

Abstract
The small molecule CCR5 inhibitors are a new class of drugs for treating infection by human immunodeficiency virus type 1 (HIV-1). They act by binding to the CCR5 co-receptor and preventing its use during HIV-1-cell fusion. Escape mutants can be raised against CCR5 inhibitors in vitro and will arise when these drugs are used clinically. Here, we have assessed the responses of CCR5 inhibitor-resistant viruses to other anti-retroviral drugs that act by different mechanisms, and their sensitivities to neutralizing antibodies (NAbs). The rationale for the latter study is that the resistance pathway for CCR5 inhibitors involves changes in the HIV-1 envelope glycoproteins (Env), which are also targets for NAbs. The escape mutants CC101.19 and D1/85.16 were selected for resistance to AD101 and vicriviroc (VVC), respectively, from the primary R5 HIV-1 isolate CC1/85. Each escape mutant was cross resistant to other small molecule CCR5 inhibitors (aplaviroc, maraviroc, VVC, AD101 and CMPD 167), but sensitive to protein ligands of CCR5: the modified chemokine PSC-RANTES and the humanized MAb PRO 140. The resistant viruses also retained wild-type sensitivity to the nucleoside reverse transcriptase inhibitor (RTI) zidovudine, the non-nucleoside RTI nevirapine, the protease inhibitor atazanavir and other attachment and fusion inhibitors that act independently of CCR5 (BMS-806, PRO-542 and enfuvirtide). Of note is that the escape mutants were more sensitive than the parental CC1/85 isolate to a subset of neutralizing monoclonal antibodies and to some sera from HIV-1-infected people, implying that sequence changes in Env that confer resistance to CCR5 inhibitors can increase the accessibility of some NAb epitopes. The need to preserve NAb resistance may therefore be a constraint upon how escape from CCR5 inhibitors occurs in vivo.
Introduction
New classes of drugs to treat infection with human immunodeficiency virus type 1 (HIV-1) are still being developed, not least because of the propensity of the virus to develop resistance to the reverse transcriptase inhibitors (RTI) and protease inhibitors (PI) that have been the mainstay of therapy for the past decade (Hammer et al., 2006). The new classes include the small molecule CCR5 inhibitors; maraviroc (MVC) has now been approved for clinical use, vicriviroc (VVC) is in Phase III trials, and several others are at earlier stages in the drug-development process (Kuhmann and Hartley, 2008). These compounds act by binding to the CCR5 co-receptor and altering its conformation in a way that impedes its recognition by the HIV-1 gp120 glycoprotein during virus-cell fusion (Dragic et al., 2000; Kondru et al., 2007; Seibert et al., 2006; Tsamis et al., 2003; Watson et al., 2005). HIV-1 is, however, able to mutate to escape the inhibitory effects of the small molecule CCR5 inhibitors, a process that involves altering the conformation of the gp120 protein to allow its binding to the inhibitor-CCR5 complex (Pugach et al., 2007; Westby et al., 2007). The resistant viruses can also use the free CCR5 co-receptor for entry, so they are not drug-dependent (Baba et al., 2007; Marozsan et al., 2005; Trkola et al., 2002; Westby et al., 2007). The resistance phenotype is stable in vitro, in that the escape mutants are fit and they do not rapidly revert to sensitivity when cultured in the absence of the selecting compound (Anastassopoulou et al., 2007; Trkola et al., 2002; Westby et al., 2007). Whether this also applies in vivo remains to be determined, as multiple selection pressures on the HIV-1 Env glycoproteins may work together to compromise fitness under those conditions.
Details are now emerging about how resistance to the small molecule CCR5 inhibitors arises at a molecular level. The natural interaction between gp120 and CCR5 appears to involve two principal points of contact; the V3 region and the bridging sheet of gp120 bind to the second extracellular loop (ECL-2) and the tyrosine-sulfated N-terminus (Nt) of CCR5, respectively (Cormier and Dragic, 2002; Huang et al., 2007). In the escape mutants, the sequence changes in gp120 may disrupt the former interaction, rendering the virus much more dependent on the binding of the bridging sheet to the CCR5 Nt (our unpublished results). Genetically, this is usually achieved by the introduction of sequence changes within V3 (Baba et al., 2007; Kuhmann et al., 2004; Ogert et al., 2008; Westby et al., 2007). However, at least one VVC-resistant clone has no V3 sequence changes, which implies the existence of alternative genetic pathways to the same phenotype (Marozsan et al., 2005).
All the above observations were made using escape mutants that were generated in cell culture, but early clinical studies of the small molecule CCR5 inhibitors suggest that resistant viruses generated in vivo have broadly similar properties (Mori et al., 2007; Strizki et al., 2006). We have therefore used two different CCR5 inhibitor-resistant viruses to address two questions of relevance to the clinical use of these new drugs: Do the changes in gp120 that confer resistance to CCR5 inhibitors affect how the virus is neutralized by antibodies that target the viral envelope gp120/gp41 glycoprotein complex? Are the resistant viruses still sensitive to inhibitors with different mechanisms of action, including PIs and RTIs and other fusion/entry inhibitors that target different steps in the fusion process? The former sub-study is particularly relevant to understanding how CCR5 inhibitor resistance might evolve in vivo, because most recipients will have plasma antibodies against the HIV-1 envelope glycoproteins. To enable it to persist in the face of humoral immunity, HIV-1 has evolved multiple natural defensive mechanisms that shield the vulnerable regions of its Env complex from antibody binding, including the CCR5 binding site (Burton et al., 2004; Labrijn et al., 2003). Thus, the V3 region of most primary viruses is not well exposed for antibody binding (Hartley et al., 2005). The question therefore arises as to whether the sequence changes in the CCR5 inhibitor-escape mutants disturb any of the antibody-resistance mechanisms possessed by wild-type viruses such as their parental strain. If so, then there will be additional constraints on how CCR5 inhibitor-resistant viruses arise in vivo; the escape mutants must not only acquire resistance to the drug, but also retain their natural resistance to NAbs.
Our observations are that the escape mutants are moderately more sensitive than the parental CC1/85 isolate to some sera from HIV-1-infected people and also to a subset of neutralizing monoclonal antibodies (MAbs). These findings imply that acquisition of resistance to CCR5 inhibitors can affect how the Env complex resists the action of NAbs, which may be relevant to how resistance to these drugs develops in vivo. We also show that the small molecule CCR5 inhibitor-resistant viruses are generally cross resistant within this drug class but retain wild-type sensitivity to RTIs, PIs and various attachment and entry inhibitors that act independently of CCR5. However, as reported previously, the escape mutants remain sensitive to protein ligands of CCR5 such as the modified chemokine PSC-RANTES and anti-CCR5 MAbs that act by different mechanisms (Ji et al., 2007; Pugach et al., 2007; Westby et al., 2007).
Results
Responses of the CCR5 inhibitor-resistant isolates to NAbs
To determine whether the AD101 and VVC escape mutants CC101.19 and D1/85.16 had altered responses to antibodies against the HIV-1 envelope glycoproteins, we tested their sensitivity to four broadly reactive NAbs, b12, 2G12, 2F5 and 4E10, in a PBMC-based multi-cycle replication assay, using the CC1/85 parental isolate and the CCcon.19 passage control isolate as reference standards (Fig.1, Table 1). To reduce the variation commonly observed with primary cells, we pooled PBMCs from two different donors for each experiment (Ketas et al., 2007).
Figure 1. Inhibition of CCR5-inhibitor-resistant and control isolates by MAbs.
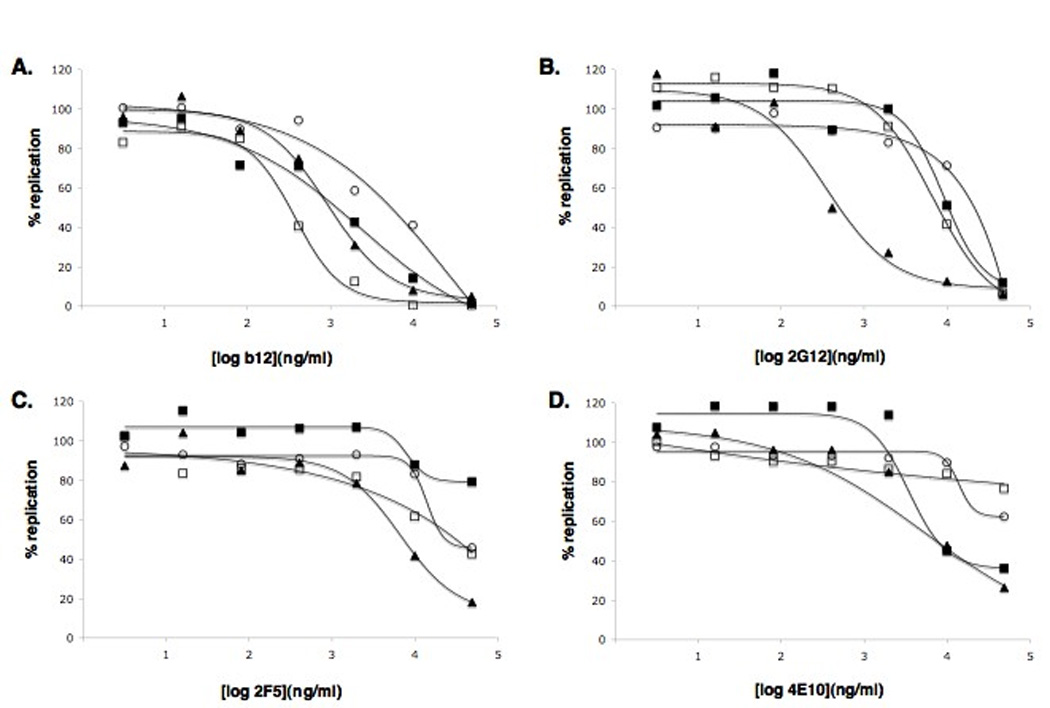
HIV-1 isolates CC1/85 (open circles), CC101.19 (filled squares), D1/85.16 (filled triangles) and CCcon.19 (open squares) were tested for their sensitivity in primary CD4+ T cells to A) b12; B) 2F5; C) 2G12; D) 4E10. The extent of virus replication is presented as a percentage of p24 antigen production in the control culture (defined as 100%). The data presented represent average values derived from three independent experiments. Error bars representing standard errors of the mean have been omitted for clarity, but are typically ± 10% of the mean value. Curve fitting was performed with nonlinear regression analysis using Prism software.
Table 1.
Neutralization of the parental and CCR5 inhibitor-resistant viruses by MAbs
| CC1/85 | CCcon.19 | CC101.19 | D1/85.16 | |
|---|---|---|---|---|
| b12 | 4.5 | 0.3 | 1.3 | 1.1 |
| 2G12 | 23 | 9.0 | 11 | 0.4 |
| 2F5 | 44 | 23 | >50 | 7.7 |
| 4E10 | >50 | >50 | 9.3 | 5.2 |
The values recorded are the MAb concentrations (µg/ml) that reduce the replication of each test virus by 50% in a PBMC-based assay. The values highlighted in bold differ by > 5-fold for the resistant viruses compared to the parental and passage control isolates. The data presented represent average values derived from three independent experiments.
We have previously reported that prolonged adaptation of the CC1/85 isolate to PBMC culture renders it more sensitive to inhibitors of the gp120-CD4 interaction, probably because of a selection pressure for increased CD4 affinity that promotes target cell infection (Pugach et al., 2004). This finding was confirmed in the present experiment: CCcon.19 was markedly (15-fold) more sensitive than CC1/85 to b12, a MAb to the CD4-binding site (CD4bs) on gp120. The two CCR5-inhibitor escape mutants had intermediate sensitivities to b12, with ~4-fold reductions in IC50 compared to CC1/85. These modest effects could arise from either in vitro passage during the resistance selection process, and/or any additional effects of becoming CCR5 inhibitor resistant.
The VVC-resistant isolate D1/85.16 was substantially more sensitive to NAb 2G12 against a glycan-dependent gp120 epitope, with a 50-fold decrease in the IC50 value compared to CC1/85. However, the AD101-resistant and passage control isolates had unchanged sensitivities to 2G12. The increase in the 2G12 sensitivity of D1/85.16 is therefore a consequence of the non-V3 sequence changes that arise as the virus becomes VVC resistant, but may not be obligatorily linked to resistance.
The 2F5 and 4E10 NAbs recognize epitopes in the membrane-proximal external region (MPER) of gp41 (Zwick et al., 2001). The D1/85.16 isolate was moderately (~6-fold) more sensitive to 2F5 than the parental isolate, whereas 2F5 did not detectably inhibit CC101.19. Both CCR5 inhibitor-resistant viruses were >5-fold more sensitive than the parental and passage control isolates to 4E10 (IC50 ~10 µg/ml); the magnitude of the sensitivity increase is hard to judge because neither CC1/85 nor CCcon.19 was sensitive to this MAb (IC50 >50 µg/ml). Nonetheless, CCR5 inhibitor resistance does appear to be associated with a modestly increased sensitivity to MPER-targeted MAbs, with the exception of 2F5 against CC101.19.
We also assessed whether the resistant viruses had altered sensitivities to three V3 MAbs (447-52D, F425 and 39F) and to MAb 17b to a CD4i epitope that overlaps the CCR5 binding site. These studies were inconclusive; none of the control or resistant viruses was sensitive to any of the tested MAbs at concentrations up to 50 µg/ml (data not shown).
Binding assays using monomeric gp120 do not predict changes in NAb sensitivity of the CCR5 inhibitor-resistant isolates
Changes in neutralization sensitivity to a gp120-targeted MAb could arise because of alterations in the structure of its epitope at the level of the gp120 subunit, or because of changes in how the epitope is presented on the functional Env trimer (Moore et al., 1995). To assess which mechanism applied to the CCR5 inhibitor resistant isolates, we measured the binding of various MAbs and CD4-IgG2 to gp120 proteins present in detergent-inactivated preparations of the control and resistant viruses. Titration curves were generated, with half-maximal MAb binding concentrations calculated and presented (Table 2).
Table 2.
MAb binding to gp120s from the parental and CCR5 inhibitor-resistant viruses
| CC1/85 | CC101.19 | D1/85.16 | |
|---|---|---|---|
| CD4-IgG2 | 0.08 | 0.12 | 0.06 |
| 2G12 | 0.19 | 0.24 | 0.09 |
| b12 | 0.22 | 0.20 | 0.17 |
| 447-52D | 0.29 | 0.37 | 0.30 |
| F425 | 0.01 | 0.02 | 0.01 |
| 39F | 0.01 | N.A. | 0.01 |
The values recorded are the MAb or CD4-IgG2 concentrations (µg/ml) that give 50% binding to gp120 proteins that were derived from the corresponding viruses by detergent treatment. N.A. = no detectable binding. The data presented represent average values of two independent wells at each antibody concentration and are the results from a representative experiment.
The binding of b12 and CD4-IgG2 to each of the gp120 proteins was comparable (± 2-fold, the precision limit of this type of assay) (Table 2). Hence, the ~4-fold increased b12 neutralization sensitivity of the resistant isolates may arise because of alterations in the exposure of the CD4bs and its associated epitopes on the Env trimer. Although we have no direct evidence for this supposition, a similar, but more dramatic, effect was previously observed with CCcon.19. This passage control virus was 100-fold more sensitive to neutralization by soluble CD4 without any increase in soluble CD4 binding to monomeric gp120 (Pugach et al., 2004).
There was only a modest (<3-fold) increase in the binding of 2G12 to D1/85.16 gp120 compared to CC1/85 gp120 (Table 2), which is unlikely to be sufficient to account for the ~50-fold increase in the relative neutralization sensitivity of D1/85.16 (Fig.1, Table 1). Structural changes in the presentation of the 2G12 epitope on the Env trimer are probably relevant, although again we have no direct evidence that this is the case.
All three V3 MAbs bound strongly to gp120 from CC1/85 (Table 2). Their inability to neutralize this virus therefore arises because their epitopes are poorly exposed on the Env trimer, as is commonly observed with V3 MAbs (Moore et al., 1995). Since the V3 sequence of D1/85.16 is identical to that of CC1/85, there was, as expected, no difference in the binding of the three V3 MAbs to these two gp120s (Table 2). CC101.19, in contrast to D1/85.16, has 4 sequence changes in V3, which account for its CCR5 inhibitor resistance (Kuhmann et al., 2004). There was no detectable binding of MAb 39F to CC101.19 gp120, presumably because the V3 sequence changes destroy its epitope. However, both 447-52D and F425 did still bind CC101.19 gp120 (Table 2). The failure of these MAbs to neutralize CC101.19, and the failure of all three to neutralize D1/85.16, suggests that the alteration in how the resistant viruses recognize CCR5 does not involve a gross change in how the V3 region is exposed to antibodies on the native trimer.
Sensitivities of the CCR5 inhibitor-resistant isolates to neutralizing sera
To further characterize the neutralization phenotype of the resistant viruses, we assessed their sensitivities to sera from 15 different HIV-1-infected individuals, and to a preparation of HIVIg, again using a PBMC-based replication assay. Our general experience with this type of assay is that 50% neutralization titer differences of < 3-fold are not meaningful, because of experimental variation. Applying this ad hoc cut-off, a subset of sera neutralized the CCR5 inhibitor-resistant viruses more potently than the CC1/85 parental and CCcon.19 passage control viruses. In general, D1/85.16 was the most neutralization sensitive isolate, albeit not profoundly so. More specifically, 6/16 sera neutralized D1/85.16 at higher than expected titers, whereas 2/16 sera did so against CC101.19 (Table 3). However, the increases were generally modest in magnitude (<10-fold).
Table 3.
Neutralization of CCR5 inhibitor and control isolates by sera from HIV-1-infected individuals
| CC1/85 | CCcon.19 | CC101.19 | D1/85.16 | |
|---|---|---|---|---|
| HIVIG | 270 | 185 | 285 | 600 |
| 20316 | 70 | 60 | 85 | 270 |
| 20422 | 120 | 110 | 215 | 455 |
| 20507 | 30 | 70 | 20 | 115 |
| 20451 | 320 | 200 | 160 | 530 |
| 20777 | 240 | 55 | 60 | 285 |
| 20207 | 800 | 2640 | 1900 | 2730 |
| 20394 | 1245 | 1870 | 8000 | 2550 |
| 20608 | 3800 | 2400 | 11800 | 3660 |
| 20479 | 1900 | 1630 | 3630 | 2240 |
| 20555 | 1730 | 3200 | 680 | 3130 |
| 20699 | 3725 | 1900 | 1815 | 3150 |
| 170 | 55 | N.D. | 150 | 340 |
| 214 | 75 | N.D. | 185 | 850 |
| 371 | 20 | N.D. | 40 | 110 |
| 494 | 80 | N.D. | 130 | 270 |
The values listed are the reciprocal serum dilutions that reduce virus production by 50% for each serum/virus combination. The sera designated 20XXX were obtained from the MACS cohort via Dr. Steven Wolinsky, the other four sera were provided by Dr. Robert Doms. The values highlighted in bold for the two VVC-resistant isolates CC101.19 and D1/85.16 differ by >3-fold from the mean values for the parental and passage control isolates CC1/85 and CCcon.19 (or, for the last 4 listed sera, from the value for CC1/85). N.D. = Not Done, due to a shortage of these sera. The data presented represent average values derived from two or three independent experiments, depending on the availability of the sera.
The CCR5 inhibitor-resistant isolates are cross-resistant to other small molecule CCR5 inhibitors but sensitive to inhibitors that act elsewhere in the HIV-1 replication cycle
Both the CC101.19 and D1/85.16 isolates were highly resistant to five different small molecule inhibitors: AD101, VVC, MVC, aplaviroc and CMPD-167 (Fig.2). This cross-resistance does not extend, however, to inhibitors that target CCR5 via a different mechanism, as we have previously reported that CC101.19 and D1/85.16 retain wild-type sensitivity to chemokines and MAbs that bind to CCR5 and inhibit HIV-1 replication (Pugach et al., 2007; Trkola et al., 2002).
Figure 2. Sensitivity of CCR5-inhibitor-resistant and control isolates to inhibitors of HIV-1 replication.
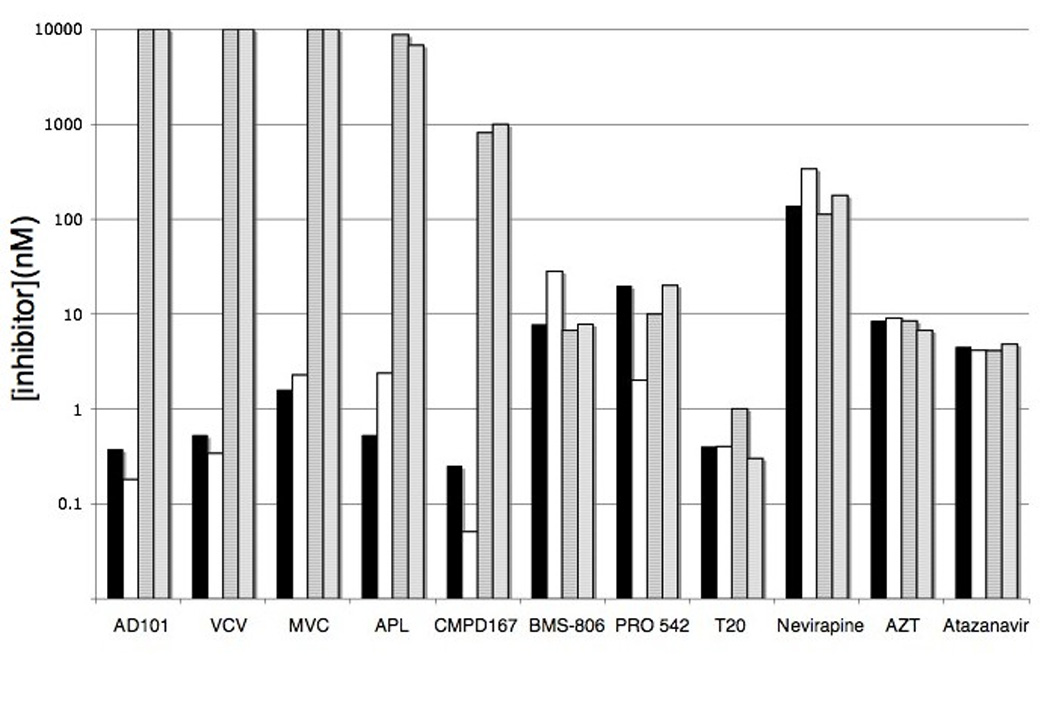
HIV-1 isolates CC1/85 (filled bars), CCcon19 (open bars), CC101.19 (hatched bars) and D1/85.16 (gray bars) were tested in primary CD4+ T cells for their sensitivity to the various inhibitors listed on the x-axis. The inhibitor concentrations required to inhibit replication by 50% are shown. The data presented represent average values derived from two independent experiments.
To assess whether resistance to small molecule CCR5 inhibitors affects the response to other anti-retroviral drugs that act at other stages of the HIV-1 replication cycle, we used BMS-806, PRO-542 (CD4-IgG2) and enfuvirtide (T-20) as representatives of attachment/fusion inhibitors, zidovudine (AZT) and nevirapine as RTIs and atazanavir as a PI. Neither CC101.19 nor D1/85.16 differed by ± 2-fold from CC1/85 and CCcon19 in their sensitivities to any of these inhibitors (Fig.2). The CCcon19 passage control virus was modestly (10-fold) more sensitive than the others to PRO-542 and, conversely, 4-fold less sensitive to BMS-806 (Fig.2). These altered sensitivities to inhibitors of gp120-CD4 binding are consistent with the increase in CD4 affinity that occurred during the prolonged in vitro passage of this virus (Pugach et al., 2004).
Discussion
One goal of this study was to assess whether the clinical use of CCR5 inhibitors to treat HIV-1 infection might drive the virus down a resistance pathway that altered its sensitivity to plasma NAbs. Both CCR5 inhibitors and NAbs affect envelope glycoprotein functions, and both cause resistance mutations to emerge in these proteins. Hence the potential for reinforcing (or interfering) actions clearly exists (Moore and Doms, 2003). A particularly interesting scenario is that the principal resistance pathway for a CCR5 inhibitor might render HIV-1 more sensitive to neutralization by disrupting some of the natural defenses against NAb binding that have evolved in its Env protein complex. Another, related possibility is that the sequence changes producing a resistant virus also create neo-epitopes that drive the production of new NAbs to which the virus is sensitive, thereby creating a new selection pressure. We have not yet addressed the latter eventuality; doing so will require testing whether any resistant viruses that emerge in vivo have altered sensitivities to longitudinal, autologous serum samples. We have, however, used two different small molecule CCR5 inhibitor-resistant viruses that were generated in vitro, to assess whether the acquisition of resistance is associated with altered sensitivity to MAbs and sera from infected individuals. The outcome of these experiments is that there is indeed such a linkage, albeit not a dramatic one.
Our two resistant variants were both derived from the same R5 primary isolate, CC1/85, using two different CCR5 inhibitors, AD101 and VVC. Although the phenotypes of the two escape mutants are generally very similar, two different genetic pathways to resistance were followed. The AD101-resistant variant, CC101.19, has 4 sequence changes in V3 that appear to disrupt the binding interaction between this region of gp120 and ECL-2 of CCR5, rendering the escape mutant more dependent on the binding of the gp120 bridging sheet to the CCR5 N-terminus (our unpublished results). In contrast, the VVC-resistant virus D1/85.16 has no sequence changes in V3, and probably relies on other sequence changes to create a broadly similar rearrangement of the components of its CCR5 binding site (we are still in the process of determining which particular Env changes in D1/85.16 are responsible for resistance) (Marozsan et al., 2005). For both viruses, the eventual outcome of the genetic changes is the ability to use the inhibitor-CCR5 complex as well as free CCR5 for entry (Marozsan et al., 2005; Trkola et al., 2002).
The two resistant variants, CC101.19 and D1/85.16, were each moderately more sensitive to the CD4bs-associated MAb b12, but to a lesser extent than the passage control isolate CCcon.19. This virus adapted to extended growth in PBMC culture by increasing its affinity for CD4 (Pugach et al., 2004), an outcome that also modestly affects its inhibition by PRO 542 (CD4-IgG2) and BMS-806 (Fig.2) In contrast, both escape mutants had unchanged sensitivities to CD4-IgG2 or BMS-806, so the principal driving force behind their slightly greatly susceptibilities to b12 is probably not related to changes in CD4 binding. However, sequence changes that arose during either the in vitro adaptation process or the development of CCR5 inhibitor resistance could be involved.
The D1/85.16 variant, but not CC101.19, was markedly (~50-fold) more sensitive to 2G12, a MAb that binds a discontinuous epitope comprising mannose moieties from at least three N-linked glycans (Sanders et al., 2002; Scanlan et al., 2002). There are no sequence changes in D1/85.16 that directly affect the known glycan contributors to the 2G12 epitope, but there is a G354P change in the C3 region adjacent to residue F353 that is known to influence 2G12 binding, albeit in the genetic context of HIV-1 JR-FL (Scanlan et al., 2002). Because 2G12 did not have a significantly higher affinity for gp120 derived from D1/85.16, the most likely explanation of the increased 2G12 sensitivity is an alteration in the conformation or accessibility of the 2G12 epitope on the functional Env trimer. Whether, and if so how, this is linked to CCR5 inhibitor resistance remains to be determined. However, because of the unique nature of the 2G12 MAb and its epitope, it seems unlikely that the increased sensitivity of a CCR5 inhibitor-resistant virus to 2G12 will have much relevance to what may arise during the clinical use of these drugs.
There were also modest increases in the sensitivity of the resistant viruses to the MPER-directed MAbs 4E10 and, in the case of D1/85.16, also to 2F5. The only sequence change in close proximity to the continuous epitopes for these MAbs was E662A in D1/85.16 (a change not present in CC101.19). This is a common polymorphism in the first position of the 2F5 epitope, E/ALDKWAS, that was also present in one clone from the CC1/85 parental isolate. It could account for the modest (~6-fold) increase in the 2F5 neutralization sensitivity of D1/85.16, particularly as 2F5 did not neutralize CC101.19 detectably. There is no obvious sequence-based explanation for why either escape mutant is moderately (>5-fold) more sensitive to 4E10, as is often the case (Binley et al., 2004). A reduction in the rate of fusion can increase sensitivity to neutralization by 2F5 and 4E10, by increasing the time available for these MAbs to bind to gp41 (Reeves et al., 2005). Hence we are now investigating if resistance to CCR5 inhibitors is associated with alterations in fusion rates.
Given how we believe the resistant viruses acquire their ability to use the CCR5-inhibitor complex, by becoming more dependent on the bridging sheet region for a binding interaction with the CCR5 N-terminus, perhaps the most interesting possibility for an alteration in their neutralization sensitivity would involve an increase in V3 exposure on the native Env trimer. In vivo, such an occurrence could render the escape mutant more sensitive to existing V3-directed antibodies and/or create a new, and perhaps more immunogenic, V3 configuration for the generation of NAbs specific to the escape mutant. The V3 region is normally poorly exposed on R5 primary viruses (Hartley et al., 2005; Moore et al., 1995); accordingly, none of the V3-directed MAbs we tested could significantly neutralize the parental CC1/85 virus, even when their epitope was present on the gp120 monomer. We found that neither escape mutant had acquired any sensitivity to the available V3 MAbs. The 4 sequence changes within V3 destroyed the epitope for MAb 39F on CC101.19, but the epitopes for two other MAbs, 447-52D and F425, were unaltered, and this was true of all three V3 MAbs on D1/85.16. The preservation of resistance to these V3 MAbs on the resistant viruses therefore implies that the V3-dependent and V3–independent changes that create CCR5 inhibitor resistance do not necessarily alter how V3 is exposed to antibodies on the functional Env trimer.
Some modest and sporadic increases in sensitivity of D1/85.16, but not CC101.19, to sera from HIV-1-infected people were noted. These changes, of course, reflect heterologous neutralization via unknown specificities, but they are generally consistent with the observations made using MAbs to known epitopes. A more relevant pattern of data will emerge as and when in vivo-generated escape mutants are tested using heterologous plasma. Nonetheless, the existing results do suggest that CCR5 inhibitor-resistant viruses are likely to be somewhat more sensitive to neutralization than their parental viruses, implying that the humoral immune system could indeed exert additional selection pressures during the escape process (Moore and Doms, 2003).
We also confirmed earlier reports that the acquisition of resistance to small molecule CCR5 inhibitors does not cause HIV-1 to become resistant to drugs with different mechanisms of action, such as RTIs, PIs and attachment/fusion inhibitors that act independently of CCR5 (Marozsan et al., 2005; Westby et al., 2007). Hence resistant variants that arise in vivo can still be treated with other drug classes. Moreover, the escape mutants were still sensitive to MAbs and chemokines targeting regions of CCR5 that are spatially distinct from the binding site for the small molecule CCR5 inhibitors, as has been reported previously (Dragic et al., 2000; Pugach et al., 2007; Trkola et al., 2002; Tsamis et al., 2003). In principle, therefore, small molecule CCR5 inhibitors could therefore be usefully combined with CCR5 MAbs that are now being evaluated clinically, particularly as the two categories of CCR5 inhibitor act synergistically to inhibit HIV-1 entry (Murga et al., 2006).
Methods
Antibodies and reagents
Sera from HIV-1-infected individuals were provided by Dr. Robert Doms (University of Pennsylvania, Philadelphia, PA) and Dr. Steven Wolinsky (Chicago MACS, Northwestern University, Chicago, IL). AZT, Nevirapine, Atazanvir, MAb 447-52D, MAb F425 and HIVIG were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. CD4-IgG2 (PRO-542), APL and T-20 were donated by Dr. William Olson (Progenics Pharmaceuticals Inc, Tarrytown, NY). MAbs 2G12, 2F5, 4E10 were gifts from Dr. Hermann Katinger (Polymun Scientific Inc, Vienna, Austria), MAb b12 from Dr. Dennis Burton (Scripps Research Institute, La Jolla, CA) and MAb 39F from Dr. James Robinson (Tulane University, New Orleans, LA). VVC and AD101 were provided by Dr. Julie Strizki (Schering-Plough Research Institute, Kenilworth, NJ), CMPD 167 was from Dr. Marty Springer (Merck Research Labs, Rahway, NJ), BMS-378806 from Richard Colonno (Bristol Myers Squibb, Wallingford, CT) and MVC from Dr. Chris Hitchcock (Pfizer, Sandwich, UK).
Gp120 binding assays
Stocks of infectious HIV-1 were inactivated with 1% Nonidet-P40 (NP-40) detergent and used as a source of gp120 proteins for determination of antibody binding by ELISA. The gp120s were captured onto plastic plates coated with the sheep polyclonal antibody D7324 to the C-terminus of gp120, then the binding of MAbs or CD4-IgG2 was detected as described elsewhere (Moore and Sodroski, 1996; Trkola et al., 1995). The background OD490 signal (no gp120) was subtracted from the signal derived using gp120 at each input MAb concentration.
Virus production
Generation of HIV-1 isolates was described previously (Marozsan et al., 2005; Trkola et al., 2002). Clonal, replication-competent, chimeric NL4-3/env viruses were prepared by transfecting 15 µg of the full-length proviral plasmid into 293T cells using Lipofectamine 2000 (Invitrogen), according to the manufacturer’s instructions.
HIV-1 infection of primary CD4+ T cells
Mitogen-activated, CD8+ T cell-depleted PBMC were prepared from leukopacks pooled from the blood of 2 healthy volunteers. The pooled fractions were treated with the “RosetteSep CD8+ depletion cocktail” (StemCell Technologies, Vancouver, BC, Canada) according to the manufacturer’s instructions, before Ficoll density gradient separation of leukocytes to deplete CD8+ T cells from the PBMC by rosette formation (Trkola et al., 2002). The cells were split into two cultures that were stimulated for three days with 5 µg/ml of phytohemagglutinin (PHA; Sigma) or surface-immobilized anti-CD3 MAb (clone OKT3). All PBMC cultures were maintained in PBMC culture medium (RPMI 1640; Invitrogen) with 10% FBS, 1X PenStrep and 100 U/ml interleukin-2 (IL-2; ARRRP, donated by Hoffmann-La Roche, Inc.).
The activated PBMC from the two stimulation cultures were then pooled at a 1:1 ratio and seeded (2 × 105 per well) into a 96-well culture plate. The cells were then treated with inhibitors for 1h at 37°C before addition of 50 TCID50 of a HIV-1 isolate. Production of the viral p24 antigen after 7–10 days of culture was quantified using an in-house ELISA (Trkola et al., 1995). In each assay, each data point was derived from triplicate wells. The amount of p24 produced was corrected by subtracting the residual p24 remaining from the inoculum virus. HIV-1 replication in the presence of inhibitors is expressed as a percentage of what occurred in their absence.
Acknowledgments
This work was supported by NIH grants T32 AI07621 (PP), AI36082 and AI41420 and by the IAVI Neutralizing Antibody Consortium. We thank Shawn Kuhmann and PJ Klasse for comments on the manuscript. We are grateful to Robert Doms, Steven Wolinsky, William Olson, Hermann Katinger, Dennis Burton, James Robinson, Julie Strizki, Marty Springer, Richard Colonno, Chris Hitchcock and donors to the NIH AIDS Research and Reference Reagent Program for gifts of reagents.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anastassopoulou CG, Marozsan AJ, Matet A, Snyder AD, Arts EJ, Kuhmann SE, Moore JP. Escape of HIV-1 from a small molecule CCR5 inhibitor is not associated with a fitness loss. PLoS Pathog. 2007;3(6):e79. doi: 10.1371/journal.ppat.0030079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M, Miyake H, Wang X, Okamoto M, Takashima K. Isolation and characterization of human immunodeficiency virus type 1 resistant to the small-molecule CCR5 antagonist TAK-652. Antimicrob Agents Chemother. 2007;51(2):707–715. doi: 10.1128/AAC.01079-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binley JM, Wrin T, Korber B, Zwick MB, Wang M, Chappey C, Stiegler G, Kunert R, Zolla-Pazner S, Katinger H, Petropoulos CJ, Burton DR. Comprehensive cross-clade neutralization analysis of a panel of anti-human immunodeficiency virus type 1 monoclonal antibodies. J Virol. 2004;78(23):13232–13252. doi: 10.1128/JVI.78.23.13232-13252.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton DR, Desrosiers RC, Doms RW, Koff WC, Kwong PD, Moore JP, Nabel GJ, Sodroski J, Wilson IA, Wyatt RT. HIV vaccine design and the neutralizing antibody problem. Nat Immunol. 2004;5(3):233–236. doi: 10.1038/ni0304-233. [DOI] [PubMed] [Google Scholar]
- Cormier EG, Dragic T. The crown and stem of the V3 loop play distinct roles in human immunodeficiency virus type 1 envelope glycoprotein interactions with the CCR5 coreceptor. J Virol. 2002;76(17):8953–8957. doi: 10.1128/JVI.76.17.8953-8957.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragic T, Trkola A, Thompson DA, Cormier EG, Kajumo FA, Maxwell E, Lin SW, Ying W, Smith SO, Sakmar TP, Moore JP. A binding pocket for a small molecule inhibitor of HIV-1 entry within the transmembrane helices of CCR5. Proc Natl Acad Sci U S A. 2000;97(10):5639–5644. doi: 10.1073/pnas.090576697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer SM, Saag MS, Schechter M, Montaner JS, Schooley RT, Jacobsen DM, Thompson MA, Carpenter CC, Fischl MA, Gazzard BG, Gatell JM, Hirsch MS, Katzenstein DA, Richman DD, Vella S, Yeni PG, Volberding PA. Treatment for adult HIV infection: 2006 recommendations of the International AIDS Society--USA panel. Top HIV Med. 2006;14(3):827–843. [PubMed] [Google Scholar]
- Hartley O, Klasse PJ, Sattentau QJ, Moore JP. V3: HIV's switch-hitter. AIDS Res Hum Retroviruses. 2005;21(2):171–189. doi: 10.1089/aid.2005.21.171. [DOI] [PubMed] [Google Scholar]
- Huang CC, Lam SN, Acharya P, Tang M, Xiang SH, Hussan SS, Stanfield RL, Robinson J, Sodroski J, Wilson IA, Wyatt R, Bewley CA, Kwong PD. Structures of the CCR5 N terminus and of a tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science. 2007;317(5846):1930–1934. doi: 10.1126/science.1145373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji C, Brandt M, Dioszegi M, Jekle A, Schwoerer S, Challand S, Zhang J, Chen Y, Zautke L, Achhammer G, Baehner M, Kroetz S, Heilek-Snyder G, Schumacher R, Cammack N, Sankuratri S. Novel CCR5 monoclonal antibodies with potent and broad-spectrum anti-HIV activities. Antiviral Res. 2007;74(2):125–137. doi: 10.1016/j.antiviral.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Ketas TJ, Schader SM, Zurita J, Teo E, Polonis V, Lu M, Klasse PJ, Moore JP. Entry inhibitor-based microbicides are active in vitro against HIV-1 isolates from multiple genetic subtypes. Virology. 2007;364(2):431–440. doi: 10.1016/j.virol.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Kondru R, Zhang J, Ji C, Mirzadegan T, Rotstein D, Sankuratri S, Dioszegi M. Molecular Interactions of Ccr5 with Major Classes of Small-Molecule Anti Hiv Ccr5 Antagonists. Mol Pharmacol. 2007 doi: 10.1124/mol.107.042101. [DOI] [PubMed] [Google Scholar]
- Kuhmann SE, Hartley O. Targeting chemokine receptors in HIV: a status report. Annu Rev Pharmacol Toxicol. 2008;48:425–461. doi: 10.1146/annurev.pharmtox.48.113006.094847. [DOI] [PubMed] [Google Scholar]
- Kuhmann SE, Pugach P, Kunstman KJ, Taylor J, Stanfield RL, Snyder A, Strizki JM, Riley J, Baroudy BM, Wilson IA, Korber BT, Wolinsky SM, Moore JP. Genetic and phenotypic analyses of human immunodeficiency virus type 1 escape from a small-molecule CCR5 inhibitor. J Virol. 2004;78(6):2790–2807. doi: 10.1128/JVI.78.6.2790-2807.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrijn AF, Poignard P, Raja A, Zwick MB, Delgado K, Franti M, Binley J, Vivona V, Grundner C, Huang CC, Venturi M, Petropoulos CJ, Wrin T, Dimitrov DS, Robinson J, Kwong PD, Wyatt RT, Sodroski J, Burton DR. Access of antibody molecules to the conserved coreceptor binding site on glycoprotein gp120 is sterically restricted on primary human immunodeficiency virus type 1. J Virol. 2003;77(19):10557–10565. doi: 10.1128/JVI.77.19.10557-10565.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marozsan AJ, Kuhmann SE, Morgan T, Herrera C, Rivera-Troche E, Xu S, Baroudy BM, Strizki J, Moore JP. Generation and properties of a human immunodeficiency virus type 1 isolate resistant to the small molecule CCR5 inhibitor, SCH-417690 (SCH-D) Virology. 2005;338(1):182–199. doi: 10.1016/j.virol.2005.04.035. [DOI] [PubMed] [Google Scholar]
- Moore JP, Cao Y, Qing L, Sattentau QJ, Pyati J, Koduri R, Robinson J, Barbas CF, 3rd, Burton DR, Ho DD. Primary isolates of human immunodeficiency virus type 1 are relatively resistant to neutralization by monoclonal antibodies to gp120, and their neutralization is not predicted by studies with monomeric gp120. J Virol. 1995;69(1):101–109. doi: 10.1128/jvi.69.1.101-109.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JP, Doms RW. The entry of entry inhibitors: a fusion of science and medicine. Proc Natl Acad Sci U S A. 2003;100(19):10598–10602. doi: 10.1073/pnas.1932511100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JP, Sodroski J. Antibody cross-competition analysis of the human immunodeficiency virus type 1 gp120 exterior envelope glycoprotein. J Virol. 1996;70(3):1863–1872. doi: 10.1128/jvi.70.3.1863-1872.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori J, Mosley M, Lewis M, Simpson P, Toma J, Huang W, Whitcomb J, Claramella G, Westby M. Barbados: XVI International HIV Drug Resistance Workshop; Characterization of maraviroc resistance in patients failing treatment with CCR5-tropic HIV-1 in MOTIVATE 1 and MOTIVATE 2. 2007
- Murga JD, Franti M, Pevear DC, Maddon PJ, Olson WC. Potent antiviral synergy between monoclonal antibody and small-molecule CCR5 inhibitors of human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2006;50(10):3289–3296. doi: 10.1128/AAC.00699-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogert RA, Wojcik L, Buontempo C, Ba L, Buontempo P, Ralston R, Strizki J, Howe JA. Mapping resistance to the CCR5 co-receptor antagonist vicriviroc using heterologous chimeric HIV-1 envelope genes reveals key determinants in the C2-V5 domain of gp120. Virology. 2008;373(2):387–399. doi: 10.1016/j.virol.2007.12.009. [DOI] [PubMed] [Google Scholar]
- Pugach P, Kuhmann SE, Taylor J, Marozsan AJ, Snyder A, Ketas T, Wolinsky SM, Korber BT, Moore JP. The prolonged culture of human immunodeficiency virus type 1 in primary lymphocytes increases its sensitivity to neutralization by soluble CD4. Virology. 2004;321(1):8–22. doi: 10.1016/j.virol.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Pugach P, Marozsan AJ, Ketas TJ, Landes EL, Moore JP, Kuhmann SE. HIV-1 clones resistant to a small molecule CCR5 inhibitor use the inhibitor-bound form of CCR5 for entry. Virology. 2007;361(1):212–228. doi: 10.1016/j.virol.2006.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves JD, Lee FH, Miamidian JL, Jabara CB, Juntilla MM, Doms RW. Enfuvirtide resistance mutations: impact on human immunodeficiency virus envelope function, entry inhibitor sensitivity, and virus neutralization. J Virol. 2005;79(8):4991–4999. doi: 10.1128/JVI.79.8.4991-4999.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders RW, Venturi M, Schiffner L, Kalyanaraman R, Katinger H, Lloyd KO, Kwong PD, Moore JP. The mannose-dependent epitope for neutralizing antibody 2G12 on human immunodeficiency virus type 1 glycoprotein gp120. J Virol. 2002;76(14):7293–7305. doi: 10.1128/JVI.76.14.7293-7305.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlan CN, Pantophlet R, Wormald MR, Ollmann Saphire E, Stanfield R, Wilson IA, Katinger H, Dwek RA, Rudd PM, Burton DR. The broadly neutralizing anti-human immunodeficiency virus type 1 antibody 2G12 recognizes a cluster of alpha1-->2 mannose residues on the outer face of gp120. J Virol. 2002;76(14):7306–7321. doi: 10.1128/JVI.76.14.7306-7321.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibert C, Ying W, Gavrilov S, Tsamis F, Kuhmann SE, Palani A, Tagat JR, Clader JW, McCombie SW, Baroudy BM, Smith SO, Dragic T, Moore JP, Sakmar TP. Interaction of small molecule inhibitors of HIV-1 entry with CCR5. Virology. 2006;349(1):41–54. doi: 10.1016/j.virol.2006.01.018. [DOI] [PubMed] [Google Scholar]
- Strizki JM, Qiu P, Murgola N, Greaves WRL, J W. Characterization of HIV envelope clones from patients with reduced susceptibility to vicriviroc reveals patient specific mutational patterns in gp120; 7th Annual Symposium on Antiviral Drug Resistance; Chantilly, VA. 2006. [Google Scholar]
- Trkola A, Kuhmann SE, Strizki JM, Maxwell E, Ketas T, Morgan T, Pugach P, Xu S, Wojcik L, Tagat J, Palani A, Shapiro S, Clader JW, McCombie S, Reyes GR, Baroudy BM, Moore JP. HIV-1 escape from a small molecule, CCR5-specific entry inhibitor does not involve CXCR4 use. Proc Natl Acad Sci U S A. 2002;99(1):395–400. doi: 10.1073/pnas.012519099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trkola A, Pomales AB, Yuan H, Korber B, Maddon PJ, Allaway GP, Katinger H, Barbas CF, 3rd, Burton DR, Ho DD, et al. Cross-clade neutralization of primary isolates of human immunodeficiency virus type 1 by human monoclonal antibodies and tetrameric CD4-IgG. J Virol. 1995;69(11):6609–6617. doi: 10.1128/jvi.69.11.6609-6617.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsamis F, Gavrilov S, Kajumo F, Seibert C, Kuhmann S, Ketas T, Trkola A, Palani A, Clader JW, Tagat JR, McCombie S, Baroudy B, Moore JP, Sakmar TP, Dragic T. Analysis of the mechanism by which the small-molecule CCR5 antagonists SCH-351125 and SCH-350581 inhibit human immunodeficiency virus type 1 entry. J Virol. 2003;77(9):5201–5208. doi: 10.1128/JVI.77.9.5201-5208.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson C, Jenkinson S, Kazmierski W, Kenakin T. The CCR5 receptor-based mechanism of action of 873140, a potent allosteric noncompetitive HIV entry inhibitor. Mol Pharmacol. 2005;67(4):1268–1282. doi: 10.1124/mol.104.008565. [DOI] [PubMed] [Google Scholar]
- Westby M, Smith-Burchnell C, Mori J, Lewis M, Mosley M, Stockdale M, Dorr P, Ciaramella G, Perros M. Reduced maximal inhibition in phenotypic susceptibility assays indicates that viral strains resistant to the CCR5 antagonist maraviroc utilize inhibitor-bound receptor for entry. J Virol. 2007;81(5):2359–2371. doi: 10.1128/JVI.02006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwick MB, Labrijn AF, Wang M, Spenlehauer C, Saphire EO, Binley JM, Moore JP, Stiegler G, Katinger H, Burton DR, Parren PW. Broadly neutralizing antibodies targeted to the membrane-proximal external region of human immunodeficiency virus type 1 glycoprotein gp41. J Virol. 2001;75(22):10892–10905. doi: 10.1128/JVI.75.22.10892-10905.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]