

Abstract
Non-nucleoside inhibitors of HIV-1 reverse transcriptase (NNRTIs) are part of the combination therapy currently used to treat HIV infection. Based on analogy with known HIV-1 NNRT inhibitors, eighteen novel alkenyldiarylmethanes (ADAMs) containing 5-chloro-2-methoxyphenyl, 3-cyanophenyl or 3-fluoro-5-trifluoromethylphenyl groups were synthesized and evaluated as HIV inhibitors. Their stabilities in rat plasma have also been investigated. Although introducing 5-chloro-2-methoxyphenyl, or 3-fluoro-5-trifluoromethylphenyl groups into alkenyldiarylmethanes does not maintain the antiviral potency, the structural modification of alkenyldiarylmethanes with a 3-cyanophenyl substituent can be made without a large decrease in activity. The oxazolidinonyl group was introduced into the alkenyldiarylmethane framework and found to confer enhanced metabolic stability in rat plasma.
Introduction
Acquired immunodeficiency syndrome (AIDS) was responsible for an estimated 3 million deaths in 2004, with an estimated 39.4 million people living with HIV, which includes the 4.9 million people who acquired HIV in 2004.1 The reverse transcriptase (RT) of the human immunodeficiency virus type 1 (HIV-1) plays an essential and central role in the viral replication cycle by conversion of the single-stranded RNA genome of HIV-1 into a double-stranded DNA chain that subsequently is incorporated into the DNA of the infected host cell. HIV-1 RT is a multifunctional heterodimer consisting of a 66-kDa subunit and a 51-kDa subunit that, as a proteolytic product of the p66 subunit, has the same sequence as the corresponding region of p66 subunit but adopts a different conformation.
As an essential viral enzyme, HIV-1 RT is one of the major targets of the antiretroviral drug therapies that are used in the treatment of AIDS. Non-nucleoside inhibitors of HIV-1 reverse transcriptase (NNRTIs) inhibit the enzyme by occupation of an induced allosteric binding site very close to the active site.2,3 However, the emergence of resistant HIV viral strains is a limitation for all therapeutic classes. The emerging cross-resistance among the approved drugs (nevirapine, delavirdine, and efavirenz) has particularly limited the use of the NNRTI class. Therefore, the development of new NNRTIs with more favorable side effect profiles and improved characteristics influencing drug compliance will be essential to the future management of HIV infection.
The alkenyldiarylmethanes (ADAMs) are a unique class of non-nucleoside reverse transcriptase inhibitors.4–13 A number of HIV-1 strains containing AZT resistance mutations have shown increased sensitivity to some of the ADAMs, indicating a possible therapeutic role for the ADAMs in combination with AZT.6 Certain ADAMs have been found to inhibit the cytopathic effect of HIV-1 in cell culture at low nanomolar concentrations. For example, the oxazolidinone ADAM analog 1 displays an EC50 of 0.02 μM for inhibition of the cytopathic effect of HIV-1RF in CEM-SS cells, and it inhibits HIV-1 RT with rCdG as the template primer with an IC50 of 0.499 μM.9 The dimethoxyphenyl compound 2 inhibits the cytopathic effect of HIV-1RF in CEM-SS cell culture with an EC50 of 0.21 μM, and inhibits HIV-1 RT with an IC50 of 0.074 μM, which is the most potent RT inhibitor in the ADAM series to date.11 The main impetus for the present study stems from the fact that the methyl ester moieties present in the biologically active ADAMs are hydrolyzed to biologically inactive acids in blood plasma. A general effort has therefore been initiated to find metabolically stable replacements for the methyl ester moieties that would still preserve the desired antiviral activity. We envisaged that compound 3, with the replacement of methyl ester moiety of the ADAM 2 with a 2-oxoxazolidinyl group, might retain the anti-HIV potency of the parent compound. The isomeric compound 4 might also result in the retention of anti-HIV activity. It might be expected that the cyclic carbamate present in 3 and 4 would be more metabolically stable than the corresponding ester of 2. Therefore, compounds 3 and 4 were selected as targets for synthesis.
Recently, benzophenones 5 and 6 containing 5-chlorophenyl, 3-cyanophenyl or 3-fluoro-5-trifluoromethylphenyl groups were reported as NNRTIs of HIV-1. These inhibitors displayed IC50 values of ≤ 2 nM against wild type HIV-1 and <10 nM against 16 mutants.14 Since these NNRTIs are structurally related to the ADAMs, we were intrigued by the possibility that the substituted phenyl rings of the ADAMs could be replaced by those of 5 and 6. More specifically, the analogy that we had considered would view the halogenated rings of 5 and 6 as equivalent to the two aromatic rings of the ADAMs, while the remaining part would correspond to the alkenyl side chain of the ADAMs. Alkenyldiarylmethanes 7–22, with 5-chloro-2-methoxyphenyl, 3-cyanophenyl or 3-fluoro-5-trifluoromethylphenyl groups, were therefore designed and have been synthesized via the Sonogashira and Stille cross-coupling reactions. The isoxazole rings of compounds 19–22 were conceived as metabolically stable replacements for the 4-methoxy-3-methoxycarbonyl substituents found in many of the active ADAMs. In this paper, we report the synthesis of alkenyldiarylmethanes 3, 4, and 7–22, their anti-HIV activities, and their metabolic stabilities in rat blood plasma.
Chemistry
Alkenyldiarylmethanes 3 and 4 were prepared according to the procedures shown in Scheme 1. Commercially available 3-butyn-1-ol (23) was converted to the corresponding tosylate 24, which reacted with 2-oxazolodinone to afford the alkylated intermediate 25. The Sonogashira coupling of the terminal alkyne 25 with the aryl iodide 29 or 33 yielded the disubstituted alkynes 30 or 34. The hydrostannation of 30 or 34 with tri-n-butyltin hydride in the presence of Pd(PPh3)4 afforded the regiochemically and stereochemically defined vinylstannanes 31 or 35, that underwent the Stille cross-coupling with the aryl iodides 33 or 29 to afford the desired target compounds 3 or 4.
Scheme 1a.
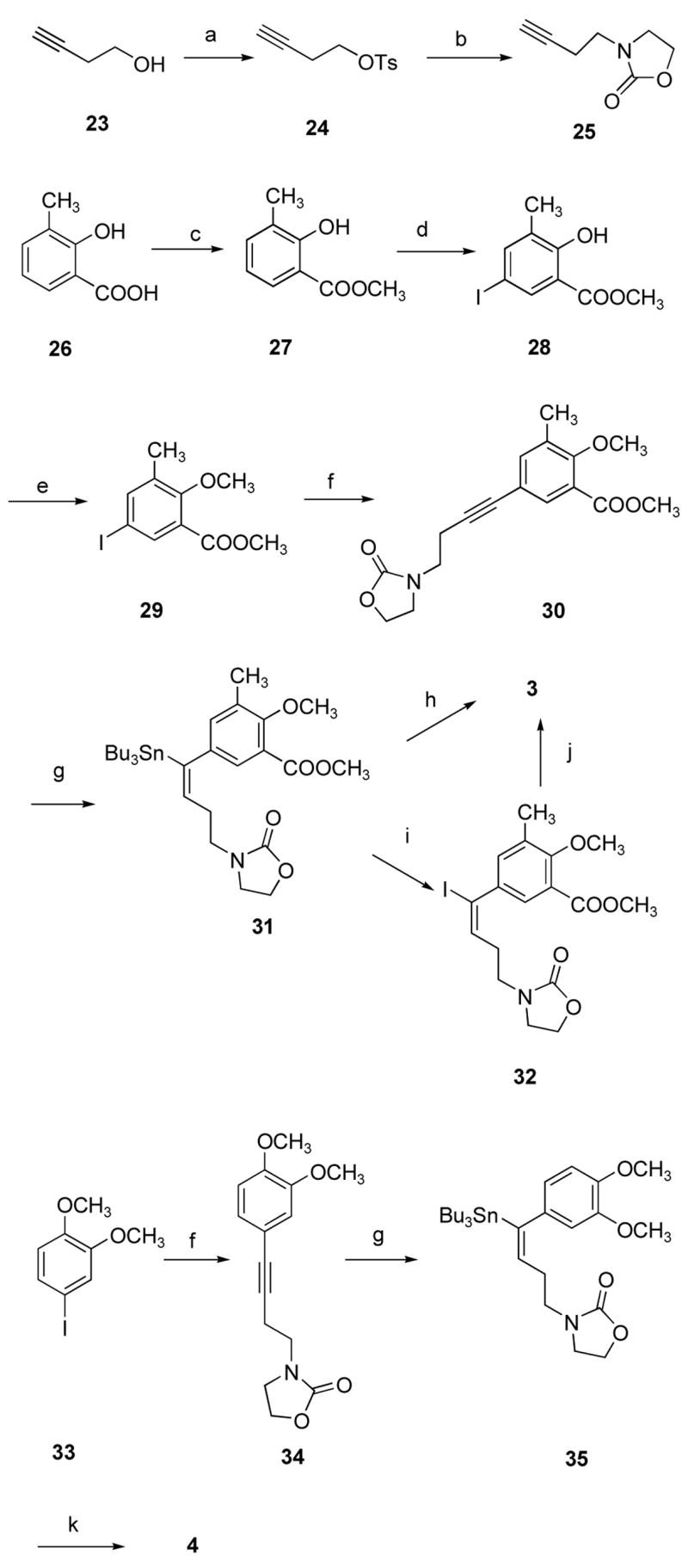
aReagents and conditions: (a) TsCl/pyridine, <20 °C; or KOH, THF-H2O, room temperature; (b) 2-oxazolidinone, K2CO3, Bu4NBr, toluene, reflux; (c) TMSCHN2, MeOH, benzene, room temperature; or dimethyl sulfate, K2CO3, Bu4NBr, CH2Cl2-H2O, room temperature; (d) NaI, NaOH, NaOCl, 0–3 °C; (e) dimethyl sulfate, K2CO3, acetone, reflux; or dimethyl sulfate, NaOH, Bu4NBr, CH2Cl2-H2O, room temperature; (f) compound 25, PdCl2(PPh3)2, Cu(I)I, Et3N, THF, room temperature; (g) Bu3SnH, Pd(PPh3)4, THF, room temperature; (h) compound 33, Pd2(dba)3, AsPh3, CuI, NMP, 80 °C; (i) I2, CH2Cl2, room temperature; (j) 3,4-dimethoxyphenylboronic acid, Pd(OAc)2, 2-(di-t-butylphosphine)biphenyl, KF, THF, 60 °C; (k) compound 29, Pd(PBut3)2, toluene, reflux.
3-Butynyl-1-tosylate (24) was previously synthesized in 83% yield from 3-butyn-1-ol (23) with p-toluenesulfonyl chloride in the presence of pyridine according to a literature procedure.15 However, there are some limitations for this reaction. First, since no solvent was used for the reaction, it is difficult to stir the reaction mixture, which sometimes leads to low yield. Second, it is time consuming to remove the pyridine after the reaction. Therefore, it would be advantageous to have an alternative method to synthesize compound 24 that would allow the reaction to be scaled up. This was accomplished by performing the reaction in aqueous THF with sodium hydroxide as the base, which allowed 24 to be synthesized on a 27-gram scale. This reaction occurs rapidly at room temperature and the work-up is very easy.
It was previously reported that the reaction of 4-bromo-1-butene with 2-oxoazolidinone using cesium carbonate as the base in acetone gave 3-but-3-enyl-1,3-oxazolidin-2-one in very good yield.9 However, an attempted synthesis of 3-but-3-ynyl-1,3-oxazolidin-2-one (25) from 3-butyn-1-bromide or 3-butyn-1-iodide and 2-oxazolidinone failed due to an elimination reaction. In order to minimize the competitive elimination reaction, the halide leaving group was changed to a tosylate, which favors nucleophilic substitution over elimination when a competition between SN2 and E2 processes is involved.16 Since 2-oxoazolidinone has tautomers under certain conditions, the alkyation may give rise to N-alkyl derivatives or O-alkyl derivatives. Although it is known that alkylation of 2-oxoazolidinone with 4-bromo-1-butene results in N-alkylation, it is still difficult to estimate the effect of the reaction medium, the alkylator and other factors on the regiochemistry of the process. Phase-transfer catalysis is widely used as a way to influence the product ratio in alkylation of ambident anions.17,18 Taking this into account, the reaction of 2-oxazolidinone with 3-butyl-1-tosylate in the presence of tetrabutylammonium bromide and potassium carbonate was examined in dichloromethane and water at reflux temperature, but no product was formed. Eventually the alkylation of 2-oxazolidinone with 3-butynyl-1-tosylate (24) in the presence of tetrabutylammonium bromide in toluene was carried out successfully to afford 3-but-3-ynyl-1,3-oxazolidin-2-one (25) in 83% yield.
5-Iodo-3-methyl-2-methoxybenzoate (29) was previously synthesized from methyl 2-hydroxy-5-iodo-3-methylbenzoate (28) with dimethyl sulfate and potassium carbonate in refluxing acetone.10 Methyl 2-hydroxy-5-iodo-3-methylbenzoate (28) was prepared from 3-methyl salicylic acid (26).10 In the reported route, 3-methyl salicylic acid (26) was converted into its methyl ester 27 using (trimethylsilyl)diazomethane in a mixture of methanol and benzene. The product 27 was then iodinated with sodium iodide in the presence of sodium hypochlorite and sodium hydroxide. The overall yield was very good. However, an excess of (trimethylsilyl)diazomethane had to be added to complete the transformation of 26 to 27.
In order to avoid using the expensive reagent (trimethylsilyl)diazomethane, a new synthetic method has been developed to synthesize 29. 3-Methyl salicylic acid (26) was converted into its methyl ester 27 using dimethyl sulfate and potassium carbonate in the presence of tetrabutylammonium bromide in a mixture of dichloromethane and water at room temperature. The methyl ester 27 was iodinated with sodium iodide in the presence of sodium hypochlorite and sodium hydroxide to afford methyl 2-hydroxy-5-iodo-3-methylbenzoate (28) in 98% overall yield. 5-Iodo-3-methyl-2-methoxybenzoate (29) was synthesized in 91% yield from methyl 2-hydroxy-5-iodo-3-methylbenzoate (28) with dimethyl sulfate in the presence of tetrabutylammonium bromide and sodium hydroxide in a mixture of dichloromethane and water at room temperature. The new method using phase transfer catalysis is superior to the established method. First of all, the very expensive reagent (trimethylsilyl)diazomethane and anhydrous solvents, methanol and benzene, are avoided. Second, these reactions occur rapidly at room temperature, and the work-up is very easy. Therefore, these reactions can be scaled up easily.
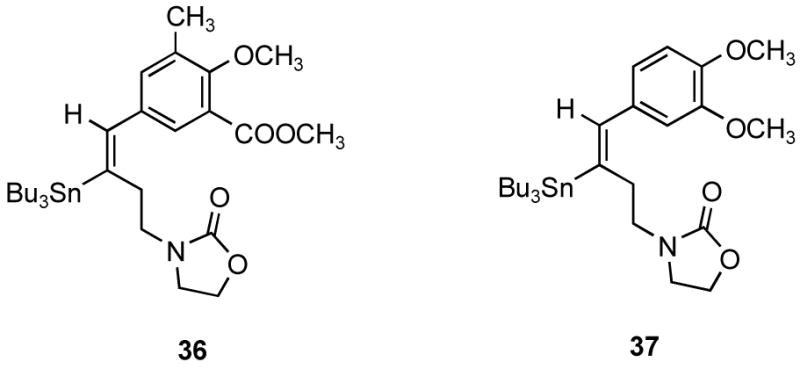
The Sonogashira reaction of compound 25 with iodo compounds 29 or 33 afforded alkynes 30 or 34 in a good yield. In our initial attempts, the hydrostannation of the alkynes 30 or 34 in the presence of Pd(PPh3)4 to afford the regio- and stereodefined vinylstannane 31 or 35 proceeded with low conversions. The process of Pd-catalyzed addition of trialkyl-stannanes to triple bonds is generally best for terminal alkynes, and yields and selectivity suffer with sterically hindered internal alkynes. A general problem is that the activated Pd-H intermediate Bu3Sn-Pd-H can proceed down an alternate path by irreversible generation of H2, dimerization of the stannane, and precipitation of the Pd as palladium black. Attempts were made to optimize this reaction by using Pd(PPh3)2Cl2 as the catalyst and employing slow addition of the Bu3SnH. The results were still disappointing. This undesired side reaction could be minimized by maintaining a low concentration of tin hydride, which is more readily achieved by dropwise addition as mentioned in the literature.19 The concentration of catalyst and the temperature might also be manipulated to minimize the side reactions. Therefore, the hydrostannations were preformed with low concentrations of the alkynes 30 or 34 by a very slow dropwise addition of tributyltin hydride with low load of catalyst at 0 °C to afford the regio- and stereodefined vinylstannanes 31 or 35 in excellent yields. Two side products 36 and 37 were also isolated as minor products.
The palladium-catalyzed cross-coupling reaction between aryl or vinyl halides and triflates with organostannanes, well known as the Stille coupling,20 has been developed in the recent decades into an extremely popular synthetic tool for the construction of carbon-carbon bonds.21,22 The three-step catalytic cycle including oxidative addition, transmetallation, and reductive elimination has been proposed for the Stille reaction.23 It is known that the transmetallation is very often the rate-determining step of the Stille reaction. Unfortunately, use of the Stille coupling of aryl iodide and vinylstannane to reach the final products 3 and 4 was unsuccessful by using different catalysts [Pd2(dba)3, or Pd(PPh3)4], solvents (toluene, dioxane, THF), additives (CsF, PBut3 or PPh3) and temperatures (room temperature or reflux temperature). The aryl iodide decomposed and the vinylstannane was partly recovered. Based on these initial results, improving the rate of the transmetallation step of the Stille coupling became crucial in the successful execution of this reaction. It has been shown that under certain conditions, copper iodide reacts with organostannanes to produce transient organocopper intermediates that are presumably more reactive than organostannanes towards transmetallation to palladium.24 However, the reaction of vinylstannane 31 with aryl iodide 33 in the presence of CuI with CsF and AsPh3 in THF at reflux temperature only led to the recovery of starting materials. Compound 3 was finally synthesized in about 43% yield in the highly polar solvent 1-methyl-2-pyrrolidinone (NMP) at 80 °C. The Suzuki coupling22,25 of the vinyl iodide 32 with 3,4-dimethoxyphenylboronic acid in the presence of palladium acetate and 2-(di-t-butylphosphine)biphenyl also afforded compound 3 in 62% optimized yield. However, the synthesis of compound 4 using a similar method did not work very well. Compound 4 was finally synthesized in 77% yield from the vinylstannane 35 with aryl iodide 29 in the presence of Pd(PBut3)2 with CsF in toluene at reflux temperature. These conditions are very general and practical and have been employed to synthesize all of ADAMs as shown in the following schemes.
The stereoselective syntheses of alkenyldiarylmethanes 7, 8 and 9 with trifluoromethyl compounds 38 or 39 are outlined in Scheme 2. The synthesis relies on the Stille coupling of 1-bromo-3-fluoro-5-trifluoromethylbenzene (38) or 2-bromo-4-fluoro-1-trifluoromethylbenzene (39) with the tributyltin derivatives 31 or 35 in the presence of Pd(PBut3)2 with CsF in toluene at reflux temperature.
Scheme 2a.
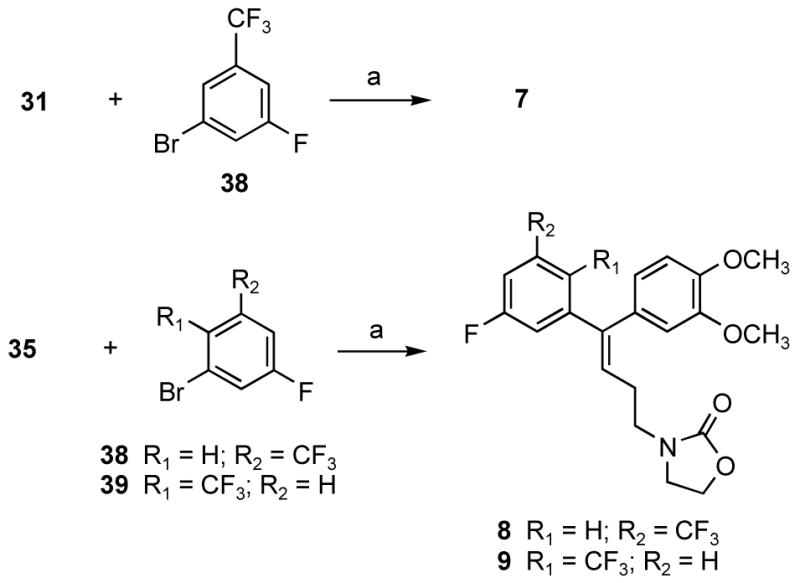
aReagents and conditions: (a) Pd(PBut3)2, toluene, reflux.
Employing the same strategies used to synthesize alkenyldiarylmethanes (ADAMs) having nonidentical aromatic substituents, we synthesized vinylstannanes 43, 44, 51, and 52. As outlined in Scheme 3, the methylation of the phenol group of 45 using dimethyl sulfate in the presence of sodium hydroxide and tetrabutylammonium bromide as a phase-transfer catalyst in a mixture of dichloromethane and water at room temperature gave 2-bromo-4-chloro-1-methoxybenzene (47) in very good yield. Methyl 5-bromo-2-methoxy-3-methylbenzoate (48) was synthesized by O-alkylation of 5-bromo-3-methylsalicylic acid (46) with dimethyl sulfate in acetone at reflux temperature, utilizing potassium carbonate as the base. The Sonogashira coupling of methyl 5-hexynoate with substituted aromatic bromides 38, 40, 47, and 48, followed by hydrostannation with tributyltin hydride, yielded vinyl stannanes 43, 44, 51, and 52 in very good yield.
Scheme 3a.
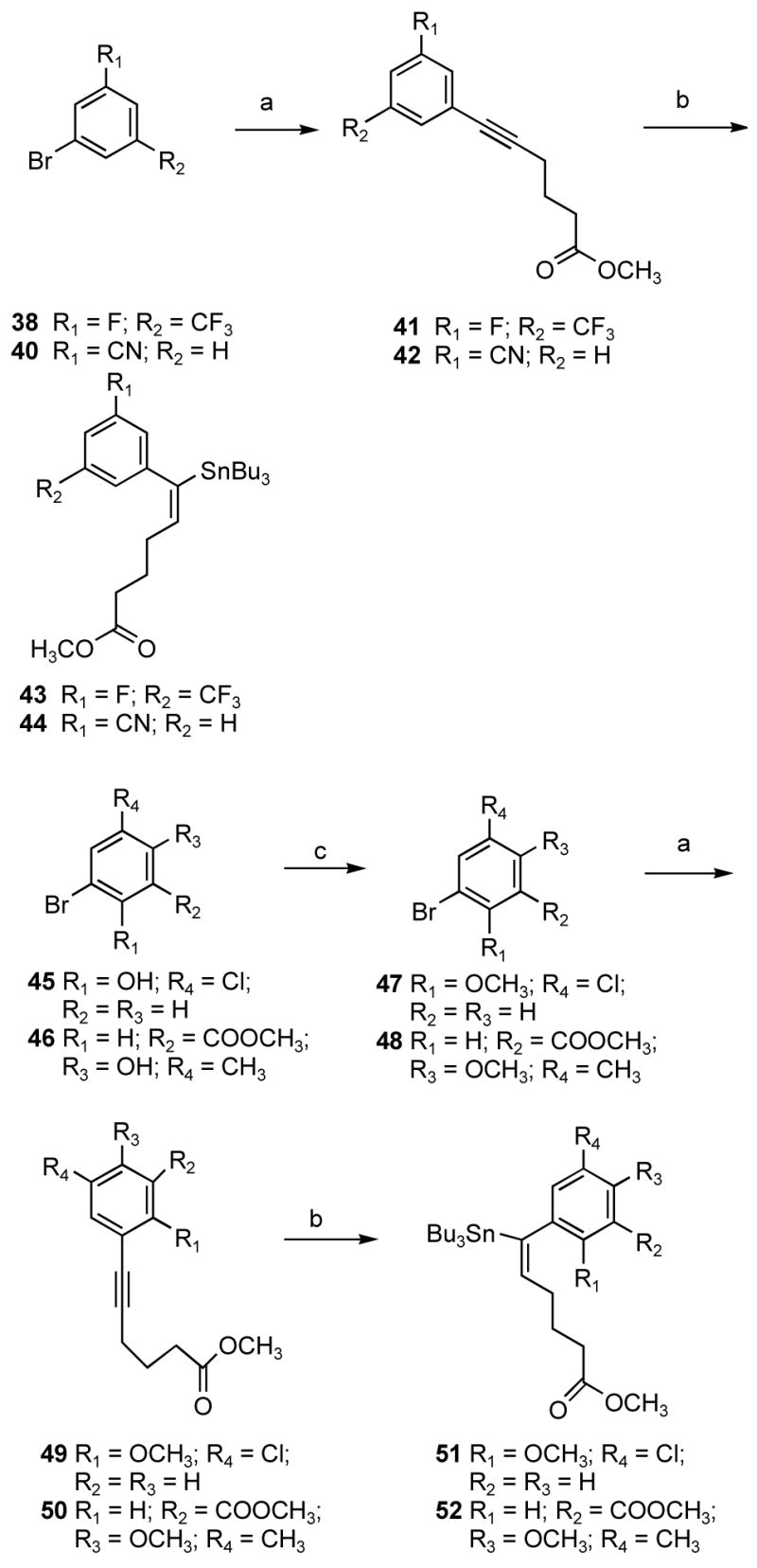
aReagents and conditions: (a) methyl 5-hexynoate, PdCl2(PPh3)2, Cu(I)I, Et3N, THF; (b) Bu3SnH, Pd(PPh3)4, THF, room temperature; (c) for 45: dimethyl sulfate, NaOH, Bu4NBr, CH2Cl2-H2O, room temperature; for 46: dimethyl sulfate, potassium carbonate, acetone, reflux.
As outlined in Scheme 4, methylation of both the phenol and carboxylic acid groups of 53 using dimethyl sulfate in the presence of sodium hydroxide and tetrabutylammonium bromide as the phase-transfer catalyst in a mixture of dichloromethane and water at room temperature gave methyl 5-iodo-2-methoxybenzoate 54. However, chlorination of the aromatic ring at the C3 position of compound 54 with SO2Cl2 in dichloromethane did not give reproducible results.10 Therefore, the reaction was carried out without solvent at 50 °C to afford the ester 55 in very good yield. The Stille approach was also employed for the synthesis of ADAMs having nonidentical aromatic rings as shown in Scheme 4. The Stille coupling of vinyl stannanes 43, 44, 51, and 52 with different halo aromatic derivatives in the presence of Pd(PBut3)2 with CsF in toluene at reflux temperature afforded alkenyldiarylmethanes 10–18.
Scheme 4a.
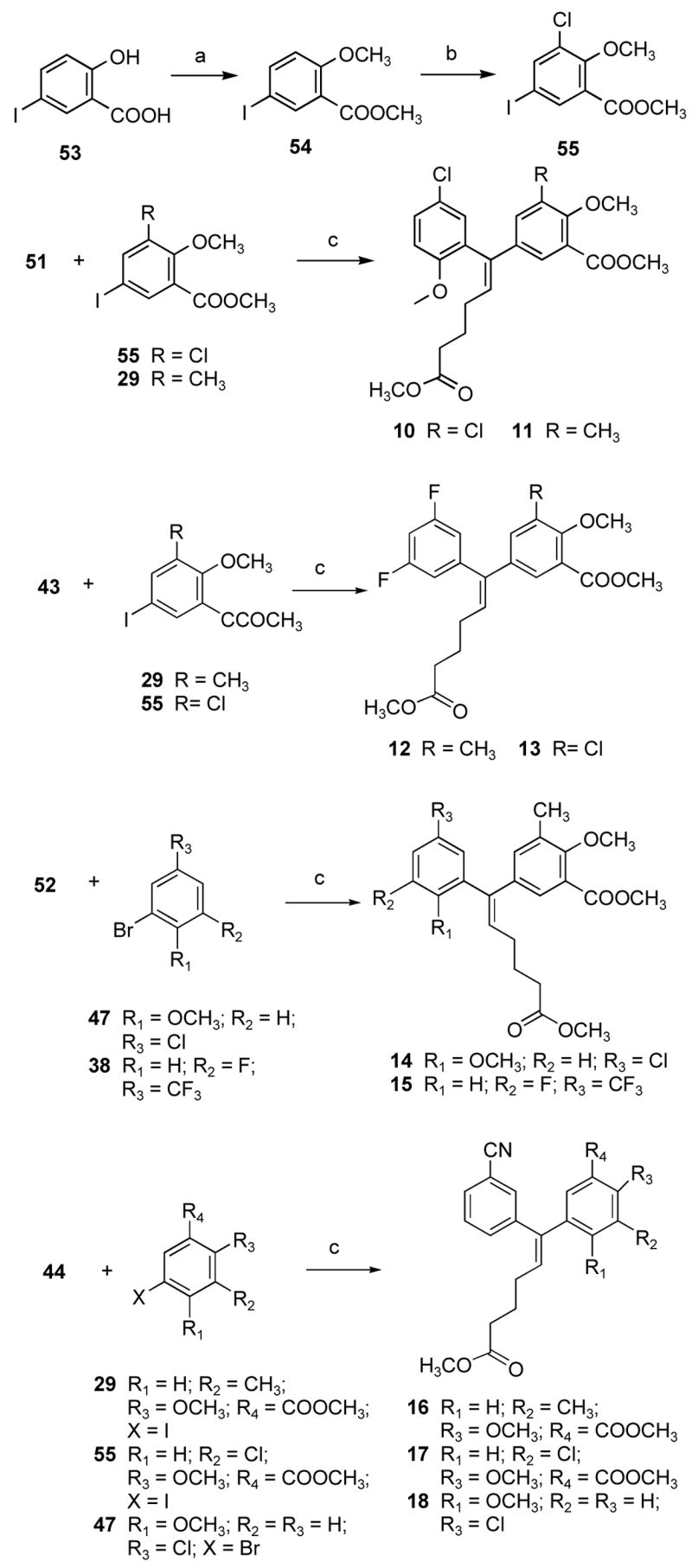
aReagents and conditions: (a) dimethyl sulfate, NaOH, TBAB, CH2Cl2-H2O, room temperature; (b) SO2Cl2, 50 °C; (c) Pd(PBut3)2, toluene, reflux.
A methoxylated econazole replacement for the ester group and the neighboring methoxyl group on the aromatic ring in compounds 12 and 16 was proposed in ADAMs 19 and 20. In these compounds, the nitrogen of the isoxazole mimics the carbonyl oxygen of the ester, and the methoxy group on the isoxazole mimics the methoxy group on the methyl ester. This type of methyl ester replacement was previously developed to substitute for the methyl ester present in norarecoline.26 These compounds are obviously conformationally constrained ester mimics that may provide information about the biologically active conformation of the corresponding methyl ester in the ADAMs. As outlined in Scheme 5, 2,N-dihydroxy-5-iodo-3-methylbenzamide (56) was prepared from methyl 2-hydroxy-5-iodo-3-methylbenzoate (28) and hydroxylamine hydrochloride in very good yield. 5-Iodo-7-methylbenzo[d]isoxazol-3-one (57) was synthesized from 2,N-dihydroxy-5-iodo-3-methylbenzamide (56) and carbonyldiimidazole in almost quantitative yield.27 Methylation of 5-iodo-7-methylbenzo[d]isoxazol-3-one (57) with iodomethane afforded 5-iodo-2,7-dimethyl-benzo[d]isoxazol-3-one (58) and 5-iodo-3-methoxy-7-methylbenzo[d]isoxazole (59). The structure of 5-iodo-2,7-dimethyl-benzo[d]isoxazol-3-one (58) was confirmed by X-ray crystallography.
Scheme 5a.
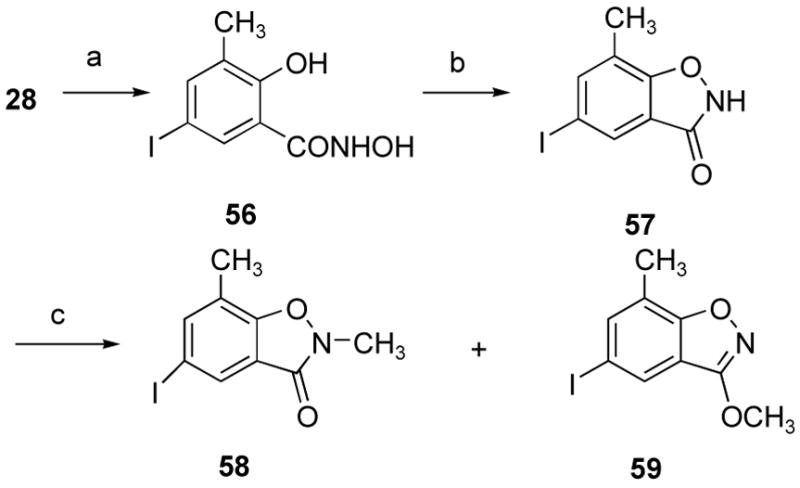
aReagents and conditions: (a), N2HOH.HCl, KOH, MeOH; (b), carbonyldiimidazole, THF, reflux; (c), K2CO3, DMSO, CH3I.
The Stille approach was employed for the synthesis of ADAMs 19–22 having isoxazole as shown in Scheme 6. The Stille coupling of vinyl stannanes 43, 44, and 51 with different halo aromatic derivatives in the presence of Pd(PBut3)2 with CsF in toluene at reflux temperature afforded alkenyldiarylmethanes 19–22. Compound 22 was recrystallized from a mixture of ethyl acetate and hexanes to yield colorless needles. The structure of compound 22 was confirmed by X-ray crystallography. Since the Stille coupling leads to the retention of the stereochemical integrity of the coupling partners, the E stereochemistry of the alkene 43 confirmed the regioselective cis addition during the hydrostannation reaction of the alkyne 41.
Scheme 6a.
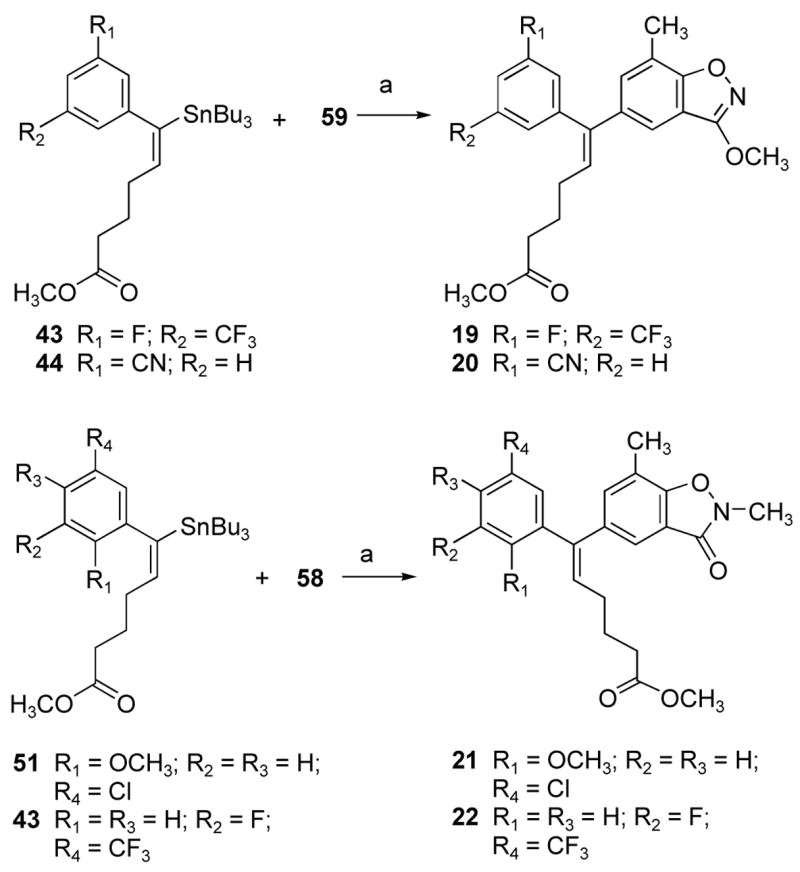
aReagents and conditions: (a) Pd(PBut3)2, toluene, reflux.
Biological Results and Discussion
The 18 new ADAMs synthesized in this study were evaluated for prevention of the cytopathic effect of HIV-1RF in CEM-SS cells and for cytotoxicity in uninfected CEM-SS cells. The biological data are listed in Table 1. The ADAMs were also tested for their ability to inhibit HIV-1 RT, and the resulting IC50 values are also included in Table 1. Ten analogues were found to inhibit HIV-1RT with poly(rC)·oligo(dG) as the template primer with IC50 values ranging from 0.75 to 97.8 μM. Four ADAMs also prevented the cytopathic effect of HIV-1RF with EC50 values ranging from 7.0 to 8.3 μM. Sixteen ADAMs were tested for inhibition of cytopathic effects of both HIV-1IIIB and HIV-2ROD in MT-4 cells, and the resulting EC50 values are also listed in Table 1, along with their cytotoxicities (CC50 values) in uninfected MT-4 cells. Five compounds displayed EC50 versus HIV-1IIIB between 2.54 to 12.8 μM. In addition, the metabolic stabilities of the ADAMs were investigated in rat plasma, and the resulting half-lives of the compounds are also summarized in Table 1.
Table 1.
Anti-HIV Activities, Cytotoxicities, and Metabolic Stabilities of ADAM Analogues
| Comp. | IC50(μM)a | EC50(μM)b |
CC50(μM)c |
Rat Plasma t1/2(min±SD) | |||
|---|---|---|---|---|---|---|---|
| HIV-1RF | HIV-1IIIB | HIV-2ROD | CEM-SS Cells | MT-4 Cells | |||
| 1 | 0.499 | 0.02 | NTd | NTd | 1.95 | NTd | NTd |
| 2 | 0.074 | 0.21 | NTd | NTd | 1.14 | NTd | NTd |
| 3 | 1.0 | >100 | >1.05 | >1.05 | 0.49 | 1.05 | 5.76 ± 0.68 |
| 4 | >100 | >100 | >33.8 | >35.4 | 4.79 | 82.5 | 0.79 ± 0.10 |
| 7 | 15.7 | 8.30 | 9.60 | >22.6 | 15.0 | 24.7 | 9.39 ± 0.73 |
| 8 | 97.8 | >100 | >18.5 | >13.4 | 14.5 | 18.5 | NHe |
| 9 | >100 | >100 | NTd | NTd | 26.7 | NTd | NHe |
| 10 | >100 | >100 | >5.41 | >5.41 | 9.3 | 5.41 | 0.77 ± 0.08 |
| 11 | >100 | >100 | >13.8 | >13.8 | 15.5 | 13.8 | 0.98 ± 0.02 |
| 12 | >100 | >5.0 | >5.51 | >5.89 | >5.0 | 5.70 | NTd |
| 13 | >100 | >100 | NTd | NTd | 16.1 | NTd | NTd |
| 14 | >100 | >100 | >18.8 | >14.5 | 8.3 | 18.8 | 0.47 ± 0.02 |
| 15 | 55.3 | 7.30 | 2.54 | 10.6 | 12.0 | 113 | 4.35 ± 0.41 |
| 16 | 0.84 | >100 | 12.8 | >169 | 23.3 | 153 | 0.71 ± 0.01 |
| 17 | 0.75 | 7.1 | 8.55 | >223 | 20.2 | 213 | 0.44 ± 0.05 |
| 18 | 7.58 | 7.0 | 9.11 | >67.6 | 16.3 | ≥45.2 | 0.42 ± 0.02 |
| 19 | 0.9 | >100 | >5.76 | >5.60 | 2.1 | 5.76 | 4.26 ± 0.01 |
| 20 | 5.7 | >100 | >2.05 | >5.63 | 0.8 | 4.69 | 1.46 ± 0.08 |
| 21 | 8.2 | >100 | >28.8 | >28.8 | 13.0 | >28.8 | 0.48 ± 0.06 |
| 22 | >100 | >100 | >35.7 | >43.2 | 16.8 | 39.8 | 0.87 ± 0.12 |
Inhibitory activity vs HIV-1 reverse transcriptase with rCdG as the template primer.
EC50 is the 50% inhibitory concentration for inhibition of cytopathicity of HIV-1RF, HIV-1IIIB or HIV-2ROD.
The CC50 is the 50% cytotoxic concentration for mock-infected CEM-SS cells or MT-4 cells.
Not tested.
Not hydrolysized. All data are derived from triplicate tests with the variation of the mean averaging 10%.
Based on the data in the Table 1, ADAM 3 that replaces the methyl ester moiety of ADAM 2 with a 2-oxoxazolidinyl group still retained the enzyme inhibitory potency of the parent compound. However, it was generally cytotoxic and lost the ability to inhibit the cytopathic effect of the virus in cells at non-cytotoxic concentrations. Compound 4, with exchanged the aromatic rings relative to compound 3, lost both enzyme inhibitory and antiviral activities. Introducing 5-chloro-2-methoxyphenyl or 3-fluoro-5-trifluoromethylphenyl groups into alkenyldiarylmethanes does not retain the antiviral potency, except in the case of ADAMs 15 and 18. The alkenyldiarylmethanes 16–18 with a 3-cyanophenyl group displayed both enzyme inhibitory and antiviral activities.
The stabilities of ADAMs 3, 4, 8–11, 14–18, 20, 21 in rat plasma were investigated. The ADAMs displayed a range of metabolic stabilities in rat plasma, with half-lives ranging from 0.4 to 9.4 min, except compounds 8 and 9. Compounds 8 and 9, which lack any methyl ester moieties, had not been hydrolyzed at 37 °C after three days. This demonstrates that the oxazolidinonyl group is stable in rat plasma and that the metabolic instabilities of the ADAMs are likely to be due to the presence of methyl esters.
Conclusions
The main conclusions and significant accomplishments resulting from the present study may be summarized in the following points: (1) The structural modifications introduced in the present series of ADAMs document the fact that a 3-cyanophenyl can be incorporated into the ADAM system with retention of anti-HIV activity. In addition, the hydrolysis data in rat plasma indicate that the oxazolidinonyl group is stable in the ADAMs. These studies will facilitate the future development of novel metabolically stable ADAMs with enhanced bioavailabilities and improved therapeutic potentials for the treatment of AIDS. (2) An efficient route was devised to prepare intermediate 29 on large scale, and general methods were developed to synthesize ADAMs using metal-catalyzed reactions (Sonogashira reaction, hydrostannation, Stille coupling and Suzuki coupling). In particular, the protocol used for hydrostannation of sterically hindered internal alkynes is an improvement over the literature procedures. The general flexibility of the route opens up the opportunity to synthesize the other alkenyldiarylmethanes (ADAMs) having nonidentical aromatic substituents with optimized biological activities. (3) This investigation demonstrates the use of phase-transfer catalyzed reactions to synthesize several intermediates, such as 25, 27, 29, 47, 54, in good yields. This offers several advantages, such as operational simplicity, mild reaction conditions in aqueous media, environmental benefits, and suitability for large-scale reactions. (4) The structures of a representative ADAM compound 22, as well as the intermediate 58, were determined by X-ray crystallography. The crystal structure of 58 establishes the outcome of the alkylation reaction used to prepare both 58 and 59, while the structure of 22 confirms the expected regiochemistry and stereochemistry of hydrostannation. (5) The inactivity of ADAM 12 indicates that its hypothetical structural analogy to the very potent NNRTI 514 is weak, at least as far as its enzyme inhibitory activity is concerned. It is likely that the ADAMs bind to the enzyme differently than NNRTIs 5 and 6. On the other hand, the concept did eventually result in the active cyanophenyl ADAMs 16–18. In addition, the ADAM 19, having meta F and CF3 groups, is a submicromolar RT inhibitor, although it proved to be too cytotoxic to be of value as an anti-HIV agent. In the case of ADAM 15, which is an isomer of 12, anti-HIV activity was observed, although the weak activity of 15 vs. RT and the activity of the compound vs. HIV-2ROD would argue that the antiviral activity of compound 15 is not due to inhibition of RT. (6) A reduction in the total number of ester groups in the molecule does not necessarily result in a more metabolically stable ADAM, even in compounds with similar structures. An example of this is provided by comparison of the half-lives of 14 (t1/2 0.47 min), 18 (t1/2 0.42 min), and 21 (t1/2 0.48 min). It appears that the terminal ester on the alkenyl side chain is a more important contributor to metabolic instability than the ester on the aromatic ring. (7) The structures of the lactim ether 19 and the amide 22 are very similar, but differences in their enzyme inhibitory activities are striking. ADAM 19 displays an IC50 of 0.9 μM vs. HIV-1 RT, while 22 is inactive. Also, compound 19 is more stable in plasma (t1/2 4.26 min) than 22 (t1/2 0.87 min) even though the structural change is relatively remote from the labile ester. (8) The stereochemical orientation of the oxazolidinonyl side chain in ADAMs 3 and 4 has a significant effect on the hydrolysis rate of the methyl ester. Whereas the ester 3 has a half-life of 5.76 min in rat blood plasma, the half-life of its isomer 4 is 0.79 min. It appears that the oxazolidinonyl side chain that is cis to the methyl ester in compound 3 may retard its hydrolysis due to a steric effect. (8) The long-range objective of designing and synthesizing a metabolically stable ADAM analogue that retains significant antiviral activity or has enhanced antiviral activity was not realized in the present series of compounds.
Experimental Section
NMR spectra were obtained at 300 MHz (1H) and 75 MHz (13C) in CDCl3 using CHCl3 as internal standard. Flash chromatography was performed with 230–400 mesh silica gel. TLC was carried out using Baker-flex silica gel IB2-F plates of 2.5 mm thickness. Melting points are uncorrected. Unless otherwise stated, chemicals and solvents were of reagent grade and used as obtained from commercial sources without further purification. Tetrahydrofuran (THF) was freshly distilled from sodium/benzophenone ketyl radical prior to use. Dichloromethane was freshly distilled from calcium hydride prior to use. Lyophilized rat plasma (lot 052K7609) was obtained from Sigma Chemical Co., St. Louis, MO. All yields given refer to isolated yields.
(E)-6-[1-3,4-Dimethoxyphenyl]-4-(2-oxo-oxazolidin-3-yl)-but-1-enyl]-2-methoxy-3-methyl-benzoic Acid Methyl Ester (3)
Method I
A mixture of the vinylstannane 31 (100 mg, 0.164 mmol), aryl iodide 33 (44.7 mg, 0.169 mmol), triphenylarsine (21.3 mg, 0.068 mmol), copper(I) iodide (10.0 mg, 0.053 mmol) and tris(dibenzylideneacetone)dipalladium (15 mg, 0.016 mmol) in 1-methyl-2-pyrrolidinone (NMP) (4 mL) was heated at 80 °C under argon atmosphere for 14 h, cooled to room temperature, filtered through a pad of Celite, and washed with ethyl acetate and dichloromethane. The filtrate was concentrated to afford a residue. The residue was purified by column chromatography on silica gel (50 g) using ethyl acetate in hexanes (0–30%) to afford the product 3 (32 mg) in 43% yield.
Method II
A mixture of the vinyl iodide 32 (353 mg, 0.793 mmol), palladium acetate (9.0 mg, 0.039 mmol), 2-(di-t-butylphosphine)biphenyl (24.2 mg, 0.080 mmol), potassium fluoride (147 mg, 2.48 mmol) and 3,4-dimethoxyphenylboronic acid (321 mg, 1.764 mmol) in THF (4.0 mL) was stirred at room temperature for 14 h and at 60 °C for 8.5 h. The reaction mixture was cooled to room temperature. Ethyl ether (30 mL) was added to dilute the mixture. The mixture was washed with aqueous 10% potassium hydroxide (2 × 20 mL). The organic phase was collected and washed with brine (30 mL). The aqueous solution was extracted with ethyl acetate (3 × 15 mL), washed with brine (20 mL), and then combined with the ether solution, dried over anhydrous sodium sulfate and concentrated. The residue was purified by column chromatography on silica gel (40 g) using ethyl acetate in hexanes (0–50%) to afford the product 3 (0.224 g) as an oil in 62% yield. IR (KBr film): 2926, 2851, 1751, 1729, 1599, 1580, 1513, 1481, 1438, 1378, 1262, 1142, 1025, 804, 762 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.42 (d, J = 2.4 Hz, 1 H), 7.12 (d, J = 1.5 Hz, 1 H), 6.80 (d, J = 2.1 Hz, 1 H), 6.74 (d, J = 8.4 Hz, 1 H), 6.63 (dd, J = 2.1 Hz, J = 8.4 Hz, 1 H), 5.92 (t, J = 7.5 Hz, 1 H), 4.23 (t, J = 8.1 Hz, 2 H), 3.88 (s, 3 H), 3.86 (s, 3 H), 3.37 (t, J = 6.9 Hz, 2 H), 3.32 (t, J = 8.1 Hz, 2 H), 2.37 (m, 2 H), 2.30 (s, 3 H); 13C NMR (75 MHz, CDCl3) δ 166.8, 158.5, 157.5, 148.6, 142.7, 136.3, 135.1, 135.0, 132.8, 130.2.124.4, 120.3, 110.7, 110.5, 61.6, 56.0, 55.9, 52.2, 44.2, 44.0, 27.9, 16.1; ESIMS m/z (rel intensity) 478 (MNa+, 100). Anal. (C25H29NO7) C, H, N.
General Procedure for Synthesis of Alkenyldiarylmethanes by the Cross-Coupling Reaction of Vinylstannanes with Aromatic Iodides or Bromides
A mixture of vinylstannane (1 equiv), iodide or bromide (1.2–1.5 equiv), cesium fluoride (3.0–4.5 equiv), and Pd(PBut3)2 (10 mol%) in toluene (1 mL) under argon was stirred at room temperature for 7.3–24 h and at 90–110 °C for 9.5–43 h. The reaction mixture was cooled to room temperature, filtered through a short column of silica gel (5 g), and the column washed with ethyl acetate. The organic solution was concentrated.
(Z)-5-[1-(3,4-Dimethoxyphenyl)-4-(2-oxo-oxazolidin-3-yl)-but-1-enyl]-2-methoxy-3-methyl-benzoic Acid Methyl Ester (4)
The general procedure was followed using the vinylstannane derivative 35 (367.7 mg, 0.649 mmol), iodide 29 (209.6 mg, 0.685 mmol), cesium fluoride (306 mg, 2.0 mmol), and Pd(PBut3)2 (53 mg, 0.102 mmol) in toluene (3 mL). After the mixture was stirred at room temperature for 22 h, and at 80 °C for 5.5 h, compound 29 (103.5 mg, 0.34 mmol) and Pd(PBut3)2 (9.7 mg, 0.019 mmol) were added. The mixture was heated 80 °C for another 16 h. The residue was purified by column chromatography on silica gel (40 g) using ethyl acetate in hexanes (0–50%) to afford the product 4 (227 mg) as an oil in 77% yield. IR (KBr) 2932, 1751, 1579, 1580, 1513, 1481, 1437, 1320, 1257, 1138, 1027, 804, 762 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.43 (d, J = 2.4 Hz, 1 H), 7.17 (d, J = 2.1 Hz, 1 H), 6.87 (d, J = 8.4 Hz, 1 H), 6.71 (dd, J = 2.1 Hz, J = 8.1 Hz, 1 H), 6.61 (d, J = 1.8 Hz), 5.92 (t, J = 7.5 Hz, 1 H), 4.22 (t, J = 8.1 Hz, 2 H), 3.90 (s, 3 H), 3.86 (s, 3 H), 3.82 (s, 3 H), 3.79 (s, 3 H), 3.36 (t, J = 6.9 Hz, 2 H), 3.32 (t, J = 7.2 Hz, 2 H), 2.40 (m, 2 H), 2.24 (s, 3 H); 13C NMR (75 MHz, CDCl3) δ 166.7, 158.1, 157.2, 148.5, 148.0, 142.5, 137.6, 133.6, 132.1, 131,5, 127.4, 124.9, 124.0, 121.8, 112.5, 110.7, 61.4, 61.2, 55.7, 55.6, 51.9, 44.0, 43,7, 27.6, 15.9; ESIMS m/z (rel intensity) 478 (MNa+, 100). Anal. (C25H29NO7) C, H, N.
(E)-5-[1-(3-Fluoro-5-trifluoromethylphenyl)-4-(2-oxo-oxazolidin-3-yl)-but-1-enyl]-2-methoxy-3-methylbenzoic Acid Methyl Ester (7)
The general procedure was followed using the vinylstannane 31 (323 mg, 0.531 mmol), bromide 38 (219 mg, 0.874 mmol), cesium fluoride (280 mg, 1.825 mmol) and Pd(PBut3)2 (28.4 mg, 0.054 mmol) in toluene (1 mL). The mixture was stirred at room temperature for 24 h, at 60 °C for 25 h and at 110 °C for 22 h. The residue was purified by column chromatography on silica gel (30 g), eluting with EtOAc-hexanes (0–50%) to afford the product 7 (82.5 mg) as an oil in 32% yield. IR (KBr) 2952, 1752, 1600, 1482, 1438, 1351, 1264, 1236, 1205, 1171, 1128, 1094, 1034, 1008, 939, 875, 801, 762, 700 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.40 (d, J = 2.4 Hz, 1 H), 7.17 (d, J = 8.1 Hz, 1 H), 7.09 (d, J = 1.8 Hz, 1 H), 7.03 (d, J = 9.6 Hz, 1 H), 6.08 (t, J = 7.5 Hz, 1 H), 4.24 (t, J = 7.8 Hz, 2 H), 3.89 (s, 3 H), 3.84 (s, 3 H), 3.40-3.31 (m, 4 H), 2.38 (m, 2 H), 2.31 (s, 3 H); 13C NMR (75 MHz, CDCl3) δ 166.5, 158.4, 157.9, 145.2, 140.9, 136.0, 133.5, 130.1, 128.8, 124.8, 119.7, 118.0, 111.9, 61.6, 52.4, 44.4, 43.8, 28.1, 16.2; ESIMS m/z (rel intensity) 504.09 (MNa+, 50). Anal. (C24H23F4NO5) C, H, F, N.
(E)-3-[4-(3-Fluoro-5-trifluoromethylphenyl)-4-(3,4-dimethoxyphenyl)-but-3-enyl]-oxazolidin-2-one (8)
The general procedure was followed using the vinylstannane 35 (178.4 mg, 0.315 mmol), bromide 38 (130 mg, 0.519 mmol), cesium fluoride (179 mg, 1.17 mmol) and Pd(PBut3)2 (17.8 mg, 0.034 mmol) in toluene (1 mL). After the mixture was stirred at room temperature for 18 h and at 90 °C for 4 h. More bromide 38 (147.6 mg, 0.589 mmol) and Pd(PBut3)2 (8.8 mg, 0.017 mmol) were added. The mixture was heated at 90 °C for 22.5 h and at 115 °C for 26 h. The residue was purified by column chromatography on silica gel (25 g) using ethyl acetate in hexanes (0–50%) to afford the product 8 (45.2 mg) as an oil in 33% yield. IR (KBr) 2917, 2849, 1751, 1600, 1582, 1514, 1484, 1466, 1441, 1350, 1255, 1232, 1200, 1169, 1130. 1093, 1027, 971, 927, 868, 814, 763, 700 cm–1; 1H NMR (300 MHz, CDCl3) δ 7.25 (s, 1 H), 7.17 (d, J = 8.1 Hz, 1 H), 7.10 (d, J = 9.9 Hz, 1 H), 6.89 (d, J = 8.4 Hz, 1 H), 6.71 (dd, J = 2.1 Hz, J = 8.1 Hz, 1 H), 6.60 (d, J = 1.8 Hz, 1 H), 6.064 (t, J = 7.5 Hz, 1 H), 4.22 (t, J = 7.8 Hz, 2 H), 3.91 (s, 3 H), 3.82 (s, 3 H), 3.38 (t, J = 6.9 Hz, 2 H), 3.32 (t, J = 7.8 Hz, 2 H), 2.43 (q, J = 7.2 Hz, J = 6.9 Hz, 2 H); 13C NMR (75 MHz, CDCl3) δ 164.0, 160.7, 158.3, 149.0, 148.6, 145.8, 141.9, 130.6, 127.7, 122.1, 119.7, 117.8, 117.5, 112.5, 111.4, 111.1, 61.5, 55.9, 55.8, 44.2, 43.8, 27.9; ESIMS m/z (rel intensity) 440 (MH+, 48). Anal. (C22H21F4NO4) C, H, F, N.
(E)-3-[4-(5-Fluoro-2-trifluoromethylphenyl)-4-(3,4-dimethoxyphenyl)-but-3-enyl]-oxazolidin-2-one (9)
The general procedure was followed using the vinylstannane 35 (84 mg, 0.148 mmol), bromide 39 (0.032 mL, 0.224 mmol), cesium fluoride (84 mg, 0.55 mmol), and Pd(PBut3)2 (8.9 mg, 0.017 mmol) in toluene (1 mL). The mixture was stirred at room temperature for 19 h and at 90 °C for 22 h. The residue was purified by column chromatography on silica gel (25 g) using ethyl acetate in hexanes (0–50%) to afford the product 9 (14.7 mg) as an oil in 23% yield and the starting materials 35 (16.3 mg) in 19.4% yield. IR (KBr) 2929, 1751, 1611, 1583, 1514, 1428, 1364, 1310, 1260, 1234, 1160, 1137, 1104, 1047, 1026, 826, 763 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.62 (dd, J = 5.4 Hz, J = 8.7 Hz, 1 H), 7.06-6.96 (m, 2 H), 6.81 (d, J = 8.1 Hz, 1 H), 6.72 (dd, J = 2.1 Hz, J = 8.4 Hz, 1 H), 6.65 (d, J = 1.8 Hz, 1 H), 5.63 (t, J = 7.2 Hz, 1 H), 4.22 (t, J = 7.8 Hz, 2 H), 3.86 (s, 3 H), 3.80 (s, 3 H), 3.40-3.31 (m, 4 H), 2.57 (q, J = 7.2 Hz, J = 6.9 Hz, 2 H); ESIMS m/z (rel intensity) 440 (MH+, 100). Anal. (C22H21F4NO4) C, H, N.
(Z)-3-Chloro-5-[1-(5-chloro-2-methoxyphenyl)-5-methoxycarbonyl-pent-1-enyl]-2-methoxybenzoic Acid Methyl Ester (10)
The general procedure was followed using the vinylstannane 51 (283.8 mg, 0.509 mmol), iodide 55 (254.4 mg, 0.779 mmol), cesium fluoride (355 mg, 2.314 mmol) and Pd(PBut3)2 (32.2 mg, 0.062 mmol) in toluene (1 mL). The mixture was stirred at room temperature for 7.3 h and at 110 °C for 24 h. The residue was purified by column chromatography on silica gel (25 g), eluting with EtOAc-hexanes (0–5%) to afford the product 10 (28 mg) as an oil in 12% yield. IR (KBr) 2950, 1735, 1487, 1436, 1248, 1208, 1130, 1000, 810 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.52 (d, J = 2.4 Hz, 1 H), 7.27 (dd, J = 2.7 Hz, J = 8.7 Hz, 1 H), 7.25 (d, J = 2.4 Hz, 1 H), 6.99 (d, J = 2.7 Hz, 1 H), 6.85 (d, J = 9.0 Hz, 1 H), 6.10 (t, J = 7.5 Hz, 1 H), 3.88 (s, 6 H), 3.67 (s, 3 H), 3.60 (s, 3 H), 2.25 (t, J = 7.5 Hz, 2 H), 2.02-1.95 (m, 2 H), 1.77-1.70 (m, 2 H); 13C NMR (75 MHz, CDCl3) δ 173.8, 166.0, 155.6, 154.4, 138.0, 135.5, 131.6, 130.8, 129.2, 128.9, 127.1, 126.4, 125.5, 112.4, 61.9, 55.7, 52.4, 51.5, 33.3, 29.3, 24.3; ESIMS m/z (rel intensity) 466.78/468.83 (MH+, 85/51). Anal. (C23H24Cl2O6) C, H, Cl.
(Z)-5-[1-(5-Chloro-2-methoxyphenyl)-5-methoxycarbonyl-pent-1-enyl]-2-methoxy-3-methylbenzoic Acid Methyl Ester (11)
The general procedure was followed using the vinylstannane 51 (260 mg, 0.466 mmol), iodide 29 (219 mg, 0.715 mmol), cesium fluoride (222 mg, 1.447 mmol) and Pd(PBut3)2 (21.7 mg, 0.042 mmol) in toluene (1 mL). The mixture was stirred at room temperature for 7.3 h and at 110 °C for 24 h. The residue was purified by column chromatography on silica gel (20 g), eluting with EtOAc-hexanes (0–5%) to afford the product 11 (29 mg) as an oil in 14% yield. IR (KBr) 2949, 1731, 1486, 1436, 1248, 1121, 1008, 884, 810 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.47 (d, J = 2.4 Hz, 1 H), 7.27 (dd, J = 1.2 Hz, J = 10.2 Hz, 1 H), 7.09 (d, J = 2.4 Hz, 1 H), 7.00 (d, J = 2.7 Hz, 1 H), 6.86 (d, J = 8.7 Hz, 1 H), 6.08 (t, J = 7.5 Hz, 1 H), 3.87 (s, 3 H), 3.78 (s, 3 H), 3.68 (s, 3 H), 3.61 (s, 3 H), 2.27 (t, J = 7.5 Hz, 2 H), 2.23 (s, 3 H), 2.02-1.94 (q, 2 H), 1.78-1.68 (m, 2 H); 13C NMR (75 MHz, CDCl3) δ 173.9, 167.0, 157.3, 155.7, 136.6, 136.4, 132.8, 132.3, 130.8, 130.2, 129.8, 128.5, 126.7, 125.4, 124.2, 112.3, 61.5, 55.8, 52.2, 51.5, 33.4, 29.3, 24.5, 16.2; ESIMS m/z (rel intensity) 469.12 (MNa+, 100), 471.04 (MNa+, 34). Anal. (C24H27ClO6) C, H, Cl.
(Z)-5-[1-(3-Fluoro-5-trifluoromethylphenyl)-5-methoxycarbonyl-pent-1-enyl]-2-methoxy-3-methylbenzoic Acid Methyl Ester (12)
The general procedure was followed using the vinylstannane 43 (250 mg, 0.43 mmol), iodide 29 (201.8 mg, 0.66 mmol), cesium fluoride (230 mg, 1.5 mmol) and Pd(PBut3)2 (25 mg, 0.048 mmol) in toluene (1 mL). The mixture was stirred at room temperature for 7 h and at 110 °C for 24 h. The residue was purified by column chromatography on silica gel (20 g), eluting with EtOAc-hexanes (0–10%) to afford the product 12 (118 mg) as an oil in 58% yield. IR (KBr) 2953, 1734, 1599, 1481, 1437, 1375, 1320, 1264, 1234, 1209, 1170, 1130, 1090, 1008, 937, 882, 800, 770, 702 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.42 (d, J = 2.4 Hz, 1 H), 7.28 (m, 1 H), 7.19 (s, 1 H), 7.05-7.02 (m, 2 H), 6.04 (t, J = 7.5 Hz., 1 H), 3.88 (s, 3 H), 3.80 (s, 3 H), 3.61 (s, 3 H), 2.28 (t, J = 7.5 Hz, 2 H), 2.24 (s, 3 H), 2.13-2.06 (q, 2 H), 1.82-1.72 (m, 2 H); 13C NMR (75 MHz, CDCl3) δ 173.6, 166.8, 164.0, 160.7, 157.7, 142.8, 139.2, 136.6, 133.6, 132.8, 130.9, 127.5, 124.6, 122.3, 120.4, 120.1, 111.9, 111.6, 61.5, 52.2, 51.5, 33.3, 29.0, 24.8, 16.1; ESIMS m/z (rel intensity) 491.13 (MNa+, 100). Anal. (C24H24F4O5) C, H, F.
(E)-3-Chloro-5-[1-(3-fluoro-5-trifluoromethylphenyl)-5-methoxycarbonyl-pent-1-enyl]-2-methoxy-benzoic Acid Methyl Ester (13)
The general procedure was followed using the vinylstannane 43 (337 mg, 0.58 mmol), iodide 55 (289 mg, 0.885 mmol), cesium fluoride (300 mg, 1.96 mmol) and Pd(PBut3)2 (31 mg, 0.059 mmol) in toluene (1 mL). The mixture was stirred at room temperature for 23 h, at 60 °C for 24 h and at 100 °C for 24 h. The residue was purified by column chromatography on silica gel (20 g), eluting with EtOAc-hexanes (0–5%) to afford the product 13 (13.2 mg) as an oil in 5% yield. IR (KBr) 2953, 1737, 1599, 1478, 1438, 1374, 1314, 1264, 1207, 1171, 1131, 1093, 1000, 929, 878, 797, 744, 702 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.46 (d, J = 2.4 Hz, 1 H), 7.30 (m, 2 H), 7.26 (d, J = 7.26, 1 H), 7.18 (s, 1 H), 6.09 (t, J = 7.5 Hz, 1 H), 3.91 (s, 3 H), 3.87 (s, 3 H), 3.60 (s, 3 H), 2.28 (t, J = 7.5 Hz, 2 H), 2.14-2.07 (q, 2 H), 1.82-1.72 (m, 2 H); ESIMS m/z (rel intensity) 511.18 (MNa+, 100). Anal. (C23H21ClF4O5) C, H, Cl, F.
(E)-5-[1-(5-Chloro-2-methoxyphenyl)-5-methoxycarbonyl-pent-1-enyl]-2-methoxy-3-methylbenzoic Acid Methyl Ester (14)
The general procedure was followed using the vinyl tributylstannane 52 (166 mg, 0.279 mmol), bromide 47 (111 mg, 0.50 mmol), cesium fluoride (122 mg, 0.787 mmol) and Pd(PBut3)2 (13.2 mg, 0.025 mmol) in toluene (1 mL). The mixture was stirred at room temperature for 2 h and at 110 °C for 27.5 h. The residue was purified by column chromatography on silica gel (25 g), eluting with EtOAc-hexanes (0–5%) to afford the product 14 (50.1 mg) as an oil in 40% yield. IR (KBr) 2950, 1732, 1591, 1484, 1436, 1366, 1291, 1252, 1231, 1195, 1170, 1124, 1010, 883, 808 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.39 (d, J = 2.1 Hz, 1 H), 7.16 (dd, J = 2.7 Hz, J = 8.7 Hz, 1 H), 7.10 (d, J = 2.7 Hz, 1 H), 7.07 (d, J = 2.1 Hz, 1 H), 6.72 (d, J = 8.7 Hz, 1 H), 3.87 (s, 3 H), 3.79 (s, 3 H), 3.61 (s, 3 H), 3.55 (s, 3 H), 2.29 (m, 2 H), 2.26 (s, 3 H), 2.19 (m, 2 H), 1.75 (m, 2 H); ESIMS m/z (rel intensity) 469 (MNa+, 100), 471 (MNa+, 39). Anal. (C24H27ClO6) C, H, Cl.
(E)-5-[1-(3-Fluoro-5-trifluoromethylphenyl)-5-methoxycarbonyl-pent-1-enyl]-2-methoxy-3-methylbenzoic Acid Methyl Ester (15)
The general procedure was followed using the vinylstannane 52 (365 mg, 0.613 mmol), bromide 38 (268 mg, 1.07 mmol), cesium fluoride (358 mg, 2.33 mmol) and Pd(PBut3)2 (34.2 mg, 0.066 mmol) in toluene (1 mL). The mixture was stirred at room temperature for 5.5 h, at 60 °C for 16 h and at 100 °C for 26 h. The residue was purified by column chromatography on silica gel (25 g), eluting with EtOAc-hexanes (0–5%) to afford the product 15 (210.5 mg) as an oil in 73% yield. IR (KBr) 2952, 1734, 1600, 1437, 1350, 1257, 1201, 1169, 1128, 1094, 1009, 873 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.38 (d, J = 2.1 Hz, 1 H), 7.28 (s, 1 H), 7.16 (d, J = 8.1 Hz, 1 H), 7.07 (d, J = 2.1 Hz, 1 H), 6.96 (d, J = 9.9 Hz, 1 H), 6.09 (d, J = 7.5 Hz, 1 H), 3.89 (s, 3 H), 3.86 (s, 3 H), 3.61 (s, 3 H), 2.31 (s, 3 H), 2.27 (m, 2 H), 2.14 (m, 2 H), 1.77 (m, 2 H); ESIMS m/z (rel intensity) 491 (MNa+, 100). Anal. (C24H24F4O5) C, H, F.
(E)-5-[5-Carboxy-1-(3-cyanophenyl)-pent-1-enyl]-2-methoxy-3-methyl benzoic Acid Methyl Ester (16)
The general procedure was followed using the vinylstannane 44 (212 mg, 0.409 mmol), iodide 29 (197.5 mg, 0.645 mmol), cesium fluoride (190 mg, 1.24 mmol) and Pd(PBut3)2 (23 mg, 0.044 mmol) in toluene (1 mL). The mixture was stirred at room temperature for 65 h, at 65 °C for 8.5 h and at 110 °C for 22 h. The residue was purified by column chromatography on silica gel (25 g), eluting with EtOAc-hexanes (0–10%) to afford the product 16 (98 mg) as an oil in 59% yield. IR (KBr) 2951, 2230, 1731, 1645, 1480, 1436, 1318, 1263, 1201, 1124, 1007, 770 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.58 (d, J = 7.8 Hz, 1 H), 7.46 (t, J = 7.8 Hz, 1 H), 7.40-7.34 (m, 3 H), 7.03 (d, J = 2.4 Hz, 1 H), 6.02 (t, J = 7.5 Hz, 1 H), 3.84 (s, 3 H), 3.77 (s, 3 H), 3.58 (s, 3 H), 2.25 (t, J = 7.5 Hz, 2 H), 2.21 (s, 3 H), 2.06 (q, J = 7.5 Hz, 2 H), 1.78-1.69 (m, 2 H); 13C NMR (75 MHz, CDCl3) δ 173.5, 166.6, 157.5, 140.7, 139.2, 136.8, 134.2, 133.5, 133.1, 132.6, 130.8, 130.5, 129.2, 127.4, 126.6, 124.4, 118.6, 112.5, 61.4, 52.1, 51.4, 33.2, 28.9, 24.7, 16.0; ESIMS m/z (rel intensity) 407.58 (MH+, 46). Anal. (C24H25NO5) C, H, N.
(E)-3-Chloro-5-[1-(3-cyanophenyl)-5-methoxycarbonyl-pent-1-enyl]-2-methoxybenzoic Acid Methyl Ester (17)
The general procedure was followed using the vinylstannane 44 (247 mg, 0.477 mmol), iodide 55 (246.7 mg, 0.756 mmol), cesium fluoride (220 mg, 1.45 mmol) and Pd(PBut3)2 (25.2 mg, 0.048 mmol) in toluene (1 mL). The mixture was stirred at room temperature for 18 h, at 60 °C for 23.5 h and at 110 °C for 23 h. The residue was purified by column chromatography on silica gel (15 g), eluting with EtOAc-hexanes (0–5%) to afford the product 17 (60 mg) as an oil in 29% yield. IR (KBr) 2952, 2230, 1736, 1597, 1576, 1477, 1436, 1403, 1362, 1303, 1264, 1201, 1162, 1095, 999, 969, 884, 849, 800, 744, 706, 634 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.58 (dt, J = 1.5 Hz, J = 7.8 Hz, 1 H), 7.45 (t, J = 7.6 Hz, 1 H), 7.40-7.37 (m, 2 H), 7.32 (dt, J = 1.5 Hz, J = 7.8 Hz, 1 H), 7.20 (m, 1 H), 6.04 (t, J = 7.5 Hz, 1 H), 3.86 (s, 3 H), 3.84 (s, 3 H), 3.57 (s, 3 H), 2.23 (t, J = 7.2 Hz, 2 H), 2.04 (q, J = 7.5 Hz, 2 H), 1.76-1.66 (m, 2 H); 13C NMR (75 MHz, CDCl3) δ 173.5, 165.7, 154.9, 140.0, 138.3, 138.1, 134.2, 133.1, 132.3, 132.0, 131.2, 129.5, 128.0, 126.7, 118.5, 112.9, 62.0, 52.5, 51.6, 33.3, 29.1, 24.7; ESIMS m/z (rel intensity) 450.13/452.11 (MNa+, 86/33), 427.94/429.94 (M+, 37/12). Anal. (C23H22ClNO5) C, H, Cl, N.
(E)-6-(5-Chloro-2-methoxyphenyl)-6-(3-cyanophenyl)-hex-5-enoic Acid Methyl Ester (18)
The general procedure was followed using the vinylstannane 44 (250 mg, 0.48 mmol), bromide 47 (185 mg, 0.835 mmol), cesium fluoride (235 mg, 1.532 mmol) and Pd(PBut3)2 (27.6 mg, 0.053 mmol) in toluene (1 mL). The mixture was stirred at room temperature for 13 h, at 60 °C for 23.5 h and at 110 °C for 9.5 h. The residue was purified by column chromatography on silica gel (15 g), eluting with EtOAc-hexanes (0–5%) to afford the product 18 (67 mg) as an oil in 38% yield. IR (KBr) 2949, 2230, 1735, 1644, 1484, 1319, 1240, 1127, 1027, 807, 707 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.52-7.49 (m, 1 H), 7.42-7.37 (m, 3 H), 7.21 (dd, J = 2.4 Hz, J = 8.4 Hz, 1 H), 7.15 (d, J = 2.4 Hz, 1 H), 6.71 (d, J = 8.7 Hz, 2 H), 5.84 (t, J = 7.5 Hz. 1 H), 3.62 (s, 3 H), 3.52 (s, 3 H), 2.30 (t, J = 7.5 Hz, 2 H), 2.23-2.15 (q, J = 7.2-7.8 Hz, 2 H), 1.82-1.72 (m, 2 H); 13C NMR (75 MHz, CDCl3) δ 173.6, 155.5, 141.4, 137.4, 133.4, 132.5, 130.3, 128.6, 125.4, 118.9, 112.5, 111.9, 55.6, 51.5, 33.3, 28.5, 24.8; ESIMS m/z (rel intensity) 391.95/393.96 (MNa+, 39/12). Anal. (C21H20ClNO3) C, H, Cl, N.
(Z)-6-(3-Fluoro-5-trifluoromethylphenyl)-6-(3-methoxy-7-methylbenzo[d] isoxazol-5-yl)-hex-5-enoic Acid Methyl Ester (19)
The general procedure was followed using the vinylstannane 43 (305 mg, 0.526 mmol), iodide 59 (200 mg, 0.692 mmol), cesium fluoride (245 mg, 1.6 mmol) and Pd(PBut3)2 (28 mg, 0.054 mmol) in toluene (1 mL). The mixture was stirred at room temperature for 19 h, at 60 °C for 8 h and at 100 °C for 22 h. The residue was purified by column chromatography on silica gel (20 g), eluting with EtOAc-hexanes (0–2%) to afford the product 19 (112 mg) as an oil in 47% yield. IR (KBr) 2949, 1736, 1599, 1549, 1496, 1438, 1376, 1329, 1220, 1171, 1130, 1047, 934, 874, 765, 701 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.29 (d, J = 7.8 Hz, 1 H), 7.20 (s, 1 H), 7.13 (d, J = 8.4 Hz, 2 H), 7.05 (d, J = 8.7 Hz, 1 H), 6.06 (t, J = 7.5 Hz, 1 H), 4.12 (s, 3 H), 3.62 (s, 3 H), 2.45 (s, 3 H), 2.30 (t, J = 7.5 Hz, 2 H), 2.17-2.09 (q, J = 7.2-7.5 Hz, 2 H), 1.84-1.74 (m, 2 H); 13C NMR (75 MHz, CDCl3) δ 173.6, 167.3, 164.0, 162.8, 160.7, 143.3, 139.7, 137.3, 132.8, 130.9, 130.2, 122.3, 120.8, 120.4, 120.1, 116.6, 113.8, 111.9, 111.6, 57.3, 51.5, 33.3, 29.1, 24.8, 14.7; ESIMS m/z (rel intensity) 451.97 (MH+, 100). Anal. (C23H21F4NO4) C, H, F, N.
E)-6-(3-Cyanophenyl)-6-(3-methoxy-7-methylbenzo[d]isoxazol-5-yl)-hex-5-enoic Acid Methyl Ester (20)
The general procedure was followed using the vinylstannane 44 (232 mg, 0.448 mmol), iodide 59 (195 mg, 0.675 mmol), cesium fluoride (245 mg, 1.6 mmol) and Pd(PBut3)2 (22 mg, 0.043 mmol) in toluene (1 mL). The mixture was stirred at room temperature for 19 h, at 60 °C for 8 h and at 100 °C for 43 h. The residue was purified by column chromatography on silica gel (20 g), eluting with EtOAc-hexanes (0–20%) to afford the product 20 (72 mg) as an oil in 41% yield. IR (KBr) 2949, 2230, 1738, 1615, 1548, 1495, 1394, 1315, 1169, 1047, 910, 804, 765, 695 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.61 (dt, J = 1.5 Hz, J = 7.8 Hz, 1 H), 7.48 (t, J = 7.48 Hz, 1 H), 7.43 (br, 1 H), 7.38 (dt, J = 1.5 Hz, J = 7.8 Hz, 1 H), 7.13 (br, 1 H), 7.09 (br, 1 H), 6.06 (t, J = 7.5 Hz, 1 H), 4.11 (s, 3 H), 3.61 (s, 3 H), 2.43 (s, 3 H), 2.29 (t, J = 7.5 Hz, 2 H), 2.14-2.07 (q, J = 7.2-7.8 Hz, 2 H), 1.82-1.72 (m, 2 H); 13C NMR (75 MHz, CDCl3) δ 173.6, 167.3, 162.8, 141.2, 139.9, 137.5, 134.3, 133.2, 130.9, 130.6, 130.2, 129.3, 120.8, 118.6, 116.6, 113.7, 112.7, 57.3, 51.6, 33.3, 29.1, 24.9, 14.7; ESIMS m/z (rel intensity) 391.03 (MH+, 100). Anal. (C23H22N2O4) C, H, N.
(Z)-6-(5-Chloro-2-methoxyphenyl)-6-(2,3-dihydro-2,7-dimethyl-3-oxo-benzo[d]isoxazol-5-yl)-hex-5-enoic Acid Methyl Ester (21)
The general procedure was followed using the vinylstannane 51 (360 mg, 0.645 mmol), iodide 58 (272 mg, 0.941 mmol), cesium fluoride (349 mg, 2.27 mmol) and Pd(PBut3)2 (36 mg, 0.07 mmol) in toluene (1 mL). The mixture was stirred at room temperature for 22 h, at 60 °C for 23.5 h and at 110 °C for 23.4 h. The residue was purified by column chromatography on silica gel (25 g), eluting with EtOAc-hexanes (0–20%) to afford the product 21 (91 mg) as an oil in 33% yield. 1H NMR (300 MHz, CDCl3) δ 7.17 (d, 2 H), 7.13 (dd, J = 2.7 Hz, J = 9.0 Hz, 1 H), 6.87 (d, J = 2.7 Hz, 1 H), 6.72 (d, J = 9.0 Hz, 1 H), 5.97 (t, J = 7.5 Hz, 1 H), 3.53 (s, 3 H), 3.50 (s, 3 H), 3.48 (s, 3 H), 2.19 (s, 3 H), 2.12 (t, J = 7.5 Hz, 2 H), 1.90-1.84 (m, 2 H), 1.66-1.56 (m, 2 H); ESIMS m/z (rel intensity) 452.09 (MNa+, 88), 454.09 (MNa+, 28). Anal. (C23H24ClNO5) C, H, Cl, N.
(Z)-6-(2,3-Dihydro-2,7-dimethyl-3-oxo-benzo[d]isoxazol-5-yl)-6-(3-fluoro-5-trifluoromethylphenyl)-hex-5-enoic Acid Methyl Ester (22)
The general procedure was followed using the vinylstannane 43 (310 mg, 0.535 mmol), the iodide 58 (239 mg, 0.827 mmol), cesium fluoride (262 mg, 1.71 mmol) and Pd(PBut3)2 (29 mg, 0.056 mmol) in toluene (1 mL). The mixture was stirred at room temperature for 18 h, at 60 °C for 8 h and at 100 °C for 45 h. The residue was purified by column chromatography on silica gel (20 g), eluting with EtOAc-hexanes (0–20%) to afford the product 22 (101 mg) as a white solid in 42% yield. The solid was recrystallized with ethyl acetate and hexanes to afford a colorless needle crystal for X-ray crystallography: mp 97.5–98.5 °C. IR (KBr) 2952, 1738, 1694, 1616, 1599, 1492, 1470, 1438, 1377, 1318, 1228, 1171, 1131, 1090, 1001, 934, 875, 854, 772, 786, 712, 698, 631 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.35 (d, J = 1.2 Hz, 2 H), 7.28 (dt, J =8.4 Hz, 1 H), 7.21 (br, 1 H), 7.17 (br, 1 H), 7.02 (dt, J = 8.7 Hz, 1 H), 6.54 (s, 1 H), 6.06 (t, J = 7.5 Hz, 1 H), 3.64 (s, 3 H), 3.60 (s, 3 H), 2.33 (s, 3 H), 2.29 (t, J = 7.5 Hz, 2 H), 2.15-2.08 (q, J = 7.2-7.5 Hz, 2 H), 1.82-1.72 (m, 2 H); 13C NMR (75 MHz, CDCl3) δ 173.6, 164.1, 162.9, 158.3, 143.0, 139.3, 137.5, 132.9, 131.2, 122.3, 120.3, 120.0, 116.1, 111.7, 51.5, 33.3, 32.7, 29.1, 24.8, 14.1; ESIMS m/z (rel intensity) 451.99 (MH+, 100). Anal. (C23H21F4NO4) C, H, F, N.
Determination of the Structure of (Z)-6-(2,3-Dihydro-2,7-dimethyl-3-oxo-benzo[d]isoxazol-5-yl)-6-(3-fluoro-5-trifluoromethylphenyl)-hex-5-enoic Acid Methyl Ester (22) by X-ray Crystallography
DATA COLLECTION
A colorless needle of C23H21F4NO4 having approximate dimensions of 0.44 × 0.29 × 0.13 mm was mounted on a glass fiber in a random orientation. Preliminary examination and data collection were performed Mo Kα radiation (λ = 0.71073 Å on a Nonius KappaCCD equipped with a graphite crystal, incident beam monochromator.
Cell constants for data collection were obtained from least-squares refinement, using the setting angles of 12884 reflections in the range 2 < θ < 27°. The triclinic cell parameters and calculated volume are: a = 8.8822(7), b = 9.7444(9), c = 12.9675(17) Å, a = 76.306(4), b =72.480(6), g = 89.873(5) °, V = 1036.98(19) Å3. For Z = 2 and F.W. = 451.42 the calculated density is 1.45 g/cm3. The refined mosaicity from DENZO/SCALEPACK was 0.39 ° indicating good crystal quality. The space group was determined by the program ABSEN.29 There were no systematic absences; the space group was determined to be P-1(#2). The data were collected at a temperature of 150 (1)K. Data were collected to a maximum 2θ of 55.8 °.
DATA REDUCTION
A total of 12884 reflections were collected, of which 4884 were unique. Lorentz and polarization corrections were applied to the data. The linear absorption coefficient is 1.2/cm for Mo Ka radiation. An empirical absorption correction using SCALEPACK30 was applied. Transmission coefficients ranged from 0.940 to 0.986. Intensities of equivalent reflections were averaged. The agreement factor for the averaging was 3.8% based on intensity.
STRUCTURE SOLUTION AND REFINEMENT
The structure was solved by direct methods using SIR2002.36 The remaining atoms were located in succeeding difference Fourier syntheses. Hydrogen atoms were included in the refinement but restrained to ride on the atom to which they are bonded. The structure was refined in full-matrix least-squares where the function minimized was Σw(|Fo|2−|Fc|2)2 and the weight w is defined as 1/[σ2(Fo2)+(0.0722P)2+0.0000P] where P=(Fo2 +2Fc2)/3. Scattering factors were taken from the “International Tables for Crystallography”.32 4884 reflections were used in the refinements. However, only the 3461reflections with Fo2>2σ(Fo2) were used in, calculating R1. The final cycle of refinement included 292 variable parameters and converged (largest parameter shift was <0.01 times its su) with unweighted and weighted agreement factors of:
The standard deviation of an observation of unit weight was 1.03. The highest peak in the final difference Fourier had a height of 0.27 e/A3. The minimum negative peak had a height of −0.36 e/A3. Refinement was performed on a LINUX PC using SHELX-97.33 Crystallographic drawings were done using programs ORTEP34 and PLUTON.35
Butynyl-1-tosylate (24)
Method I
A mixture of p-toluenesulfonyl chloride (62.21 g, 0.323 mol) and pyridine (31 mL, 0.383 mol) was warmed to get a colorless solution, and then cooled to get small crystals. 3-Butyn-1-ol (23) (23 mL, 0.29 mol) was added dropwise by syringe during about 20 min with stirring at 15 °C. The resulting mixture was stirred below 20 °C under nitrogen atmosphere for 20 h. Water was added with cooling. The mixture was extracted with ethyl acetate (4 × 120 mL). The organic solution was washed with 5% aqueous sulfuric acid (3 × 120 mL), water (100 mL), 10% aqueous sodium hydrogen carbonate, brine, dried over sodium sulfate and concentrated. The crude product was purified by flash column chromatography on silica gel (800 g), eluting with ethyl acetate-hexanes (0–10%) to afford the tosylate 24 (56.13 g) as colorless oil in 85% yield.11 1H NMR (300 MHz, CDCl3) δ 7.79 (d, J = 8.4 Hz, 2 H), 7.33 (d, J = 8.1 Hz, 2 H), 4.08 (t, J = 6.9–7.2 Hz, 2 H), 2.54 (dt, J = 2.7 Hz, J = 6.9–7.2 Hz, 2 H), 2.43 (s, 3 H), 1.94 (t, J = 2.7 Hz, 1 H).
Method II
A solution of sodium hydroxide (22.53 g, 0.563 mol) in water (200 mL) was added to a mixture of 3-butyn-1-ol (23) (26.76 g, 0.370 mol) and p-toluenesulfonyl chloride (86.3 g, 0.448 mol) in THF (500 mL). The resulting mixture was stirred at room temperature for 50.5 h and concentrated to remove the organic solvent. The residue was extracted with ethyl acetate (3 × 100 mL). The combined organic solution was washed with brine, dried over Na2SO4 and concentrated to afford a residue. The residue was purified by column chromatography on silica gel (200 g), eluting with 0–10% EtOAc in hexanes, to afford the tosylate 24 (68.70 g) as an oil in 77% yield.
3-But-3-ynyl-1,3-oxazolidin-2-one (25)
A flask was charged with 2-oxazolidinone (1.721 g, 19.17 mmol), tetrabutylammonium bromide (715 mg, 2.20 mmol), potassium carbonate (19.09 g, 138 mmol) and toluene (90 mL). 3-Butynyl-1-tosylate (24) (11.72 g, 52.26 mmol) was added. After the resulting mixture was stirred at 100 °C for 4 h, more tosylate 24 (10.62 g, 47.35 mmol) was added. After stirring overnight, more potassium carbonate (7.52 g, 54.41 mmol) and tosylate 24 (11.94 g, 53.24 mmol) were added. The resulting mixture was stirred at 105 °C for 5.5 h, and potassium carbonate (10.00 g, 72.35 mmol) and tosylate 24 (11.312 g, 50.44 mmol) were added. The mixture was heated at 105 °C overnight, and more tosylate 24 (10.70 g, 47.71 mmol) and potassium carbonate (9.29 g, 67.22 mmol) were added. The mixture was heated to reflux for another 5 h and then cooled to room temperature. Water was added to quench the reaction. The mixture was extracted ethyl acetate (4 × 200 mL). The organic solution was washed with brine, dried over Na2SO4 and concentrated to afford a residue, which was purified by column chromatography on silica gel (40 g), eluting with EtOAc-hexanes (0–30%), to afford the product 25 as an oil (2.197 g) in 83% yield. IR (KBr film) 3286, 2919, 2118, 1744, 1485, 1429, 1366, 1271, 1236, 1093, 1042, 970, 763 cm−1; 1H NMR (300 MHz, CDCl3) δ 4.32 (t, J = 7.8 Hz, 2 H), 3.70 (t, J = 7.8 Hz, 2 H), 3.42 (t, J = 6.6 Hz, 2 H), 2.45 (dt, J = 2.7 Hz, J = 6.6 Hz, 2 H), 2.00 (t, J = 2.7 Hz, 1 H); 13C NMR (75 MHz, CDCl3) δ 157.9, 80.6, 70.0, 61.5, 44.5, 42.5, 17.4; ESIMS m/z (rel intensity) 139.98 (100). Anal. (C7H9NO2) C, H, N.
2-Hydroxy-5-iodo-3-methylbenzoic Acid Methyl Ester (28).10
Tetrabutylammonium bromide (0.9 g, 2.764 mmol) was added to a stirred solution of 3-methylsalicylic acid 26 (4.634 g, 29.85 mmol) in dichloromethane (50 mL). A solution of potassium carbonate (13.433 g, 97.34 mmol) in water (25 mL) was added. Dimethyl sulfate (6.0 mL, 62.77 mmol) was added to afford a clear solution. The resulting solution was stirred at room temperature for 4 h. The organic layer was separated and the aqueous layer was diluted with water (40 mL) and extracted with dichloromethane (2 × 20 mL). The combined organic solutions were washed with sat. ammonium chloride (30 mL), brine (2 × 30 mL), dried over sodium sulfate and concentrated to afford crude methyl ester 27 as an oil. 1H NMR (300 MHz, CDCl3) δ 10.99 (s, 1 H), 7.67 (dd, J = 1.5 Hz, J = 8.1 Hz, 1 H), 7.30 (d, J = 7.5 Hz, 1 H), 6.76 (t, J = 7.5 Hz, 1 H), 3.94 (s, 3 H), 2.25 (s, 3 H). The crude methyl ester 27 was dissolved in methanol (80 mL), sodium iodide (5.526 g, 36.86 mmol) and sodium hydroxide (1.489 g, 36.86 mmol) were added, and the solution was cooled to 0 °C. Aqueous sodium hypochlorite (62.5 mL, 36.86 mmol, = 4%) was added dropwise. The resulting brown mixture was stirred for 4.5 h at 0–3 °C and then treated with 10% sodium thiosulfate (60 mL). The pH of the mixture was adjusted to 5–6 using 1 N HCl. Ether (200 mL) was added and the layers were separated. The aqueous layer was extracted with ether (3 × 200 mL). The combined organic solution was washed with brine (300 mL), dried over anhydrous Na2SO4 and concentrated to afford crude 28 (8.52 g) as colorless crystals in 98% yield: mp 94–95.5 °C (lit.10 mp 82–84 °C). 1H NMR (300 MHz, CDCl3) δ 10.80 (s, 1 H), 7.83 (d, J = 2.4 Hz, 1 H), 7.43 (d, J = 2.4 Hz, 1 H), 3.80 (s, 3 H), 2.07 (s, 3 H).
Methyl 5-Iodo-2-methoxy-3-methylbenzoate (29)
The methyl ester (28) (8.52 g, 29.17 mmol) was dissolved in dichloromethane (100 mL). Then tetrabutylammonium bromide (958 mg, 2.94 mmol) was added. A solution of sodium hydroxide (3.4 g, 85 mmol) in water (50 mL) was added, followed by dimethyl sulfate (5.5 mL, 57.55 mmol). The resulting solution was stirred at room temperature overnight and quenched with solid ammonium chloride (6 g), and the pH was adjust to 5–6 with 1 N HCl. The organic layer was separated and the aqueous layer was extracted with dichloromethane (3 × 100 mL). The combined organic solution was washed with brine (200 mL), dried over sodium sulfate and concentrated to afford a solid, which was purified by column chromatography on silica gel (40 g) using hexanes and 5% ethyl acetate in hexanes to afford 29 (7.69 g) as white crystals in 86% yield: mp 66–67.5 °C (lit.10 mp 55–57 °C). 1H NMR (300 MHz, CDCl3) δ 7.93 (d, J = 2.4 Hz, 1 H), 7.66 (d, J = 2.4 Hz, 1 H), 3.90 (s, 3 H), 3.81 (s, 3 H), 2.27 (s, 3 H).
2-Methoxy-3-methyl-5-[4-(2-oxo-oxazolidin-3-yl)-but-1-ynyl]benzoic Acid Methyl Ester (30)
Triethylamine (1.35 mL, 9.69 mmol) and Pd(PPh3)Cl2 (139 mg, 0.19 mmol) were added to a mixture of the iodide 29 (1.310 g, 4.28 mmol) and the alkyne 25 (536 mg, 3.86 mmol) in THF (25 mL) at room temperature, and then Cu(I)I (75 mg, 0.40 mmol) was added. The resulting mixture was stirred at room temperature for 22 h. The reaction was quenched with water (20 mL) and concentrated to remove the organic solvents. The residue was extracted with ethyl acetate (3 × 50 mL). The organic layer was separated and washed with brine (60 mL), dried over sodium sulfate and concentrated. The residue was purified by column chromatography on silica gel (35 g), eluting with EtOAc-hexanes (0–50%) to afford the product 30 (981 mg) as brown oil in 80% yield. 1H NMR (300 MHz, CDCl3) δ 7.63 (d, J = 2.1 Hz, 1 H), 7.34 (d, J = 1.5 Hz, 1 H), 4.33 (t, J = 7.8 Hz, 2 H), 3.89 (s, 3 H), 3.80 (s, 3 H), 3.73 (t, J = 7.8 Hz, 2 H), 3.50 (t, J = 6.6 Hz, 2 H), 2.66 (t, J = 6.6 Hz, 2 H), 2.26 (s, 3 H); ESIMS m/z (rel intensity) 340.10 (MNa+, 100). Anal. Calcd for (C17H19NO5) C, H, N.
2-Methoxy-3-methyl-5-[1-(tributylstannanyl)-4-(2-oxo-oxazolidin-3-yl)-but-1-enyl]benzoic Acid Methyl Ester (31)
Compound 30 (945 mg, 0.945 mmol) was dissolved in THF (20 mL), and then tetrakis(triphenylphosphine)palladium (7.0 mg, 6.05 μmol) was added. The mixture was cooled to 0 °C, degassed by gently bubbling argon through for 15 min, and then tributyltin hydride (0.4 mL, 1.44 mmol) was added dropwise over 60 min. After the mixture was stirred at room temperature for 3 h, it was concentrated to yield a residue. The residue was purified by column chromatography on silica gel (40 g) using hexanes and EtOAc-hexanes (5–15%) to afford the vinylstannanes 31 (437 mg) as an oil in 76% yield and 36 (53 mg) in 9% yield. Spectral data of 31: IR (KBr) 2926, 1756, 1435, 1257, 1197, 1126 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.15 (d, J = 2.4 Hz, 1 H), 6.86 (d, J = 2.1 Hz, 1 H), 5.71 (t, J = 6.9 Hz, 1 H), 4.22 (t, J = 7.8 Hz, 2 H), 3.87 (s, 3 H), 3.79 (s, 3 H), 3.34 (t, J = 7.8 Hz, 2 H), 3.27 (t, J = 7.2 Hz, 2 H), 2.26 (m, 5 H), 1.42-1.34 (m, 6 H), 1.29-1.17 (m, 6 H), 0.88-0.78 (m, 9 H); ESIMS m/z (rel intensity) 633.33 (MNa+, 77), 632.20 (MNa+, 100). Anal. (C29H47NO5Sn) C, H, N, Sn.
2-Methoxy-3-methyl-5-[2-(tributylstannanyl)-4-(2-oxo-oxazolidin-3-yl)-but-1-enyl]benzoic Acid Methyl Ester (36)
IR (KBr) 2955, 2926, 2871, 1756, 1732, 1480, 1424, 1377, 1360, 1315, 1263, 1232, 1201, 1128, 1102, 1051, 1010, 881, 799, 762, 695 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.47 (d, J = 2.1 Hz, 1 H), 7.24 (d, J = 2.1 Hz, 1 H), 6.61 (s, 1 H), 4.21 (t, J = 7.8 Hz, 2 H), 3.90 (s, 3 H), 3.81 (s, 3 H), 3.42 (t, J = 7.8 Hz, 2 H), 3.30 (t, J = 7.2 Hz, 2 H), 2.70 (t, J = 7.2 Hz, 2 H), 2.31 (s, 3 H), 1.58-1.47 (m, 6 H), 1.39-1.27 (m, 6 H), 1.00-0.96 (m, 6 H), 0.89 (t, J = 7.2 Hz, 9 H); ESIMS m/z (rel intensity) 552.17 ([M-Bu]+, 38.5), 608.09 (M+, 12), 610.07 (M+, 18). Anal. (C29H47NO5Sn) C, H, N, Sn.
5-[1-Iodo-4-(2-oxo-oxazolidin-3-yl)-but-1-enyl]-2-methoxy-3-methylbenzoic acid Methyl Ester (32)
The vinyl tributylstannane 31 (157 mg, 0.258 mmol) was dissolved in dry CH2Cl2 (6 mL). Finely divided I2 (80 mg, 0.315 mmol) was added, and the mixture was stirred vigorously at room temperature for 50 min. Saturated aqueous Na2S2O3 (15 mL) was added, the phases were separated, and the aqueous phase was extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were dried (Na2SO4) and concentrated. The residue was purified by flash chromatography on silica gel (20 g, 30% EtOAc-hexanes as eluent) to afford the vinyl iodide as an oil (114 mg, 99%): 1H NMR (CDCl3) δ 7.46 (d, J = 2.1 Hz, 1 H), 7.20 (d, J = 1.5 Hz, 1 H), 6.40 (t, J = 7.5 Hz, 1 H), 4.23 (t, J = 7.8 Hz, 2 H), 3.86 (s, 3 H), 3.78 (s, 3 H), 3.36 (t, J = 7.8 Hz, 2 H), 3.24 (t, J = 6.9 Hz, 2 H), 2.25 (s, 3 H), 2.19 (t, J = 7.2 Hz, 2 H); 13C NMR (CDCl3) δ 166.1, 158.2, 158.0, 139.4, 136.5, 134.9, 133.1, 128.8, 124.1, 95.5, 61.5, 61.4, 52.2, 44.5. 43.1, 30.1, 16.0; ESIMS m/z (rel intensity) 467.85 (MNa+, 100). Anal. (C17H20INO5) C, H, I, N.
3-[4-(3,4-Dimethoxyphenyl)-but-3-ynyl]-1,3-oxazolidin-2-one (34)
Triethylamine (0.08 mL, 0.574 mmol) and Pd(PPh3)Cl2 (8.0 mg, 0.011 mmol) were added to a mixture of 3,4-dimethoxyphenyl iodide (33) (64 mg, 0.242 mmol) and alkyne 25 (34 mg, 0.245 mmol) in THF (3 mL) at room temperature. Then Cu(I)I (5 mg, 0.026 mmol) was added. The resulting mixture was stirred at room temperature for 4 h. The reaction was quenched with water (10 mL) and concentrated to remove the organic solvents. The residue was diluted with ethyl acetate (15 mL). The organic layer was separated and washed with brine (2 × 10 mL), dried over sodium sulfate and concentrated. The residue was purified by column chromatography on silica gel (25 g), eluting with EtOAc-hexanes (10–50%) to afford the product 34 (54 mg) as solid in 80% yield: mp 78–79 °C. 1H NMR (300 MHz, CDCl3) δ 6.95 (dd, J = 2.1 Hz, J = 8.4 Hz, 1 H), 6.88 (d, J = 1.8 Hz, 1 H), 6.76 (d, J = 8.4 Hz, 1 H), 4.32 (t, J = 7.8 Hz, 2 H), 3.86 (s, 3 H), 3.85 (s, 3 H), 3.73 (t, J = 7.8 Hz, 2 H), 3.51 (t, J = 6.6 Hz, 2 H), 2.67 (t, J = 6.6 Hz, 2 H); ESIMS m/z (rel intensity) 276.05 (MH+, 94), 298.05 (MNa+, 98). Anal. (C15H17NO4) C, H, N.
3-[4-(Tributylstannanyl)-4-(3,4-dimethoxyphenyl)-but-3-enyl]-1,3-oxazolidin-2-one (35)
The intermediate 34 (254 mg, 0.923 mmol) was dissolved in THF (20 mL), and then tetrakis(triphenylphosphine)palladium (9.4 mg, 8.13 μmol) was added. The mixture was cooled to 0 °C, degassed by gently bubbling argon through for 15 min and then tributyltin hydride (0.4 mL, 1.44 mmol) was added dropwise over 70 min. After the mixture was stirred at room temperature for 3.5 h, it was concentrated to yield a residue. The residue was purified by column chromatography on silica gel (30 g) using hexane and EtOAc-hexane (0–30%) to afford the vinylstannanes 35 (415 mg) in 79% yield and 37 as an oil (39.2 mg) in 8% yield. Spectral data of 35: IR (KBr) 2954, 2926, 1755, 1508, 1463, 1254, 1233, 1136, 1028, 865, 761 cm−1; 1H NMR (300 MHz, CDCl3) δ 6.77 (m, 1 H), 6.45-6.42 (m, 2 H), 5.68 (t, J = 6.9 Hz, 1 H), 4.19 (t, J = 8.1 Hz, 2 H), 3.84 (s, 3 H), 3.82 (s, 3 H), 3.32 (t, J = 8.1 Hz, 2 H), 3.27 (t, J = 7.2 Hz, 2 H), 2.32 (m, 2 H), 1.49-1.38 (m, 6 H), 1.34-1.18 (m, 6 H), 0.95-0.76 (m, 14 H); 13C NMR (75 MHz, CDCl3) δ 158.1, 148.6, 148.4, 146.4, 137.1, 136.8, 118.5, 111.0, 110.1, 61.4, 55.7, 55.6, 44.1, 43.8, 28.8, 27.1, 13.5, 9.8; ESIMS m/z (rel intensity) 588.45 (MNa+, 80.4), 590.19 (MNa+, 100). Anal. (C27H45NO4Sn) C, H, N, Sn.
3-[3-(Tributylstannanyl)-4-(3,4-dimethoxyphenyl)-but-3-enyl]-1,3-oxazolidine-2-one (37)
1H NMR (300 MHz, CDCl3) δ 6.83 (m, 2 H), 6.74 (s, 1 H), 6.64 (s, 1 H), 4.20 (t, J = 7.5–8.4 Hz, 2 H), 3.89 (s, 3 H), 3.87 (s, 3 H), 3.38 (t, J = 7.5–8.4 Hz, 2 H), 3.31 (t, J = 7.5–8.1 Hz, 2 H), 2.74 (t, J = 7.5–8.1 Hz, 2 H), 1.59–1.49 (m, 6 H), 1.40-1.28 (m, 6 H), 1.02-0.96 (m, 6 H), 0.090 (t, J = 7.1 Hz, 9 H); ESIMS m/z (rel intensity) 564.30 (M+, 12), 566.25 (M+, 23), 568.26 (M+, 28), 586.32 (MNa+, 42), 588.31 (MNa+, 77), 590.26 (MNa+, 100).
6-(3-Fluoro-5-trifluoromethylphenyl)-hex-5-ynoic Acid Methyl Ester (41)
Pd(PPh3)2Cl2 (236 mg, 0.336 mmol) was added to a mixture of bromide 38 (1.677 g, 6.70 mmol) and methyl 5-hexynoate (1.025 g, 8.12 mmol) in triethylamine (5.0 mL) at room temperature. Cu(I)I (136 mg, 0.714 mmol) was added. The resulting mixture was stirred at room temperature for 1.5 h and at 80 °C for 22.5 h. The reaction mixture was cooled to room temperature, filtered through a short column of silica gel (5 g), and the column was washed with ethyl acetate. The organic solution was concentrated. The residue was purified by column chromatography on silica gel (40 g), eluting with EtOAc-hexanes (2%) to afford the product 41 (1.564 g) as a white solid in 81% yield: mp 44–45 °C. IR (KBr) 3084, 2955, 2848, 2238, 1740, 1619, 1599, 1467, 1439, 1363, 1253, 1240, 1224, 1171, 1133, 1093, 1046, 995, 973, 924, 911, 875, 695 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.40 (s, 1 H), 7.21 (m, 2 H), 3.66 (s, 3 H), 2.47 (t, J = 7.2 Hz, 4 H), 1.90 (m, 2 H); 13C NMR (75 MHz, CDCl3) δ 173.3, 163.7, 160.4, 132.8, 132.3, 126.8, 126.7, 124.7, 124.3, 121.8, 121.5, 112.3, 111.9, 79.1, 51.6, 32.7, 23.5, 18.7; ESIMS m/z (rel intensity) 288.96 (MH+, 51). Anal. (C14H12F4O2) C, H, F.
6-(3-Cyanophenyl)-hex-5-ynoic Acid Methyl Ester (42)
Pd(PPh3)2Cl2 (223 mg, 0.315 mmol) was added to a mixture of 3-bromobenzonitrile (40) (832 mg, 6.43 mmol) and methyl 5- hexynoate (970 mg, 7.69 mmol) in triethylamine (4.5 mL) at room temperature, and then Cu(I)I (122 mg, 0.64 mmol) was added. The resulting mixture was stirred at room temperature for 1 h and at 80 °C for 22 h. The reaction mixture was cooled to room temperature, filtered through a short column of silica gel (5 g), and the column washed with ethyl acetate. The organic solution was concentrated. The residue was purified by column chromatography on silica gel (30 g), eluting with EtOAc-hexanes (3–5%) to afford the product 42 (1.117 g) as an oil in 76% yield. IR (KBr) 3069, 2952, 2232, 1737, 1597, 1572, 1479, 1436, 1416, 1370, 1316, 1222, 1160, 1057, 894, 799, 684 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.64 (m, 1 H), 7.58 (dt, J = 1.2 Hz, J = 7.8 Hz, 1 H), 7.53 (dt, J = 1.2 Hz, J = 7.8 Hz, 1 H), 7.38 (t, J = 7.5 Hz, 1 H), 3.68 (s, 3 H), 2.48 (m, 4 H), 1.92 (m, 2 H); 13C NMR (75 MHz, CDCl3) δ 173.4, 135.6, 134.9, 130.9, 129.1, 125.3, 118.1, 112.6, 91.7, 79.3, 51.6, 32.8, 23.6, 18.8; EIMS m/z (rel intensity) 227 (M+, 29), CIMS m/z (rel intensity) 228 (MH+, 100). Anal. (C14H13NO2) C, H, N.
6-Tributylstannanyl-6-(3-fluoro-5-trifluoromethylphenyl)-hex-5-enoic Acid Methyl Ester (43)
Alkyne 41 (1.545 g, 5.36 mmol) was dissolved in THF (220 mL), and then tetrakis(triphenylphosphine)palladium (56 mg, 48 μmol) was added. The mixture was cooled to 0 °C, degassed by gently bubbling argon through for 20 min, and then tributyltin hydride (2.2 mL, 7.93 mmol) was added dropwise over 120 min. After the mixture was stirred at room temperature for 3 h, it was concentrated to yield a residue. The residue was purified by column chromatography on silica gel (50 g) using hexanes and EtOAc-hexanes (0–1%) to afford the vinylstannane 43 (2.651 g) as an oil in 90% yield. IR (KBr) 2957, 2928, 2873, 2854, 1743, 1617, 1593, 1464, 1434, 1369, 1327, 1246, 1224, 1170, 1133, 1090, 978, 903, 877, 865, 695, 706 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.05 (d, J = 8.7 Hz, 1 H), 6.92 (s, 1 H), 6.76 (t, J = 9.3 Hz, 1 H), 5.76 (t, J = 7.2 Hz, 1 H), 3.59 (s, 3 H), 2.23 (t, J = 7.5 Hz, 2 H), 2.01 (m, 2 H), 1.68 (m, 2 H), 1.45-1.35 (m, 6 H), 1.30-1.18 (m, 6 H), 0.89-0.81 (m, 9 H); 13C NMR (75 MHz, CDCl3) δ 173.7, 163.9, 160.6, 148.8, 148.7, 144.6, 142.2, 132.4, 132.0, 119.4, 117.0, 116.7, 109.2, 108.8, 51.4, 33.2, 29.4, 28.9, 27.2, 24.6, 13.5, 10.0; ESIMS m/z (rel intensity) 523.15 (M-Bu+, 64). Anal. (C26H40F4O2Sn) C, H, F, Sn.
6-(Tributylstannanyl)-6-(3-cyanophenyl)-hex-5-enoic Acid Methyl Ester (44)
Alkyne 42 (787 mg, 3.46 mmol) was dissolved in THF (150 mL) and then tetrakis(triphenylphosphine)palladium (40 mg, 34.6 μmol) was added. The mixture was cooled to 0 °C, degassed by gently bubbling argon through for 15 min, and then tributyltin hydride (1.5 mL, 5.41 mmol) was added dropwise over 60 min. After the mixture was stirred at 0 °C for 15 min and at room temperature for 6 h, the mixture was concentrated to yield a residue. The residue was purified by column chromatography on silica gel (20 g) using hexanes and EtOAc-hexanes (5%) to afford the vinylstannane 44 (1.696 g) as an oil in 95% yield. IR (KBr) 2955, 2927, 2871, 2853, 2230, 1740, 1607, 1590, 1571, 1474, 1457, 1436, 1417, 1376, 1313, 1292, 1247, 1217, 1161, 1074, 1048, 1074, 1022, 1000, 960, 911, 875, 841, 804, 770, 697, 677 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.37 (dt, J = 1.5 Hz, J = 7.8 Hz, 1 H), 7.34 (q, J = 7.8 Hz, 1 H), 7.14 (m, 1 H), 7.08 (dt, J = 1.5 Hz, J = 7.5 Hz, 1 H), 5.75 (t, J = 7.2 Hz, 1 H), 3.61 (s, 3 H), 2.20 (t, J = 7.5 Hz, 2 H), 1.97 (q, J = 7.5 Hz, 2 H), 1.64 (m, 2 H), 1.47-1.35 (m, 6 H), 1.28-1.16 (m, 6 H), 0.87-0.79 (m, 9 H); 13C-NMR (75 MHz, CDCl3) δ 173.7, 146.4, 144.6, 142.1, 131.3, 130.0, 128.9, 128.5, 119.0, 112.1, 51.4, 33.2, 29.3, 28.8, 27.2, 24.6, 13.6, 9.9; ESIMS m/z (rel intensity) 515.96 (M+, 31), 517.99 (M+, 49), 519.98 (M+, 61). Anal. (C26H41NO2Sn) C, H, N, Sn.
2-Bromo-4-chloro-1-methoxybenzene (47)
A solution of sodium hydroxide (836 mg, 20.9 mmol) in water (8.0 mL) was added to a solution of 2-bromo-4-chlorophenol (45) (2.1 g, 9.92 mmol) and tetrabutylammonium bromide (336.4 mg, 1.03 mmol) in dichloromethane. Dimethyl sulfate (1.5 mL, 15.69 mmol) was added. The resulting mixture was stirred at room temperature for 22 h and then 1 N aq HCl (about 2 mL) was added to quench the reaction. The organic phase was separated and the aqueous phase was extracted with dichloromethane (2 × 15 mL). The combined organic phase was washed with brine, dried over sodium sulfate and concentrated to afford an oil. The crude product was purified by column chromatography on silica gel (10 g) using EtOAc-hexanes (5%) to afford the product 47 as a colorless oil (2.17 g) in 99% yield.28 1H NMR (300 MHz, CDCl3) δ 7.51 (d, J = 2.4 Hz, 1 H), 7.22 (dd, J = 2.4 Hz, J = 8.7 Hz, 1 H), 6.80 (d, J = 8.7 Hz, 1 H), 3.86 (s, 3 H).
Methyl 5-Bromo-2-methoxy-3-methylbenzoate (48)
5-Bromo-3-methylsalicylic acid (46) (6.6 g, 28.57 mmol),13 potassium carbonate (19.2 g) and acetone (150 mL) were added to an oven-dried 250 mL round-bottom flask equipped with a stirring bar. Dimethyl sulfate (8.55 mL, 88.94 mmol) was then added to the flask via syringe. The reaction mixture was heated at reflux temperature for 26 h. The mixture was cooled to room temperature, filtered, and the inorganic salts were washed with methylene chloride (20 mL). The solution was evaporated to yield the crude product. The crude product was purified by flash column chromatography on silica gel (60 g) using 3–30% ethyl acetate in hexanes as the eluent to yield the product 48 as a crystallizing oil (7.4 g) in quantitative yield: mp 57–58.5 °C (lit.13 mp 59–60 °C). 1H NMR (300 MHz, CDCl3) δ 7.76 (d, J = 2.58 Hz, 1 H), 7.47 (d, J = 2.58 Hz, 1 H), 3.91 (s, 3 H), 3.81 (s, 3 H), 2.30 (s, 3 H).
6-(5-Chloro-2-methoxyphenyl)-hex-5-ynoic Acid Methyl Ester (49)
Pd(PPh3)2Cl2 (76 mg, 0.106 mmol) was added to a mixture of bromide 47 (498 mg, 2.25 mmol) and methyl 5-hexynoate (440 mg, 3.48 mmol) in triethylamine (3.0 mL) at room temperature, and then Cu(I)I (45 mg, 0.235 mmol) was added. The resulting mixture was stirred at room temperature for 8 h, and at 80 °C for 21 h. The reaction mixture was cooled to room temperature, filtered through a short column of silica gel (5 g), and the column washed with ethyl acetate. The organic solution was concentrated. The residue was purified by column chromatography on silica gel (25 g), eluting with EtOAc-hexanes (3–5%) to afford the product 49 (589 mg) as a slightly yellow oil in 98% yield. IR (KBr) 2950, 2844, 2234, 1736, 1591, 1490, 1460, 1439, 1397, 1370, 1313, 1287, 1266, 1229, 1180, 1161, 1138, 1095, 1026, 935, 882, 807, 709, 647 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.30 (d, J = 2.4 Hz, 1 H), 7.17 (dd, J = 2.7 Hz, J = 8.7 Hz, 1 H), 6.74 (d, J = 8.0 Hz, 1 H), 3.83 (s, 3 H), 3.67 (s, 3 H), 2.52 (m, 4 H), 1.92 (m, 2 H); ESIMS m/z (rel intensity) 289.04 (MNa+, 4.5). Anal. (C14H15ClO3) C, H, Cl.
6-Tributylstannanyl-6-(5-chloro-2-methoxyphenyl)-hex-5-enoic Acid Methyl Ester (51)
Alkyne 49 (1.217 g, 4.63 mmol) was dissolved in THF (200 mL), and then tetrakis(triphenylphosphine)palladium (45 mg, 39 μmol) was added. The mixture was cooled to 0 °C, degassed by gently bubbling argon through for 15 min, and then tributyltin hydride (1.9 mL, 6.85 mmol) was added dropwise over 60 min. After the mixture was stirred at 0 °C for 15 min and at room temperature for 2 h, it was concentrated to yield a residue. The residue was purified by column chromatography on silica gel (45 g) using hexanes and EtOAc-hexanes (3%) to afford the vinylstannane 51 (2.269 g) as an oil in 88% yield. IR (KBr) 2954, 2926, 2871, 2852, 1740, 1481, 1462, 1438, 1395, 1375, 1290, 1240, 1173, 1128, 1079, 1030, 878, 804 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.04 (dd, J = 2.4 Hz, J = 8.7 Hz, 1 H), 6.80 (d, J = 2.4 Hz, 1 H), 6.68 (d, J = 8.7 Hz, 1 H), 5.70 (t, J = 6.9 Hz, 1 H), 3.70 (s, 3 H), 3.59 (s, 3 H), 2.23 (t, J = 7.5 Hz, 2 H), 2.04 (m, 2 H), 1.66 (m, 5 H), 1.40 (m, 6 H), 1.29-1.17 (m, 6 H), 0.85-0.76 (m, 9 H); 13C NMR (75 MHz. CDCl3) δ 174.1, 153.9, 142.0, 141.0, 134.9, 127.7, 125.8, 125.0, 110.9, 55.2, 51.4, 33.3, 29.5, 28.9, 27.7, 27.3, 26.9, 24.7, 13.7, 10.3; ESIMS m/z (rel intensity) 501.14 (M-Bu+, 100). Anal. (C26H43ClO3Sn) C, H, Cl, Sn.
Methyl 6-(Tributylstannyl)-6-[4-methoxy-5-methoxycarbonyl-3-methylphenyl]-hex-5-enoate (52)
Aryl bromide 48 (2.25 g, 8.68 mmol), methyl 5-hexynoate (1.329 g, 10.54 mmol) and dichlorobis(triphenylphosphine)palladium(II) (307 mg, 0.437 mmol) were added to a flask under an argon atmosphere. Triethylamine (6.5 mL) was added to the flask. Cuprous iodide (168 mg, 0.88 mmol) was then added to the flask. The reaction mixture was stirred at room temperature for 0.5 h and heated at 80 °C for 21.5 h. The reaction mixture was cooled to room temperature, filtered through a short column of silica gel (5 g), and the column was washed with ethyl acetate. The organic solution was concentrated. The residue was purified by column chromatography on silica gel (40 g), eluting with EtOAc-hexanes (2%) to afford the alkyne 50 as a crude red oil. The crude oil was dissolved in dry THF (400 mL) and added to a flask under an argon atmosphere. Tetrakis(triphenylphosphine)palladium(0) (100 mg, 0.0865 mmol) was added to the flask. The mixture was cooled to 0 °C, degassed by gently bubbling argon through for 20 min, and then tributyltin hydride (3.6 mL, 12.98 mmol) was added dropwise over 110 min. After the mixture was stirred at room temperature for 2 h, it was concentrated to yield a residue. Solvent was removed in vacuo to yield a crude black oil. The oil was purified by flash column chromatography using silica gel (60 g) and a gradient eluant from 0 to 2% ethyl acetate in hexanes to yield the product 52 as a colorless oil (4.64 g, 90%, over two steps).13 1H NMR (500 MHz, CDCl3) δ 7.15 (d, J = 2.06 Hz, 1 H), 6.85 (d, J = 2.06 Hz, 1 H), 5.69 (t, J = 6.91 Hz, 1 H), 3.87 (s, 3 H), 3.79 (s, 3 H), 3.60 (s, 3 H), 2.26 (s, 3 H), 2.23 (t, J = 7.68 Hz, 2 H), 2.04 (q, J = 7.17 Hz, 2 H), 1.66 (q, J = 7.46 Hz, 2 H), 1.45-1.20 (m, 11 H), 0.86-0.69 (m, 16 H).
Methyl 5-Iodo-2-methoxybenzoate (54)
To a solution of 5-iodosalicyclic acid (53) (25.20 g, 90.67 mmol) in dichloromethane (200 mL) was added tetrabutylammonium bromide (3.1 g, 9.52 mmol). A solution of sodium hydroxide (14.66 g, 0.367 mol) in water (60 mL) was added, followed by an addition of dimethyl sulfate (26 mL, 0.272 mol). The resulting mixture was stirred at room temperature for 53 h. 1 N HCl was added until the pH was about 5 to quench the reaction. The organic phase was separated and the aqueous phase was extracted with dichloromethane (60 mL). The combined organic solution was washed with brine (80 mL), dried over anhydrous sodium sulfate and concentrated. The residue was purified by column chromatography on silica gel (100 g) using hexanes and 1% ethyl acetate in hexanes to afford the product 54 (20.02 g) as a white solid in 76% yield: mp 57–57.5 °C (lit.25 mp 48–50 °C). 1H NMR (300 MHz, CDCl3) δ 8.04 (d, J = 2.1 Hz, 1 H), 7.70 (dd, J = 8.7, 2.4 Hz, 1 H), 6.73 (d, J = 8.7 Hz, 1 H), 3.86 (s, 6 H).
Methyl 3-Chloro-5-iodo-2-methoxybenzoate (55)
A mixture of the iodide 54 (4.331 g, 14.83 mmol) and SO2Cl2 (5.1 mL, 61.58 mmol) was heated at 50 °C for 20 h and then cooled to room temperature. It was poured into ice (20 g) and extracted with CH2Cl2 (3 × 80 mL). The CH2Cl2 extracts were combined, washed with brine, dried over Na2SO4 and evaporated in vacuo. The residue was further purified by flash chromatography on silica gel (50 g, hexanes and 3% EtOAc in hexanes as eluent) and was recrystallized with ethyl acetate and hexanes to afford white crystals (4.60 g, 95%): mp 48–48.5 °C (lit.7 mp 41–43 °C); 1H NMR (CDCl3) δ 7.97 (d, J = 2.1 Hz, 1 H), 7.83 (d, J = 2.1 Hz, 1 H), 3.91 (s, 6 H).
2,N-Dihydroxy-5-iodo-3-methylbenzamide (56)
A mixture of ester 28 (2.0 g, 6.86 mmol), hydroxylamine hydrochloride (967 mg, 13.74 mmol) and potassium hydroxide (1.91 g, 30.0 mmol) in methanol (40 mL) was heated to reflux for 6 h, and then cooled to room temperature. The mixture was acidified with acetic acid until the pH was about 6, and concentrated to remove the solvents. The residue was mixed with EtOAc (100 mL), and water (80 mL) was added to get a clear solution. The organic solution was separated and the aqueous solution was extracted with EtOAc (2 × 50 mL). The combined organic solution was washed with brine, dried over anhydrous sodium sulfate and concentrated to afford a white solid residue, which was purified by column chromatography on silica gel (35 g) using EtOAc-hexanes (0–20%) to afford the product 56 (1.688 g) in 84% yield: mp 151–152.5 °C (dec). 1H NMR (300 MHz, acetone-d6) δ 12.60 (s, 1 H), 11.12 (s, 1 H), 8.61 (s, 1 H), 7.81 (d, J = 1.8 Hz, 1 H), 7.59 (d, J = 1.8 Hz, 1 H), 2.20 (s, 3 H); ESIMS m/z (rel intensity) 293.87 (MH+, 45); negative ion ESIMS m/z (rel intensity) 292.03 (M-H+, 100). Anal. (C8H8INO3) C, H, I, N.
5-Iodo-7-methylbenzo[d]isoxazol-3-one (57)
A solution of carbonyldiimidazole (2.243 g, 13.83 mmol) in THF (40 mL) was added to a boiling solution of 56 (2.022 g, 6.9 mmol) in THF (20 mL). The resulting solution was then heated under reflux for 2 h, cooled and evaporated. The residue was mixed with water to afford a precipitate. The precipitate was collected and washed with water, and then dissolved in ethyl acetate (150 mL), washed with brine (3 × 50 mL) dried over Na2SO4, and concentrated. The residue was recrystallized with ethyl acetate to afford the product 57 (1.879 g) as a white solid in 99% yield: mp 232–233 °C. IR (KBr) 2927, 2760, 2672, 2613, 1726, 1702, 1608, 1564, 1510, 1465, 1384, 1345, 1296, 1260, 1198, 1176, 1122, 1019, 967, 742, 864, 806, 763, 721, 612, 559, 503 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 12.22 (bs, 1 H), 7.90 (s, 1 H), 7.71 (s, 1 H), 2.39 (s, 3 H); ESIMS m/z (rel intensity) 275.95 (MH+, 28); negative ion ESIMS m/z (rel intensity) 274.10 (M-H+, 100). Anal. (C8H6INO2) C, H, I, N.
5-Iodo-2,7-dimethyl-benzo[d]isoxazol-3-one (58) and 5-Iodo-3-methoxy-7-methylbenzo[d]isoxazole (59)
Iodomethane (0.34 mL, 5.46 mmol) was added to a mixture of 57 (746 mg, 2.7 mmol) and potassium carbonate (1.225 g, 8.86 mmol) in DMSO (5 mL). The resulting mixture was stirred at room temperature for one day. Water (50 mL) was added to quench the reaction. The mixture was extracted with ethyl acetate (3 × 40 mL). The organic solution was washed with brine (2 × 50 mL), dried over anhydrous sodium sulfate and concentrated to afford a residue. The residue was purified by column chromatography on silica gel (25 g) using ethyl acetate in hexanes (0–20%) to afford 58 (350 mg) as a white solid in 45% yield and 59 (298 mg) as white solid in 38% yield. The product 58 was recrystallized with ethyl acetate and hexanes to afford crystals for X-ray crystallography: mp 107–108 °C. IR (KBr) 1694, 1398, 1368, 1297, 1267, 1218, 1007, 966, 871, 850, 831, 752, 705, 649, 595, 561, 549, 483 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.91 (s, 1 H), 7.61 (s, 1 H), 3.63 (s, 3 H); 13C NMR (75 MHz, CDCl3) δ 161.1, 158.2, 142.0, 130.2, 122.7, 118.2, 86.1, 32.6, 13.7; ESIMS m/z (rel intensity) 289.94 (MH+, 100). Anal. (C9H8INO2) C, H, I, N.
5-Iodo-3-methoxy-7-methylbenzo[d]isoxazole (59)
mp 79 °C. IR (KBr) 2921, 1603, 1546, 1481, 1453, 1422, 1407, 1366, 1311, 1260, 1228, 1205, 1038, 986, 951, 908, 866, 760 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.76 (s, 1 H), 7.56 (s, 1 H), 4.13 (s, 3 H); 13C NMR (75 MHz, CDCl3) δ 166.1, 162.8, 139.1, 127.0, 123.2, 115.9, 85.7, 57.5, 14.3; ESIMS m/z (rel intensity) 289.94 (MH+, 24). Anal. (C9H8INO2) C, H, I, N.
Determination of the Structure of 5-Iodo-2,7-dimethyl-benzo[d]isoxazol-3-one (58) by X-ray Crystallography
DATA COLLECTION
A colorless needle of C9H8INO2 having approximate dimensions of 0.50 × 0.19 × 0.10 mm was mounted on a glass fiber in a random orientation. Preliminary examination and data collection were performed Mo Ka radiation (λ = 0.71073Å) on a Nonius KappaCCD equipped with a graphite crystal, incident beam monochromator. Cell constants for data collection were obtained from least-squares refinement, using the setting angles of 6370 reflections in the range 2 < θ < 27°. The monoclinic cell parameters and calculated volume are: a = 4.1515(5), b = 16.0162(12); c = 14.1406(11) Å, b = 97.411(5)°, V = 932.37(15)Å3. For Z = 4 and F.W. = 289.07 the calculated density is 2.06 g/cm3. The refined mosaicity from DENZO/SCALEPACK was 0.70° indicating moderate crystal quality. The space group was determined by the program ABSEN.29 From the systematic presences of: h0l l=2n; 0k0 k=2n, and from subsequent least-squares refinement, the space group was determined to be P21/c(# 14). The data were collected at a temperature of 150(1)K. Data were collected to a maximum 2θ of 55.7°.
DATA REDUCTION
A total of 6370 reflections were collected, of which 2194 were unique. Lorentz and polarization corrections were applied to the data. The linear absorption coefficient is 33.6/cm for Mo Ka radiation. An empirical absorption correction using SCALEPACK30 was applied. Transmission coefficients ranged from 0.623 to 0.715. Intensities of equivalent reflections were averaged. The agreement factor for the averaging was 3.7% based on intensity.
STRUCTURE SOLUTION AND REFINEMENT
The structure was solved using the structure solution program PATTY in DIRDIF99.31 The remaining atoms were located in succeeding difference Fourier syntheses. Hydrogen atoms were included in the refinement but restrained to ride on the atom to which they are bonded. The structure was refined in full-matrix least-squares where the function minimized was Σw(|Fo|2−|Fc|2)2 and the weight w is defined as 1/[σ2(Fo2)+(0.0384P)2+0.7511P] where P=(Fo2 +2Fc2)/3. Scattering factors were taken from the “International Tables for Crystallography”.32 2194 reflections were used in the refinements. However, only the 1876 reflections with Fo2>2σ(Fo2) were used in, calculating R1. The final cycle of refinement included 120 variable parameters and converged (largest parameter shift was <0.01 times its su) with unweighted and weighted agreement factors of:
The standard deviation of an observation of unit weight was 1.07. The highest peak in the final difference Fourier had a height of 0.62 e/A3. The minimum negative peak had a height of −1.58 e/A3. Refinement was performed on a LINUX PC using SHELX-97.33 Crystallographic drawings were done using programs ORTEP34 and PLUTON.35
RT Inhibition Assay
Analysis of the effects of the compounds on recombinant HIV-1 RT enzyme (p66/51 dimer) was performed as previously described.37 Briefly, inhibition of purified recombinant reverse transcriptase enzyme was measured by the incorporation of [32P]GTP into poly(rC)/oligo(dG) (rCdG) homopolymer template primers.5
In Vitro Antiviral Assays
Evaluation of the antiviral activity of compounds against HIV-1RF infection in CEM-SS cells was performed using the MTS cytoprotection assay as previously described.38 Evaluation of the antiviral activity of the compounds against HIV-1 strain IIIB and HIV-2 strain (ROD) in MT-4 cells was performed using the MTT assay as previously described.13,39
In vitro Hydrolytic Stability Study in Rat Plasma
The alkenyldiarylmethanes 3, 4, 8–11, 14–18, 20, 21 (4.3–9.3 mg) (1,1-diphenylethylene (2.1–5.1 mg) or benzophenone (2.1 mg) as internal standard) were tested for their hydrolytic stability, utilizing rat plasma in vitro using methods as previously described.13
Supplementary Material
Elemental analyses of compounds 3, 4, 7-22, 25, 30-32, 34-36, 41-44, 49, 51, 56-59. Xray crystal structures of 22 and 58 in CIf format and elemental analyses. This material is available free of charge via the Internet at http://pubs.acs.org.
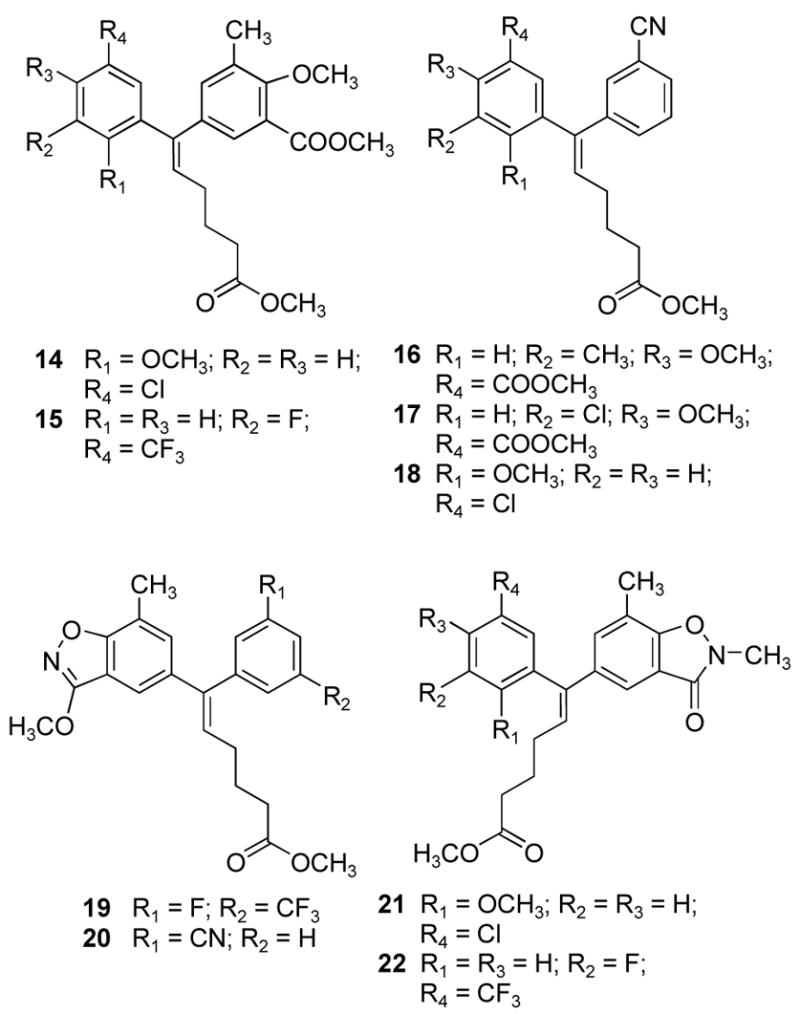
Acknowledgments
This investigation was made possible by Grant RO1-AI-43637, awarded by the National Institutes of Health, DHHS. This research was conducted in a facility constructed with support from Research Facilities Improvement Program Grant Number C06-14499 from the National Center for Research Resources of the National Institutes of Health.
References
- 1.UNAIDS/World Health Organization. AIDS Epidemic Update, December 2004. UNAIDS/World Health Organization; Geneva: 2004. [Google Scholar]
- 2.De Clercq E. Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs): Past, Present, and Future. Chem Biodiversity. 2004;1:44–64. doi: 10.1002/cbdv.200490012. [DOI] [PubMed] [Google Scholar]
- 3.Esnouf R, Ren J, Ross C, Jones Y, Stammers D, Stuart D. Mechanism of Inhibition of Reverse Transcriptase by Nonnucleoside Inhibitors. Nat Struct Biol. 1995;2:303–308. doi: 10.1038/nsb0495-303. [DOI] [PubMed] [Google Scholar]
- 4.Cushman M, Golebiewski M, Buckheit RW, Jr, Graham L, Rice WG. Synthesis and Biological Evaluation of an Alkenyldiarylmethane (ADAM) which Acts as a Novel Non-nucleoside HIV-1 Reverse Transcriptase Inhibitor. Bioorg Med Chem Lett. 1995;5:2713–2716. [Google Scholar]
- 5.Cushman M, Golebiewski WM, Graham L, Turpin JA, Rice WG, Fliakas-Boltz V, Buckheit RW., Jr Synthesis and Biological Evaluation of Certain Alkenyldiarylmethanes as Anti-HIV-1 Agents Which Act as Non-Nucleoside Reverse Transcriptase Inhibitors. J Med Chem. 1996;39:3217–3227. doi: 10.1021/jm960082v. [DOI] [PubMed] [Google Scholar]
- 6.Cushman M, Casimiro-Garcia A, Hejchman E, Ruell JA, Huang M, Schaeffer CA, Williamson K, Rice WG, Buckheit RW., Jr New Alkenyldiarylmethanes with Enhanced Potencies as Anti-HIV Agents Which Act as Non-Nucleoside Reverse Transcriptase Inhibitors. J Med Chem. 1998;41:2076–2089. doi: 10.1021/jm9800595. [DOI] [PubMed] [Google Scholar]
- 7.Cushman M, Casimiro-Garcia A, Williamson K, Rice WG. Synthesis of a Non-Nucleoside Reverse Transcriptase Inhibitor in the Alkenyldiarylmethane (ADAM) Series with Optimized Potency and Therapeutic Index. Bioorg Med Chem Lett. 1998;8:195–198. doi: 10.1016/s0960-894x(97)10214-1. [DOI] [PubMed] [Google Scholar]
- 8.Casimiro-Garcia A, Micklatcher M, Turpin JA, Stup TL, Watson K, Buckheit RW, Cushman M. Novel Modifications in the Alkenyldiarylmethane (ADAM) Series of Non-Nucleoside Reverse Transcriptase Inhibitors. J Med Chem. 1999;42:4861–4874. doi: 10.1021/jm990343b. [DOI] [PubMed] [Google Scholar]
- 9.Xu G, Micklatcher M, Silvestri M, Hartman TL, Burrier J, Osterling MC, Wargo H, Turpin JA, Buckheit RW, Jr, Cushman M. The Biological Effects of Structural Variation at the Meta Position of the Aromatic Rings and at the End of the Alkenyl Chain in the Alkenyldiarylmethane Series of Non-Nucleoside Reverse Transcriptase Inhibitors. J Med Chem. 2001;44:4092–4113. doi: 10.1021/jm010212m. [DOI] [PubMed] [Google Scholar]
- 10.Xu G, Loftus TL, Wargo H, Turpin JA, Buckheit RW, Cushman M. Solid Phase Synthesis of the Alkenyldiarylmethane (ADAM) Series of Non-Nucleoside Reverse Transcriptase Inhibitors. J Org Chem. 2001;66:5958–5964. doi: 10.1021/jo0100291. [DOI] [PubMed] [Google Scholar]
- 11.Xu G, Hartman TL, Wargo H, Turpin JA, Buckheit RW, Jr, Cushman M. Synthesis of Alkenyldiarylmethane (ADAM) Non-Nucleoside HIV-1 Reverse Transcriptase Inhibitors with Non-Identical Aromatic Rings. Bioorg Med Chem. 2002;10:283–290. doi: 10.1016/s0968-0896(01)00282-6. [DOI] [PubMed] [Google Scholar]
- 12.Silvestri M, Miles D, Rothwell AP, Wood KV, Cushman M. The “Apparent” Hydrolysis of Alkyl Esters During Electrospray Ionization. Rapid Commun Mass Spectrom. 2003;17:1703–1708. doi: 10.1002/rcm.1104. [DOI] [PubMed] [Google Scholar]
- 13.Silvestri MA, Nagarajan M, De Clercq E, Pannecouque C, Cushman M. Design, Synthesis, Anti-HIV Activities, and Metabolic Stabilities of Alkenyldiarylmethane (ADAM) Non-nucleoside Reverse Transcriptase Inhibitors. J Med Chem. 2004;47:3149–3162. doi: 10.1021/jm049916x. [DOI] [PubMed] [Google Scholar]
- 14.Chan JH, Freeman GA, Tidwell JH, Romines KR, Schaller LT, Cowan JR, Gonzales SS, Lowell GS, Andrews CW, III, Reynolds DJ, St Clair M, Hazen RJ, Ferris RG, Creech KL, Roberts GB, Short SA, Weaver K, Koszalka GW, Boone LR. Novel Benzophenones as Non-nucleoside Reverse Transcriptase Inhibitors of HIV-1. J Med Chem. 2004;47:1175–1182. doi: 10.1021/jm030255y. [DOI] [PubMed] [Google Scholar]
- 15.Eglinton G, Whiting MC. Researchs on Acetylenic Compounds 27. The Preparation and Properties of the Toluene-para-Sulphonates of Acetenic Alcohols. J Chem Soc. 1950:3650–3656. [Google Scholar]
- 16.a) Veeravagu P, Arnold RT, Eigenmann GW. Competitive Elimination-Substitution Reactions. Some Dramatic Difference between Bromide and Tosylate. J Am Chem Soc. 1964;86:3072–3075. [Google Scholar]; b) Chatti S, Bortolussi M. Loupy, A Synthesis of New Diols Derived from Dianhydrohexitols Ethers under Microwave-Assisted Phase Transfer Catalysis. Tetrahedron. 2000;56:5877–5883. [Google Scholar]
- 17.Dehmlow EV, Dehmlow SS. Phase-Transfer Catalysis. 2 Weinheim; Chemie: 1983. [Google Scholar]
- 18.Dehmlow EV. In: Phase-Transfer Catalysis. Mechanisms and Synthesis. Halpern ME, editor. Washington; Am. Chem. Soc: 1996. pp. 108–122. [Google Scholar]
- 19.Smith ND, Mancuso J, Lautens M. Metal-Catalyzed Hydrostannations. Chem Rev. 2000;100:3257–3282. doi: 10.1021/cr9902695. [DOI] [PubMed] [Google Scholar]
- 20.Stille JK. The Palladium-Catalyzed Cross-Coupling Reactions of Organotin Reagents with Organic Electrophiles. Angew Chem Int Ed. 1986;25:508–523. [Google Scholar]
- 21.Farina V, Krishnamurthy V, Scoot WJ. Org React. 1997;50:1–652. [Google Scholar]
- 22.Littke AF, Fu GC. Palladium-Catalyzed Coupling Reactions of Aryl Chlorides. Angew Chem Int Ed. 2002;41:4176–4211. doi: 10.1002/1521-3773(20021115)41:22<4176::AID-ANIE4176>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 23.Espinet P, Echavarren AM. The Mechanisms of the Stille Reaction. Angew Chem Int Ed. 2004;43:4704–4734. doi: 10.1002/anie.200300638. [DOI] [PubMed] [Google Scholar]
- 24.Farina V, Kapadia S, Krishnan B, Wang C, Liebeskind LS. On the Nature of the “Copper Effect” in the Stille Cross-Coupling. J Org Chem. 1994;59:5905–5911. [Google Scholar]
- 25.a) Miyaura N, Suzuki A. Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem Rev. 1995;95:2457–2483. [Google Scholar]; b) Suzuki A. Recent Advances in the Cross-Coupling Reactions of Organoboron Derivatives with Organic Electrophiles, 1995–1998. J Organomet Chem. 1999;576:147–168. [Google Scholar]
- 26.Lenz SM, Meier E, Pedersen H, Frederiksen K, BøgesØ KP, Krogsgarrd-Larsen P. Muscarinic Agonists. Syntheses and Structure-Activity Relationahips of Bicyclic Isoxazole Ester Bioisosteres of Norarecoline. Eur J Med Chem. 1995;30:263–270. [Google Scholar]
- 27.Friary R, Sunday BR. A Direct Preparation of 3-Hydroxy-1,2-benzisoxazoles. J Heterocyclic Chem. 1979;16:1277. [Google Scholar]
- 28.Dischino DD, Gribkoff VK, Hewawasam P, Luke GM, Rinehart JK, Spears TL, Starrett JE., Jr Synthesis of 3H and 14C Labeled (S)-3-(5-Chloro-2-methoxyphenyl)-1,3-dihydro-3-fluoro-6-(trifluoromethyl)-2H-indol-2-one, MaxipostTM. An Agent for Post-Stroke Neuroprotection. J Label Compd Radiopharm. 2003;46:139–149. [Google Scholar]
- 29.McArdle P. ABSEN-a PC Computer Program for Listing Systematic Absences and Apace-Group Determination. J Appl Cryst. 1996;29(3):306. [Google Scholar]
- 30.Otwinowski Z, Minor W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 31.Burla MC, Camalli M, Carrozzini B, Cascarano GL, Giacovazzo C, Polidori G, Spagna R. SIR2002: the Program. J Appl Cryst. 2003;36:1103. [Google Scholar]
- 32.International Tables for Crystallography. C. Kluwer Academic Publishers; Dordrecht, The Netherlands: 1992. Tables 4.2.6.8 and 6.1.1.4. [Google Scholar]
- 33.Sheldrick GM. SHELXL97 A Program for Crystal Structure Refinement. Univ. of Gottingen; Germany: 1997. [Google Scholar]
- 34.Johnson CK. ORTEPII, Report ORNL-5138. Oak Ridge National Laboratory; Tennessee, USA: 1976. [Google Scholar]
- 35.Spek AL. PLUTON Molecular Graphics Program. Univ. of Ultrecht; The Netherlands: 1991. [Google Scholar]
- 36.Beurskens PT, Beurskens G, deGelder S, Garcia-Granda R, Gould RO, Israel R, Smits JMM. The DIRDIF-99 Program System. Crystallography Laboratory, Univ. of Nijmegen; The Netherlands: 1999. [Google Scholar]
- 37.Buckheit RWJ, Fliakas-Boltz V, Decker WD, Robertson JL, Stup TL, Pyle CA, White EL, McMahon JB, Currens MJ, Boyd MR, Bader JP. Comparitive Anti-HIV Evaluation of Diverse HIV-1-Specific Reverse Transcriptase Inhibitor-Resistant Virus Isolates Demonstrates the Existence of Distinct Phenotypic Subgroups. Antiviral Res. 1995;26:117–132. doi: 10.1016/0166-3542(94)00069-k. [DOI] [PubMed] [Google Scholar]
- 38.Rice WG, Bader JP. Discovery and in Vitro Development of AIDS Antiviral Drugs as Biopharmaceuticals. Adv Pharmacol (San Diego) 1995;6:389–438. doi: 10.1016/s1054-3589(08)60675-4. [DOI] [PubMed] [Google Scholar]
- 39.Pauwels R, Balzarini J, Baba M, Snoeck R, Schols D, Herdewijn P, Desmyter J, De Clercq E. Rapid and Automated Tetrazolium-based Colorimetric Assay for Detection of Anti-HIV Compounds. J Virol Methods. 1988;20:309–321. doi: 10.1016/0166-0934(88)90134-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Elemental analyses of compounds 3, 4, 7-22, 25, 30-32, 34-36, 41-44, 49, 51, 56-59. Xray crystal structures of 22 and 58 in CIf format and elemental analyses. This material is available free of charge via the Internet at http://pubs.acs.org.