΔ51–122flgNΔ1–65 (fliT 1–150::flgN 198–420) | This study |
Table 2.
Oligonucleotides.
| Primer | Sequence (5′–3′) | Site |
|---|---|---|
| N1 | GAGTAAATAAGCATATGACTCGTTTG | NdeI |
| N2 | GCCTACAAGGGATCCAGGCCGGAAAGG | BamHI |
| N3 | GGATTATCTGGATCCACAGCGCCATATGGAGCAGAATG | NdeI–BamHI |
| N(ups) | GACAATGGCGCAGGACATCAACGC | |
| N(ds) | CGATGCGGGTGGTCAAACAGATGG | |
| T1 | GTCTCGCATATGACCTCAACCGTGGAGTTTATC | NdeI |
| T2 | GAGACGGGATCCTATTGAGGCGCCAAGGCGC | BamHI |
| T3 | CAAAGTATTGAAACGCATATGGATCCGCAAACT | NdeI |
| T4 | ATTGAAACGGTGATGGATCCGCAAACTCCACCG | BamHI |
| T5 | CCCGGTGGAGTTTGCGGATCCATCACCGTT | BamHI |
| T(ups) | GCGTTCAAACATATGGCGGAAATTGGCATC | |
| T(ds) | CTCAATCCATTCTTTATAAAACCAGGC | XbaI |
Mismatch nucleotides are shown in bold; start or termination codons are underlined.
Analysis of total cell and supernatant proteins
Wild-type, late exponential phase cells were fractionated using a modification of a previous protocol (Koronakis et al., 1991). To create spheroplasts and release periplasmic contents, cell pellets were resuspended in one volume of 0.5 M sucrose, 40 mM Tris-HCl, pH 7.4, 5 mM EDTA before the addition of lysozyme (final concentration 40 μg ml−1) and one volume of sterile dH2O. To stabilize the cytoplasmic membranes, MgCl2 (final concentration 20 mM) was added to the suspension of spheroplasts before being pelleted by centrifugation at 60 000 g for 20 min (the supernatant being the periplasmic fraction). The pellet was resuspended in a hypotonic solution of 20 mM Tris-HCl, pH 7.4, 5 mM EDTA, 1 mM Pefabloc; DNase was added and the suspension agitated until viscosity decreased. To pellet membranes, MgCl2 (final concentration 20 mM) was added, and the suspension was centrifuged (the supernatant being the cytosolic fraction). The pellet was resuspended in urea–SDS loading buffer. Proteins in culture supernatants of wild-type and mutant cell cultures were concentrated as described previously (Fraser et al., 1999) before SDS–PAGE. Chaperone stability was assessed as described previously (Claret and Hughes, 2000).
Expression of recombinant flagellar proteins
E. coli BL21 (DE3) cells carrying the appropriate plasmid were grown to mid-exponential phase (OD600 of 0.7), and T7-controlled expression of protein was induced by the addition of IPTG to 1 mM (Studier and Moffat, 1986). Cultures were incubated for a further 3 h, after which cells (OD600 of 1.0) were harvested by centrifugation. Induction of S. typhimurium cells carrying pBAD18 derivatives was achieved by adding 0.1% (w/v) l-arabinose.
Generation of chromosomal null mutants
Approximately 1 kb of 5′ or 3′ DNA flanking flgN or fliT was amplified by PCR using primer combinations Nups/Nds and Tups/Tds, respectively (Table 2), and each product was blunt end ligated into pBluescript-II SK. A unique BamHI site at codon 50 of fliT was created by PCR mutagenesis (contained within primer T5) and used to insert a chloramphenicol omega (Ω) cassette that contained both transcriptional and translational terminators (derived from pUTmTn5; Fellay et al., 1987). Deletion of codons 1–74 of flgN by digestion with Eco47III and subsequent religation created a unique Eco47III restriction site for the insertion of a Ω cassette. Disrupted loci were inserted separately into the suicide vector pCVD442 and maintained in the donor strain E. coli SM10::λpir (Miller and Mekalanos, 1988). After conjugal transfer of suicide vector into S. typhimurium SJW1103 (a spontaneous spectinomycin mutant), transconjugants were selected on LB agar (50 μg ml−1 spectinomycin and chloramphenicol). After replica plating to LB agar containing 100 μg ml−1 ampicillin, transconjugants that failed to grow, thus confirming loss of the pCVD442 vector, were analysed by Southern blotting using PCR-amplified flgN or fliT and Ω cassette as probes. Inactivation of flgN or fliT was confirmed by immunoblotting with anti-FlgN or anti-FliT antisera.
Affinity purification of histidine- and GST-tagged proteins
To isolate His-tagged proteins, BL21 (DE3) cells carrying pETN or pETT were induced for 3 h and pelleted by centrifugation (7000 g for 10 min at 4°C), resuspended in a 1:25 culture volume of binding buffer (BB; 20 mM Tris-HCl, pH 7.4, 500 mM NaCl, 1 mM Pefabloc, 10 mM imidazole) and lysed in a French pressure cell (SLM Instruments) (700 p.s.i., three passages). After centrifugation (39 000 g for 30 min), soluble extract was filtered through a 0.45 μm filter (Schleicher and Schuell) and loaded onto a HisBind Quick 900 cartridge (Novagen), pre-equilibrated with BB. The cartridge was first washed with 10 column volumes (c.v.) of BB, followed by 5 c.v. of BB containing 30 mM imidazole before bound proteins were eluted with 2 c.v. of BB containing 400 mM imidazole. His(6)FlgK and His(6)FliD were purified from inclusion bodies using a HisBind Quick 300 cartridge (Novagen) under denaturing conditions, according to the manufacturer's instructions, and each was eluted using 8 M urea, 20 mM Tris-HCl, pH 7.4, 500 mM NaCl, 400 mM imidazole, 1 mM Pefabloc. To purify GST-N66–140, E. coli BL21 carrying pGSTN1Δ1-65 was pelleted by centrifugation (7000 g for 10 min at 4°C), resuspended in PBS and lysed in a French pressure cell. Filtered soluble extracts were incubated with glutathione Sepharose 4B (Pharmacia Biotech) for 2 h at room temperature. The beads were then washed three times in PBS, and bound protein was eluted with 10 mM reduced glutathione (Sigma).
Affinity blotting
E. coli BL21 (DE3) carrying pETK, pETL or pETD was grown to mid-exponential phase (A600 0.7), and T7-controlled expression of protein was induced by the addition of IPTG to 1 mM (Studier and Moffat, 1986). Cultures were incubated for a further 3 h, after which the cells were centrifuged and resuspended in 8 M urea SDS–PAGE loading buffer. Proteins separated by SDS–PAGE (15%) were transferred to nitrocellulose and preincubated for 2 h at room temperature in PBS containing 5% (w/v) skimmed milk. Purified FlgN or GST-N66–140 was added directly to the preincubation solution to a final concentration of 2 μg ml−1. Membranes were incubated for 16 h at room temperature before immunoblotting with anti-FlgN polyclonal antisera.
Gel filtration chromatography
Soluble cell extract (200 μl; in 20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM Pefabloc) from 2.5 ml of LB broth culture (OD600 of 1.0) was loaded onto a pre-equilibrated Superose 12 HR 10/30 column (Pharmacia) and developed at 0.5 ml min−1 by fast protein liquid chromatography (FPLC; Waters). Purified His(6)FlgN, His(6)FliT, His(6)FliD, His(6)FlgK and/or preformed chaperone–susbtrate complexes were analysed by chromatography using a 1 ml bed volume Sephacryl S-200 HR column (Pharmacia). The sample (< 5% bed volume) was developed by gravity. In both cases, proteins in eluted fractions were precipitated by adding 10% (w/v) trichloroacetic acid (TCA). Columns were size calibrated with ferritin (440 kDa), catalase (232 kDa), aldolase (158 kDa), BSA (67 kDa), ovalbumin (43 kDa), chymotrypsinogen A (25 kDa) and ribonuclease A (13.7 kDa).
In vitro formation of chaperone–substrate complexes
Purified His(6)FlgK was diluted 2:1 in 20 mM Tris-HCl, pH 7.4, and centrifuged at 350 000 g for 10 min to remove anyinsoluble aggregates. Solubilized protein (4 M urea, 20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 200 mM imidazole) was dialysed (42 kDa membrane) against 4 M urea, 20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA for 4 h at room temperature. Purified His(6)FlgN was dialysed against 20 mM Tris-HCl, pH 7.4, 150 mM NaCl for 4 h at 4°C, and soluble protein was separated by centrifugation (607 000 g for 30 min at 4°C). Protein concentration was determined using the Bradford assay and adjusted to 1 mg ml−1. Isolation of a soluble complex between His(6)FlgK and His(6)FlgN was achieved by diluting (1:100) His(6)FlgK (86 or 43 pmol) in 20 mM Tris-HCl, pH 7.4, 150 mM NaCl containing purified His(6)FlgN, such that the molar ratio of His(6)FlgN to His(6)FlgK was either 2:1 or 1:2, in a total volume of < 200 μl. The mixture was incubated for 20 min at room temperature, and insoluble proteins were removed by centrifugation at 607 000 g for 10 min at 16°C. Purified His(6)FlgN, His(6)FlgK or a preformed complex of these were analysed by gel filtration chromatography using a 1 ml bed volume Sephacryl S-200 HR column (Pharmacia). To isolate a stable FliT–FliD complex, soluble His(6)FliD (20 mM Tris-HCl, pH 7.4, 500 mM NaCl) was added at an equimolar concentration to His(6)FliT (20 mM Tris-HCl, pH 7.4, 500 mM NaCl).
Acknowledgements
We thank Emma McGhie and Donald Tipper for comments on the manuscript. This work was supported by a Wellcome Trust Programme grant (C.H.) and Wellcome Trust (J.C.Q.B.), MRC (G.F.) and BBSRC (J.T.) studentships.
References
- Abe A, de Grado M, Pfuetzner RA, Sanchez-Sanmartin C, Devinney R, Puente JL, et al. Enteropathogenic Escherichia coli translocated intimin receptor, Tir, requires a specific chaperone for stable secretion. Mol Microbiol. 1999;33:1162–1175. doi: 10.1046/j.1365-2958.1999.01558.x. [DOI] [PubMed] [Google Scholar]
- Aizawa S-I. Flagellar assembly in Salmonella typhimurium. Mol Microbiol. 1996;19:1–5. doi: 10.1046/j.1365-2958.1996.344874.x. [DOI] [PubMed] [Google Scholar]
- Bennett JCQ, Hughes C. From flagellum assembly to virulence: the extended family of type III export chaperones. Trends Microbiol Sci. 2000;8:202–204. doi: 10.1016/s0966-842x(00)01751-0. [DOI] [PubMed] [Google Scholar]
- Cheng LW, Schneewind O. Yersinia enterocolitica type III secretion. On the role of SycE in targeting YopE into HeLa cells. J Biol Chem. 1999;274:22102–22108. doi: 10.1074/jbc.274.31.22102. [DOI] [PubMed] [Google Scholar]
- Claret L, Hughes C. Rapid turnover of FlhD and FlhC, the flagellar regulon transcriptional activator proteins during Proteus swarming. J Bacteriol. 2000;182:833–836. doi: 10.1128/jb.182.3.833-836.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnenberg MS, Kaper JB. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect Immun. 1991;59:4310–4317. doi: 10.1128/iai.59.12.4310-4317.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Economou A. Bacterial protein translocase: a unique molecular machine with an army of substrates. FEBS Lett. 2000;476:18–21. doi: 10.1016/s0014-5793(00)01662-8. [DOI] [PubMed] [Google Scholar]
- Elliott SJ, Hutcheson SW, Dubois MS, Mellies JL, Wainwright LA, Batchelor M, et al. Identification of CesT, a chaperone for the type III secretion of Tir in enteropathogenic Escherichia coli. Mol Microbiol. 1999;33:1176–1189. doi: 10.1046/j.1365-2958.1999.01559.x. [DOI] [PubMed] [Google Scholar]
- Fahrner KA, Block SM, Krishnaswamy S, Parkinson JS, Berg HC. A mutant hook-associated protein (HAP3) facilitates torsionally in transformations of the flagellar filament of Escherichia coli. J Mol Biol. 1994;238:173–186. doi: 10.1006/jmbi.1994.1279. [DOI] [PubMed] [Google Scholar]
- Fan F, Macnab RM. Enzymatic characterization of FliI. An ATPase involved in flagellar assembly in Salmonella typhimurium. J Biol Chem. 1996;271:31981–31988. doi: 10.1074/jbc.271.50.31981. [DOI] [PubMed] [Google Scholar]
- Fellay R, Frey J, Krisch H. Interposon mutagenesis of soil and water bacteria: a family of DNA fragments designed for in vitro insertional mutagenesis of Gram-negative bacteria. Gene. 1987;52:147–154. doi: 10.1016/0378-1119(87)90041-2. [DOI] [PubMed] [Google Scholar]
- Fraser GM, Bennett JCQ, Hughes C. Substrate-specific binding of hook-associated proteins by FlgN and FliT, putative chaperones for flagellum assembly. Mol Microbiol. 1999;32:569–580. doi: 10.1046/j.1365-2958.1999.01372.x. [DOI] [PubMed] [Google Scholar]
- Frithz-Linsten E, Rosqvist R, Johansson L, Forsberg A. The chaperone-like protein YerA of Yersinia pseudotuberculosis stabilizes YopE in the cytoplasm but is dispensable for targeting to the secretion loci. Mol Microbiol. 1995;16:635–647. doi: 10.1111/j.1365-2958.1995.tb02426.x. [DOI] [PubMed] [Google Scholar]
- Guzman L-M, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation and high-level expression by vectors containing the arabinose pBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gygi D, Fraser G, Dufour A, Hughes C. A motile but non-swarming mutant of Proteus mirabilis lacks FlgN, a facilitator of FliC export and flagella assembly. Mol Microbiol. 1997;25:597–604. doi: 10.1046/j.1365-2958.1997.5021862.x. [DOI] [PubMed] [Google Scholar]
- Harshey RM, Matsuyama T. Dimorphic transition in Escherichia coli and Salmonella typhimurium: Surface-induced differentiation into hyperflagellate swarmer cells. Proc Natl Acad Sci USA. 1994;91:8631–8635. doi: 10.1073/pnas.91.18.8631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma M, DeRosier DJ, Macnab RM. Flagellar hook and hook-associated proteins of Salmonella typhimurium and their relationship to other axial components of the flagellum. J Mol Biol. 1990;213:819–832. doi: 10.1016/S0022-2836(05)80266-9. [DOI] [PubMed] [Google Scholar]
- Ikeda T, Oosawa K, Hotani H. Self-assembly of the filament capping protein, FliD, of bacterial flagella into an annular structure. J Mol Biol. 1996;259:679–686. doi: 10.1006/jmbi.1996.0349. [DOI] [PubMed] [Google Scholar]
- Jones CJ, Macnab RM, Okino H, Aizawa S-I. Stoichiometric analysis of the flagellar hook-(basal body) complex of Salmonella typhimurium. J Mol Biol. 1990;212:377–387. doi: 10.1016/0022-2836(90)90132-6. [DOI] [PubMed] [Google Scholar]
- Karlinsey JE, Lonner J, Brown KL, Hughes KT. Translation/secretion coupling by type III secretion systems. Cell. 2000;180:5384–5397. doi: 10.1016/s0092-8674(00)00053-2. [DOI] [PubMed] [Google Scholar]
- Koronakis V, Hughes C, Koronakis E. Energetically-distinct early and late stages of HlyB/HlyD-dependent protein secretion across both E. coli membranes. EMBO J. 1991;10:3263–3272. doi: 10.1002/j.1460-2075.1991.tb04890.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubori T, Shimamoto N, Yamaguchi S, Namba K, Aizawa S. Morphological pathway of flagellar assembly in Salmonella typhimurium. J Mol Biol. 1992;226:433–446. doi: 10.1016/0022-2836(92)90958-m. [DOI] [PubMed] [Google Scholar]
- Kumamoto CA, Gannon PM. Effects of Escherichia coli secB mutation on pre-maltose binding protein conformation and export kinetics. J Biol Chem. 1988;263:11554–11558. [PubMed] [Google Scholar]
- Lupas A, Van Dyke M, Stock J. Predicting coiled-coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- Ménard R, Sansonetti P, Parsot C, Vasselon T. Extracellular association and cytoplasmic partitioning of the IpaB and IpaC invasins of S. flexneri. Cell. 1994;79:515–525. doi: 10.1016/0092-8674(94)90260-7. [DOI] [PubMed] [Google Scholar]
- Miller VL, Mekalanos JJ. A novel suicide vector and its uses in constructional insertion mutants: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J Bacteriol. 1988;170:2575–2583. doi: 10.1128/jb.170.6.2575-2583.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamino T, Macnab RM. Components of the Salmonella flagellar export apparatus and classification of export substrates. J Bacteriol. 1999;181:1388–1394. doi: 10.1128/jb.181.5.1388-1394.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz T, Francis N, Morgan DG, DeRosier DJ. Size of the export channel in the flagellar filament of Salmonella typhimurium. Ultramicroscopy. 1993;48:417–425. doi: 10.1016/0304-3991(93)90247-u. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd edn. Cold Spring Harbor, NY: Cold Spring Harbour Laboratory Press; 1989. [Google Scholar]
- Smith DB, Johnson KS. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- Studier FW, Moffatt BA. Use of bacteriophage T7 DNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- Vinson CR, Sigler PB, McKnight SL. Scissors-grip model for DNA recognition by a family of leucine zipper proteins. Science. 1989;246:911–916. doi: 10.1126/science.2683088. [DOI] [PubMed] [Google Scholar]
- Vonderviszt F, Imada K, Furukawa Y, Uedaira H, Taniguchi H, Namba K. Mechanism of self-association and filament capping by flagellar HAP2. J Mol Biol. 1998;284:1399–1416. doi: 10.1006/jmbi.1998.2274. [DOI] [PubMed] [Google Scholar]
- Wainwright LA, Kaper JB. EspB and EspD require a specific chaperone for proper secretion from enteropathogenic E. coli. Microbiol. 1998;27:1247–1260. doi: 10.1046/j.1365-2958.1998.00771.x. [DOI] [PubMed] [Google Scholar]
- Wattiau P, Cornelis GR. SycE, a chaperone-like protein of Yersinia enterocolitica involved in the secretion of YopE. Mol Microbiol. 1993;8:123–131. doi: 10.1111/j.1365-2958.1993.tb01209.x. [DOI] [PubMed] [Google Scholar]
- Wattiau P, Woestyn S, Cornelis GR. Customized secretion chaperones in pathogenic bacteria. Mol Microbiol. 1996;20:255–262. doi: 10.1111/j.1365-2958.1996.tb02614.x. [DOI] [PubMed] [Google Scholar]
- Woestyn S, Sory M-P, Boland A, Lequenne O, Cornelis GR. The cytosolic SycE and SycH chaperones of Yersinia protect the region of YopE and YopH involved in translocation across eukaryotic cell membranes. Mol Microbiol. 1996;20:1261–1271. doi: 10.1111/j.1365-2958.1996.tb02645.x. [DOI] [PubMed] [Google Scholar]
- Yan H, Lim JTE, Contillo LG, Krolewski JJ. Glutathione S-transferase fusion proteins mimic receptor dimerization in permeabilised cells. Anal Biochem. 1995;231:455–458. doi: 10.1006/abio.1995.0080. [DOI] [PubMed] [Google Scholar]
- Yokoseki T, Kutsukake K, Ohnishi K, Iino T. Functional analysis of the flagellar genes in the fliD operon of Salmonella typhimurium. Microbiology. 1995;141:1715–1722. doi: 10.1099/13500872-141-7-1715. [DOI] [PubMed] [Google Scholar]