

Abstract
An understanding of the molecular basis of drug action provides opportunities for refinement of drug properties and for development of more potent and selective molecules that act at the same biological target. In this work, we have identified the active enantiomers in racemic mixtures of structurally related benzophenone derivatives of 1,5-benzodiazepines, representing both antagonist and agonist ligands of the type A cholecystokinin receptor. The parent compounds of the 1,5-benzodiazepine CCK receptor photoaffinity ligands were originally prepared in an effort to develop orally active drugs. The enantiomeric compounds reported in this study selectively photoaffinity-labeled the CCK receptor, resulting in the identification of a site of attachment for the photolabile moiety of the antagonist probe deep within the receptor’s membrane-spanning region at Leu88, a residue within transmembrane segment two. In contrast, the agonist probe labeled a region including extracellular loop one and a portion of transmembrane segment three. The antagonist covalent attachment site to the receptor served as a guide in the construction of theoretical three-dimensional molecular models for the antagonist-receptor complex. These models provided a means for visualization of physically plausible ligand-receptor interactions in the context of all currently available biological data that address small molecule interactions with the CCK receptor. Our approach, featuring the use of novel photolabile compounds targeting the membrane-spanning receptor domain to probe the binding site region, introduces powerful tools and a strategy for direct and selective investigation of non-peptidyl ligand binding to peptide receptors.
Keywords: cholecystokinin receptor, G protein-coupled receptor, photoaffinity labeling, ligand binding, receptor structure, active site
INTRODUCTION
Understanding of the molecular basis of drug action is extremely important, contributing to refinement of the specificity, affinity, and potency of lead compounds, and even to the rational design of new drugs. Insights into this can come from the complementary approaches of receptor mutagenesis and photoaffinity labeling. Loss of function in a receptor mutant can be explained either by direct or allosteric effects, while clean photoaffinity labeling of specific residues is direct and unambiguous, but can be quite difficult to achieve. Typical challenges include identification of a small molecule ligand that can accommodate both a photolabile functional group and a radiolabel, while retaining receptor binding affinity and biological activity.
We have previously reported the development of structurally related 1,5-benzodiazepine ligands that act as agonists and antagonists at the type A cholecystokinin receptor.1 This is a guanine nucleotide-binding protein (G protein)-coupled receptor in the rhodopsin-β-adrenergic receptor family.2 It normally binds and responds to a peptide hormone that is important in the regulation of gallbladder contraction, exocrine pancreatic secretion, gastric emptying, enteric transit, and satiety.3 We have been able to incorporate photolabile benzophenone and diazirine moieties into these non-peptidyl CCK receptor ligands.1 We have also been successful in radiolabeling them with 14C, without changing their agonist or antagonist properties and with maintenance of adequate binding affinity.1 In spite of the extremely low specific radioactivity of these probes (0.05 Ci/mmol), representing less than one thirty-thousandth of the activity of the radioiodinated peptide probes we have previously used in affinity labeling this receptor,4–7 we have been successful in using these unique ligands to photoaffinity label the CCK receptor.1
In this work, through the use of resolved enantiomeric CCK-A ligands, we have been able to extend the insights possible with the racemic compounds that we initially described in a prior publication.1 The previously reported racemic photolabile benzophenone-derivatives of the 1,5-benzodiazepine agonist (mixture of compounds 5 and 6, Figure 1) and antagonist (mixture of compounds 7 and 8, Figure 1) were separated into their enantiomeric components and each chiral compound was then assayed for biological activity, binding affinity and CCK receptor labeling efficiency. For the subsequent photoaffinity labeling work, the racemic intermediate amines (shown in Synthetic scheme 1) (1, 2) and (3, 4) were resolved into their respective enantiomers and converted into 14C analogs of each of the four separated enantiomers (5, 6, 7 and 8).
Fig 1.
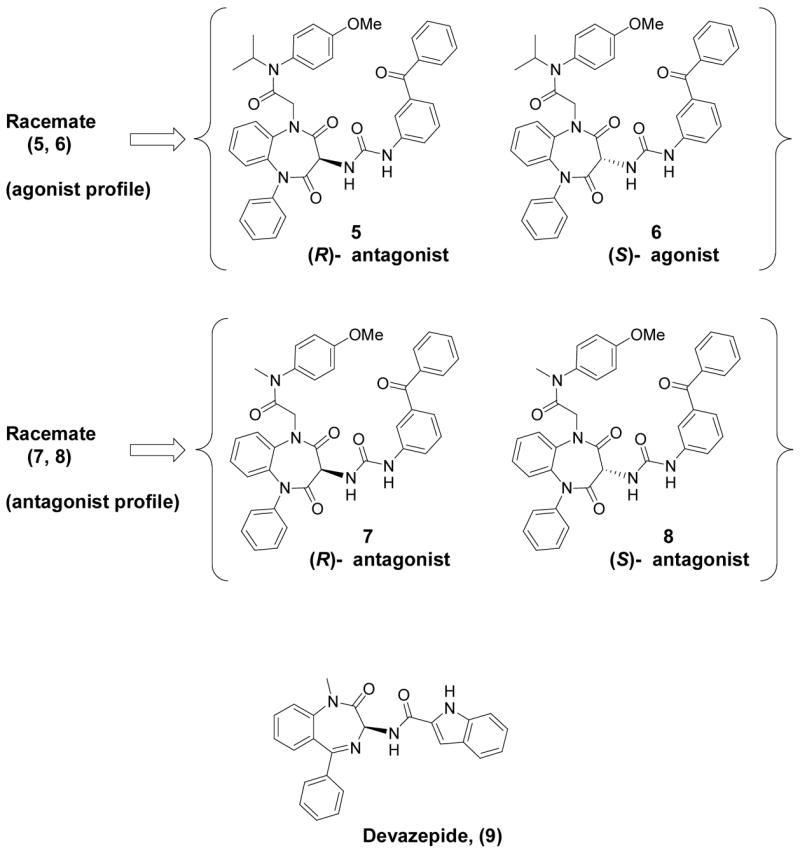
Chemical structures of non-peptidyl CCK receptor ligands used in the current series of studies. The racemic mixture of compounds 5 and 6 having an agonist profile was separated into its components (5, (R)- form with no agonist activity and 6, (S)- form with agonist activity), and the racemic mixture of compounds 7 and 8 having an antagonist profile was similarly separated into its components (7, (R)- form and 8, (S)- form). Also illustrated is the structure of a related 1,4-benzodiazepine CCK receptor antagonist that was developed by Merck, initially called L-364,718 and subsequently referred to as devazepide (9). This was used in pharmacological competition assays and molecular model-building studies.
Scheme 1.

(a) C6H5NHNH2, EtOH, H2O, HCl; (b) PCl5, CCl4; (c) bromoacetyl bromide, DIPEA, DCM; (d) N-phenyl phenylenediamine, NaH, DMF; (e) THF, ≤4 °C; (f) Zn dust, AcOH
When the racemic mixture demonstrating agonist activity was separated into its components, only the putative (S)-enantiomer (6) displayed biological agonist activity, and it was also the isomer that bound more tightly to the receptor. Therefore, the corresponding (R)-enantiomer in this series (5) actually represents a functional antagonist. Of interest, this (S)-enantiomer (6) was substantially less efficient in covalent labeling of the CCK receptor than was the (R)-enantiomer (5). In contrast, both antagonist enantiomers (7 and 8) had similar binding affinities for the CCK receptor, but the proposed (S)-enantiomer (8) was substantially more efficient in covalent labeling of the receptor than the corresponding (R)-enantiomer (7). Covalent attachment sites for these structurally similar agonist and antagonist probes were characterized, and the results indicate that agonist and antagonist ligands covalently labeled distinct regions within the CCK receptor. The site labeled by the antagonist was determined definitively with proteolytic peptide mapping, followed by Edman degradation sequencing of the labeled fragment. The antagonist was covalently attached to Leu88 within the second transmembrane helical segment, clearly establishing a non-peptidyl antagonist binding site that is distinct from the extramembranous receptor regions involved in binding the natural peptide agonist ligand.8,9 The non-peptidyl agonist ligand covalently labeled a different portion of the receptor, believed to represent the cyanogen bromide fragment that includes the first extracellular loop and the upper portion of the third transmembrane segment. The receptor fragments covalently labeled by agonist and antagonist probes were distinct, but co-incubation of the receptor preparation with a competing concentration (1 μM) of the endogenous peptide ligand, CCK, along with the benzodiazepine probes, did not block photoaffinity labeling by either probe. However, competitive co-incubation with the non-peptidyl antagonist, devazepide (also referred to in the literature as L-364,718 and MK329) (9), blocked photoaffinity labeling by both agonist and antagonist probes in a concentration-dependent manner. Based on these results, we propose that both agonist and antagonist probes probably bind to receptor sites that are composed of partially overlapping regions within the CCK receptor membrane-spanning domains.
The data obtained from photoaffinity labeling studies with these compounds were used to construct three-dimensional molecular models for ligand-receptor complexes. Small molecule probes containing a fused benzodiazepine ring system have far less conformational flexibility than peptide ligands. Therefore, they proved amenable to conformational search strategies that are impractical for peptide ligands. For example, methods were applied that evaluated various conformations of the ligands within the context of the putative receptor binding site. The predominantly alpha-helical transmembrane domain where the non-peptidyl ligands bind also possesses far fewer degrees of conformational freedom than the flexible extracellular loops shown to be important for peptide binding.8,9 Therefore, we were able to generate plausible three-dimensional models for these ligand-receptor complexes using only the photoaffinity labeling results, along with mutagenesis data from earlier studies for the closely related antagonist, devazepide.
RESULTS
Synthetic Scheme 1 illustrates the strategy utilized for the preparation of the key racemic amine intermediates (1, 2) and (3, 4) to be used in the synthesis of the photolabile chiral probes (5, 6, 7 and 8). The known bromoacetamides (12 and 13) were prepared in high yield from bromoacetyl bromide and the corresponding N-methyl or N-isopropyl secondary amine using the general procedure as described in Aquino et al.10 Subsequent alkylation of commercially available N-phenylphenylenediamine with the appropriate bromoacetamide yielded the substituted phenylenediamine intermediates (14 and 15). The known 2-(phenylhydrazono-propanedioyldichloride (17) was prepared in two steps from the commercially available ketomalonic acid using the procedure outlined in Aquino et al.,10 and condensed with the phenylenediamine intermediates (14 and 15) to afford the corresponding hydrazones (18 and 19). Zinc reduction of the hydrazones yielded the racemic amines (1, 2) and (3, 4).
With the amine racemates in hand, we approached the synthesis of the final resolved chiral photoaffinity labeling probes (5, 6, 7 and 8) using two complementary routes, as shown in Scheme 2. Both methods provide pure, separated photoaffinity ligands with similar synthetic efficiency and enantiopurity.
Scheme 2.

(a) phosgene, 3-aminobenzophenone, base, <4 °C; (b) Chiral reverse-phase HPLC purification.
As illustrated by the first method shown in Synthetic Scheme 2, initially for the bio-activity and binding experiments, the racemic amine intermediates were cleanly converted to the racemic benzophenone ureas (5, 6) and (7, 8) using a phosgene equivalent to couple the amines with commercially available 3-aminobenzophenone. Subsequently, chiral chromatography resolved each of these two racemic benzophenone-1,5-benzodiazepine urea pairs into the final four enantiomers (5, 6, 7, and 8) in high enantiomeric excess and complete baseline peak separation under chiral HPLC conditions.
For further exploration with modeling analysis, and in the absence of being able to obtain adequate crystallographic data on the resolved enantiomers, we provisionally assigned the absolute configurations of our compounds based on comparison to reported X-ray structures for highly analogous CCK ligands. Within the racemate originally displaying agonist activity, (racemate 5, 6), after enantiomer separation, the antagonist compound 5 was putatively assigned the (R)- configuration based on its strong structural homology with devazepide (9), as well as other benzodiazepine CCK antagonists having X-ray structures reported in the literature.11–13 The only agonist ligand in the set, compound 6, was correspondingly assigned the (S)- configuration. Comparing, under the same stationary and mobile phase conditions (see experimental section), the relative chiral HPLC retention times of these two compounds (41.3 for compound 5 and 48.3 minutes for compound 6) with those observed for the two antagonist enantiomers (49.9 minutes for 7 and 82.2 minutes for 8) allowed for the provisional assignment of compounds 7 and 8 to be (R)- and (S)-, respectively.
As can be seen in Figure 2, after separating the racemic mixture that demonstrated agonist activity, only the putative (S)-enantiomer (6) displayed intrinsic agonist activity (EC50 150±36 nM). The corresponding (R)-enantiomer (5) showed no ability to stimulate an increase in pancreatic acinar cell amylase secretion in concentrations as high as 1 μM. These two enantiomers also displayed distinct binding affinities, with compound 6 having a higher affinity than compound 5 (IC50’s of 61±17 nM for compound 6 and 710±420 nM for compound 5). The data for compound 5 are consistent with the pharmacological profile of a type A CCK receptor antagonist. Figure 2 also shows binding data for the pair of compounds separated from the antagonist racemic mixture. Both compounds 7 and 8 had similar apparent binding affinities for the CCK receptor, with IC50’s for inhibition of the binding of the CCK-like peptide radioligand of 490±55 nM for the proposed (S)-enantiomer (8) and 470±66 nM for the corresponding (R)-enantiomer (7).
Fig 2.

Binding and biological activity of CCK receptor ligands. Shown are curves representing the abilities of different concentrations of CCK receptor ligands (the racemic mixture having agonist action and its enantiomeric components in the top left panel, and the racemic mixture having antagonist action and its enantiomeric components in the bottom left panel) to compete for the saturable binding of 125I-D-Tyr-Gly-[(Nle28,31)CCK-26–33]. 100% represents total saturable binding of this CCK-like radioligand in the absence of competitor and 0% represents the binding in the presence of 1 μM CCK, with this value always less than 10% of total bound radioactivity. Shown in the right panels are biological activity curves, representing the abilities of these compounds to stimulate amylase secretion from dispersed rat pancreatic acini. 100% represents the maximal response of these cells to CCK, occurring at 0.1 nM CCK. Shown are means±S.E.M. of the values from three independent experiments.
In order to minimize the use of radioactivity during the synthesis and purification of the chiral photoaffinity probes, we employed the second method shown in Synthetic Scheme 2 whereby the racemic amine intermediates were separated into their respective enantiomers prior to phosgene coupling with the 3-aminobenzophenone. This approach ultimately allowed facile incorporation of 14C into the molecules during the last possible step, thus providing more efficient handling and purification of radioactive compounds.
However, to validate the ultimate enantiopurity of this synthetic methodology, each chiral amine was first cleanly converted into its respective chiral urea (5, 6, 7 and 8) using non-radioactive phosgene and 3-aminobenzophenone. The purified compounds synthesized using this approach were compared via chiral HPLC analysis to the corresponding separated enantiomeric final products previously obtained using method 1, and each was determined to be >96.5% enantiomeric excess (ee). Using this methodology, the enantiopurity of the final ureas synthesized from the resolved amine intermediates closely matched the enantiopurities of the respective intermediates themselves, indicating no significant reduction in enantiomeric excess during the final urea formation and purification. Further experiments, in particular the treatment of the separated enantiomers under basic conditions (with TEA or DIPEA), caused rapid racemization and the appearance of both parent peaks in the chiral HPLC traces, yet no degradation of compound to other species by HPLC, NMR or MS analysis. Method 2 was then applied directly to the synthesis of the 14C incorporated photoaffinity probes (5, 6, 7 and 8), and the previously synthesized and purified non-radioactive, “cold” analogs were then used as comparative standards during the synthesis and purification of the radiolabeled versions.
Each of the four 14C-tagged probes labeled the CCK receptor, as demonstrated by the covalent labeling of a band migrating at Mr=85,000–95,000 (Figure 3). This band had a core protein migrating at Mr=42,000 (data not shown). These match the established electrophoretic migrations of this receptor glycoprotein and its deglycosylated core protein, as well as the broad nature of those bands resolved under these conditions, as previously reported.14 Each probe labeled the receptor with distinct efficiencies, as reflected by the relative intensities of the bands on autoradiographs, as seen and quantified in Figure 3. Compound 8 (the proposed (S)-enantiomer antagonist) reproducibly generated the strongest receptor signal in experiments utilizing the same amounts of receptor-bearing membrane and similar amounts of probes having similar specific radioactivity.
Fig 3.

Efficiency of photoaffinity labeling of the CCK receptor. Shown in the upper panel is a typical autoradiograph of an SDS-polyacrylamide gel used to separate the products of affinity labeling of the CCK receptor expressed on CHO-CCKR cell membranes. The arrowhead marks the typical migration of the labeled CCK receptor. Shown in the lower panel is the relative densitometric quantitation of the covalent labeling of the CCK receptor by these probes, with the labeling by compound 8 always found to be the darkest, and considered to represent 100 percent. Values reflect means±S.E.M. of results from four independent experiments.
Inhibition of affinity labeling of the CCK receptor by each of the four probes was attempted with various concentrations of CCK and devazepide (9). Of note, no inhibition was achieved for any probe after competitive co-incubation with concentrations of the endogenous peptide ligand, CCK, as high as 1 μM (data not shown). In contrast, co-incubation with the 1,4-benzodiazepine, devazepide, effectively inhibited the covalent labeling of this receptor by each of the 1,5-benzodiazepine probe compounds (Figure 4), suggesting some shared commonality of binding site between the non-peptide ligands.
Fig 4.

Inhibition of affinity labeling of the CCK receptor using devazepide (9). Shown are representative autoradiographs of SDS-polyacrylamide gels used to separate products of photoaffinity labeling experiments performed with each of the 14C-tagged CCK receptor probes, using as competitor in the incubation the noted molar concentrations of devazepide.
The CCK receptor bands labeled by the agonist (6) and the corresponding (S)- enantiomer antagonist (8) were cleaved using cyanogen bromide (Figure 5). The resultant labeled receptor fragments were both small, migrating on the gel below the 6 kDa protein standard. Based on differential electrophoretic migration, these fragments appeared to be distinct peptide segments.
Fig 5.

Identification of the regions of the CCK receptor that were labeled with (S)-1,5-benzodiazepine compounds (6 and 8). Shown is a representative autoradiograph of an SDS-polyacrylamide gel used to resolve the cyanogen bromide fragments of the CCK receptor that were covalently labeled using two photolabile (S)- enantiomeric CCK receptor probes representing agonist (6) and antagonist (8). These bands were clearly distinct, based on their electrophoretic migration.
Only the antagonist-labeled receptor fragment was recovered in sufficient quantities for detailed sequence characterization. After purification by reversed-phase capillary HPLC, this fragment was sequenced by Edman degradation (Figure 6). The sequencing results show definitively that the antagonist covalently labels a peptide fragment contained within transmembrane segment two (residues Arg73 through Met89 of the mature receptor). This sequence was verified in two independent preparations. Of particular interest, this sequence was clean and absolute, until it ended abruptly after residue Asp87, suggesting that the labeled residue was either Leu88 or Met89. However, the Met89 residue cannot be the site of labeling, since its modification would interfere with cyanogen bromide cleavage at this site. On the other hand, covalent attachment of the photoaffinity label to Leu88 would result in the release of a residue in that cycle of Edman degradation that would not be recognized as a standard amino acid, which supports the cleavage results observed experimentally. The presumptive labeling of Leu88 was not directly confirmed by radiochemical sequencing, due to the extremely low specific radioactivity of the probe.
Fig 6.

Localization of the site of labeling with the (S)-1,5-benzodiazpine antagonist (compound 8). Shown is a diagram of the rat type A CCK receptor, with the separations of the peptide chain representing sites of cyanogen bromide cleavage. The cyanogen bromide fragment of the receptor that was covalently labeled by compound 8 (residues 73 through 89) is highlighted in black circles with white lettering. After this was resolved by SDS-polyacrylamide gel (shown in Figure 5 above), it was eluted and further resolved by microbore C-18 reversed-phase HPLC. The entire eluent from this separation was collected directly onto a moving PVDF membrane that was exposed to autoradiography. The single dominant radioactive peak was sequenced by Edman degradation to yield the expected residues from Arg73 through Asp87, where the sequence stopped abruptly. This is consistent with Leu88 having its migration and identification altered by the covalent attachment of the probe to this residue. The receptor fragment (residues 96 through 121) that was covalently labeled by compound 6 is also illustrated in this diagram.
The agonist (6) probe labeled a much larger receptor fragment, and analysis of cyanogen bromide cleavage sites within the receptor suggest this fragment is most likely either a peptide segment extending from Pro96 to Met121 (loop one and top of transmembrane segment 3) or a segment comprised of Gln206 to Met225 (transmembrane segment 5). Partial sequencing of this fragment, furthermore, clearly established the first ten residues as Pro96 through Lys105, although it was not possible to sequence the entire fragment since the signal blended into background after that point. However, since the covalent label attachment site would have to occur after Lys105, these results indicate that the agonist probe (6) labels the receptor at a residue near the top of transmembrane segment 3.
Molecular models were constructed for receptor complexes with both the agonist and antagonist photoaffinity labeling probes, using previous models for the receptor-peptide complex as a starting point.15 There are more extensive experimental constraint data for the antagonist complex, due to earlier mutagenesis studies for the closely-related antagonist, devazepide, that implicate CCK-A receptor residues Cys94, Met121, Ser124, Val125, Ile330, Asn334, Leu357, and Ser364 in ligand binding,16–19 and the successful identification of the covalent attachment site (Leu88) for compound 8 reported in this study. Initial automated docking attempts for the 1,5-benzodiazepines with our unmodified starting receptor structure were unsuccessful, since the cavities within the membrane-spanning domain were not quite large enough to accommodate these ligands. Therefore, initial refinements of the membrane-spanning domain required the use of interactive molecular graphics techniques to manually dock the closely related antagonist, devazepide, using the aforementioned mutagenesis data available from binding studies with this molecule to guide the docking process (Figure 7). The receptor backbone atoms were initially held fixed in our starting model, and amino acid side chain conformations were adjusted using a well-documented rotamer library 16 to create a binding pocket for devazepide, as suggested by the mutagenesis data (see Methods). The location of this putative antagonist-binding site correlates well with our photoaffinity labeling data, as the Leu88 side chain projects directly into this pocket. After manual model building, the complexes were refined with energy minimization and molecular dynamics. All residues implicated in previously reported mutagenesis studies featuring the devazepide antagonist were co-localized in a pocket within the membrane-spanning domain. These amino acids experienced subtle conformational changes that were necessary in the initial refinement of this binding site to accommodate small molecule ligands. The addition of a non-disruptive hydrogen bonding interaction between the hydroxyl of Ser124 and the backbone carbonyl of Phe120 was the only observed change in intramolecular hydrogen bonding as a result of these initial refinements. The resulting receptor complex structure (Figure 7) differed from our previously reported model 15 by only 0.97 Å RMSD in the membrane-spanning region.
Fig 7.

Devazepide (9)-refined type A CCK receptor model. Amino acid residues implicated in mutagenesis studies (referenced above) with contributions to the small molecule binding site are labeled near their alpha carbon positions in the membrane-spanning helical coils (translucent green). Non-polar residues are shown in orange; Polar residues appear in red. The left panel is a view of the binding site from the extracellular loops that are clipped away for a clear view of the small molecule binding site. The right panel is a view of the same ligand-receptor complex from the space between membrane-spanning helices two (left of ligand) and three (right of ligand).
Automated docking calculations for the agonist compound produced many equally plausible docked conformers with high scores that were all consistent with labeling of a receptor fragment composed of portions of the third transmembrane helix and the first extracellular loop regions. The agonist models were not refined further due to the lack of an identified site of covalent attachment for the agonist photoaffinity ligand. Based on the limited nature of the current data, we cannot rule out a possible attachment site for the agonist photoaffinity probe in the first extracellular loop of the receptor, although the failure of co-incubation with the peptide agonist to block photolabeling by the 1,5-benzodiazepine agonist probe would appear to preclude a direct interaction in the first extracellular loop.
The benzophenone photophore reacts with the receptor through a bond-insertion mechanism that targets electron-rich C-H bonds such as those found in residues with tertiary centers (isoleucine, leucine, and valine) or next to heteroatoms, for example, peptide backbone Cα-H bonds, glycine Cα-H bonds, and the Cγ-H bonds of methionine.20 The 25 best-scoring complexes from both shape-based and energy-based DOCK 4.0 scoring functions were examined and those that were consistent with the antagonist photoaffinity labeling data were retained as starting points for further consideration. None of the structures generated by automated docking positioned the 1,5-benzodiazepine in perfect position to form a covalent bond with the target receptor residue, Leu88. However, one of the top-scoring docked complexes did orient the ligand such that the benzophenone substituent was properly aligned for covalent insertion in the Cγ-H bond of Leu88, although the interatomic separation was too long (10 Å) to be consistent with the benzophenone reaction mechanism. This complex was further refined with manual adjustments and restrained molecular dynamics simulations.
The refined structure displays a 3.5 Å separation between the benzophenone carbonyl carbon and the reactive Cγ-H bond of Leu88 (Figure 8), quite consistent with the labeling results and the photochemistry involved. In addition, two intermolecular hydrogen bonds are formed in this docked complex with residues Asn334 and Ser364 (Figure 8) that were both previously implicated in a mutagenesis study by Smeets et al.21 Furthermore, favorable van der Waals interactions are maintained with all above referenced residues from mutagenesis studies that were used in the initial refinement of the membrane-spanning region of our model using devazepide. The refined receptor complex structure for the putative (S)-1,5-benzodiazepine antagonist probe (8) (Figure 8) differed from the starting structure (Figure 7) by 2.57 Å RMSD in the membrane-spanning regions. This difference reflects some minor repacking of the model in this region. The only significant changes observed were a bending of the second transmembrane helix at Pro96 to form favorable packing interactions with the (S)-1,5-benzodiazepine antagonist probe (8) and a disruption of one turn of the sixth transmembrane helix by the formation of a hydrogen bond between the hydroxyl of Ser360 and the backbone carbonyl of Ile356. The docked complex was subsequently refined at two different temperatures, 20K (not shown) and 300K (Figure 8) and both of the features detailed above were present in both of the resulting models, which differed by only 1.2 Å RMSD in their membrane-spanning regions. Therefore, it is likely that these features of our refined model are mostly due to the presence of the larger (S-)-antagonist photoprobe ligand (8) that is now docked into a previously poorly refined region of the small molecule binding site. Of note, docking of the (R)-antagonist photoprobe ligand (7) using similar protocols (data not shown) did not result in the formation of significant intermolecular hydrogen bonds with polar residues or produce favorable van der Waals contacts with a majority of the binding site residues implicated by previous mutagenesis studies, as did the docking of the experimentally more efficient (S)-antagonist photoprobe (8).
Fig 8.

(S)-1,5-benzodiazpine antagonist (8)-refined type A CCK receptor model. For comparison with Figure 7, the left and right panel views correspond to those of the compound 9 ligand-receptor complex (shown above). In our refined model, the antagonist benzophenone moiety is positioned only 3.5 Å away from the Cγ-H bond of Leu88 (the photoaffinity label attachment site), as shown by the white dashed line. Intermolecular hydrogen bonds are formed during structural refinement between the sidechain amide group of Asn334 (in helix six) and the methoxy group of the (S)-antagonist photoprobe, as well as between the hydroxyl group of Ser364 (in helix seven) and a carbonyl group of the diazepine ring moiety of the (S)-antagonist photoprobe. Hydrogen bonds are highlighted with pink, dashed lines. Key binding site residues are labeled and colored as described for Figure 7.
DISCUSSION
G protein-coupled receptors are remarkable for the structural diversity of natural ligands that bind and activate these molecules, ranging from photons, odorants, and small biogenic amines to larger peptides, proteins, and even viral particles.22 The CCK receptor is a member of the rhodopsin/β-adrenergic receptor family of G protein-coupled receptors.2 Its natural ligand is a small linear peptide hormone. A series of receptor mutagenesis and photoaffinity labeling studies have localized the peptide-binding domain of this receptor to the extracellular surface of the membrane, where the peptide ligand interacts with regions of the receptor amino terminus and loops.6, 7, 9, 23 We have previously generated and refined three-dimensional CCK peptide-receptor complex models that are consistent with all existing structure-activity data and direct biophysical measurements, including photoaffinity labeling and fluorescence resonance energy transfer experiments.9, 15
We have reported previously that the bovine rhodopsin crystal structure 24 is probably not an ideal template for homology modeling of peptide receptors like the type A CCK receptor, since the extracellular loops that comprise the peptide hormone binding site probably adopt quite different conformations in rhodopsin and CCK.9 Therefore, we have used de novo modeling techniques, together with constraint data from mutagenesis and biophysical experiments, to generate three-dimensional models for peptide complexes with the CCK receptor.9 The new photoaffinity labeling data reported here for non-peptidyl CCK ligands is especially useful, as these data provide specific constraint information for regions of the type A CCK receptor that were not well defined in previous models.
Many previous studies of small molecule complexes with type A CCK receptor relied primarily on site-directed mutagenesis and other indirect structural probes to investigate ligand-receptor complexes.19 These data are quite useful, but interpretation of results can be difficult, and it is generally not possible to build unique structural models based on indirect data alone. Nonetheless, these data have provided important clues about the location and nature of the small molecule binding site in the type A CCK receptor. For example, residues His381 and Val354 in the rat type B CCK receptor align with type A receptor residues Leu357 and Ile330. In type B CCK receptor mutant proteins, changes at these positions to the corresponding type A residues shifted the pharmacological profile of devazepide substantially toward that of the type A CCK receptor.18, 19 Previous alanine-scanning mutagenesis experiments implicated the polar residues Ser124, Asn334, and Ser364 in binding to devazepide.21 Mutation of residues Cys94, Met121, and Val125 in the type A CCK receptor abolished binding for the non-peptidyl agonist, SR-146,131.17 All these residues implicated in previous mutagenesis studies are co-localized in a cavity within the membrane-spanning domain of our previous type A CCK receptor model. The key Leu88 residue covalently labeled by our antagonist probe is also present in this cavity, and our initial manual docking studies were guided and constrained by these observations. Both the devazepide antagonist ligand 9 (Figure 7) and the 1,5-benzodiazepine antagonist photoaffinity probe (Figure 8) form plausible interactions with these binding pocket residues in our refined receptor complex models. These current models are also fully compatible with all previously reported structure-activity and photoaffinity labeling data for the type A CCK receptor. However, this current study provides the first direct experimental evidence for a specific ligand contact with a CCK receptor residue deep inside the transmembrane domain.
The methodological difficulties and uniqueness of this effort should be noted. Photoaffinity probes require the incorporation of both photolabile and indicator moieties. In peptide and protein ligands, it is often feasible to incorporate a photolabile amino acid derivative and a site for radioiodination that can be utilized in autoradiography. Indeed, a single radioiodine per probe molecule provides a specific radioactivity of 2000 Ci/mmol. Small drug candidates are often much less able to accommodate extraneous groups. We were fortunate that structure-activity considerations allowed the incorporation of a benzophenone moiety to confer photolability, without having substantial negative impact on either biological activity or binding affinity. However, for practical reasons, we were able to incorporate only a single 14C into this compound, yielding a specific radioactivity of 0.05 Ci/mmol. This resulted in a specific radioactivity that was thirty-thousand-fold lower than that of any of the peptide photoaffinity labeling probes previously utilized to label this receptor and its subdomains and residues.4–7, 9, 23 Nonetheless, we were still able to demonstrate specific photoaffinity labeling of this receptor with the benzodiazepine probes, and we were also able to localize the site of covalent attachment for the antagonist probe that labeled this receptor.
While it is well recognized that different G protein-coupled receptors bind their natural ligands in different ways and in different domains, correlating with the distinct chemical characteristics of their ligands,22 our data present a clear, direct demonstration of ligand binding to two distinct domains within the same receptor. Our current study has directly established an intramembranous binding site with moderately high affinity for the putative (S)-1,5-benzodiazpine antagonist (compound 8) that is clearly distinct from the site of action of the natural peptide ligand. Unfortunately, the same robust results could not be definitively confirmed for the structurally related agonist compound (6). At this stage, we cannot define a precise binding position or mode for the agonist, since the current constraint data for probe attachment spans a 15 amino acid residue fragment (Asp106-Met121). This may be due in large measure to the low efficiency of photoaffinity labeling by the agonist probe molecule, and could easily reflect unfavorable interatomic distances or directionality between the binding site residues and the photo-reactive ketone of the benzophenone moiety. This result underscores the importance of localization of the site of probe molecule covalent attachment to a single receptor amino acid. Recent reports on agonist-induced conformational changes in the β2-adrenergic receptor indicated that even structurally distinct agonists of the same receptor could stabilize different receptor conformational states, and that a full agonist may stabilize multiple receptor states that have various kinetic lifetimes.25,26 Additionally, fluorescence lifetime data from the adrenergic receptor studies suggest that high affinity agonist-receptor complexes would likely be the shortest lived of two observed states.20 These reports may explain the difficulty encountered in the current work to obtain high efficiency receptor labeling with the biologically active, (S)-agonist probe (compound 6).
Evidence exists in the literature to support distinct binding sites in the type A CCK receptor for non-peptidyl agonists and antagonists,27 however, our results do not distinguish between the possibility for the small molecule agonist to bind at a site that includes residues from the first extracellular loop versus a binding site contained entirely within the transmembrane domain. Our results suggest that the small molecule agonist binding site may not overlap with the peptide agonist binding site, since micromolar concentrations of the endogenous peptide failed to competitively block the affinity labeling observed with the (S)-agonist photoaffinity probe (compound 6), a compound that retained full biological activity at reasonably low concentrations (EC50 ~ 150 nM). Therefore, some questions remain regarding whether the same peptide receptor may be similarly activated by binding of different types of ligands to distinct receptor regions. Certainly, the small molecule, antagonist-binding site is clearly within the membrane-spanning helical bundle domain, while the natural peptide ligand binds to the ectodomain of this receptor. Our photoaffinity labeling data confirm this for the (S)-1–5-benzodiazepine antagonist (compound 8).
In conclusion, we have added to our understanding of the multiple ways that ligands can bind to the type A CCK receptor by reporting direct evidence for a small molecule antagonist binding site deep within the transmembrane domain. Additionally, the photoaffinity labeling data reported here for non-peptidyl CCK ligands provide specific constraint information for regions of the type A CCK receptor that were not well defined in previous receptor models.
However, much remains to be explained regarding the molecular basis for differences between small molecule agonist and antagonist ligand binding. We have taken an important initial step in this direction with refinement of the transmembrane domain of our previous CCK receptor model structures. Here, we have employed a conformationally constrained small molecule antagonist with a well-defined point of interaction in the receptor membrane-spanning domain as a tool in the refinement of our evolving model of the CCK receptor. This type of direct biophysical data, collected using novel molecular probes, will likely serve as an important tool for gaining further insights into the structures of membrane-spanning domains of G protein-coupled receptors that bind peptide hormones.
EXPERIMENTAL SECTION
Reagents
All chemicals and solvents are reagent grade unless otherwise specified. Synthetic sulfated cholecystokinin octapeptide (CCK) was purchased from Peninsula Chemical Co. The radioiodinatable CCK analogue, D-Tyr-Gly-[(Nle28,31)CCK-26–33], was synthesized in our laboratory as we have described.4,28 The CCK antagonist, devazepide, was provided by Merck Research Laboratories (West Pointe, PA). Cyanogen bromide (CNBr) and the solid-phase oxidant, N-chloro-benzenesulfonamide (Iodobeads) were from Pierce Chemical Co. (Rockville, IL); wheat germ agglutinin-agarose was from E-Y Laboratories (San Mateo, CA); purified type II collagenase and soybean trypsin inhibitor (STI) were from Worthington Biochemicals (Lakewood, NJ); and formic acid was from Fluka Chemical Co. (Milwaukee, WI). Endoglycosidase F was prepared in our laboratory, as we have described.29 The following solvents and reagents have been abbreviated: acetic acid (AcOH), dimethyl sulfoxide (DMSO), dimethylformamide (DMF), dichloromethane (DCM), diisopropylethylamine (DIPEA), ethanol (EtOH), ethyl ether (Et2O), ethyl acetate (EtOAc), isopropanol (IPA), methanol (MeOH), tetrahydrofuran (THF), trifluroacetic acid (TFA), triethylamine (TEA). Non-radioactive phosgene was purchased commercially as a 20% solution in toluene from Fluka Chemical Co, while 14C labeled phosgene was available from American Radiolabeled Chemicals, Inc. (St. Louis, MO). Reactions were monitored by thin-layer chromatography (TLC) on 0.25 mm silica gel plates (60F-254, E. Merck) and visualized with UV light.
Analytical Purity was assessed by RP-HPLC using a Waters 600E system equipped with a Waters 990 diode-array spectrophotometer. The stationary phase was a Vydac C-18 column (4.6 mm × 200 mm), the mobile phase employed 0.1% aqueous TFA with acetonitrile and the flow rate was maintained at 1.0 or 1.5 mL/min (t0 = 3 min).
1H NMR spectra were recorded on a Varian Unity-300 instrument. Chemical shifts are reported in parts per million (ppm, δ units). Coupling constants are reported in units of Hertz (Hz). Splitting patterns are designated as s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; b, broad. Low resolution mass spectra (MS) were recorded on a JEOL JMS-AX505HA, a JOEL SX-102 or a SCIEX-APIiii spectrometer. Mass spectra were acquired in either positive ion mode or negative ion mode under electrospray ionization (ESI) or atmospheric pressure chemical ionization conditions (APCI). Combustion analyses were performed by Atlantic Microlabs, Inc., Norcross, GA, and the results reported in the form “Anal. (C39H33N5O6) C, H, N” where the observed results were within ±0.4% of calculated values.
Synthesis and separation of individual non-peptidyl enantiomers for use as photolabile radioligand probes
Representative Procedure for Formation of Bromoacetamides (12 & 13); 2-Bromo-N-isopropyl-N-(4-methoxyphenyl)-acetamide (12)
Procedure was followed as described in Aquino et al,10 wherein N-isopropyl-N-(4-methoxyphenyl)-amine (10) was prepared by reductive animation of 4-methoxyphenylamine with acetone, employing the method of Abdel-Magid et al.30, 31 Subsequent reaction of compound 10 with bromoacetyl bromide at ≤4 °C cleanly provided the previously described compound 2-bromo-N-isopropyl-N-(4-methoxyphenyl)-acetamide (12).10
2-Bromo- N-(4-methoxyphenyl)-N-methyl- acetamide (13)
Using the same procedure for the preceding example, reaction of bromoacetyl bromide with commercially available N-(4-methoxyphenyl)-N-methyl-amine (Sigma-Aldrich) provided 2-bromo-N-(4-methoxyphenyl)-N-methyl-acetamide (13) in 85% overall yield after chromatography: 1H-NMR (300 MHz, CDCl3) δ 3.22 (s, 3H), 3.61 (s, 2H), 3.79 (s, 3H), 6.89 (d, 2H, J = 8.8 Hz), 7.15 (d, 2H, J = 8.8 Hz); 13C-NMR (100.58 MHz, CDCl3) δ 26.89, 38.20, 55.52, 115.01, 128.11, 135.68, 159.33, 166.83; LRMS (APCI) m/z 279.9, 281.8 (Br isotopes) [M+Na]+; TLC Rf = 0.68 (EtOAc:Hex, 1:2)
2-(Phenylhydrazono)-malonic acid (16) & 2-(Phenylhydrazono)-propanedioyl dichloride (17)
To a vigorously stirred suspension of ketomalonic acid disodium salt (5.0 g, 30.9 mmol) cooled to ≤4 °C in an ice bath, was added drop wise a solution of hydrochloric acid (1.0 N, 62.0 mL, 62.0 mmol). After the suspension had dissolved, ethanol (20 mL) was added, and the reaction proceeded as outlined in Aquino et al.10
2-[2,4-Dioxo-5-phenyl-3-(phenyl-hydrazono)-2,3,4,5-tetrahydrobenzo[b][1,4]diazepin-1-yl]-N-isopropyl-N-(4-methoxy-phenyl)-acetamide (18)
As per the modified procedure of Hirst and coworkers,32 commercially available N-phenyl-l,2-phenylenediamine (5.0 g, 27.1 mmol) was dissolved in DMF (25 mL) and stirred at room temperature. 1.4 molar equivalents of bromoacetamide (12) were added to this solution. Solid K2CO3 (8.0 g, 57.9 mmol) was then added and the resulting suspension stirred vigorously overnight at room temperature. Upon completion as monitored by TLC (1:2, EtOAc:Hex), the DMF was evaporated and the resulting oil partitioned into ice water/EtOAc. The organic layer was washed twice with ice water then once with brine, and the organic layer was then dried over MgSO4 and evaporated to a reddish oil. The crude intermediate thus obtained was pure enough to use directly for the formation of the 1,5-benzodiazepine ring scaffold. Solutions of compound 14 (6.0 g, 16.6 mmol) in THF (40 mL) and compound 17 (5.1 g, 20.8 mmol) in THF (40 mL) were added concomitantly drop wise with cooling in an ice/methanol bath over 30 min. The solution was allowed to warm to room temperature and stirred 16 h. Upon completion of reaction, as monitored by TLC, the solution was evaporated down to a yellow solid that was then resuspended in EtOAc and washed twice with 0.02 N sodium hydroxide solution and once with brine. The organic layer was dried over MgSO4 and the solvent removed. The resulting solid was purified by flushing through a pad of silica gel with (1:1) EtOAc:Hex as eluent After drying under vacuum overnight, the corresponding compounds 18 were obtained as yellow solids in 90% yield (8.4 g) and high purity (as approx. 1:1 mixtures of syn- and anti- hydrazone). 1H-NMR (300 MHz, CDCl3) δ 1.09 (m, 6H), 3.88 (s, 3H), 3.90 (m, 1H), 4.20–4.60 (m, 2H), 6.90–7.65 (m. 18H), 10.80 (s, 0.5H), 11.54 (s, 0.5H]; LRMS (APCI) m/z 584.0 [M + Na]+; TLC Rf = 0.27 (EtOAc:Hex, 1:2). 1:1 Mixture of cis-and trans- hydrazones.
2-[2,4-Dioxo-5-phenyl-3-(phenyl-hydrazono)-2,3,4,5-tetrahydrobenzo[b][1,4]diazepin-1-yl]-N-(4-methoxy-phenyl)-N-methyl-acetamide (19)
Prepared using the procedure described for (18) to provide 90% yield (8.0 g) of a light yellow solid (19). 1H-NMR (300 MHz, CDCl3) δ 3.39 (m, 3H), 3.89 (s, 3H), 4.30–4.65 (m, 2H), 6.90–7.65 (m, 18H).10.80 (s, 0.5H). 11.54 (s, 0.5H); LRMS (APCI) m/z 556.3 [M + Na]+; TLC Rf = 0.12 (EtOAc:Hex, 1:2). 1:1 Mixture of cis- and trans- hydrazones.
Procedure for Cleavage of Hydrazone to Provide Racemic Benzodiazepine Amines
2-(3-Amino-2,4-dioxo-5-phenyl-2,3,4,5-tetrahydro-benzo[b][1,4]diazepin-1-yl)-N-isopropyl-N-(4-methoxy-phenyl)-acetamide (Racemate 1, 2)
To a vigorously stirred solution of hydrazone 18 (4.27 g, 8.0 mmol) in glacial acetic acid (50 mL) at room temperature, was added anhydrous zinc dust (4.20 g, 64.3 mmol) portion wise. After addition, the solution was stirred 3–5 h until the color of the slurry changed from green to yellow. The zinc was removed by filtration through celite and the cake washed with EtOAc (2 × 75 mL). The filtrate was concentrated, adsorbed onto silica gel and washed with EtOAc:Hex (1:2) to remove more polar impurities. The product was then eluted with 10% methanol in DCM and concentrated in vacuo to give a yellowish-orange oil which was dried under high vacuum to provide the corresponding racemic amine intermediate (1, 2) in good yield (3.38 g product as a white amorphous solid, 93.7% yield). 1H-NMR (300 MHz, CD3OD) δ 1.09 (m, 6H), 3.87 (s, 3H), 4.37 (d, 1H, J = 21.0 Hz), 4.60 (d, 1H, J = 21.0 Hz), 4.35 (m, 1H), 4.85 (m, 1H), 6.80–7.50 (m, 13H); LCMS (APCI) m/z 473.3 [M +H]+; TLC Rf = 0.45 (EtOAc:Hex, 1:2); Anal. (C27H28N4O4) C, H, N.
2-(3-Amino-2,4-dioxo-5-phenyl-2,3,4,5-tetrahydro-benzo[b][1,4]diazepin-1-yl)-N-(4-methoxy-phenyl)-N-methyl-acetamide (Racemate 3, 4)
Using the procedure for the racemate (1, 2) above and starting with 8 mmol hydrazone 19, 3.22 g racemate (3, 4) product (95.3% yield) was obtained. 1H-NMR (300 MHz, CD3OD) δ 3.20 (s, 3H), 3.87 (s, 3H), 4.45 (d, 1H, J = 21.0 Hz), 4.72 (d, 1H, J = 21.0 Hz), 4.75 (b, 1H), 6.70–7.50 (m, 13H); LRMS (APCI) m/z 445.2 [M +H]+; TLC Rf = 0.47 (EtOAc:Hex, 1:2); Anal. (C25H24N4O4) C, H, N.
Method 1: General Procedure for Racemic Benzophenone Urea Formation and Subsequent Chiral Resolution;
2-{3-[3-(4-Benzoyl-phenyl)-ureido]-2,4-dioxo-5-phenyl-2,3,4,5-tetrahydro-benzo[b][1,4]diazepin-1-yl}-N-isopropyl-N-(4-methoxy-phenyl)-acetamide (Racemate 5, 6)
Under anhydrous conditions, triphosgene (0.5 mmol) was dissolved in anhydrous acetonitrile (1 mL) and the stirring solution cooled to ≤4 °C under dry N2. A solution containing commercially available 3-aminobenzophenone (0.10 g, 0.5 mmol) and diisopropylethylamine, (DIPEA, 0.1 mL) in anhydrous acetonitrile (1 mL) was added drop wise to the stirring reaction. The reaction was allowed to stir under dry N2 at ≤4 °C for 30 min, at which time a second solution containing the 1,5-benzodiazepine amine racemate (1, 2) (0.5 mmol) and DIPEA (0.1 mL) in dry acetonitrile (2 mL) was added drop-wise to the cooled, stirring reaction. Stirring continued at ≤4 °C for 3–6 h until the reaction was complete as monitored by TLC. The product urea precipitated out of acetonitrile during the course of the reaction and was purified by filtration. Subsequent further precipitation of product from filtrate occurred upon standing for several hours at room temperature. The white solid was washed thoroughly with acetonitrile and dried under vacuum to provide 350 mg white solid racemate (5, 6). No further purification was required as the solids isolated were observed to be pure by HPLC, NMR and combustion analysis. 1H-NMR (300 MHz, CDCl3) δ 0.91 (d, 3H, J = 6.8 Hz), 0.94 (d, 3H, J = 6.8 Hz), 3.78 (s, 3H), 4.16 (d, 1H, J = 16.8 Hz), 4.58 (d, 1H, J = 16.8 Hz), 4.74 (septet, 1H, J = 6.8 Hz), 4.99 (d, 1H, J = 8.0 Hz), 6.91 (t, 2H, J = 8.8 Hz), 7.43 (d, 2H, J = 8.8 Hz), 7.19–7.58 (m, 15H), 7.65 (t, 1H, J = 7.2 Hz), 7.68 (d, 2H, J = 7.2 Hz), 7.79 (s, 1H), 9.44 (s, 1H); LRMS (APCI) m/z 717.9 [M + Na]+, 694.3 [M]−; Anal. (C41H37N5O6) C, H, N.
Chiral resolution procedure & results for resolved compound 5 (and 6)
The isopropyl racemic mixture of (5) and (6) was separated into the resolved enantiomers using semi-preparative chiral RP-HPLC and monitored by UV absorbance (Dynamax C-8 column, 25cm × 4.1 mm, 8 mL/min flow rate, ramping from [75% CH3CN/25% (H2O with 0.1% TFA)] up to 100% CH3CN over 30 min). 100 Mg of the parent N-isopropyl racemate (5, 6) was separated into its purified component enantiomers, which were isolated as white solids (30 mg of compound 5, and 25 mg of compound 6). Enantiomer purity and enantiomeric excess were confirmed by chiral RP-HPLC analysis using an alternate isocratic analytical method (Analytical Chiralpak AD C-18 column, with mobile phase (65:35, Hex:IPA) eluting at 1 mL/min flow rate). Compound 5 eluted at 41.28 minutes, while compound 6 eluted at 48.33 minutes (showing good baseline peak separation); co-injections with each other and with the parent racemate confirmed chiral purity. Compound 5 (and 6) 1H-NMR (300 MHz, CDCl3) δ 0.92 (d, 3H, J = 6.8 Hz), 0.95 (d, 3H, J = 6.8 Hz), 3.78 (s, 3H), 4.17 (d, 1H, J = 16.8 Hz), 4.58 (d, 1H, J = 16.8 Hz), 4.76 (septet, 1H, J = 6.8 Hz), 5.00 (d, 1H, J = 8.0 Hz), 6.92 (t, 2H, J = 8.8 Hz), 7.43 (d, 2H, J = 8.8 Hz), 7.19–7.58 (m, 15H), 7.65 (t, 1H, J = 7.2 Hz), 7.68 (d, 2H, J = 7.2 Hz), 7.80 (s, 1H), 9.44 (s, 1H); LRMS (APCI) m/z 718.1 [M + Na]+, 694.3 [M]−; Anal. (C41H37N5O6) C, H, N.
2-{3-[3-(4-Benzoyl-phenyl)-ureido]-2,4-dioxo-5-phenyl-2,3,4,5-tetrahydro-benzo[b][1,4]diazepin-1-yl}-N-(4-methoxy-phenyl)-N-methyl-acetamide (Racemate 7, 8)
Reaction with commercially available 3-aminobenzophenone and the intermediate amine racemate (3, 4), using the procedure outlined for racemate (5, 6), provided 300 mg of the racemic N-methyl analogs (7, 8) as a white solid that was pure by NMR, LCMS and combustion analysis. 1H-NMR (300 MHz, CDCl3) δ 3.10 (s, 3H), 3.77 (s, 3H), 4.35 (d, 1H, J = 16.8 Hz), 4.64 (d, 1H, J = 16.8 Hz), 5.01 (d, 1H, J = 8.0 Hz), 6.88 (d, 1H, J = 8.0 Hz), 6.92 (d, 1H, J = 7.6 Hz), 7.03, (d, 1H, J = 8.0 Hz), 7.21 (d. 1H, J = 7.6 Hz), 7.24 (d, 1H, J = 7.6 Hz), 7.30–7.48 (m, 13H), 7.51 (t, 1H, J = 7.6 Hz); 7.63 (t, 1H, J = 7.2 Hz), 7.68 (d, 2H, J = 7.2 Hz), 7.79 (s, 1H), 9.43 (s, 1H); LRMS (APCI) m/z 689.9 [M + Na]+, 666.7 [M]−; Anal. (C39H33N5O6) C, H, N.
Chiral resolution procedure & results for resolved compound 7 (and 8)
Following the procedure for the resolved enantiomers (5) and (6), 100 mg of the parent N-methyl racemate (7, 8) was separated into its purified component enantiomers, which were isolated as white solids (35 mg of compound 7, and 25 mg of compound 8). Enantiomer purity and enantiomeric excess were confirmed by chiral RP-HPLC analysis using an alternate isocratic analytical method (Analytical Chiralpak AD C-18 column, with mobile phase (65:35, Hex:IPA) eluting at 1 mL/min flow rate). Compound 7 eluted at 49.94 minutes, while compound 8 eluted at 82.17 minutes (showing good baseline peak separation); co-injections with each other and with the parent racemate confirmed chiral purity. Compound 7 (and 8): 1 3.09 (s, 3H), 3.76 (s, 3H), H-NMR (300 MHz, CDCl3) δ 4.37 (d, 1H, J = 16.8 Hz), 4.67 (d, 1H, J = 16.8 Hz), 5.02 (d, 1H, J = 8.0 Hz), 6.88 (d, 1H, J = 8.0 Hz), 6.92 (d, 1H, J = 7.6 Hz), 7.04, (d, 1H, J = 8.0 Hz), 7.21 (d. 1H, J = 7.6 Hz), 7.24 (d, 1H, J = 7.6 Hz), 7.30–7.48 (m, 13H), 7.50 (t, 1H, J = 7.6 Hz); 7.63 (t, 1H, J = 7.2 Hz), 7.69 (d, 2H, J = 7.2 Hz), 7.79 (s, 1H), 9.43 (s, 1H); LRMS (APCI) m/z 689.9 [M + Na]+, 666.7 [M]−; Anal. (C39H33N5O6) C, H, N.
Method 2: Resolve the Enantiomeric Amine Intermediates Prior to Forming Chiral Ureas
Resolved (R)- and (S)-2-(3-Amino-2,4-dioxo-5-phenyl-2,3,4,5-tetrahydro-benzo[b][1,4]diazepin-1-yl)-N-isopropyl-N-(4-methoxy-phenyl)-acetamide (1) and (2)
For use in the synthesis of 14C-labeled (R)- and (S)- photoaffinity probes, the isopropyl racemic mixture of amines 1 and 2 were then separated into the resolved enantiomers using semi-preparative chiral RP-HPLC and monitored by UV absorbance, showing good baseline peak separation (Dynamax C-8 column, 25cm × 4.1 mm, 8 mL/min flow rate, ramping from [40% CH3CN/60% (H2O with 0.1% TFA)] up to 100% CH3CN over 30 min). Each enantiomer was collected in a flask containing at least one equivalent of acid (HCl) in order to prevent epimerization of the purified enantiomers upon sample concentration. Chiral HPLC purification of the original 500 mg parent racemate yielded 202 mg of compound 1 and 165 mg of compound 2. Under standard achiral NMR conditions and LCMS conditions, both enantiomers of each pair gave identical spectra to each other as well as to that of the parent racemate (1, 2) provided above. Enantiomer purity and enantiomeric excess were confirmed by chiral RP-HPLC analysis, showing good baseline peak separation using an alternate isocratic analytical method (Analytical Chiralpak AD C-18 column, with mobile phase (3:1, Hex:IPA) eluting at 1 mL/min flow rate). Compound 1 eluted at 20.60 minutes, while compound 2 eluted at 31.76 minutes; co-injections with the parent racemate confirmed chiral purity. Compound 1 (and 2) 1H-NMR (300 MHz, CD3OD) δ 1.10 (m, 6H), 3.87 (s, 3H), 4.35 (d, 1H, J = 21.0 Hz), 4.61 (d, 1H, J = 21.0 Hz), 4.35 (m, 1H), 4.87 (m, 1H), 6.80–7.50 (m, 13H); LCMS (APCI) m/z 473.3 [M +H]+; TLC Rf = 0.45 (EtOAc:Hex, 1:2); Anal. (C27H28N4O4) C, H, N.
Resolved (R)- and (S)-2-(3-Amino-2,4-dioxo-5-phenyl-2,3,4,5-tetrahydro-benzo[b][1,4]diazepin-1-yl)-N-(4-methoxy-phenyl)-N-methyl-acetamide (3) and (4)
The methyl racemate (3, 4) was separated similarly into its respective enantiomeric amines (3) and (4) using the procedure outlined for racemate (1, 2). Chiral HPLC purification of the original 500 mg parent racemate yielded 185 mg of (3) and 242 mg of (4). As with the isopropyl analogs (1) and (2), under standard achiral NMR conditions and LCMS conditions, both enantiomers of each pair gave identical spectra to each other as well as to that of the parent racemate (1, 2) provided above. Enantiomer purity and enantiomeric excess were confirmed by chiral RP-HPLC analysis using an alternate isocratic analytical method (Analytical Chiralpak AD C-18 column, with mobile phase (3:1, Hex:IPA) eluting at 1 mL/min flow rate). Compound 3 eluted at 36.44 minutes, while compound 4 eluted at 42.03 minutes; co-injections with the parent racemate confirmed chiral purity. Compound 3 (and 4): δ 1H-NMR (300 MHz, CD3OD) δ 3.15 (s, 3H), 3.82 (s, 3H), 4.41 (d, 1H, J = 21.0 Hz), 4.69 (d, 1H, J = 21.0 Hz), 4.75 (b, 1H), 6.70–7.50 (m, 13H). LRMS (APCI) m/z 445.2 [M +H]+; TLC Rf = 0.47 (EtOAc:Hex, 1:2); Anal. (C25H24N4O4) C, H, N.
Representative procedure for chiral urea formation;
2-{3-[3-(4-Benzoyl-phenyl)-ureido]-2,4-dioxo-5-phenyl-2,3,4,5-tetrahydro-benzo[b][1,4]diazepin-1-yl}-N-isopropyl-N-(4-methoxy-phenyl)-acetamide (5)
3-Amino-benzophenone (0.016 g, 0.08 mmol) was dissolved in anhydrous CH3CN (1mL) and stirred under N2. DIPEA (0.010g, 0.08 mmol, 14 μL) was then added directly into the reaction solution and the reaction solution was cooled to 4 °C in an ice-water bath. Phosgene (20% in toluene, 0.08 mmol, 42.1 μL) was quickly syringed directly into the reaction solution and the reaction stirred at ≤4 °C under N2 for 1 h. Drop wise the chiral amine hydrochloride salt of compound 1 (0.038g, 0.08 mmol) in 2 mL dry CH3CN was added to the stirring reaction solution. After 2–3 min, 1 equivalent of DIPEA (0.010g, 0.08 mmol, 14 μL) was added to the ≤4 °C solution while stirring under N2. After an additional 2 min, the final equivalent of DIPEA (0.010g, 0.08 mmol, 14 μL) was added and the reaction allowed to stir at ≤4 °C under N2 for 2 h.
For each of the isopropyl compounds (5) and (6), the product precipitated out of solution and was filtered and washed with dry acetonitrile, then dried to constant weight under vacuum. Analytical data for the isolated products matched the data obtained for the same materials synthesized via method 1.
For the methyl compounds (7) and (8), the solvent was evaporated using a stream of dry N2, then the resulting oil was chromatographed on silica gel using (55% EtOAc/45% Hex) as eluent. The purified material was evaporated to a white solid, and then dried to constant weight under vacuum. As with the isopropyl compounds, the analytical data for the isolated products matched the data obtained for the same materials synthesized via method 1 previously shown. In all four examples (5, 6, 7 and 8), pure white solids were typically obtained in 80% yields. This procedure was used for subsequent 14C synthesis of the enantiomers 5, 6, 7 and 8, merely substituting 14C labeled phosgene solution for the non-radioactive phosgene used above.
The enantiopurity of the final compounds was verified using reverse phase HPLC (Analytical Chiralpak AD C-18 column, isocratic method with 1 mL/min flow rate, eluent = 65% hexane/35% IPA). Retention times for the isolated enantiomers obtained by using method 2 are given below:
| Compound # | Retention time | Purity | Enantiomeric excess |
| 5 | 41.3 minutes | >99% | > 99% |
| 6 | 48.3 minutes | >99% | > 99% |
| 7 | 49.9 minutes | >99% | 97.0% |
| 8 | 82.2 minutes | >99% | 96.5% |
Enantiomer Racemazation Conditions
The purified enantiomer (1, 2, 3, 4, 5, 6, 7 or 8) (0.1g) was dissolved in DCM and 1.5 equivalents of TEA or DIPEA added at room temperature. The reaction was allowed to stir at room temperature in open to air for several h, and the conversion back to the parent racemate was followed by chiral HPLC, using the conditions described previously for the individual enantiomers. In all cases, conversion was allowed to proceed until approx. 1:1 ratio of stereoisomers was achieved. The residual solvent and base were then removed in vacuo, and the newly formed racemate analyzed for degradation products. In all cases, each newly formed racemate was clean of impurities, and provided the identical analytical results to those obtained for the original parent racemate given above.
Receptor-bearing cell line
The Chinese hamster ovary cell line stably expressing the type A rat CCK receptor (CHO-CCKR) that has previously been fully characterized was utilized as source of receptor for the biochemical and receptor domain-mapping studies.14 This cell line was cultured in Ham’s F-12 medium supplemented with 5% fetal clone-2 (Hyclone Laboratories, Logan, UT) at 37 °C in an environment of 5% CO2 in Costar tissue culture plasticware. Cells were harvested mechanically and passaged twice weekly.
Radioligand binding studies
Membranes were prepared from the CHO-CCKR cells, as we have previously described.14 For binding characterization, membranes (3–5 μg) were incubated with a constant amount of the CCK-like radioligand, 125I-D-Tyr-Gly-[(Nle28,31)CCK-26–33] (2–5 pM), in the absence or presence of variable concentrations of unlabeled competing ligand. Incubations were performed to attain steady state, achieved after incubation for 3 h at 30 °C in Krebs-Ringers-HEPES (KRH) medium (25 mM HEPES, pH 7.4, 104 mM NaCl, 5 mM KCl, 1.2 mM MgSO4, 2 mM CaCl2, 1 mM KH2PO4, 0.2% bovine serum albumin, 0.01% soybean trypsin inhibitor and 1 mM phenylmethylsulfonyl fluoride). Bound and free radioligand were separated using a Skatron cell harvester (Molecular Devices, Sunnyvale, CA) with receptor-binding filtermats that had been pre-soaked with 0.2% bovine serum albumin. Bound radioactivity was quantified in a gamma–spectrometer. Non-specific binding was determined in the presence of 1 μM CCK and represented less than 10% of total bound radioactivity.
Affinity labeling studies
For covalent labeling studies, substantially greater amounts of receptor-bearing membranes (100–200 μg) were incubated with 1–10 μM radioligand, representing the 14C-labeled compounds described above, in the absence or presence of competing unlabeled CCK receptor ligands in KRH medium for 3 h at 30 °C. Tubes were exposed to photolysis for 30 min at 4 °C in a Rayonet photochemical reactor equipped with 3,500 Å lamps (Southern New England Ultraviolet, Hamden, CT). Membranes were then isolated by centrifugation, washed twice in KRH medium, and solubilized with 1% NP-40 in KRH for 18 h at 4 °C with constant mixing.
Solubilized affinity labeled receptor was diluted with an equal volume of KRH medium and one-tenth volume of a 50% slurry of WGA-agarose, with the incubation allowed to proceed for 24 h at 4 °C with constant mixing. The WGA-agarose was pelleted and washed with a solution containing 0.5 M NaCl and 0.1% NP-40, followed by a water wash. In preparation for electrophoresis, reducing sample buffer was added to the pelleted WGA-agarose beads and this was allowed to sit for at least 30 min at room temperature. Samples were applied to a 10% SDS-polyacrylamide gel for electrophoresis using the conditions described by Laemmli.33 At the completion of electrophoresis the receptor band was excised for further study or the gel was dried in preparation for autoradiography. Dried gels were placed into film cassettes containing BioMax MS X-ray film and a BioMax TranScreen-LE (Eastman Kodak Company, Rochester, NY). Cassettes were kept at −80 °C for 1–6 weeks before the film was developed.
Gel purified affinity labeled receptor was deglycosylated with endoglycosidase F and/or cleaved with cyanogen bromide using conditions we have previously reported, with minor modifications.34 Gel regions containing receptor bands were excised, diluted with water, and treated with dounce homogenization. Gel pellets were removed by centrifugation and the supernatants containing the labeled receptor or its fragments were lyophilized to dryness. Detergent was removed from the samples by precipitation with 85% ethanol at −20 °C for 18 h. Precipitates were pelleted, dried, and then resuspended in 70% formic acid containing 2.5 mg cyanogen bromide. Cleavage was allowed to proceed for 18 h under nitrogen at room temperature. Acid was then removed from the samples by three cycles of water washes and vacuum centrifugation. Sample buffer was added to tubes in preparation for electrophoresis on a 10% NuPAGE gel (Invitrogen, Carlsbad, CA) with MES running buffer. A small portion of the sample was run in a separate lane so it could be dried and used for autoradiography to identify the position of the cleavage fragment. The remaining portion of the gel was frozen and, once the position of the labeled fragment of interest was identified, that region of the gel could be excised and the sample eluted for further purification by HPLC.
HPLC purification and sequence analysis
Prior to having components resolved by HLPC, the samples were applied to a Dispo-Biodialyzer (The Nest Group, Inc., Southborough, MA) with a molecular weight cutoff of 1 kDa, and samples were dialyzed for 24 h against 2 liters of water. Capillary HPLC was performed using a Perkin-Elmer/Brownlee reversed-phase C18 column, measuring 0.5 × 150 mm with 5 μm beads and a pore size of 300 Å. The buffer composition was 0.1% TFA as buffer A and 0.085% TFA/80% acetonitrile as buffer B. The sample was injected at 10% B and the system was allowed to run for 20 min before beginning a 230 min gradient to 90% B. The flow rate was 5 μl/min and samples were collected directly on polyvinylidene difluoride (PVDF) membranes. The UV detector monitored absorbance at 210 nm. The PVDF sample membrane was exposed to BioMax MS X-ray film and a BioMax TranScreen-LE. The major radioactive spot was localized and excised from the PVDF membrane and was then subjected to Edman degradation sequencing using an Applied Biosystems automated instrument.
Biological activity studies
Rat pancreatic acini were freshly prepared from 80–100 g male Sprague Dawley rats. All studies were reviewed and approved by the Mayo Clinic Animal Care and Use Committee. Pancreatic tissue was harvested and placed in iced, oxygenated KRH medium. The pancreas was trimmed of fat and connective tissue, and injected with 600 units of purified collagenase diluted to 6 mL with KRH medium. The tissue was minced, oxygenated and incubated at 37 °C for 5 min in a shaking water bath. Additional digestion took place with 10 min of shaking by hand before the acini were washed and filtered through 200 μm nytex mesh. The health of the acini was assessed morphologically and the cells were suspended in KRH medium for use.
One mL aliquots of cells were added to tubes containing various concentrations of secretagogues. The samples were incubated for 30 min at 37 °C in a shaking water bath. The secretion assay was terminated by centrifugation of an aliquot of the cell suspension over Nyosil oil (William F. Nye, Inc., New Bedford MA). Amylase measurement utilized the Phadebas amylase reagent (Pharmacia Diagnostics, Uppsala, Sweden). Tablets were dissolved in assay buffer (0.02 M NaH2PO4, 0.05 M NaCl, 0.02% NaN3, pH 7.0). Two mL of Phadebas suspension was added to aliquots of secretion samples in lysis buffer (0.01 M NaH2PO4, pH 7.8, containing 0.1% SDS and 0.1% bovine serum albumin). Samples were incubated at 37 °C for 15 min before the incubation was terminated by addition of 0.5 M NaOH. Samples were measured with a spectrophotometer at 620 nm and values were expressed as percentages of amylase release in response to maximal stimulation with CCK (0.1 nM CCK).
Molecular modeling
Our current model for the high affinity agonist-bound type A CCK receptor served as a starting point for the construction of molecular models that incorporated the photoaffinity labeling data reported in this study.15 The receptor’s small molecule binding site was manually adjusted via interactive computer graphics techniques to accommodate small molecule ligands within the membrane-spanning domain. These adjustments were performed using the most probable sidechain rotamer conformations from the backbone-dependent amino acid rotamer library of Dunbrack and Karplus,16 without alteration of the positions of protein backbone atoms. We targeted stable, low energy structure modifications by choosing only combinations of rotamers with the three highest probabilities based on a statistical analysis of experimentally observed conformations in the above library of sidechain dihedral angles for each of the following amino acid sidechains (Thr118, Ser124, Asn334, Arg337, Tyr339, Thr341, Ile356). We next performed manual docking studies for the non-peptidyl antagonist, devazepide,35 a 1,4-benzodiazepine that competitively blocks 1,5-benzodiazepine agonist and antagonist photoaffinity labeling. Manual docking of devazepide (compound 9) was guided by mutagenesis data from previous ligand-binding studies described above.
Limited energy minimization and constrained molecular dynamics simulations (5 ps at 20K) of the devazepide ligand-receptor complex were performed in vacuo with a distance dependent dielectric constant to relieve some minor steric and conformational strain introduced during initial ligand docking. In this initial refinement of the membrane-spanning region, previously published constraints were used to maintain the overall conformation of the receptor starting structure.15 Snapshots were taken from the molecular dynamics trajectories; each molecular dynamics snapshot was energy minimized; and then the devazepide antagonist ligand (9) was removed to generate target receptor structures for automated ligand docking calculations for the 1,5-benzodiazepines (5, 6, 7, and 8).
The setup for DOCK 4.036 calculations involved the generation of a negative image of potential ligand binding sites in the receptor by filling the area within the confluence of membrane-spanning helices with spheres. Boundaries for agonist and antagonist binding sites were defined using photoaffinity-labeling data for the respective ligands, and sphere generation was confined to bounding boxes centered at the receptor positions for agonist and antagonist covalent labeling. Although the bounding boxes for the agonist and antagonist sites have significant overlap, the antagonist photoaffinity-labeling data clearly precludes any receptor contacts for compound 8 in the first extracellular loop. Therefore, the bounding box for the antagonist photoaffinity label was limited to the membrane-spanning receptor domain, while the bounding box (and corresponding sphere set) for the agonist photoaffinity ligand contained portions of the receptor membrane-spanning domain and the extracellular loop regions.
Docking calculations were performed using low energy conformations for each compound. Geometry optimizations were performed using ab initio Hartree-Fock calculations with a 3–21G* basis set. Quantum mechanical electrostatic potentials were calculated for each molecule using a 6–31G* basis set, and then used to fit atom-centered point charges with the RESP module of AMBER 7.37 Conformational searches in torsion angle space for all rotatable bonds of each ligand were conducted using the torsion-drive feature in DOCK 4.0.36 Most sp2-sp3 bonds were scanned at 60° increments, and all sp3-sp3 bonds were probed at −60°, +60°, and 180°. Additionally, energy minimization of the ligand conformers generated from this search were performed ‘on the fly’ in DOCK to facilitate the identification of low energy complexes for each ligand with the receptor model. Docked complexes were evaluated according to the DOCK 4.0 shape-based and energy-based scoring functions.36 Representative receptor-ligand complex structures generated with DOCK were then refined in vacuo with limited (500 steps) energy minimization. For the receptor-(S)-antagonist (8) docked complex, a limited number of additional dihedral angle adjustments were performed (following the above outlined strategy) for the following protein sidechains prior to energy refinements to relieve unfavorable van der Waals contacts around the photoaffinity-labeling site (Leu88, Cys91, Cys94, Met121, Leu357, Ser364). Molecular dynamics simulations for the (S)-antagonist (8) complex were performed in vacuo with a distance-dependent dielectric constant at temperatures of 20K and 300K for 100ps. Since our model does not include membrane lipids or solvent, weak (1–10 kcal/mol) harmonic constraints were gradually imposed for the 300K simulations over a 50ps timeframe to stabilize the conformation of the membrane-spanning helix bundle at the higher temperatures. An NMR-style energy restraint (5 kcal/mol) was also imposed on the distance between the ketone carbonyl of the photo-labile benzophenone moiety and the Cγ–H bond of Leu88 to maintain a physically realistic photo-incorporation distance consistent with the experimental data in this report.
All quantum mechanical calculations were performed with Gaussian98.38 Model structures were evaluated for quality of side chain packing, backbone geometry, and stereochemistry using the programs QPACK39 and PROCHECK.40 Manual model building and rms deviation calculations were performed with the interactive graphics program PSSHOW.41 All energy minimization and molecular dynamics calculations were performed using the AMBER 7.0 suite of programs.37 All non-database forcefield parameters for the benzodiazepine ligands were developed by analogy to database values in the parm96.dat parameter set of AMBER 7.0.37 MDDISPLAY was used for visual analysis of molecular dynamics trajectories.42 Figure generation was performed using DINO version 0.9.43
Statistical analysis
All observations were repeated at least three times in independent experiments and are expressed as the means±S.E.M. Binding and biological activity curves were analyzed and plotted using the nonlinear regression analysis routine in the Prism software package (GraphPad Software, San Diego, CA). Binding kinetics were determined by analysis with the LIGAND program.44
Acknowledgments
We acknowledge the expert contribution of Manon Villeneuve and Itzela Correa of Glaxo-SmithKline Research Laboratories, who performed the chiral separations and the analysis and radiolabeling of the compounds used in this study. We thank Dr. Maoqing Dong for his advice and help with preparing this manuscript. This work was supported by grants from the National Institutes of Health (DK32878 to LJM and NS33290 to TPL), Glaxo-SmithKline, and the Fiterman Foundation.
The abbreviations used are the following
References
- 1.Darrow JW, Hadac EM, Miller LJ, Sugg EE. Structurally similar small molecule photoaffinity CCK-A agonists and antagonists as novel tools for directly probing 7TM receptor-ligand interactions. Bioorg Med Chem Lett. 1998;8:3127–3132. doi: 10.1016/s0960-894x(98)00548-4. [DOI] [PubMed] [Google Scholar]
- 2.Ulrich CD, Ferber I, Holicky E, Hadac E, Buell G, Miller LJ. Molecular cloning and functional expression of the human gallbladder cholecystokinin A receptor. Biochemical & Biophysical Research Communications. 1993;193:204–211. doi: 10.1006/bbrc.1993.1610. [DOI] [PubMed] [Google Scholar]
- 3.Mutt V. Cholecystokinin: isolation, structure, and functions. In: Glass GBJ, editor. Gastrointestinal hormones. Raven Press; New York: 1980. pp. 169–221. [Google Scholar]
- 4.Powers SP, Fourmy D, Gaisano H, Miller LJ. Intrinsic photoaffinity labeling probes for cholecystokinin (CCK)-gastrin family receptors D-Tyr-Gly-[Nle28,31,pNO2-Phe33)CCK-26–33) J Biol Chem. 1988;263:5295–5300. [PubMed] [Google Scholar]
- 5.Klueppelberg UG, Gaisano HY, Powers SP, Miller LJ. Use of a nitrotryptophan-containing peptide for photoaffinity labeling the pancreatic cholecystokinin receptor. Biochemistry. 1989;28:3463–3468. doi: 10.1021/bi00434a047. [DOI] [PubMed] [Google Scholar]
- 6.Ji Z, Hadac EM, Henne RM, Patel SA, Lybrand TP, Miller LJ. Direct identification of a distinct site of interaction between the carboxyl-terminal residue of cholecystokinin and the type A cholecystokinin receptor using photoaffinity labeling. J Biol Chem. 1997;272:24393–24401. doi: 10.1074/jbc.272.39.24393. [DOI] [PubMed] [Google Scholar]
- 7.Hadac EM, Pinon DI, Ji Z, Holicky EL, Henne RM, Lybrand TP, Miller LJ. Direct identification of a second distinct site of contact between cholecystokinin and its receptor. J Biol Chem. 1998;273:12988–12993. doi: 10.1074/jbc.273.21.12988. [DOI] [PubMed] [Google Scholar]
- 8.Ding XQ, Miller LJ. Characterization of the type A cholecystokinin receptor hormone-binding domain: use of contrasting and complementary methodologies [Review] Peptides. 2001;22:1223–1228. doi: 10.1016/s0196-9781(01)00445-4. [DOI] [PubMed] [Google Scholar]
- 9.Ding XQ, Pinon DI, Furse KE, Lybrand TP, Miller LJ. Refinement of the conformation of a critical region of charge-charge interaction between cholecystokinin and its receptor. Mol Pharmacol. 2002;61:1041–1052. doi: 10.1124/mol.61.5.1041. [DOI] [PubMed] [Google Scholar]
- 10.Aquino CJ, Armour DR, Berman JM, Birkemo LS, Carr RAE, Croom DK, Dezube M, Dougherty RW, Jr, Ervin GN, Grizzle MK, Head JE, Hirst GC, James MK, Johnson MF, Miller LJ, Queen KL, Rimele TJ, Smith DN, Sugg EE. Discovery of 1,5-benzodiazepines with peripheral cholecystokinin (CCK-A) receptor agonist activity.1. Optimization of the agonist “Trigger”. J Med Chem. 1996;39:562–569. doi: 10.1021/jm950626d. [DOI] [PubMed] [Google Scholar]
- 11.Evans BE, Rittle KE, Bock MG, DiPardo RM, Freidinger RM, Whitter WL, Gould NP, Lundell GF, Homnick CF, Veber DF, Anderson PS, Chang RSL, Lotti VJ, Cerino DJ, Chen TB, King PJ, Kunkel KA, Springer JP, Hirshfield J. Design of nonpeptidal ligands for a peptide receptor: cholecystokinin antagonists. J Med Chem. 1987;30:1229–1239. doi: 10.1021/jm00390a019. [DOI] [PubMed] [Google Scholar]
- 12.Evans BE, Rittle KE, Bock MG, DiPardo RM, Freidinger RM, Whitter WL, Lundell GF, Veber DF, Anderson PS, Chang RS, Lotti VJ, Cerino DJ, Chen TB, Kling PJ, Kunkel KA, Springer JP, Hirshfield J. Methods for drug discovery: development of potent, selective, orally effective cholecystokinin antagonists. J Med Chem. 1988;31:2235–2246. doi: 10.1021/jm00120a002. [DOI] [PubMed] [Google Scholar]
- 13.Ursini A, Capelli AM, Carr RA, Cassara P, Corsi M, Curcuruto O, Curotto G, Dal CM, Davalli S, Donati D, Feriani A, Finch H, Finizia G, Gaviraghi G, Marien M, Pentassuglia G, Polinelli S, Ratti E, Reggiani AM, Tarzia G, Tedesco G, Tranquillini ME, Trist DG, van Amsterdam FT. Synthesis and SAR of new 5-phenyl-3-ureido-1,5-benzodiazepines as cholecystokinin-B receptor antagonists. J Med Chem. 2000;43:3596–3613. doi: 10.1021/jm990967h. [DOI] [PubMed] [Google Scholar]
- 14.Hadac EM, Ghanekar DV, Holicky EL, Pinon DI, Dougherty RW, Miller LJ. Relationship between native and recombinant cholecystokinin receptors - role of differential glycosylation. Pancreas. 1996;13:130–139. doi: 10.1097/00006676-199608000-00003. [DOI] [PubMed] [Google Scholar]
- 15.Harikumar KG, Pinon DI, Wessels WS, Dawson ES, Lybrand TP, Prendergast FG, Miller LJ. Mesurement of intermolecular distances for the natural agonist peptide docked at the cholecystokinin receptor expressed in situ using fluorescence resonance energy transfer. Mol Pharmacol. 2004;65:28–35. doi: 10.1124/mol.65.1.28. [DOI] [PubMed] [Google Scholar]
- 16.Dunbrack RL, Jr, Karplus M. Backbone-dependent rotamer library for proteins. Application to side-chain prediction. J Molec Biol. 1993;230:543–574. doi: 10.1006/jmbi.1993.1170. [DOI] [PubMed] [Google Scholar]
- 17.Escrieut C, Gigoux V, Archer E, Verrier S, Maigret B, Behrendt R, Moroder L, Bignon E, Silvente-Poirot S, Pradayrol L, Fourmy D. The biologically crucial C terminus of cholecystokinin and the non-peptide agonist SR-146,131 share a common binding site in the human CCK1 receptor - Evidence for a crucial role of Met-121 in the activation process. J Biol Chem. 2002;277:7546–7555. doi: 10.1074/jbc.M108563200. [DOI] [PubMed] [Google Scholar]
- 18.Jagerschmidt A, Guillaume-Rousselet N, Vikland ML, Goudreau N, Maigret B, Roques BP. His381 of the rat CCKB receptor is essential for CCKB versus CCKA receptor antagonist selectivity. Eur J Pharmacol. 1996;296:97–106. doi: 10.1016/0014-2999(95)00676-1. [DOI] [PubMed] [Google Scholar]
- 19.Kopin AS, McBride EW, Quinn SM, Kolakowski LF, Jr, Beinborn M. The role of the cholecystokinin-B/gastrin receptor transmembrane domains in determining affinity for subtype-selective ligands. J Biol Chem. 1995;270:5019–5023. doi: 10.1074/jbc.270.10.5019. [DOI] [PubMed] [Google Scholar]
- 20.O’Neil KT, Erickson-Viitanen S, DeGrado WF. Photolabeling of calmodulin with basic, amphiphilic alpha-helical peptides containing p-benzoylphenylalanine. J Biol Chem. 1989;264:14571–14578. [PubMed] [Google Scholar]
- 21.Smeets RL, IJzerman AP, Hermsen HP, Ophorst OJ, van Emst-de Vries SE, De Pont JJ, Willems PH. Mutational analysis of the putative devazepide binding site of the CCK(A) receptor. Eur J Pharmacol. 1997;325:93–99. doi: 10.1016/s0014-2999(97)00106-4. [DOI] [PubMed] [Google Scholar]
- 22.Ji TH, Grossmann M, Ji I. G protein-coupled receptors. I. Diversity of receptor-ligand interactions. J Biol Chem. 1998;273:17299–17302. doi: 10.1074/jbc.273.28.17299. [DOI] [PubMed] [Google Scholar]
- 23.Hadac EM, Ji ZS, Pinon DI, Henne RM, Lybrand TP, Miller LJ. A peptide agonist acts by occupation of a monomeric G protein-coupled receptor: Dual sites of covalent attachment to domains near TM1 and TM7 of the same molecule make biologically significant domain-swapped dimerization unlikely. J Med Chem. 1999;42:2105–2111. doi: 10.1021/jm980732q. [DOI] [PubMed] [Google Scholar]
- 24.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Trong ILe, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 25.Ghanouni P, Steenhuis JJ, Farrens DL, Kobilka BK. Agonist-induced conformational changes in the G-protein-coupling domain of the beta 2 adrenergic receptor. Proc Natl Acad Sci USA. 2001;98:5997–6002. doi: 10.1073/pnas.101126198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghanouni P, Gryczynski Z, Steenhuis JJ, Lee TW, Farrens DL, Lakowicz JR, Kobilka BK. Functionally different agonists induce distinct conformations in the G protein coupling domain of the beta 2 adrenergic receptor. J Biol Chem. 2001;276:24433–24436. doi: 10.1074/jbc.C100162200. [DOI] [PubMed] [Google Scholar]
- 27.Gouldson P, Legoux P, Carillon C, Delpech B, Le Fur G, Ferrara P, Shire D. The agonist SR 146131 and the antagonist SR 27897 occupy different sites on the human CCK1 receptor. Eur J Pharmacol. 2000;400:185–194. doi: 10.1016/s0014-2999(00)00414-3. [DOI] [PubMed] [Google Scholar]
- 28.Powers SP, Pinon DI, Miller LJ. Use of N, O-bis-Fmoc-D-Tyr-ONSu for introduction of an oxidative iodination site into cholecystokinin family peptides. Int J Pept Protein Res. 1988;31:429–434. doi: 10.1111/j.1399-3011.1988.tb00899.x. [DOI] [PubMed] [Google Scholar]
- 29.Pearson RK, Miller LJ, Hadac EM, Powers SP. Analysis of the carbohydrate composition of the pancreatic plasmalemmal glycoprotein affinity labeled by short probes for the cholecystokinin receptor. J Biol Chem. 1987;262:13850–13856. [PubMed] [Google Scholar]
- 30.Abdel-Magid AF, Maryanoff CA, Carson KG. Reductive amination of aldehydes and ketones by using sodium triacetoxyborohydride. Tetrahedron Lett. 1990;31:5595–5598. [Google Scholar]
- 31.Abdel-Magid AF, Carson KG, Harris BD, Maryanoff CA, Shah RD. Reductive amination of aldehydes andketones with sodium triacetoxyborohydride. Studies on direct and indirect reductive amination procedures. J Org Chem. 1996;61:3849–3862. doi: 10.1021/jo960057x. [DOI] [PubMed] [Google Scholar]
- 32.Hirst GC, Aquino C, Birkemo L, Croom DK, Dezube M, Dougherty RW, Jr, Ervin GN, Grizzle MK, Henke B, James MK, Johnson MF, Momtahen T, Queen KL, Sherrill RG, Szewczyk J, Willson TM, Sugg EE. Discovery of 1,5-benzodiazepines with peripheral cholecystokinin (CCK-A) receptor agonist activity (II): Optimization of the C3 amino substituent. J Med Chem. 1996;39:5236–5245. doi: 10.1021/jm9601664. [DOI] [PubMed] [Google Scholar]
- 33.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 34.Dong M, Wang Y, Pinon DI, Hadac EM, Miller LJ. Demonstration of a direct interaction between residue 22 in the carboxyl-terminal half of secretin and the amino-terminal tail of the secretin receptor using photoaffinity labeling. J Biol Chem. 1999;274:903–909. doi: 10.1074/jbc.274.2.903. [DOI] [PubMed] [Google Scholar]
- 35.Evans BE, Bock MG, Rittle KE, DiPardo RM, Whitter WL, Veber DF, Anderson PS, Freidinger RM. Design of potent, orally effective, nonpeptidal antagonists of the peptide hormone cholecystokinin. Proc Natl Acad Sci USA. 1986;83:4918–4922. doi: 10.1073/pnas.83.13.4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ewing TJA, Kuntz ID. DOCK 4.0. J Comput Chem. 1997;18:1175–1189. [Google Scholar]
- 37.Case DA, Pearlman DA, Caldwell JW, Cheatham TE, Wang J, Ross WS, Simmerling C, Darden T, Merz KM, Stanton RV, Cheng A, Vincent JJ, Crowley M, Tsui V, Gohlke H, Radmer R, Duan Y, Pitera J, Massova I, Seibel GL, Singh UC, Weiner P, Kollman PA. AMBER 70. University of California; San Francisco: 2002. [Google Scholar]
- 38.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery JA, Jr, Stratmann RE, Burant JC, Dapprich S, Millam JM, Daniels AD, Kudin KN, Strain MC, Farkas O, Tomasi J, Barone V, Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochterski J, Petersson GA, Ayala PY, Cui Q, Morokuma K, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Cioslowski J, Ortiz JV, Baboul AG, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Gomperts R, Martin RL, Fox DJ, Keith D, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Andres JL, Gonzalez C, Head-Gordon M, Replogle ES, Pople JA. Gaussian 98, Revision A9. Gaussian, Inc; Pittsburgh, PA: 1998. [Google Scholar]
- 39.Gregoret LM, Cohen FE. Novel method for the rapid evaluation of packing in protein structures. J Mol Biol. 1990;211:959–974. doi: 10.1016/0022-2836(90)90086-2. [DOI] [PubMed] [Google Scholar]
- 40.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 41.Swanson E. PSSHOW: Silicon Graphics 4D version. Seattle, WA: 1995. [Google Scholar]
- 42.Moth C, Callahan T, Swanson E, Lybrand TP. MDDISPLAY v30. 2002. [Google Scholar]
- 43.Ansgar Philippsen. DINO: Visualizing Structural Biology. 2003 http:www.dino3d.org.
- 44.Munson PJ, Rodbard D. LIGAND: a versatile computerized approach for characterization of ligand-binding systems. Anal Biochem. 1980;107:220–239. doi: 10.1016/0003-2697(80)90515-1. [DOI] [PubMed] [Google Scholar]