

In the past twenty years, metal-promoted SN2′ reactions with carbon nucleophiles have become valuable tools in organic synthesis.1 In that period, several protocols for stoichiometric2 and catalytic3 copper-mediated allylic substitutions with carbon nucleophiles that allow complete control over regio- and stereoselectivity of the reactions have been developed. In comparison, SN2′ substitution reactions with heteroatom nucleophiles are virtually unexplored.
Isolated examples of SN2′ reactions of allylic halides and esters with amines, alkoxides, and thiols have been known for some time.4 However, selective formation of the rearranged (SN2′) products can be achieved only if the competing SN2 reaction is sterically disfavored.5,6 The reactions of Cp*2(py)Ti=S (1, Cp*=η5-C5Me5) with allylic chlorides,7 which provide rearranged products regardless of the substitution pattern of the electrophile, are an exception. The unusual regiospecificity of this reaction was attributed to a closed transition state that is formed as the result of an interaction between titanium atom and the leaving group. Therefore, the Lewis acidity of the metal center was essential for the observed regiospecificity.
The success of substitution reactions with 1 has not been duplicated with other heteroatom nucleophiles, and general regiospecific SN2′ reactions with nitrogen nucleophiles have still not been achieved.8 In this communication, we report a new, zirconium-mediated, regio- and stereospecific allylic substitution with a nitrogen nucleophile that allows direct transformation of allylic ethers into protected allylic amines.
Preliminary experiments established that zirconocene imido complex 2 (TBS = tert-butyldimethylsilyl) reacts with allyl chloride, bromide, and iodide, at room temperature, to yield substitution products 3 in nearly quantitative yield (Equation 1).
(1).
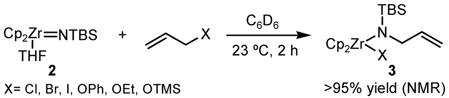
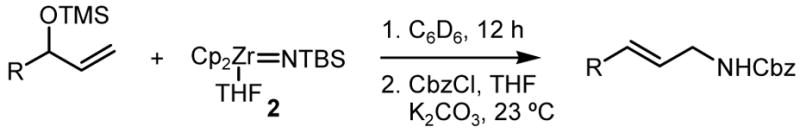
Unlike the titanocene sulfido complex 1,7 2 also reacts with alkyl, aryl, and trimethylsilyl (TMS) allyl ethers, under the same reaction conditions, to produce substitution products in high yield. These results were surprising considering the limited number of reported substitution reactions with allylic ethers9 and prompted us to further explore the scope of this tranformation. In all subsequent reactions, the products were isolated as carbobenzyloxy-protected (Cbz) amines, after in situ hydrolysis of initial zirconium amide products and Cbz protection of the free amines.
The reactions with several α-substituted TMS ethers provided the SN2′ products in good yields (Table 1). The size of the α-substituent had a significant effect on the observed diastereoselectivity and the temperature required for the reaction (Entry 2 vs. Entry 3, Table 1). The tert-butyl substituent proved to be prohibitively large and no product was observed after 12 h at elevated temperature. In reactions with both ethyl and isopropyl substituted ethers, only one regioisomer could be detected in the 1H NMR spectrum of the crude reaction mixture. The formation of the rearranged products in these reactions is in agreement with the steric bias of the substrates. Therefore, in order to assess the inherent regioselectivity of the nucleophile we explored the reactivity of substrates that should favor SN2 substitution.
Table 1.
Allylic Substitution of α-Substituted Allylic Ethers
 | ||||
|---|---|---|---|---|
| Entry | R | T (ºC) | dr (E:Z) | Isolated Yield (%) |
| 1 | H | 23 | - | 96 |
| 2 | Et | 23 | 3:1 | 92 |
| 3 | iPr | 45 | >9:1 | 89 |
| 41 | tBu | 90 | - | NR |
cyclohexane-d12 used as a solvent
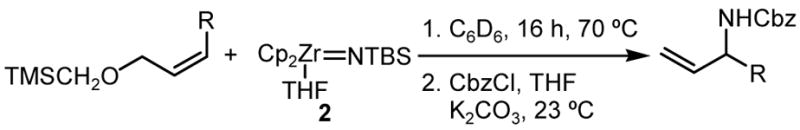
We found that a variety of ethers of Z substituted primary allylic alcohols reacts with 1 at elevated temperature. The most reactive substrates were the electron rich (trimethylsilyl)methyl ethers, which provided the products of the SN2′ substitution in up to 94% yield (Table 2).10
Table 2.
Allylic Substitution of TMS-methyl Ethers
 | ||
|---|---|---|
| Entry | Substrate | Isolated Yield (%) |
| 1 |
|
94 |
| 2 |
|
90 |
| 3 |
|
92 |
| 4 |
|
89 |
| 5 |
|
92 |
| 6 |
|
77 |
Although substituents at the 3-position of these substrates should favor the SN2 pathway, the more substituted product of an SN2′ substitution was still the only product observed in the 1H NMR spectra of the crude reaction mixtures.
The reactions with the substrates presented in Table 2 also illustrate a chemoselectivity unprecedented for the reactions of zirconium imido complexes. The substitution reaction is not affected by the presence of a variety of functional groups, including terminal alkenes, phenyl ethers, and 1,3-dithianes (Table 2, Entries 2,3, and 4). Even in the presence of an electrophilic primary chloride, the desired product was isolated in 92% yield (Table 2, Entry 5).
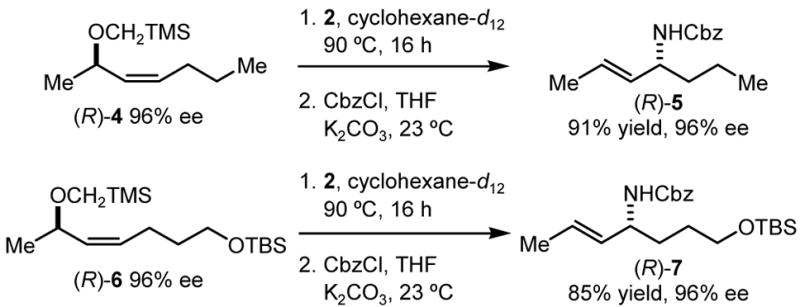
Another important feature of the substitution reactions with 2 is complete chirality transfer observed in the reactions with enantioenriched 1,3-disubstituted allylic ethers. The reaction of (R)-4 with 2 resulted in the regio- and stereospecific formation of (R)-5, in 91% yield (Scheme 1). Similarly, the reaction with (R)-6 produced only the rearranged product (R)-7, as a single diastereoisomer. These experiments demonstrated the use of the SN2′ substitution reaction in a highly stereoselective synthesis of enantioenriched allylic amines and also allowed us to determine the stereochemistry of the reaction. The absolute configuration of 5 revealed that the substitution proceeds with syn stereochemistry. Intrigued by this stereospecificity and the regiospecificity observed with all other substrates, regardless of their structure, we decided to study the mechanism of the reaction between 2 and the allyl tert-butyldimethylsilyl (TBS) ether in more detail.
Scheme 1.
The reaction with TBS allyl ether was monitored by 1H NMR spectroscopy. In the presence of excess THF and substrate, the reaction exhibits pseudo-first-order behavior with no observable intermediates.11 The first-order rate constant for the reaction (kobs=5.47±0.01×10−4 s−1, at 21.5 °C) is independent of the initial concentration of 2, indicating that the overall reaction is first order in 2. From the pseudo-first-order rate constants obtained in the presence of various concentrations of THF, we also established that the overall reaction is inverse first order in THF. Based on these results and the mechanisms of related reactions of zirconium imido complexes,12 we propose that the substitution reaction involves reversible dissociation of THF, followed by the rate limiting substitution step (Scheme 2). In agreement with the rate law (Scheme 2), we observed that at a high ratio of allylic substrate and THF concentrations, kobs becomes independent of their concentrations. By measuring kobs at different concentrations of THF and allylic ether we were able to extract values for k1=1.4±0.1×10−3 (s−1) and k2/k-1=2.2±0.2×10−2 at −7.55 °C.
Scheme 2.
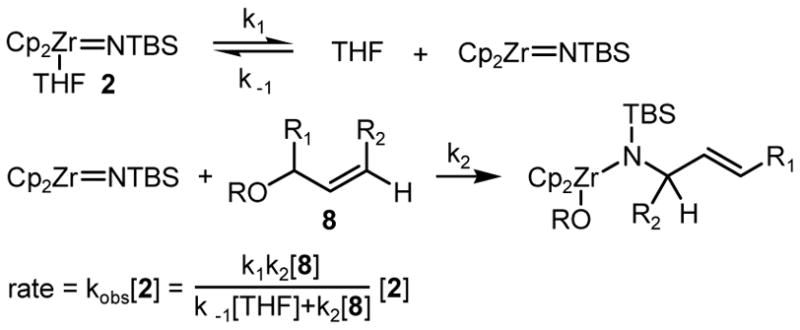
Consistent with the C-N bond formation in the rate determining step is the inverse secondary kinetic isotope effect, calculated from the measured pseudo-first-order rate constants for the reactions with TBS allyl ether and (E)-1-(tert-butyldimetylsiloxy)-3-deuterioprop-2-ene (kH/kD=0.88 ±0.01 at 20.6 °C).
More insight into the second step of the reaction can be obtained from the stereochemistry of the substitution. In the literature, syn stereochemistry of allylic substitutions has been interpreted as a strong indication of a cyclic transition state.13 Consistent with these precedents and the observed regio- and stereospecificity of the reaction, we propose 9 (Figure 1) as the transition state for this step.14 The interaction between the zirconium atom and the leaving group in the proposed transition state provides an explanation for the unusual propensity of allylic ethers to undergo the substitution reaction and is in agreement with the established Lewis acidity and oxophilicity of zirconium imido complexes.12 Furthermore, the presence of an unfavorable steric interaction between the TBS group and R3 (see Figure 1) in the proposed transition state is consistent with the lower reactivity of E-substituted allylic ethers.
Figure 1.
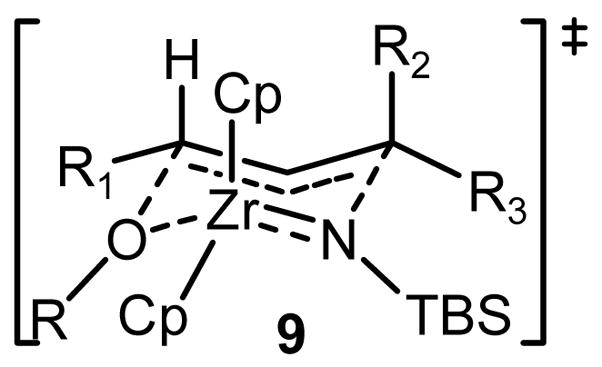
Postulated transition state for the second step of the substitution reaction.
In conclusion, we have discovered a new reaction of zirconium imido complexes that allows regio- and stereospecific transformation of allylic ethers directly into Cbz-protected allylic amines. We have also proposed a mechanism that is consistent with our studies of reaction kinetics, the measured secondary kinetic isotope effect, and the observed regio- and stereospecificity of the reaction. These results provide further insight into the reactivity of zirconium imido complexes and may facilitate the development of practical methods for the SN2′ substitution reactions.
Supplementary Material
Experimental procedures and spectral data for products (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by the NIH through Grant No. GM-25459 to R.G.B., and by the NSF through a graduate fellowship to S.A.B.
References
- 1.a) Hoveyda AH, Heron NM. In: Comprehensive Asymmetric Catalysis. Jacobsen EN, Pfaltz A, Yamamoto H, editors. Springer; Berlin, Germany: 1999. p. 431. [Google Scholar]; b) Karlström ASE, Bäckvall J-E. In: Modern Organocopper Chemistry. Krause N, editor. Wiley–VCH; Weinheim: 2001. p. 259. [Google Scholar]; c) Helmchen G, Ernst M, Paradies G. Pure Apple Chem. 2004;76:495. [Google Scholar]
- 2.a) Smitrovich JH, Woerpel KA. J Am Chem Soc. 1998;120:12998. [Google Scholar]; b) Breit B, Demel P, Studie C. Angew Chem, Int Ed Engl. 2004;43:3786. doi: 10.1002/anie.200453991. [DOI] [PubMed] [Google Scholar]; c) Harrington-Frost N, Leuser H, Calaza MI, Kneisel FF, Knochel P. Org Lett. 2003;5:2111. doi: 10.1021/ol034525i. [DOI] [PubMed] [Google Scholar]
- 3.Kacprzynski MA, Hoveyda AH. J Am Chem Soc. 2004;126:10676. doi: 10.1021/ja0478779. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 4.For an excellent review see: Magid RM. Tetrahedron. 1980;36:1901.
- 5.Lesnini DG, Buckley PD, Noyes RM. J Am Chem Soc. 1968;90:668. [Google Scholar]; b) Hemmingson JA, England BD. J Chem Soc B. 1971:1347. [Google Scholar]
- 6.Magid RM, Fruchey OS. J Am Chem Soc. 1977;99:8368. b) [Google Scholar]
- 7.Sweeney ZK, Polse JL, Andersen RA, Bergman RG. J Am Chem Soc. 1998;120:7825. [Google Scholar]
- 8.Leitner A, Shekhar S, Pouy MJ, Hartwig JF. J Am Chem Soc. ASAP and references therein. [Google Scholar]
- 9.a) Didiuk MT, Morken JP, Hoveyda AH. J Am Chem Soc. 1995;117:7273. [Google Scholar]; b) Barluenga J, Rodriguez F, Alvarez-rodrigo L, Zapico JM, Fananas FJ. Chem Eur J. 2004;10:109. doi: 10.1002/chem.200305374. [DOI] [PubMed] [Google Scholar]
- 10.Corresponding E substituted ethers provided the same product in significantly lower yield.
- 11.For details of kinetics and kinetic isotope effect experiments see supporting information
- 12.Blum SA, Walsh PJ, Bergman RG. J Am Chem Soc. 2003;125:14276. doi: 10.1021/ja037267t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Tseng CC, Paisley SD, Goering HL. J Org Chem. 1986;51:2884. [Google Scholar]; b) Young WG, Webb ID, Goering HL. 1951;73:1076. [Google Scholar]; c) ref. 2 b) and references cited therein.
- 14.An alternative mechanism that involves chelation-assisted 2+2 cycloaddition followed by the fragmentation of the bicyclic intermediate has been proposed by one of the reviewers, and is consistent with the results of the kinetics and kinetic isotope effect experiments presented in the paper.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and spectral data for products (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.